

Abstract
Multiple myeloma (MM) is a hematologic cancer arising from plasma cells. Mesenchymal stem cells (MSCs) are a heterogeneous cell population in the bone marrow microenvironment. In this study, we evaluated the regulatory effects of MSCs on the invasion and drug resistance of MM cells U266 and LP-1. Bone marrow samples from MM patients and healthy subjects were collected. MSCs were extracted from bone marrow and cultured, and their phenotypes were identified by flow cytometry. The level of CXCL13 in the supernatant of cultured MSCs was detected by ELISA. The protein expression of CXCR5 (a specific receptor of CXCL13) in U266 and LP-1 cells was detected by Western blot. The effects of MSCs on the invasion of U266 and LP-1 cells and the resistance to bortezomib were assessed by Transwell and CCK-8 assay, respectively. The mRNA and protein expressions of BTK, NF-κB, BCL-2, and MDR-1 were detected by RT-PCR and Western blot, respectively. CXCL13 was secreted by MSCs in the bone marrow microenvironment, and the level in MSCs from MM patients was significantly higher than that of healthy subjects. CXCR5 was expressed in both U266 and LP-1 cells. The resistance of MM cells to bortezomib was enhanced by MSCs through CXCL13 secretion. The invasion and proliferation of U266 and LP-1 cells were promoted, and the mRNA and protein expressions of BTK, NF-κB, BCL-2, and MDR-1 were upregulated by MSCs. The basic biological functions of MM cells U266 and LP-1 were affected by MSCs via the CXCL13-mediated signaling pathway. This study provides valuable experimental evidence for clinical MM therapy.
Keywords: Mesenchymal stem cell, multiple myeloma, invasion, proliferation, CXCL13, CXCR5, MSC, U266, LP-1, chemokine, bortezomib
INTRODUCTION
Multiple myeloma (MM) is a hematologic cancer arising from plasma cells. With a global incidence rate ranking second among all malignant hematological diseases, MM often endangers middle-aged and elderly people [1,2]. The occurrence of MM is closely related to the interaction between tumor cells and bone marrow microenvironment. The complex microenvironment in the bone marrow plays important roles in the growth, proliferation, differentiation, drug resistance, and apoptosis resistance of MM cells [3,4]. Mesenchymal stem cells (MSCs) are a heterogeneous cell population in the bone marrow microenvironment. Besides high self-renewal and multi-directional differentiation capacities, they have an important biological function in immunosuppression and chemotaxis during tissue repair [5,6].
Normal MSCs in the bone marrow can inhibit the growth of tumor cells and prevent the occurrence of tumors, whereas abnormal MSCs in the tumor microenvironment participate in the process of tumorigenesis. MSCs in the bone marrow microenvironment of MM patients undergo an obviously abnormal immunoregulation, cytokine secretion, and osteogenic differentiation [7-9]. Chemokines are a class of small-molecule peptides in the cytokine superfamily, usually consisting of 70–100 amino acids. They can induce chemotactic movement of cells, and are closely associated with the recruitment, proliferation, homing, and survival of hematopoietic stem/progenitor cells. In addition, they can specifically activate and recruit cells related to inflammation and infection. According to the motifs of the first two cysteine residues near the N-terminus, chemokines are classified into C, CC, CXC, and CX3C. As an important member of CXC-like chemokines, CXCL13 is mainly expressed and secreted by T-follicular helper cells, dendritic cells, and some stromal cells in secondary lymphoid tissues. In addition, CXCL13 allows the chemotaxis of mature B cells expressing its receptor CXCR5. When chemotactic B cells are activated, related antibodies are produced and participate in further biological processes [10]. In this study, we detected the expression level of CXCL13 in MSCs from the human bone marrow of MM patients and healthy controls, evaluated the effects of MSCs on the invasion, drug resistance, and proliferation of MM cell lines U266 and LP-1 via the CXCL13-mediated signaling pathway, and explored the underlying mechanism. Our findings provide new ideas and directions for treating MM.
MATERIALS AND METHODS
Clinical samples and cell lines
This study included 40 MM patients diagnosed by bone marrow morphological and histopathological examinations and 40 volunteers without bone marrow abnormalities. From each participant 5 ml of bone marrow blood was collected by puncture. The ratio of males to females was 1:1. The participants were aged from 39 to 71 years, with a median age of 54. The study was approved by the ethics committee of our hospital (approval number: LCPH-2016009). Written informed consents were obtained from all participants. No patient had received chemotherapy, radiotherapy, hematopoietic stem cell transplantation, or other related treatments before sample collection.
MM cell lines U266 and LP-1 were purchased from Cell Bank, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (China).
Extraction of MSCs
The donor was placed in the lateral or prone position, and the posterior superior iliac spine was selected as the puncture site (located at bony prominence on both sides of sacral vertebrae and the haunch apex). Then, 5 ml of bone marrow was collected and temporarily stored in an ethylenediaminetetraacetic acid (EDTA) anticoagulant tube. An equal volume of phosphate-buffered saline (PBS) was thoroughly mixed in a 15 ml centrifuge tube and centrifuged at 1000 rpm for 5 min to discard the fat layer and supernatant. Then, 5 ml of human lymphocyte separation solution was added into another 15 ml centrifuge tube which was tilted by 45°. Afterwards, the collected bone marrow was slowly added to the liquid surface of the lymphocyte separation solution along the tube wall with a sterile dropper. After centrifugation at 2000 rpm for 30 min at 25°C, layers of different density gradients formed in the tube, showing clear boundaries. A white cloud-like narrow band existing between the upper and middle layers was carefully removed with a sterile dropper, placed in a new sterile centrifuge tube and centrifuged at 900 rpm for 4 min to discard the supernatant. The precipitate was washed twice to three times with PBS for subsequent inoculation and culture experiments.
Culture of MSCs
After addition of 3 ml of 15% Dulbecco’s Modified Eagle’s Medium (DMEM)/F12 medium, freshly extracted human MSCs were inoculated into a 25 cm2 sterile culture flask. The cap was slightly loosened, and the flask was placed in a 37°C incubator containing 5% CO2 with saturated humidity. After 72 h of incubation, half of the medium was refreshed, and the medium was completely replaced 48 h later. The cell growth was observed at intervals of 12 h. Subsequently, the medium was changed once every 2–3 days. When the confluence reached about 90%, the cells were digested with 0.25% trypsin (containing 0.02% EDTA) for passage.
Phenotyping of MSCs
Direct immunofluorescence labeling was performed. The third-generation MSCs were trypsinized and washed with PBS once to twice, with the density adjusted to 5 × 106/ml. Then, 100 µl of suspension was taken, incubated with monoclonal antibodies against CD34-fluorescein isothiocyanate (FITC), CD45-FITC, CD14-FITC, CD105-phycoerythrin (PE), and CD29-PE for 30 min at room temperature in the dark, washed once to twice with PBS, and assessed by flow cytometry. PE-labeled isotype immunoglobulin G (IgG) was used as negative control.
Culture of MM cell lines U266 and LP-1
Frozen U266 and LP-1 cells were immediately placed in a 37°C water bath for 3 min and centrifuged at 900 rpm for 4 min to discard the supernatant. The precipitate was thereafter gently pipetted and mixed with pre-warmed culture medium. The cells were inoculated into a sterile culture flask, and culture medium was added until a volume of 3 ml. The cell morphology and growth were carefully observed under a light microscope. The inoculated cells were cultured overnight in a 37°C incubator with saturated humidity and 5% CO2, and the culture medium was then changed completely. The cells were observed once to twice daily and passaged when they grew densely or the culture medium became turbid. The cells in the culture flask were collected into a sterile centrifuge tube and centrifuged at 900 rpm for 4 min to discard the supernatant. Afterward, the precipitate was mixed with 10% fresh Roswell Park Memorial Institute (RPMI) 1640 medium by repeated pipetting. The cells were passaged in a ratio of 1:2 or 1:3.
Detection of CXCL13 level in the supernatant of cultured MSCs by enzyme-linked immunosorbent assay (ELISA)
The third-generation well growing MSCs of MM patients and healthy subjects were selected. Then, 2 ml of cells at a density of 5 × 105/ml were inoculated in a new sterile culture dish, with 3 replicate wells for each group. The experiment was repeated at least 3 times. After 48 h, the cell supernatant in each culture dish was collected into a 5 ml centrifuge tube and centrifuged at 2000 rpm for 20 min. The supernatant was collected, subpackaged into 1.5 ml EP tubes (500 µl per tube) and stored at −20°C. Subsequently, 15.6 pg/ml, 31.3 pg/ml, 62.5 pg/ml, 125 pg/ml, 250 pg/ml, 500 pg/ml, and 1000 pg/ml standards were added into 7 wells sequentially (0.1 ml each). Meanwhile, a blank well was set. The culture supernatant was diluted and then added into a microtiter plate at 0.1 ml per well. The microtiter plate was thereafter sealed and reacted for 90 min in a 37°C incubator. Afterward, the liquid was gently removed from the plate. The working solution of biotin anti-human CXCL13 antibody was added at 0.1 ml per well (except for the zeroing well). The plate was then sealed, reacted at 37°C for 60 min, washed 3 times with 1× washing buffer and soaked for about 1 min each time. The working solution of ABC was added at 0.1 ml per well (except for the zeroing well). The microtiter plate was thereafter sealed, reacted for 30 min in the 37°C incubator, washed 5 times with 1× washing buffer, and soaked for about 1–2 min each time. Subsequently, 90 µl of TMB color development solution was added to each well and reacted in the dark for 25–30 min in the 37°C incubator. Then, 0.1 ml of TMB stop buffer was added to each well, which turned the blue color into yellow. The optical density (OD) of each well was measured at 450 nm by a microplate reader. Finally, a standard curve was plotted and fitted.
Western blot
Cells were digested, collected by centrifugation, resuspended by adding radioimmunoprecipitation assay [RIPA] buffer (50 mM pH 7.5 Tris-HCl, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate [SDS]), ultrasonicated and centrifuged at 12,000 rpm and 4°C for 10 min. The resulting sample (50 µg) was subjected to SDS–polyacrylamide gel electrophoresis (PAGE) and electronically transferred onto a nitrocellulose membrane which was then blocked by 5% skimmed milk at room temperature for 1 h, incubated overnight with primary antibodies at 4°C, washed and incubated with secondary antibodies at 37°C for 1 h. The membrane was thereafter developed with enhanced chemiluminescence (ECL) reagent and scanned. Relative protein expressions were detected by Quantity One software (Bio-Rad Laboratories, CA, USA) and corrected using β-actin as the internal reference.
Evaluation of inhibitory effects of bortezomib on U266 and LP-1 cells by cell counting kit-8 (CCK-8) assay
U266 and LP-1 cells in the logarithmic growth phase were collected by centrifugation, counted, inoculated into 96-well plates at the density of 1 × 105/ml (100 µl per well), and treated with bortezomib at the concentrations of 1.0, 5.0, 10.0, 20.0, and 50.0 nmol/L, respectively. Blank control and untreated cell wells were set. Three replicate wells were set for each concentration and control well. Then, the cells were cultured in a 37°C incubator with 5% CO2 and saturated humidity for 48 h and centrifuged at 1000 rpm for 5 min to discard the supernatant. Subsequently, CCK-8 diluted 1:9 by serum-free culture medium was added (100 µl per well) in the dark, and the cells were left still in the incubator for 1 h. OD of each well was measured at 450 nm by the microplate reader, and the values of 3 replicate wells were averaged. Cell inhibition rate = (1−[ODtreated cells−ODblank]/[ODuntreated cells−ODblank]) × 100%. Inhibition rate curve was plotted to determine the drug concentration for subsequent experiments.
Co-culture of MSCs with U266 and LP-1 cells
The third- to the fifth-generation MSCs were digested with 0.25% trypsin and centrifuged at 1000 rpm for 5 min to discard the supernatant. Then, the cells were resuspended in DMEM-F12 medium containing 15% fetal bovine serum (FBS), with the density adjusted to 1 × 106/ml. Afterward, 1 ml of the cell suspension was added to each well of a 6-well plate, which was then placed in an incubator overnight at 37°C with saturated humidity and 5% CO2. Subsequent experiments were started after complete cell adherence. On the next day, the growth of MSCs in the 6-well plate was observed under an inverted microscope. Then, 15% DMEM-F12 medium was replaced by an equal amount of 10% RPMI 1640 medium. Transwell chambers with the pore diameter of 4 µm were placed on the six-well plate into which 1 ml of U266 or LP-1 cells at a density of 1 × 106/ml were added. MM + MSCs and MM + MSCs + anti-CXCL13 (a CXCL13 antagonist) groups were set for each cell line, and thereafter co-cultured for 48 h in the 37°C incubator with 5% CO2 and saturated humidity.
Evaluation of effects of MSCs on resistance of U266 and LP-1 cells to bortezomib by CCK-8 assay
As mentioned above, U266 and LP-1 cells were co-cultured with MSCs to obtain MM + MSCs and MM + MSCs + anti-CXCL13, groups. The cells were centrifuged at 900 rpm for 4 min, and 10% RPMI 1640 medium was refreshed. Three experimental groups were set for each cell line, i.e., individual MM group, MM + MSCs co-culture group, and MM + MSCs + anti-CXCL13 co-culture group. MM cells of each experimental group were resuspended by centrifugation, and bortezomib was added until a final concentration of 20 nmol/L. An appropriate area in a 96-well plate was selected, and 200 µl of medium containing 10,000 cells was added to each well. Three replicate wells were set for each experimental group. The cells were cultured in the 37°C incubator with 5% CO2 and saturated humidity for 48 h, and centrifuged at 1000 rpm for 5 min to discard the supernatant. Subsequently, CCK-8 diluted 1:9 by serum-free culture medium was added (100 µl per well) in the dark, and the cells were left still in the incubator for 1 h. OD of each well was measured at 450 nm by the microplate reader, and the values of 3 replicate wells were averaged.
Evaluation of effects of MSCs on the invasion of U266 and LP-1 cells by Transwell assay
Serum-free RPMI 1640 medium was used to starve MM cells of the three experimental groups for 12 h. Afterward, RPMI 1640 medium containing 20% FBS was added to the lower layer of the Transwell chamber in a 24-well plate (600 µl per well). Meanwhile, 200 µl of RPMI 1640 medium containing 1 × 105 cells was added to each Transwell chamber, with at least 3 replicate wells for each group. The plates were left still in the incubator for 12 h. OD of each well was detected at each time point by the microplate reader.
Evaluation of effects of MSCs on proliferation of U266 and LP-1 cells by CCK-8 assay
MSCs were co-cultured with U266 and LP-1 cells for 48 h using the same procedure as mentioned above. The two cell lines were grouped as mentioned above. The cells in each experimental group were collected by centrifugation at 900 rpm for 4 min, and RPMI 1640 medium containing 10% FBS was refreshed. CKK-8 assay was conducted at 0, 24, 48, 72, and 96 h respectively.
Evaluation of effects of MSCs on mRNA expressions of BTK, p65, BCL-2, and MDR-1 in U266 and LP-1 cells by RT-PCR
Briefly, 200 µl of chloroform was added into cells dissolved in 1000 µl of Trizol reagent, vigorously vortexed for 15 s, left still at room temperature for 3 min and centrifuged at 12,000 rpm and 4°C for 15 min. The upper aqueous layer was collected into an RNase-free EP tube, mixed with an equal volume of 4°C isopropanol by vortexing, incubated at room temperature for 10 min, and centrifuged at 4°C and 12,000 rpm for 10 min. After the supernatant was discarded, the precipitate was gently pipetted and mixed with 1 ml of pre-cooled 75% ethanol solution and centrifuged at 4°C and 7500 rpm for 5 min. Then, the supernatant was discarded, and the precipitate was redissolved with 20 µl of diethyl pyrocarbonate (DEPC)-treated water, gently pipetted and placed in an icebox for 10 min. The extracted RNA was reverse-transcribed into complementary DNA (cDNA) for agarose gel electrophoresis according to the instructions of reverse transcription polymerase chain reaction (RT-PCR) kit. The results were analyzed by the 2−△△CT method. The experiment was repeated at least 3 times. The primer sequences were as follows: Bruton’s tyrosine kinase (BTK), F: 5’-GAGAAGCTGGTGCAGTTGTAT-3’, R: 5’-GGCCGAAATCAGATACTTTAAC-3’; p65, F: 5’-ACCG CTGCATCCACAGTT-3’, R: 5’-GGATGCGCTGACTGATA GC-3’; B-cell lymphoma 2 (BCL-2), F: 5’-GTTGGCCGGAT CACCATCT-3’, R: 5’-ATATCACAGCCCCCAGGGCA-3’; multidrug resistance protein 1 (MDR-1), F: 5’-GGGTTCTT CATGAATCTGG-3’, R: 5’-CTGAATGTAAGCAGCAA CC-3’; glyceraldehyde 3-phosphate dehydrogenase (GAPDH), F: 5’-CCATGTTCGTCATGGGTGT-3’, R: 5’-TGAGTCCTTC CACGATACC-3’.
Statistical analysis
All data were analyzed by SPSS Statistics for Windows, Version 17.0. software (SPSS Inc., Chicago, IL, USA). The categorical data were expressed as mean ± standard deviation. These data were validated with the two-sided test. A value of p < 0.05 was considered statistically significant.
RESULTS
Growth and phenotyping of MSCs in bone marrow microenvironment
MSCs extracted from the human bone marrow microenvironment grew well from the second to the fifth generation, and the cell morphology was shuttle-shaped and uniform in size. There was no significant difference in cell morphology, size, and growth cycle between MSCs in healthy subjects and MM patients (Figure 1A and B). The MSC phenotyping results by flow cytometry showed that the positive expression rate of CD105, CD73 or CD90 was ≥95%, and that of CD45, CD34, CD11b or human leukocyte antigen (HLA)-DR was ≤3%.
FIGURE 1.

The morphology of bone marrow mesenchymal stem cells (MSCs) from (A) healthy subjects and (B) multiple myeloma (MM) patients. There was no significant difference in cell morphology, size, and growth cycle between MSCs from healthy subjects and MM patients; (C) CXCL13 expression in the bone marrow MSCs from MM patients and healthy subjects. The level of CXCL13 in the culture supernatant was detected by enzyme-linked immunosorbent assay (ELISA). CXCL13 level in MSCs from MM patients was significantly higher compared with healthy subjects.
CXCL13 expression in the bone marrow MSCs from MM patients was significantly higher than that of healthy subjects
MSCs were extracted from the bone marrow of MM patients and healthy subjects by density gradient centrifugation. The third-generation MSCs with a confluence of 80–90% in the logarithmic growth phase were selected and cultured for 48 h. The level of CXCL13 in the culture supernatant was detected by ELISA. CXCL13 level in MSCs from MM patients was significantly higher than that from healthy subjects [p < 0.05] (Figure 1C).
CXCR5, a specific CXCL13 receptor, was expressed in both U266 and LP-1 cells
The total proteins of U266 and LP-1 cells were extracted, and the expression level of CXCR5 protein, a specific receptor of CXCL13 chemokine, was detected by Western blot. Both U266 and LP-1 expressed CXCR5 (Figure 2).
FIGURE 2.
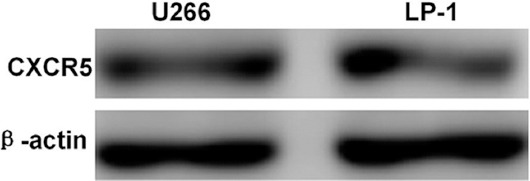
CXCR5 protein expression in U266 and LP-1 cells detected by Western blot. Both U266 and LP-1 cells expressed CXCR5.
Growth inhibitory effects of bortezomib on U266 and LP-1 cells
Different concentrations (1.0, 5.0, 10.0, 20.0, and 50.0 nmol/L) of proteasome inhibitor, bortezomib, were added to U266 and LP-1 cells. After 48 h, the growth inhibition effect of different concentrations of bortezomib on myeloma cell lines U266 and LP-1 was detected by CCK-8 assay. The 50% growth inhibitory concentrations (IC50) of U266 and LP-1 cells after 48 h of treatment with bortezomib were approximately 20 nmol/L (Figure 3A).
FIGURE 3.

(A) Growth inhibitory effects of bortezomib on U266 and LP-1 cells detected by cell counting kit-8 (CCK-8) assay. The 50% growth inhibitory concentrations (IC50) of U266 and LP-1 cells after 48 h of treatment with bortezomib was approximately 20 nmol/L; (B) inhibitory effects of anti-CXCL13 at different concentrations on bortezomib resistance of U266 and LP-1 cells co-cultured with mesenchymal stem cells (MSCs). With a rising anti-CXCL13 concentration, the growth inhibition rates of U266 and LP-1 cells also increased; (C) MSCs enhanced bortezomib resistance of U266 and LP-1 cells via the CXCL13-mediated signaling pathway, which was blocked by anti-CXCL13. After co-culture of U266 and LP-1 cells with MSCs for 48 h, three different experimental groups were set up for each cell line: individual multiple myeloma (MM) group, MM + MSCs group, and MM + MSCs + anti-CXCL13 group. Each experimental group was added bortezomib at a final concentration of 20 nmol/L. After 48 h, the growth inhibition rate was calculated. MSCs enhanced the resistance of U266 and LP-1 cells to bortezomib, which was blocked by anti-CXCL13. *p < 0.05.
Inhibitory effects of anti-CXCL13 on bortezomib resistance of U266 and LP-1 cells co-cultured with MSCs
Different concentrations (0, 200, 500, 1000, and 1500 ng/ml) of anti-CXCL13 (a CXCL13 antagonist) were added to the co-cultures of U266 and LP-1 cells with MSCs. After co-culture for 48 h, U266 and LP-1 cells of each treatment group were collected, and bortezomib at a concentration of 20 nmol/L was added to each group. The cell growth inhibition rate was detected by CCK-8 assay 48 h later. The results showed that with a rising anti-CXCL13 concentration, the growth inhibition rates of U266 and LP-1 cells also increased [p < 0.05] (Figure 3B).
MSCs enhanced bortezomib resistance of U266 and LP-1 cells via the CXCL13-mediated signaling pathway
After co-culture of U266 and LP-1 cells with MSCs for 48 h, three different experimental groups were set up for each cell line: individual MM group, MM + MSCs group, and MM + MSCs + anti-CXCL13 group. Bortezomib was added into each group until a final concentration of 20 nmol/L. After 48 h, the OD of each well in the 96-well plate was detected by CCK-8 assay, and the growth inhibition rate was calculated. The results showed that MSCs enhanced the resistance of U266 (p < 0.05) and LP-1 cells (p < 0.05) to bortezomib, which was blocked by anti-CXCL13. The OD of the experimental well containing only myeloma cells without bortezomib was set as 1 (Figure 3C).
MSCs promoted the invasion of U266 and LP-1 cells via the CXCL13-mediated signaling pathway
U266 and LP-1 cells were co-cultured with human MSCs for 48 h under appropriate conditions. Three different experimental groups were set up for each cell line: individual MM group, MM + MSCs group, and MM + MSCs + anti-CXCL13 group. The invasion ability of the myeloma cell lines U266 and LP-1 was detected by Transwell culture system. MSCs facilitated the invasion of U266 and LP-1 cells (p < 0.05), and this effect was blocked by anti-CXCL13 (Figure 4A).
FIGURE 4.
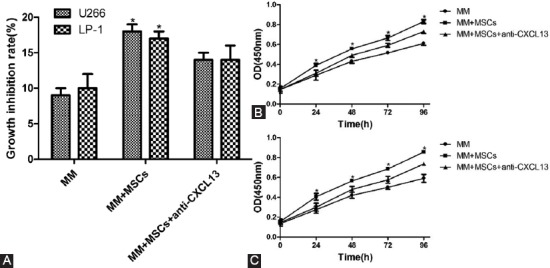
(A) Mesenchymal stem cells (MSCs) promoted invasion of U266 and LP-1 cells via the CXCL13-mediated signaling pathway, which was blocked by anti-CXCL13; MSCs facilitated proliferation of (B) U266 and (C) LP-1 cells via the CXCL13- mediated signaling pathway, which was blocked by anti-CXCL13. *p < 0.05.
MSCs facilitated the proliferation of U266 and LP-1 cells via the CXCL13-mediated signaling pathway
U266 and LP-1 cells were co-cultured with MSCs for 48 h under appropriate conditions. Three different experimental groups were set up for each cell line: individual MM group, MM + MSCs group, and MM + MSCs + anti-CXCL13 group. The proliferation of the myeloma cell lines U266 and LP-1 was detected by CCK-8 assay at different time points (0, 24, 48, 72, and 96 h). MSCs promoted the proliferation of U266 and LP-1 cells (p < 0.05), and this effect was blocked by anti-CXCL13 (Figure 4B and C).
Effects of MSCs on mRNA expressions of BTK, p65, BCL-2, and MDR-1 in U266 and LP-1 cells evaluated by RT-PCR
RT-PCR showed that after co-culture with MSCs from MM patients for 48 h, the mRNA expression levels of BTK, p65, as well as of apoptosis- and drug resistance-related BCL-2 and MDR-1 in U266 and LP-1 cells were all significantly upregulated (p < 0.05). The upregulation was blocked by adding anti-CXCL13 (Figure 5A).
FIGURE 5.
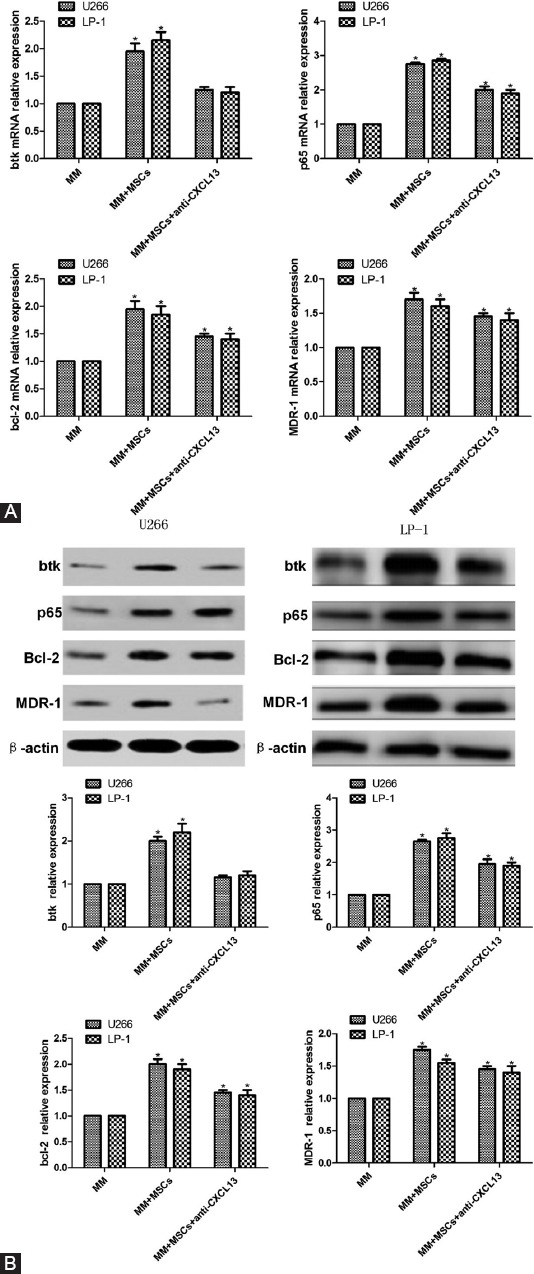
Effects of mesenchymal stem cells (MSCs) and anti-CXCL13 on (A) mRNA expressions of Bruton’s tyrosine kinase (BTK), p65, B-cell lymphoma (BCL-2) and multidrug resistance protein 1 (MDR-1) in U266 and LP-1 cells evaluated by reverse transcription polymerase chain reaction (RT-PCR) and (B) protein expressions assessed by Western blot. RT-PCR showed that after co-culture with MSCs from MM patients for 48 h, the mRNA expression levels of BTK, p65, as well as of apoptosis- and drug resistance-related BCL-2 and MDR-1 in U266 and LP-1 cells were all significantly upregulated. The upregulation was blocked by adding anti-CXCL13. Western blot showed that the protein expression levels of BTK, p65, BCL-2, and MDR-1 in U266 and LP-1 cells were significantly upregulated after co-culture with MSCs from MM patients for 48 h. The upregulation was blocked by adding anti-CXCL13. *p < 0.05.
Effects of MSCs on protein expressions of BTK, p65, BCL-2, and MDR-1 in U266 and LP-1 cells assessed by Western blot
Western blot showed that the protein expression levels of BTK, p65, BCL-2, and MDR-1 in U266 and LP-1 cells were significantly upregulated after co-culture with MSCs from MM patients for 48 h. The upregulation was blocked by adding anti-CXCL13 [p < 0.05] (Figure 5B).
DISCUSSION
The onset and progression of tumors have been closely related to the complex environment formed by the interactions between various atypical cells, i.e., the tumor microenvironment [11]. The tumor microenvironment is a special environment in which tumor cells interact with immune cells, endothelial cells and fibroblasts, and where various cytokines are secreted during tumor progression [12]. Most previous studies have focused on the characteristics of tumor cells such as gene mutations, chromosomal abnormalities, and changes in signaling pathways. In recent years, the microenvironment closely related to tumor progression has attracted widespread attention. A normal microenvironment can inhibit the growth of tumor cells, whereas an abnormal microenvironment can induce tumors [13]. The abnormal microenvironment can be caused by factors such as tissue hypoxia, interstitial hypertension, immune-related inflammatory reaction as well as production of large amounts of proteolytic enzymes and cytokines [14-17].
The bone marrow microenvironment significantly participates in the progression of hematologic diseases, maintains the hematopoiesis of bone marrow cells, promotes stem cell differentiation, and regulates the balance between osteogenesis and osteolysis [18,19]. MSCs are a group of heterogeneous cell populations with self-renewal and multi-directional differentiation potentials in the bone marrow microenvironment, which play a key role in promoting tumor onset and progression in the microenvironment [20]. On the other hand, chemokines are predominantly involved in the development, proliferation, metabolism, and drug resistance of tumor cells. Therefore, an in-depth study of chemokines is expected to provide new directions for the diagnosis and treatment of MM. It is well-documented that CXCL13/CXCR5 play key roles in the occurrence, metastasis, and invasion of various malignant tumors [21,22]. RT-PCR and immunohistochemistry were used to analyze the mRNA and protein expressions of CXCL13/CXCR5 in cancerous tissues [23], confirming that the expression level of CXCLl3/CXCR5 was closely related to tumor metastasis, stage, and recurrence. Our study demonstrated that MSCs extracted from the bone marrow of MM patients secreted higher levels of CXCL13, and they promoted the proliferation and invasion of MM cell lines U266 and LP-1 via the CXCL13-mediated signaling pathway. When this signaling pathway was blocked by anti-CXCL13, the promoting effects were significantly attenuated.
Gene mutation is a crucial mechanism by which malignant tumors progress, with the mutations of protein tyrosine kinase genes being the most common. BTK is a key member of the Tec family and capable of phosphorylating the tyrosine residues of various proteins. Mutation of BTK can block the differentiation and maturation of B lymphocytes and may also promote the transformation of normal cells into tumor cells. It affects tumor cells by regulating downstream factors such as phosphoinositide phospholipase C (PLC)γ, phosphoinositide 3-kinase (PI3K), protein kinase C (PKC), STAT5α, extracellular signal-regulated kinase (ERK), and nuclear factor-κB (NF-κB) [24,25]. NF-κB is an essential member of nuclear transcription factors. It is involved in the regulation of the transcription of various growth factors, kinases, cell adhesion molecules, and chemokines in cells, being closely associated with the onset and progression of tumors [26]. The anti-apoptotic action of malignant tumor cells is one of the important mechanisms of tumor resistance. A gross amount of research reported that the BCL-2 family acts as a main regulator of mitochondria metabolism during apoptosis [27]. Besides, Cort et al. found that tumor cells showed both the resistance to the same type of antitumor drug and cross-resistance to other antitumor agents with different structures and mechanisms of action [28]. In this study, the mRNA and protein expression levels of BTK and p65 were upregulated in U266 and LP-1 co-cultured with MSCs from MM patients. The expression levels of BCL-2 and MDR-1 were also elevated. When anti-CXCL13 was added, however, the mRNA and protein expression levels of BTK, NF-κB, BCL-2, and MDR-1 were downregulated.
CONCLUSION
In summary, the onset and progression of MM involve a variety of cytokines and signaling pathways. MSCs in the bone marrow microenvironment of MM patients expressed chemokine CXCL13 at a high level and affected the invasion, drug resistance, and proliferation of MM cell lines U266 and LP-1 via the CXCL13-mediated signaling pathway. Moreover, this pathway upregulated the mRNA and protein expression levels of BTK, NF-κB, BCL-2, and MDR-1 in U266 and LP-1 cells. After anti-CXCL13 was added to the co-culture system, the basic biological functions of these cells were significantly attenuated. This study provides new ideas and directions for future MM therapy.
Footnotes
Conflict of interest statement: The authors declare no conflict of interests
REFERENCES
- 1.Ma J, Liu S, Wang Y. MicroRNA-21 and multiple myeloma:Small molecule and big function. Med Oncol. 2014;31:94. doi: 10.1007/s12032-014-0094-5. https://doi.org/10.1007/s12032-014-0094-5. [DOI] [PubMed] [Google Scholar]
- 2.Avet-Loiseau H, Facon T. Front-line therapies for elderly patients with transplant-ineligible multiple myeloma and high-risk cytogenetics in the era of novel agents. Leukemia. 2018;32:1267–76. doi: 10.1038/s41375-018-0098-9. https://doi.org/10.1038/s41375-018-0098-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manier S, Sacco A, Leleu X, Ghobrial IM, Roccaro AM. Bone marrow microenvironment in multiple myeloma progression. J Biomed Biotechnol. 2012;2012:157496. doi: 10.1155/2012/157496. https://doi.org/10.1155/2012/157496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kawano Y, Moschetta M, Manier S, Glavey S, Görgün GT, Roccaro AM, et al. Targeting the bone marrow microenvironment in multiple myeloma. Immunol Rev. 2015;263:160–72. doi: 10.1111/imr.12233. https://doi.org/10.1111/imr.12233. [DOI] [PubMed] [Google Scholar]
- 5.Salem HK, Thiemermann C. Mesenchymal stromal cells:Current understanding and clinical status. Stem Cells. 2010;28:585–96. doi: 10.1002/stem.269. https://doi.org/10.1002/stem.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.García-Gómez I, Elvira G, Zapata AG, Lamana ML, Ramírez M, Castro JG, et al. Mesenchymal stem cells:Biological properties and clinical applications. Expert Opin Biol Ther. 2010;10:1453–68. doi: 10.1517/14712598.2010.519333. https://doi.org/10.1517/14712598.2010.519333. [DOI] [PubMed] [Google Scholar]
- 7.Xu X, Yang J, Tang Y, Li J, Zhu Y, Lu H, et al. In vitro migratory aberrancies of mesenchymal stem cells derived from multiple myeloma patients only partially modulated by bortezomib. Int J Clin Exp Pathol. 2014;7:6705–15. [PMC free article] [PubMed] [Google Scholar]
- 8.Li B, Fu J, Chen P, Zhuang W. Impairment in immunomodulatory function of mesenchymal stem cells from multiple myeloma patients. Arch Med Res. 2010;41:623–33. doi: 10.1016/j.arcmed.2010.11.008. https://doi.org/10.1016/j.arcmed.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 9.Wang X, Zhang Z, Yao C. Angiogenic activity of mesenchymal stem cells in multiple myeloma. Cancer Invest. 2011;29:37–41. doi: 10.3109/07357907.2010.496758. https://doi.org/10.3109/07357907.2010.496758. [DOI] [PubMed] [Google Scholar]
- 10.Todoerti K, Lisignoli G, Storti P, Agnelli L, Novara F, Manferdini C, et al. Distinct transcriptional profiles characterize bone microenvironment mesenchymal cells rather than osteoblasts in relationship with multiple myeloma bone disease. Exp Hematol. 2010;38:141–53. doi: 10.1016/j.exphem.2009.11.009. https://doi.org/10.1016/j.exphem.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 11.Natsuizaka M, Kinugasa H, Kagawa S, Whelan KA, Naganuma S, Subramanian H, et al. IGFBP3 promotes esophageal cancer growth by suppressing oxidative stress in hypoxic tumor microenvironment. Am J Cancer Res. 2014;4:29–41. [PMC free article] [PubMed] [Google Scholar]
- 12.Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014;2014:149185. doi: 10.1155/2014/149185. https://doi.org/10.1155/2014/149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hui L, Chen Y. Tumor microenvironment:Sanctuary of the devil. Cancer Lett. 2015;368:7–13. doi: 10.1016/j.canlet.2015.07.039. https://doi.org/10.1016/j.canlet.2015.07.039. [DOI] [PubMed] [Google Scholar]
- 14.Klemm F, Joyce JA. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 2015;25:198–213. doi: 10.1016/j.tcb.2014.11.006. https://doi.org/10.1016/j.tcb.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khawar IA, Kim JH, Kuh HJ. Improving drug delivery to solid tumors:Priming the tumor microenvironment. J Control Release. 2015;201:78–89. doi: 10.1016/j.jconrel.2014.12.018. https://doi.org/10.1016/j.jconrel.2014.12.018. [DOI] [PubMed] [Google Scholar]
- 16.Spano D, Zollo M. Tumor microenvironment:A main actor in the metastasis process. Clin Exp Metastasis. 2012;29:381–95. doi: 10.1007/s10585-012-9457-5. https://doi.org/10.1007/s10585-012-9457-5. [DOI] [PubMed] [Google Scholar]
- 17.Bohonowych JE, Hance MW, Nolan KD, Defee M, Parsons CH, Isaacs JS, et al. Extracellular hsp90 mediates an NF-κB dependent inflammatory stromal program:Implications for the prostate tumor microenvironment. Prostate. 2014;74:395–407. doi: 10.1002/pros.22761. https://doi.org/10.1002/pros.22761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamplin OJ. Chewing through roots:How leukemia invades and disrupts the bone marrow microenvironment. Cell Stem Cell. 2018;22:5–7. doi: 10.1016/j.stem.2017.12.014. https://doi.org/10.1016/j.stem.2017.12.014. [DOI] [PubMed] [Google Scholar]
- 19.Walenda T, Bork S, Horn P, Wein F, Saffrich R, Diehlmann A, et al. Co-culture with mesenchymal stromal cells increases proliferation and maintenance of haematopoietic progenitor cells. J Cell Mol Med. 2010;14:337–50. doi: 10.1111/j.1582-4934.2009.00776.x. https://doi.org/10.1111/j.1582-4934.2009.00776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schinke C, Qu P, Mehdi SJ, Hoering A, Epstein J, Johnson SK, et al. The pattern of mesenchymal stem cell expression is an independent marker of outcome in multiple myeloma. Clin Cancer Res. 2018;24:2913–9. doi: 10.1158/1078-0432.CCR-17-2627. https://doi.org/10.1158/1078-0432.ccr-17-2627. [DOI] [PubMed] [Google Scholar]
- 21.Biswas S, Sengupta S, Roy Chowdhury S, Jana S, Mandal G, Mandal PK, et al. CXCL13-CXCR5 co-expression regulates epithelial to mesenchymal transition of breast cancer cells during lymph node metastasis. Breast Cancer Res Treat. 2014;143:265–76. doi: 10.1007/s10549-013-2811-8. https://doi.org/10.1007/s10549-013-2811-8. [DOI] [PubMed] [Google Scholar]
- 22.Razis E, Kalogeras KT, Kotoula V, Eleftheraki AG, Nikitas N, Kronenwett R, et al. Improved outcome of high-risk early HER2 positive breast cancer with high CXCL13-CXCR5 messenger RNA expression. Clin Breast Cancer. 2012;12:183–93. doi: 10.1016/j.clbc.2012.03.006. https://doi.org/10.1016/j.clbc.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 23.Qi XW, Xia SH, Yin Y, Jin LF, Pu Y, Hua D, et al. Expression features of CXCR5 and its ligand, CXCL13 associated with poor prognosis of advanced colorectal cancer. Eur Rev Med Pharmacol Sci. 2014;18:1916–24. [PubMed] [Google Scholar]
- 24.Buggy JJ, Elias L. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int Rev Immunol. 2012;31:119–32. doi: 10.3109/08830185.2012.664797. https://doi.org/10.3109/08830185.2012.664797. [DOI] [PubMed] [Google Scholar]
- 25.Xie Q, Joseph RE, Fulton DB, Andreotti AH. Substrate recognition of PLCγ1 via a specific docking surface on Itk. J Mol Biol. 2013;425:683–96. doi: 10.1016/j.jmb.2012.10.023. https://doi.org/10.1016/j.jmb.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang F, Yang JL, Yu KK, Xu M, Xu YZ, Chen L, et al. Activation of the NF-κB pathway as a mechanism of alcohol enhanced progression and metastasis of human hepatocellular carcinoma. Mol Cancer. 2015;14:10. doi: 10.1186/s12943-014-0274-0. https://doi.org/10.1186/s12943-014-0274-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gross A. BCL-2 family proteins as regulators of mitochondria metabolism. Biochim Biophys Acta. 2016;1857:1243–6. doi: 10.1016/j.bbabio.2016.01.017. https://doi.org/10.1016/j.bbabio.2016.01.017. [DOI] [PubMed] [Google Scholar]
- 28.Cort A, Ozben T, Saso L, De Luca C, Korkina L. Redox control of multidrug resistance and its possible modulation by antioxidants. Oxid Med Cell Longev. 2016;2016:4251912. doi: 10.1155/2016/4251912. https://doi.org/10.1155/2016/4251912. [DOI] [PMC free article] [PubMed] [Google Scholar]