

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is characterized by an abundance of stroma. Multiple molecular classification efforts have identified a mesenchymal tumor subtype that is consistently characterized by high‐grade growth and poor clinical outcome. The relation between PDAC stroma and tumor subtypes is still unclear. Here, we aimed to identify how PDAC cells instruct the main cellular component of stroma, the pancreatic stellate cells (PSCs). We found in primary tissue that high‐grade PDAC had reduced collagen deposition compared to low‐grade PDAC. Xenografts and organotypic co‐cultures established from mesenchymal‐like PDAC cells featured reduced collagen and activated PSC content. Medium transfer experiments using a large set of PDAC cell lines revealed that mesenchymal‐like PDAC cells consistently downregulated ACTA2 and COL1A1 expression in PSCs and reduced proliferation. We identified colony‐stimulating factor 1 as the mesenchymal PDAC‐derived ligand that deactivates PSCs, and inhibition of its receptor CSF1R was able to counteract this effect. In conclusion, high‐grade PDAC features stroma that is low in collagen and activated PSC content, and targeting CSF1R offers direct options to maintain a tumor‐restricting microenvironment.
Keywords: collagen, CSF‐1, deactivation, Pancreatic ductal adenocarcinoma, pancreatic stellate cells
Subject Categories: Cancer
This study reveals that PDAC cells counteract mechanical restrictions of tumor stroma by secreting CSF‐1, which could be a potential therapeutic target.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) has a dismal prognosis and is hallmarked by a high metastatic ability and resistance to anticancer agents. It has been postulated that the stroma plays a key role in the development of metastases and treatment resistance 1, 2, 3. The stroma of PDAC is characterized by large amounts of extracellular matrix, immune cells, inflammatory cells, endothelium and pancreatic stellate cells (PSCs) 4. Activated PSCs (i.e. cancer associated fibroblasts, CAFs) contribute to the vast majority of the stroma’s cellular mass and their role in PDAC progression has been studied extensively over the past years. It has been hypothesized that factors secreted by pancreatic tumor cells, inflammatory, and endothelial cells can activate quiescent PSCs, which then obtain a more myofibroblast‐like phenotype 4, 5, 6. These activated PSCs then deposit large amounts of extracellular matrix creating a dense fibrotic, hypovascular, and hypoxic environment that hampers drug delivery and is suspected to promote metastatic dissemination [7, 8, 9, 10]. Intriguingly, in preclinical studies with genetically engineered PDAC mouse models, depletion of the tumor stroma resulted in improved chemotherapy delivery, but also more aggressive and dedifferentiated tumors with reduced survival 7, 11, 12, 13. It was postulated the composition of the tumor stroma was affected by important properties of the cancer cell fraction such as their differentiation grade 11 as well as heterogeneity of CAFs. Certain CAF subtypes are thought to have tumor‐promoting properties while others are thought to restrain tumor cell growth 5, 14, 15.
Several studies have performed molecular classification of PDAC based on gene expression 16, 17, 18, 19, 20. In general, these studies have all identified a mesenchymal (i.e., squamous/quasi‐mesenchymal/basal‐like) subtype and a group of non‐mesenchymal subtypes that are more epithelial and/or reflect the function of the organ (i.e., classical/pancreatic progenitor). Mesenchymal‐like PDAC features relatively poorly differentiated tumors (high‐grade; G3), whereas non‐mesenchymal subtypes feature relatively well‐differentiated (low‐grade; G1) tumors 16, 17, 19, 20. Recent studies have assessed the contribution of stroma to the molecular subtyping of PDAC 19, 21. Interestingly, Puleo et al revealed that mesenchymal tumor subtypes have a low stromal score while epithelial subtypes are high in stroma 21, 22. Moreover, it was found that mesenchymal tumors with very low stroma content featured the worst outcome. This suggests that a clinically relevant interplay between tumor cells and tumor stroma exists, in which the tumor cell phenotype may define the presence and characteristics of the stroma.
In the present study, we aimed to clarify whether mesenchymal‐like PDAC tumor cells instruct PSCs differently than non‐mesenchymal PDAC tumor cells do. Using primary PDAC tissue, xenografts, and organotypic co‐cultures, we found that high‐grade growth PDAC is characterized by stroma that is low in collagens and alpha‐smooth muscle actin (α‐SMA)‐positive PSCs. Subsequently, using a large set of PDAC cell lines, we show that mesenchymal‐like PDAC cells deactivate PSCs and inhibit proliferation in these cells through secretion of colony‐stimulating factor 1 (CSF‐1), and we validated these findings with immunohistochemistry in two PDAC patient cohorts. With these new insights, stroma‐targeting treatment of PDAC could be optimized in order to improve treatment outcome by fostering the tumor‐restraining properties of the stroma.
Results
High‐grade PDAC features stroma that is low in collagen and activated PSC content
To determine how epithelial‐ and mesenchymal‐like PDAC cells instruct the tumor stroma, a cohort of 15 PDAC patients (included between 2014 and 2016) was analyzed. Tumors were entirely embedded in the axial direction (Fig EV1A) and analyzed for total collagen I and III deposition using picrosirius red (PSR) staining (Fig 1A). This revealed that high‐grade, poorly differentiated PDAC (i.e., grade 3) featured a significantly lower collagen content compared to low‐grade PDAC (Fig 1B) while the tumor cell percentage between these samples was the same, suggesting that increased tumor cell expansion of high‐grade PDAC did not explain the reduced collagen deposition (Fig 1C). Subsequently, a panel of PDAC cell lines, which were classified as classical (i.e., epithelial‐like; Capan‐2 and AsPC‐1) or quasi‐mesenchymal (i.e., mesenchymal‐like; PANC‐1 and MIA PaCa‐2) using the Maupin and Broad dataset 23, 24 and the Collisson PDAssigner 20, was injected in immunodeficient mice. Tumors generated from epithelial‐like PDAC cells had markedly higher collagen content compared to tumors established from mesenchymal‐like PDAC cells (Fig 1D and E, upper panel). In silico analysis of various collagens in a panel of PDAC cell lines, which were also classified as epithelial (indicated in blue) or mesenchymal (indicated in red) using the aforementioned method, revealed that epithelial‐like PDAC cells produced equal to lower amounts of collagens than did mesenchymal‐like PDAC cells (Fig EV1B). We therefore concluded that these collagens were produced by activated PSCs. Expression of the stromal activation marker α‐SMA was assessed, and this revealed a similar pattern in which activated PSCs were present in epithelial‐like tumors, while these activated PSCs were reduced or deactivated in mesenchymal‐like PDAC tumors (Fig 1D and E, lower panel).
Figure EV1. PSCs play a role in mesenchymal‐like PDAC cell migration in 3D organotypic co‐cultures.
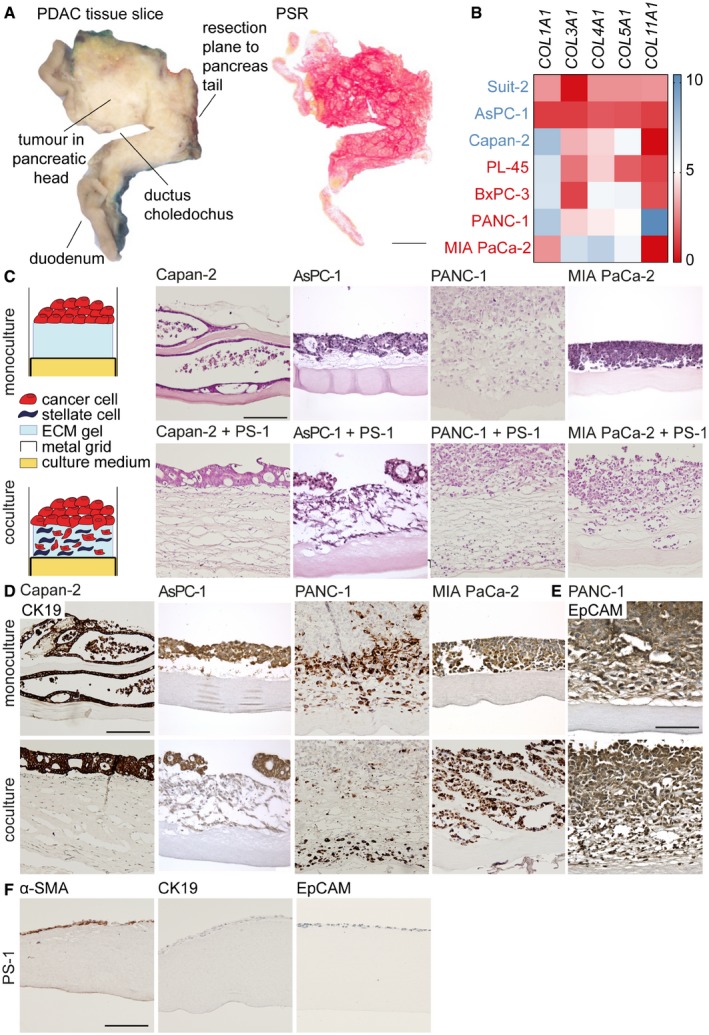
- An axial slice of the entire tumor containing pancreatic head and duodenum (left panel) was embedded in paraffin, and sections were cut and histochemically stained for PSR (right panel). Scale bar represents 1 cm.
- Gene expression of collagens in online available datasets of epithelial‐like (blue) and mesenchymal‐like (red) PDAC cell lines. Scale (0–10) represents log2 transformation.
- Schematic representation of organotypic mono and co‐culture of PDAC cells and pancreatic stellate cells (PS‐1). H&E staining was performed on organotypic cultures of indicated PDAC cell lines. Scale bar represents 100 μm.
- Organotypic mono‐ and co‐cultures were stained for CK19 with IHC. Scale bar represents 100 μm.
- Organotypic PANC‐1 mono‐ and co‐cultures were stained for EpCAM with IHC. Scale bar represents 100 μm.
- Organotypic PS‐1 monocultures were stained for α‐SMA, CK19, and EpCAM with IHC. Scale bar represents 100 μm.
Figure 1. High‐grade PDAC features stroma that is low in collagen and activated PSC content.
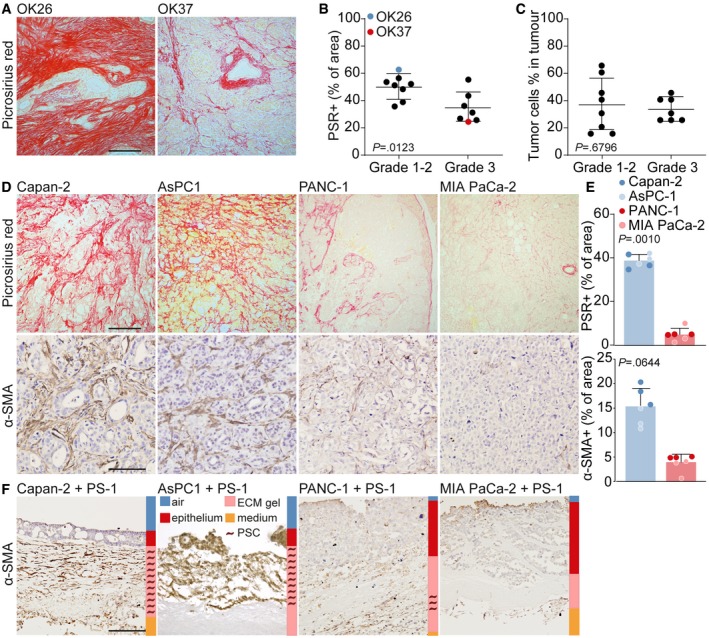
- Picrosirius red (PSR) staining of collagens (red) in PDAC tissue following surgical resection. Scale bar represents 200 μm.
- Quantification of PSR in low‐grade (1–2) and high‐grade (3) tumors indicated in panel A as percentage of area. n = 15 primary PDAC samples.
- Quantification of tumor cell percentage as scored by pathologists in low‐ and high‐grade PDAC indicated in panel A. n = 15 primary PDAC samples.
- PSR and α‐SMA staining of indicated PDAC cell lines which were injected in NSG mice. Scale bar represents 200 μm for PSR and 100 μm for α‐SMA images.
- Quantification of stainings indicated in panel D. Three tumor ROIs of a tumor derived from epithelial cell lines (Capan‐2 and AsPC‐1) and mesenchymal cell lines (MIA PaCa‐2 and PANC‐1) are displayed, and averages were pooled for analysis.
- IHC staining of α‐SMA in 3D organotypic co‐cultures of PS‐1 cells and indicated PDAC cell lines. Scale bar represents 100 μm.
To assess these findings in a controlled model system, an organotypic culture model was used 25, 26. In this model, tumor cells and primary PSCs (PS‐1) were co‐cultured on a 3D ECM gel with an air–liquid interface, to stimulate stratification of the epithelium (Fig EV1C). After three weeks of cultivation, Capan‐2 and AsPC1 cells had formed a stratified epithelium on top of the ECM gel, which was invaded by α‐SMA‐positive PS‐1 cells, while these activated PSCs were reduced in the ECM gel when co‐cultured with PANC‐1 and MIA PaCa‐2 cells (Figs 1F and EV1F). To assess the contributions of PS‐1 cells, mono‐ and co‐culture organotypics were assessed by H&E staining. This demonstrated that mono‐cultured mesenchymal‐like PANC‐1 and MIA PaCa‐2 cells grew as a stratified epithelium on top of the ECM gel (Fig EV1C, upper panel). When co‐cultured with PS‐1 cells, the architecture of the ECM gel was loosened in all PDAC cell lines (Fig EV1C, lower panel). Moreover, CK19 staining of PDAC cells revealed that epithelial‐like Capan‐2 and AsPC‐1 cells in both mono‐ and co‐culture remained on top of the ECM gel (Fig EV1D). On the contrary, CK19‐positive mesenchymal‐like PANC‐1 and MIA PaCa‐2 cells had migrated toward the nutrient gradient when co‐cultured with PS‐1 cells (Fig EV1D). Since PANC‐1 cells were only partly positive for CK19 staining, EpCAM stainings were used to demonstrate that almost all cells present in the PANC‐1 co‐culture were of epithelial origin (Fig EV1E–F; PS‐1 cells are negative for CK19 and EpCAM). Similar to MIA PaCa‐2, PANC‐1 cells had migrated into the ECM gel when co‐cultured with PS‐1 cells. This implies that, although these activated PSCs are absent after three weeks of co‐culture, PS‐1 cells initially either affect the migration capacity of the mesenchymal‐like PDAC cells or the composition of the ECM gel thereby making migration easier for these tumor cells. Together, these results suggest that epithelial‐like PDAC cells continuously instruct the activation of PSCs and deposition of collagens, while mesenchymal‐like PDAC cells either instruct PSCs to undergo apoptosis, or deactivate them to decrease the production of collagens.
Mesenchymal‐like PDAC cells deactivate pancreatic stellate cells and reduce proliferation
To confirm that epithelial‐ and mesenchymal‐like PDAC cells differently instruct PSCs, we exposed PS‐1 cells to conditioned medium (CM) of the previously described PDAC cells. Following exposure to the conditioned medium of MIA PaCA‐2 or PANC‐1 cells (mesenchymal CM), but not of Capan‐2 or AsPC‐1 cells (epithelial CM), a lower number of PS‐1 cells was found as well as a more elongated morphology (Fig 2A). Quantification of PS‐1 cell numbers using bead‐calibrated cell counting on FACS (Fig 2B), or surface area using ImageJ Software (Fig EV2A), showed a decrease in the number of PS‐1 cells in mesenchymal CM compared to epithelial CM conditions. To explain these reduced cell numbers, cell cycle analysis was performed using EdU (Fig 2C). This revealed no changes in G0/G1 phase, an accumulation of PS‐1 cells in G2/M phase and strongly reduced numbers in S phase in response to mesenchymal PDAC‐derived medium, suggesting that PSCs cell cycle progression halts at the G2/M phase (Fig 2C). This was confirmed by reduced KI‐67 (MKI67) levels in PS‐1 cells following incubation with mesenchymal PDAC cell CM (Fig EV2B) and indicated that these high‐grade PDAC cells hamper proliferation of PSCs. To determine the effects of PS‐1 cells on tumor cells, the mesenchymal cell markers were analyzed in PDAC cells after co‐culture with PS‐1 cells (separated by FACS for mCherry expression in PS‐1 cells). This revealed a substantial reduction in the mesenchymal markers N‐cadherin (CDH2), vimentin (VIM), and zinc finger E‐box‐binding homeobox 1 (ZEB1) in mesenchymal‐like PDAC cells, while this reduction was only modest in epithelial‐like cells (Fig EV2C). This implies that in our models, in contrast to the prevailing paradigm, PSCs decrease markers for invasion in high‐grade PDAC cells rather than increase them.
Figure 2. Mesenchymal PDAC cells reduce proliferation and alter the morphology of PSCs.
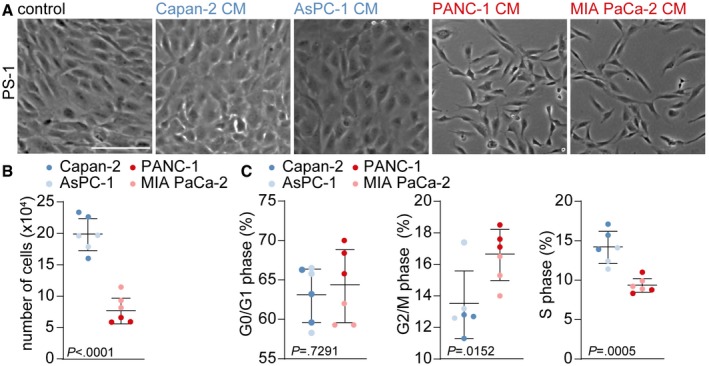
- Brightfield images of PS‐1 cells which were exposed for 72 h to CM from control (CM of PS‐1 cells), epithelial (blue) or mesenchymal (red) cell lines. Scale bar represents 100 μm.
- Quantification of bead‐calibrated cell count of PS‐1 cells after treatment indicated in panel A, using flow cytometry. n = 6 per group, 3 biological replicates for each cell line.
- EdU assay performed after treatment indicated at panel A, showing percentage of PS‐1 cells in G0/G1 phase, G2/M phase, and S phase. n = 6 per group, three biological replicates for each cell line.
Figure EV2. PSCs reduce mesenchymal markers in PDAC cells.
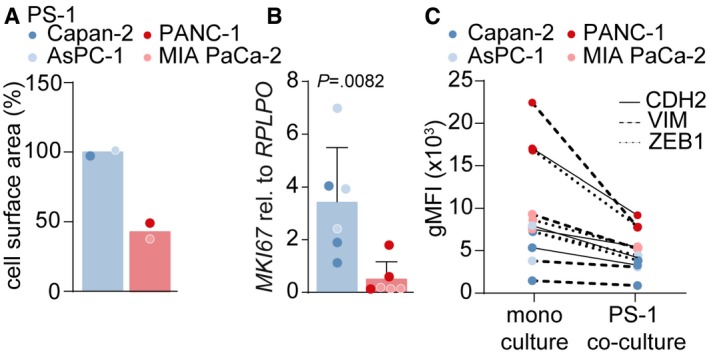
- Quantification of cell surface area of PS‐1 cells after treatment with CM from control (CM of PS‐1 cells), epithelial (blue), or mesenchymal (red) cell lines, using ImageJ. Cell surface area was normalized to average epithelial CM value. For each cell line, area was measured once.
- Gene expression level of proliferation‐marker MKi67 in PS‐1 cells after treatment indicated in panel A using qPCR. Data were normalized against control CM. Student's t‐test. n = 6 per group, 3 biological replicates for each cell line.
- Mesenchymal markers CDH2, VIM, and ZEB1 were measured in PDAC cells that were mono‐ or co‐cultured with mCherry‐expressing PS‐1 cells with flow cytometry. Representative graph of n = 2.
As we observed reduced α‐SMA‐positive fibroblasts and collagen deposition in high‐grade PDAC tumors, we exposed PS‐1 cells to tumor cell medium and measured stromal activation markers. PS‐1 cells exposed to mesenchymal CM showed a marked decrease in stromal activation markers alpha‐1 type I collagen (COL1A1) and alpha‐smooth muscle actin (ACTA2) as measured by qPCR (Fig 3A). Additional stromal activation markers secreted protein acidic and cysteine rich (SPARC), fibroblast activation protein (FAP), and fibronectin (FN1) showed a similar decrease in PS‐1 cells exposed to mesenchymal CM (Fig 3A). These findings were confirmed in a larger panel of PDAC cell lines demonstrating a clear decrease in COL1A1 and ACTA2 mRNA expression in PS‐1 cells exposed to mesenchymal PDAC cells (indicated in red) compared to epithelial PDAC cells (indicated in blue, Fig 3B). Primary PDAC cultures 67 and 53M 27, 28 conferred the same PSC phenotypes, in which 67 cells from well‐differentiated xenografts (Fig EV3C) maintained the activation status of PS‐1 cells (Fig 3B) while 53M cells from poorly differentiated tumors (Fig EV3C) deactivated PS‐1 cells as revealed by downregulation of ACTA2 and COL1A1 (Fig 3B). The epithelial state of these primary cultures was confirmed using flow cytometry, showing E‐cadherin protein expression of 67 and 53M cells to be in between the highly E‐cadherin‐positive, epithelial, Capan‐2 cells and the mesenchymal E‐cadherin‐negative MIA PaCa‐2 cells (Fig EV3A), as well as IHC in which membranous E‐cadherin expression was stronger in 67 cells (Fig EV3B). PDXs that were established from grafted tumor pieces of patients AMC‐PDAC‐67 and AMC‐PDAC‐53M in immunodeficient mice confirmed that 67 tumors were enriched in activated stroma, while the opposite was found in 53M tumors (Fig EV3D).
Figure 3. Mesenchymal‐like PDAC cells deactivate PSCs.
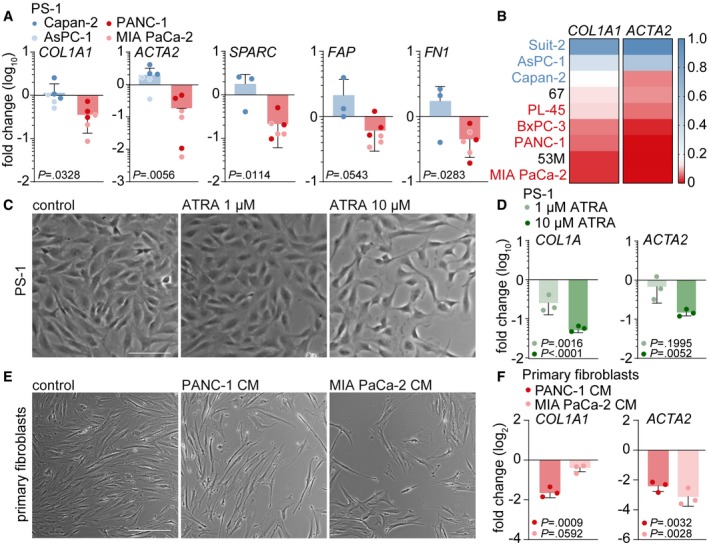
- Gene expression level of stromal activation markers COL1A1, ACTA2, SPARC, FN1, and FAP in PS‐1 cells which were exposed for 72 h to CM from control (CM of PS‐1 cells), epithelial (blue), or mesenchymal (red) PDAC cell lines using qPCR. Values were normalized to control and n = 6 per group, 3 biological replicates for each cell line.
- Heatmap representation of COL1A1 and ACTA2 relative gene expression levels measured with qPCR in PS‐1 cells after treatment indicated in panel A using a large cohort of PDAC cell lines and primary cell cultures 67 and 53 m. Values were log2 transformed and normalized to Suit‐2 expression.
- Brightfield images of PS‐1 cells exposed to 10 days of treatment with 1 and 10 μM all‐trans retinoic acid (ATRA). Scale bar represents 100 μm.
- Gene expression levels of COL1A1 and ACTA2 in PS‐1 cells after treatment indicated in panel C using qPCR, and values were normalized to control. n = 3 technical replicates per group.
- Brightfield images of primary human CAFs exposed to 72 h of treatment with CM from control (CM of primary CAFs) or mesenchymal PDAC cell lines. Scale bar represents 100 μm.
- Relative gene expression levels of COL1A1 and ACTA2 in primary CAFs after treatment indicated in panel E using qPCR, and values were normalized to control. n = 3 technical replicates per group.
Figure EV3. Grading of established primary human PDAC cultures.
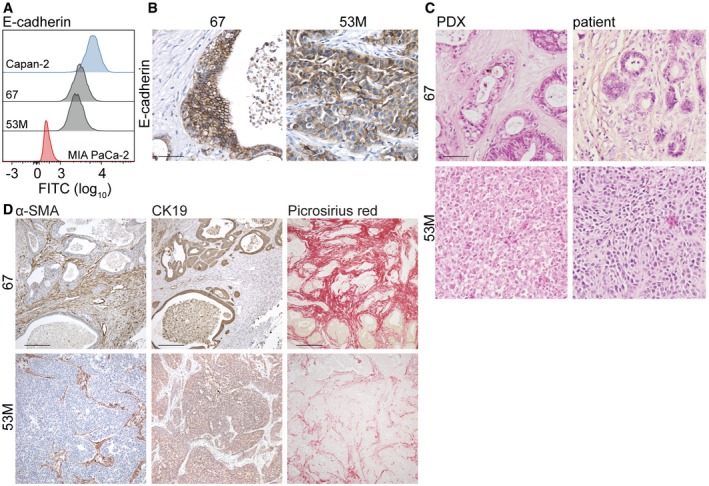
- Protein expression of E‐cadherin in indicated cell lines was assessed with flow cytometry.
- E‐cadherin was stained in established PDX tumors of 67 and 53M with IHC. Scale bar represents 50 μm.
- H&E histochemical staining was performed in established PDX tumors and matching primary PDAC tissue of patient 67 and 53M. Scale bar represents 50 μm.
- α‐SMA, CK19, and PSR were stained in established PDX tumors of 67 and 53M with (immuno)histochemistry. Scale bar represents 200 μm for α‐SMA and CK19 and 500 μm for PSR images.
To relate the PSC deactivating effects of mesenchymal‐like PDAC cells to an established method of PSCs deactivation, we treated PS‐1 cells with all‐trans retinoic acid (ATRA). Ten days of treatment with 10 μM ATRA, which is considered to be a supra‐physiologic dose 29, resulted in a more elongated morphology (Fig 3C) and decreased levels of COL1A1 and ACTA2 (Fig 3D). Of note, these deactivating effects in PS‐1 cells were comparable to the deactivating effects when exposed to three days of mesenchymal CM, suggesting that high‐grade PDAC cells are more efficient deactivators of PSCs than high doses of ATRA. Subsequently, we investigated whether mesenchymal‐like PDAC cells were also capable of deactivating other types of fibroblasts. Human primary fibroblasts derived from esophageal adenocarcinoma tissue were subjected to PANC‐1 and MIA PaCa‐2 CM for three days, after which again an altered morphology (Fig 3E) and decreased levels of COL1A1 and ACTA2 were observed (Fig 3F). Together, these results suggest that mesenchymal‐like PDAC cells secrete a factor that is able to deactivate myofibroblast‐like cells.
PSC deactivation is mediated by colony‐stimulating factor 1
Next, we aimed to identify how mesenchymal PDAC cells deactivate PSCs. We first set out to narrow down the list of potential candidates by defining their molecular size. To this end, we performed size exclusion experiments. PS‐1 cells were exposed to CM of Capan‐2, MIA PaCa‐2, and PANC‐1 cells that was either unfiltered or filtered at a molecule size of 30 and 100 kDa. As expected, the unfiltered MIA PaCa‐2 and PANC‐1 CM showed PS‐1 deactivation with decreased mRNA levels of COL1A1 and ACTA2 (Fig 4A and B). Unfiltered Capan‐2 CM did not affect morphology and increased the transcription of stromal activation markers while filtration of CM at 30 kDa was able to abolish these activating effects (Fig 4B, upper panels). PANC‐1 CM filtered at 100 kDa exerted similar effects on morphology and marker expression as did unfiltered PANC‐1 CM, whereas CM filtered at 30 kDa was unable to deactivate PS‐1 cells (Fig 4B, middle panel). MIA PaCa‐2 CM filtered at 100 kDa showed some reduced ACTA2 expression, but the deactivating effects as measured by COL1A1 and ACTA2 were abolished after 30 kDa filtration of CM (Fig 4B lower panel). This suggests that mesenchymal‐like PDAC cells secrete soluble factors larger than 30 kDa that are able to deactivate PSCs.
Figure 4. PSC deactivation is mediated by colony‐stimulating factor 1.
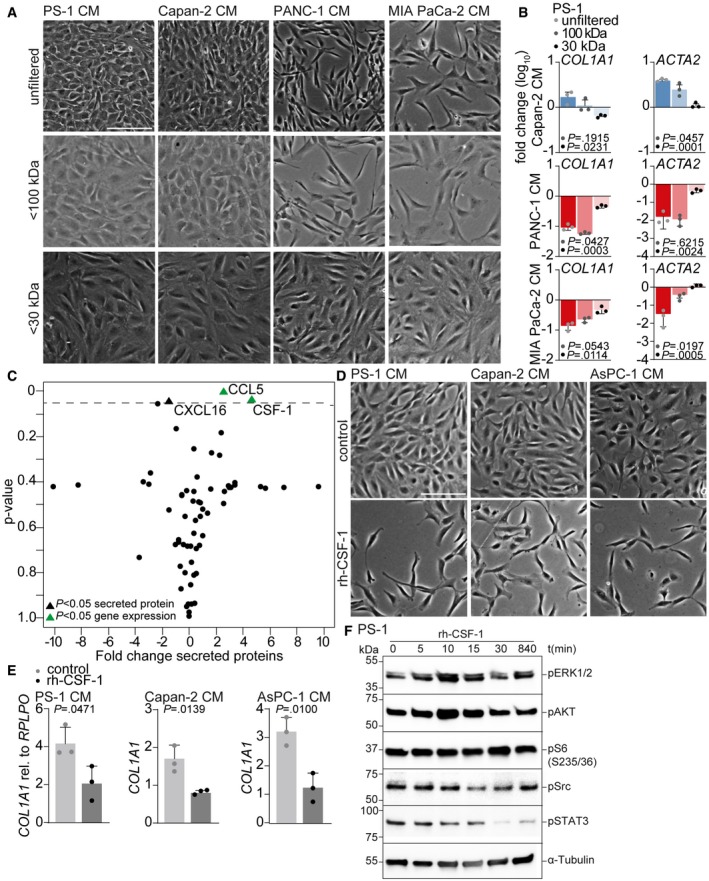
- Brightfield images of PS‐1 cells after 72 h of treatment with unfiltered, < 100 kDa or < 30 kDa filtered CM of indicated cell lines. Scale bar represents 100 μm.
- Gene expression levels of COL1A1 and ACTA2 in PS‐1 cells after treatment indicated in panel A using qPCR. Data were normalized against control CM. Statistical significance was tested against unfiltered CM samples. Graph shows n = 3 technical replicates of an example experiment that was performed three times.
- Graphical representation of 64 cyto‐ and chemokines present in CM of epithelial and mesenchymal PDAC cell lines using forward‐phase protein array. Significantly differential expression of proteins (triangles) and genes (green triangles) is identified between epithelial and mesenchymal cell lines. Dotted line indicates P = 0.05.
- Brightfield images of PS‐1 cells after 72 h of treatment with CM of control (CM of PS‐1s) or epithelial PDAC cells supplemented with recombinant human CSF‐1. Scale bar represents 100 μm.
- Gene expression levels of COL1A1 in PS‐1 cells after treatment indicated in panel D using qPCR. n = 3 biological replicates per group.
- PS‐1 cells were starved and stimulated with 500 ng/ml rh‐CSF‐1 at indicated time points. Activation of pathways involved in CSF1R signaling was analyzed using Western blot. Samples were derived from the same experiment, and blots were processed in parallel.
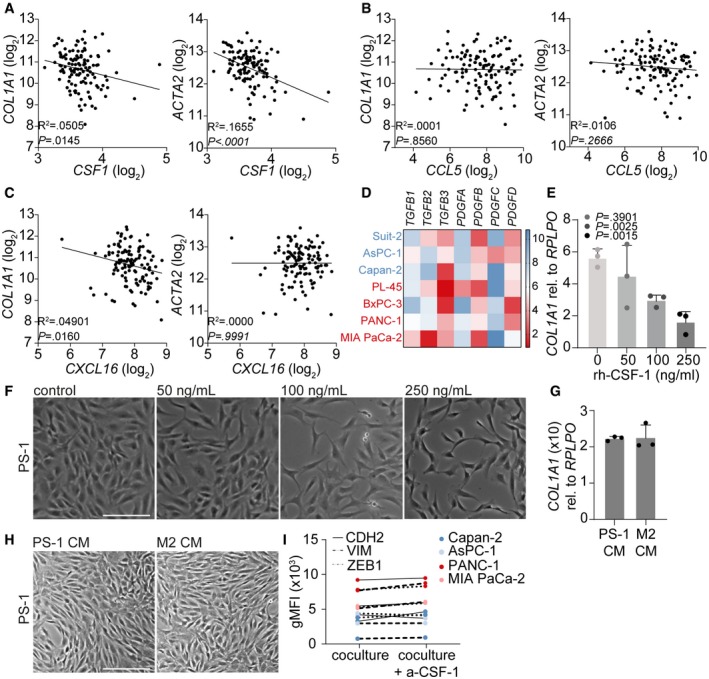
To identify the cyto‐ and chemokines differentially secreted by mesenchymal and epithelial PDAC cells, we performed a forward‐phase protein array (FPPA) for 64 proteins in the CM (Table EV1). Of these, three proteins were significantly differentially secreted between mesenchymal‐ and epithelial‐like PDAC cells: chemokine (C‐X‐C motif) ligand 16 (CXCL16), chemokine (C‐C motif) ligand 5 (CCL5), and colony‐stimulating factor 1 (CSF‐1) (Fig 4C). Of these, CCL5 and CSF1 were also differentially expressed between high‐ and low‐grade PDAC cells at the transcript level in publicly available gene expression sets 23, 24, 30. In silico assessment of additional public gene expression set containing PDAC samples (GSE62165 18) confirmed a negative correlation between CSF1 and COL1A1 and ACTA2 (Fig EV4A). CCL5 was not correlated with COL1A1 and ACTA2 (Fig EV4B), and CXCL16 was negatively correlated with COL1A1 but showed no correlation with ACTA2 (Fig EV4C). Moreover, since CXCL16 is smaller than 30 kDa this factor was not likely to induce the effects observed in PSCs. Other factors known to contribute to fibroblast activation, but not included in the FFPA, are transforming growth factor beta (TGF‐β) and platelet‐derived growth factor (PDGF). Assessment of these factors in the earlier described panel of epithelial (blue) and mesenchymal (red) PDAC cell lines using the Broad Institute (GSE36133) 24, and Maupin et al (GSE21654) 23 datasets showed no significant differences (Fig EV4D), leaving CSF‐1 as the most likely candidate.
Figure EV4. CSF1 expression correlates with COL1A1 and ACTA2 in tumors.
-
A–CCorrelation between CSF1 (A), CCL5 (B), and CXCL16 (C) gene expression and COL1A1 or ACTA2 gene expression in online available PDAC patient datasets. R 2 and P‐value were analyzed with linear regression.
-
DExpression of stromal activation markers TGFB1‐3 and PDGFA‐D in online available datasets of epithelial‐like (blue) and mesenchymal‐like (red) PDAC cell lines. Scale (2–10) represents log2 transformation.
-
ERelative gene expression of COL1A1 in PS‐1 cells exposed to indicated concentrations of recombinant human CSF‐1 using qPCR. n = 3 biological replicates per group.
-
FBrightfield images of PS‐1 cells after treatment indicated in panel E. Scale bar represents 100 μm.
-
GRelative gene expression of COL1A1 in PS‐1 cells after 72‐h treatment with control CM (PS‐1 CM) or M2 macrophages CM using qPCR. n = 3 biological replicates per group.
-
HBrightfield images of PS‐1 cells after 72‐h treatment with control CM (PS‐1 CM) or M2 macrophages CM. Scale bar represents 100 μm.
-
IMesenchymal markers CDH2, VIM, and ZEB1 were measured in PDAC cells that were co‐cultured with mCherry‐expressing PS‐1 cells with or without CSF1R inhibition using flow cytometry. Representative graph of n = 2.
To functionally demonstrate the role of CSF‐1 in PSC deactivation, we added recombinant human CSF‐1 (rh‐CSF‐1) to control medium (PS‐1), and epithelial CM (Capan‐2 and AsPC‐1) cultures. This showed that rh‐CSF‐1 was very potent at inducing a similar morphology in PS‐1 cells as compared to the effects conferred by mesenchymal CM (Fig 4D). Additionally, COL1A1 was significantly downregulated in PS‐1 cells following the addition of rh‐CSF‐1 to epithelial CM (Fig 4E) and this effect was dose dependent (Fig EV4E and F). To determine the stroma‐deactivation contributions of the immune cell best known to remodel the stroma, PS‐1 cells were treated with M2 macrophage CM 31. This did not affect morphology of PS‐1 cells nor expression levels of COL1A1 (Fig EV4G and H) and implies that M2 macrophages are not responsible for (additional) changes to stromal activation status.
To identify the mechanism by which CSF‐1 regulates PSC deactivation, activated pathways involved in CSF1R signaling (i.e., PI3K – ERK1/2, PI3K – AKT and Src – STAT3 32, 33, 34) were assessed by Western blot. Stimulation of PS‐1 cells with rh‐CSF‐1 activated both ERK1/2 and AKT as reported previously 35 (Fig 4F). However, Src and STAT3 activity was decreased in PS‐1 cells following CSF‐1 stimulation. To determine whether deactivated PSCs influence the phenotype of PDAC cells, co‐cultures of tumor cells and mCherry‐positive PS‐1 cells with or without inhibition of CSF‐1 signaling were performed revealing no differences in protein expression of CDH2, VIM, and ZEB1 in PDAC cells (Fig EV4I). This suggests that inhibition of CSF‐1 signaling in PSCs does not affect the phenotype of mesenchymal‐like PDAC cells and that the functional relevance of tumor cell secreted CSF‐1 is mainly directed against the stroma. As these mesenchymal tumor cells seem to benefit from PSC deactivation, we deduce that this hinges on structural/mechanical changes in the stroma that are pro‐tumorigenic, rather than stromal cues that impact on tumor cell phenotype.
Targeting of CSF‐1 in mesenchymal‐like PDAC cells abolishes PSC deactivation
To address whether PSC deactivation induced by mesenchymal‐like PDAC cells could be prevented, CSF‐1 inhibition experiments were performed. Selective inhibition of CSF1R (a‐CSF1R) did not affect morphology or transcription of ACTA2 and COL1A1 in PS‐1 cells exposed to PS‐1 CM compared to control (Fig 5A and B). On the contrary, treatment of PS‐1 cells with mesenchymal CM containing anti‐CSF‐1 was able to partly abolish these deactivating effects as revealed by reduced morphological changes and increased expression of stromal activation markers (Fig 5A and B). Inhibition experiments were validated by silencing CSF‐1 in PANC‐1 cells using three independent shRNA sequences targeting CSF1. PS‐1 cells exposed to CM from control scrambled vector (pLKOctrl) transduced PANC‐1 cells showed a similar elongated morphology as untransduced PANC‐1 CM (Fig 5C). Knockdown of CSF‐1 in PANC‐1 cells with all three shRNA sequences tested (#560–562) reduced these morphological effects (Fig 5C). CSF1 knockdown in PANC‐1 cells was proportionally correlated with COL1A1 expression in PS‐1 cells (Fig 5D), in which CM of PANC‐1 cells that had the lowest expression of CSF1 (i.e., shCSF1 #562) resulted in the highest expression of COL1A1 in PS‐1 cells (Fig 5D). Since full knockdown of CSF1 was not achieved despite selection, this implies that CSF‐1 secretion is an obligate feature of mesenchymal‐like PDAC cells. To further ascertain specificity that tumor cell secreted CSF‐1 is responsible for PSC deactivation, Capan‐2 cells were transduced with a lentiviral vector to overexpress CSF‐1 (Fig 5F). This revealed that CM of Capan‐2 cells that were high in CSF‐1 production were able to deactivate PSCs as measured by morphology (Fig 5E) and decreased COL1A1 and ACTA2 expression (Fig 5G) compared to CM of Capan‐2 vector control. Together, these silencing and overexpressing experiments confirm that tumor cell produced CSF‐1 is responsible for PSC deactivation and that this process can be prevented by targeting CSF‐1.
Figure 5. Targeting CSF‐1 in mesenchymal‐like PDAC cells abolishes PSC deactivation.
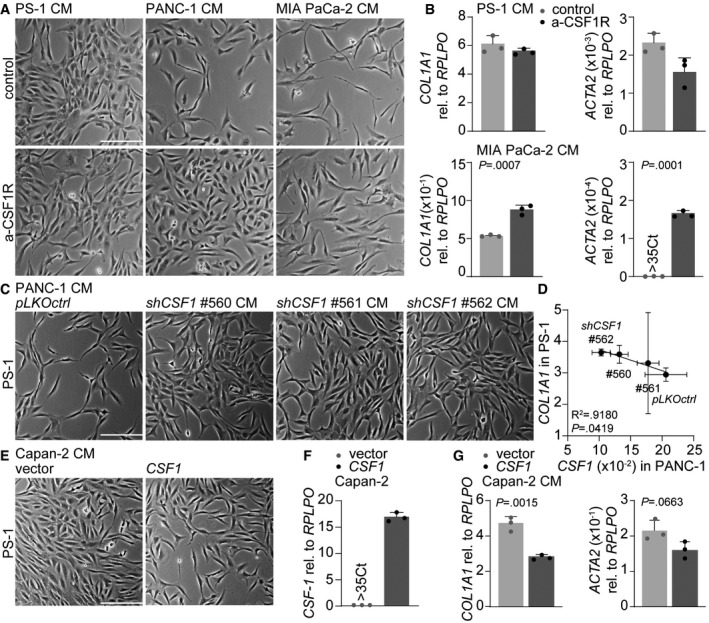
- Brightfield images of PS‐1 cells after 72 h of treatment with CM of control (CM of PS‐1s) or mesenchymal PDAC cell lines supplemented with anti‐CSF‐1. Scale bar represents 100 μm.
- Gene expression levels of COL1A1 in PS‐1 cells after treatment indicated in panel A using qPCR. Graph shows n = 3 technical replicates of an example experiment that was performed three times.
- Brightfield images of PS‐1 cells after 72 h of treatment with PANC‐1 CM of pLKOctrl or shCSF1 (#560–562). Scale bar represents 100 μm.
- Correlation graph of CSF1 expression in PANC‐1 pLKOctrl or shCSF1 (#560–562) and how these various CM affect COL1A1 expression in PS‐1 cells using qPCR, and values were normalized for RPLPO expression. n = 3 biological replicates for all individual experiments, error bars indicated SD. R 2 and P‐value were analyzed with linear regression.
- Brightfield images of PS‐1 cells after 72 h of treatment with CM of Capan‐2 cells with control vector or Capan‐2 cells overexpressing CSF1. Scale bar represents 100 μm.
- Gene expression level of CSF1 in Capan‐2 cells after transduction with control vector or overexpressing CSF1. n = 3 technical replicates per group.
- Gene expression levels of COL1A1 in PS‐1 cells after treatment indicated in panel E using qPCR. Graph shows n = 3 technical replicates of an example experiment that was performed three times.
High‐grade CSF‐1‐positive PDAC features deactivated stroma
To confirm that CSF‐1 was produced by high‐grade PDAC, the same PDAC patient cohort (n = 15, included between 2014 and 2016) used in Fig 1A was immunohistochemically stained for CSF‐1 (Fig 6A). Subsequently, for each specimen ROIs were drawn around tumor cells and CSF‐1 expression was measured in these ROIs revealing that high‐grade tumors had significantly more CSF‐1‐positive tumor cells (Fig 6A and B). To validate that tumor cell secreted CSF‐1 drives PSC deactivation resulting in stroma that is low in collagens and activated fibroblasts, an independent validation cohort of 21 PDAC patients that were included between 2001 and 2014 was composed and (immuno)histochemically stained for CSF‐1, PSR, and α‐SMA (Fig 6C). Quantification of these stainings confirmed that there was a correlation between CSF‐1 and PSR (Fig 6D) as well as CSF‐1 and α‐SMA (Fig 6E), in which PDAC tissue with a high percentage of CSF‐1‐positive tumor cells was low in collagen I and III and α‐SMA‐positive fibroblasts (Fig 6C, OK7).
Figure 6. High‐grade CSF‐1‐positive PDAC features deactivated stroma.
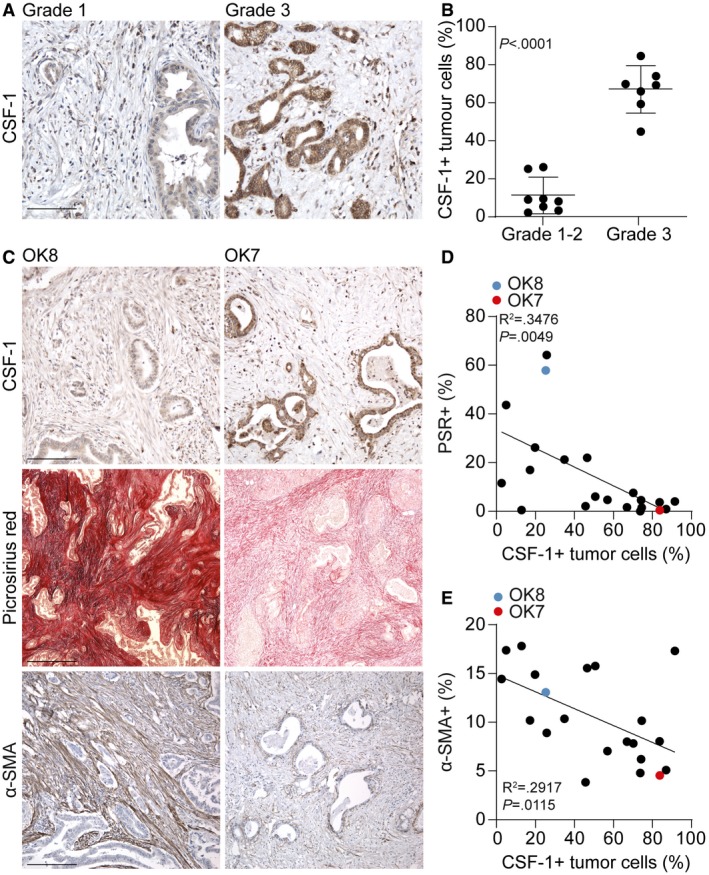
- IHC staining of CSF‐1 in PDAC tissue following surgical resection (same cohort as in Fig 1A). A representative image of CSF‐1 expression in grade 1 and grade 3 PDAC is depicted. Scale bar represents 100 μm.
- For each tumor of panel A, CSF‐1 expression in ductal adenocarcinoma cells was quantified. Student's t‐test. Shown is mean ± SD. n = 15 primary PDAC samples.
- A validation cohort of PDAC patients (n = 21) that underwent direct resection was stained for CSF‐1 (scale bar represents 100 μm), PSR (scale bar represents 500 μm), and α‐SMA (scale bar represents 200 μm). OK8 is low in CSF‐1 expression and high in PSR and α‐SMA expression, while the opposite accounts for OK7.
- Correlation of % of PSR in the tumor and CSF‐1‐expressing tumor cells, as indicated in panel C. R 2 and P‐value were analyzed with linear regression. n = 21 primary PDAC samples.
- Correlation of % of α‐SMA in the tumor and CSF‐1‐expressing tumor cells, as indicated in panel C. R 2 and P‐value were analyzed with linear regression. n = 21 primary PDAC samples.
Together, these findings imply that mesenchymal‐like PDAC cells instruct the deactivation of PSCs by the production of CSF‐1, explaining why high‐grade PDAC features tumor‐promoting stromal content that is low in collagens and activated PSCs. As this process could be counteracted by CSF1R inhibition, this offers options to maintain a tumor‐restricting microenvironment and potentially target metastatic dissemination in PDAC patients. Moreover, it could be a valuable marker to determine the differentiation grade in PDAC patients.
Discussion
Most studies on tumor–stroma crosstalk in PDAC have centered on the activation of stromal cells and how these activated stromal cells in turn instruct tumor cells. In this study however, we find that high‐grade mesenchymal PDAC is characterized by stroma that is low in collagens and activated PSCs, and that this is caused by the deactivation of PSCs through secretion of CSF‐1. In contrast, low‐grade epithelial PDAC cells maintain this activated stroma by reduced expression of CSF‐1 suggesting that these epithelial tumor cells benefit from the activated stroma‐derived signals. For mesenchymal‐like PDAC cells, however, our findings suggest that the activated stroma acts as a mechanical barrier with no beneficial signaling, prompting the rapid deactivation of surrounding stroma.
Studies on intra‐ and intertumor heterogeneity have historically focused on the tumor cell compartment. Recently, it has been postulated that also in the tumor stroma heterogenic populations of CAFs exist. Work from the Tuveson laboratory described two distinct populations of CAFs: IL1‐induced iCAFs, which are high in inflammatory markers such as IL‐6 and leukemia inhibitory factor (LIF), and the TGF‐β‐induced myCAFs, which are αSMA positive 14, 15. In addition, Neuzillet and colleagues identified by transcriptomic analysis four distinct subtypes in primary patient‐derived CAF cultures, which were all associated with a different phenotype and prognostic impact 22. Moreover, these studies suggest that these CAF populations have distinct tumor‐promoting or tumor‐constraining effects. iCAFs are hypothesized to play a role in cancer progression, while α‐SMA‐positive myCAFs are suspected to restrain tumor progression 15. This is supported by Özdemir et al 12 who showed that ablation of α‐SMA‐positive myofibroblasts from established pancreatic tumors in transgenic mice resulted in more invasive tumors, and PDAC patients with fewer myofibroblasts in the tumor had reduced survival. Additionally, it has been suggested that there is plasticity between iCAF and myCAF populations and that this CAF state is defined by the tumor‐derived cues they are exposed to 15. Based on our findings, we hypothesize that the intra‐ and intertumor heterogeneity of PSCs is determined by the subtype of the tumor cells, in which mesenchymal PDAC cells instruct activated PSCs to convert into deactivated cells. This is in agreement with work by Rhim and colleagues, suggesting that the differentiation grade of pancreatic cancers defines the resulting tumor‐promoting or suppressive effect 11. Interestingly, since tumor cell percentage is similar in low‐ and high‐grade PDAC tissue, the deactivation of PSCs and reduction of collagens does not mean that there is less stroma present in high‐grade tumors. We take this to show that the composition of the stroma is different, and that some of its constituents (e.g., hyaluronan or laminin) might be more abundantly present in high‐grade PDAC 36. Also, given the observed absence of α‐SMA‐positive fibroblasts in these high‐grade tumors, tumor‐promoting PSCs with a different phenotype (e.g., an immunologic signature that are high in IL‐6) could be part of the stroma. Future research should further elucidate the differences in the PSC and ECM signature in low‐ and high‐grade PDAC and how the signaling and mechanical contributions balance out.
In this study, we revealed that mesenchymal‐like PDAC cells reduced the proliferation, and changed the morphology, of PSCs. As STAT3 activation is required for cell proliferation, inactivation of this pathway following CSF‐1 stimulation could explain the observed reduced proliferation of PS‐1 cells 37, 38. Moreover, it has been reported that Src kinase phosphorylation impairs actin polymerization and cytoskeleton remodeling 39, 40 and is involved in E‐cadherin translocation 41. Moreover, we showed that mesenchymal‐like PDAC cells deactivate PSCs via downregulation of ACTA2 transcription. This is in agreement with Neuzillet and colleagues, showing that PS‐1 cells exposed to CM from MIA PaCa‐2 cells undergo a potential “CAF‐like” phenotypic switch in which α‐SMA expression is decreased 22. Besides the absence of activated PSCs in high‐grade PDAC tumors, we also observed a strong decrease in collagen deposition. Studies so far have only described pro‐tumorigenic roles of collagen in PDAC. This has mainly been described to occur through changes in signaling pathways as well as collagen‐induced changes to the environment (i.e., stiffness), ultimately hampering therapy delivery and promoting tumor cell invasion 42, 43, 44. We find a rapid downregulation of collagen 1 produced by PSCs following exposure to CSF‐1 secreted by mesenchymal PDAC cells, suggesting that collagens are not conductive to growth of this subtype of cells. Ongoing studies testing the effects of targeting stromal collagen in PDAC can hopefully clarify this role (NIH 1R01CA169281‐01A1 45).
The disappointing results from stroma‐targeting therapy trials in PDAC have urged researchers to reconsider the role of the tumor stroma 46. Reducing solid stress in tumors through targeting of the ECM and thus improving chemotherapy effectiveness so far has shown mixed results, and simple disruption of the dense tumor stroma in unselected patients is likely a suboptimal strategy 47, 48. It has recently been postulated that stromal normalization, rather than stromal ablation, could be one of the solutions to improve therapy outcome of PDAC patients 13, 49. In light of this, preclinical studies have successfully aimed to reprogram PSCs into a quiescent state using either calcitriol or ATRA to hamper tumor growth and prolong survival in mice 29, 50. However, our findings suggest that proliferative and α‐SMA‐positive PSCs have a tumor‐restraining effect and tumor cell instruction to convert PSCs into a deactivated state should be prevented. This is supported by several studies that reveal that high a α‐SMA content is associated with increased survival in PDAC 11, 12, 51.
As we identified CSF‐1 to be a strong inducer of PSC deactivation, inhibition of its receptor, CSF1R, could be a promising approach to maintain (or instruct) a tumor‐restraining microenvironment which is rich in collagens and activated PSCs in high‐grade PDAC tumors. As it was previously shown that PS‐1 cells have very low CSF1R protein expression 52, our CSF1 stimulation and inhibition experiments suggest that this signaling axis is very potent in deactivating PS‐1 cells. Multiple isoforms of CSF‐1 have been reported including a 44 kDa glycoprotein CSF‐1 and a > 200 kDa proteoglycan CSF‐1 53, 54, 55. This could be taken to suggest that MIA PaCa‐2 cells secrete more proteoglycan CSF‐1, while PANC‐1 cells secrete more glycoprotein CSF‐1, and that this explains how PSC deactivating effects were decreased after both 100 and 30 kDa filtration of MIA PaCa‐2 CM. Interestingly, a preclinical study testing CSF1R inhibition in PDAC with the aim of targeting macrophages showed a delay in tumor growth and improved survival in mice 56. In addition, serum levels of CSF‐1 are upregulated in pancreatic cancer, and higher levels correlate with more advanced tumor stage, which is in agreement with our IHC findings in primary PDAC tissue samples 57, 58. Monitoring CSF‐1 serum levels in PDAC patients could be a promising tool to identify patients with high‐grade disease that could benefit from stromal normalization using CSF1R targeting. Moreover, the finding that deactivated, collagen low, stroma reflects more poorly differentiated tumors gives opportunities for easier stratification of PDAC differentiation status by non‐invasive imaging or measuring blood markers for stromal activation.
Materials and Methods
Study approval
All patients included in this study signed informed consent according to the procedures approved by the Amsterdam UMC, location AMC, ethical committee (METC2013_254 (NCT01989000), Netherlands Trial Registry NTR3709, METC_A1 15.0122, METC 2018_181, METC 01/288#08.17.1042, METC2016_325, and METC2014_181) and conform the principles set out in the WMA Declaration of Helsinki and the Department of Health Services Belmont Report. All procedures which are described in this manuscript, such as the collection and expansion of tissue in xenografts and cell lines, are covered by this informed consent 27. Grafting of PDAC cell lines, breeding and maintaining of mice at the local animal facility, was performed according to the local legislation and under ethical approval of the animal ethical committee (LEX102348; LEX268AD).
Patient inclusion
Patients with pathologically confirmed resectable PDAC were included in the Amsterdam UMC between 2014 and 2016 (MIPA study; METC2013_254, NCT01989000, registered November 2013, https://clinicaltrials.gov/ct2/show/NCT01989000, and PREOPANC study; NTR3709, registered 8 November 2012, https://www.trialregister.nl/trial/3525 59). Patients reported here did not receive neo‐adjuvant therapy before surgical exploration. Upon surgical exploration, whole mount tissue sections of 15 patients were used for further histochemical analysis of collagen and tumor differentiation grade. Complete clinical follow‐up was used until September 2018. For the validation cohort, an earlier time cohort was composed containing 21 patients with pathologically confirmed PDAC that were included in the Amsterdam UMC between 2001 and 2014 retrospectively and prospectively 28 (SPACIOUS study; METC_A1 15.0122, http://www.dpcg.nl/projecten/spacious.html and BioPAN study; METC 2018_181, http://www.bijlsmalab.com/research/). Retrospective collection of paraffin blocks was in accordance with “Code for Proper Secondary Use of Human Tissue in The Netherlands” (Dutch Federation of Medical Scientific Societies). Patients underwent direct resection and had an even distribution of differentiation grades.
Cell line culture
Capan‐2, AsPC‐1, PANC‐1, MIA PaCa‐2, Suit‐2, PL‐45, and BxPC‐3 cells (ATCC, Manassas, VA) were cultured according to standard procedures in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 8% fetal bovine serum, L‐glutamine (2 mmol/l), penicillin (100 units/ml), and streptomycin (500 μg/ml, Lonza, Basel, Switzerland). Cells were monitored for mycoplasma monthly, and STR profiled no more than 6 months prior to performing experiments (Promega PowerPlex).
Establishment of primary cultures
Primary human PDAC cultures 53M and 67 were established as previously described and maintained in fully supplemented DMEM 27. Pancreatic stellate cells (PS‐1, kindly provided by Hemant Kocher) were established and immortalized as previously described and maintained in fully supplemented Advanced DMEM/F12 (Gibco, Waltham, MA) containing puromycin (1 μg/ml, Sigma, Saint Louis, MO) 25, 52. For co‐cultures, PS‐1 cells were transduced with a lentiviral gene ontology (pLeGO) vector expressing mCherry (pLeGO‐C2, #27339; Addgene, Cambridge, MA 60) as described earlier 61 and plated with tumor cells in a ratio 1:1. Primary human CAFs were derived from an esophageal adenocarcinoma biopsy, isolated as described previously 62, and maintained in fully supplemented IMDM.
Grafting of tumor cells
PANC‐1, MIA PaCa‐2, and Capan‐2 cells were injected orthotopically into the pancreas of NOD.Cg‐Prkdc scid Il2rg tm1Wjl/SzJ (NSG) mice (JAX 005557). For orthotopic tumor cell inoculation, mice received analgesics pre‐operative (meloxicam, 1 mg/kg) and were operated under isoflurane anesthesia (0.5–2.5% in 100% oxygen). In short, a small incision was made in the abdominal skin and peritoneal wall and the pancreas was gently pulled out and 106 cells in 50 μl PBS + 5% Matrigel were injected using a 25G needle. The inoculated pancreas was placed back into the abdomen, and the muscle and skin layers were closed using sutures. AsPC‐1 cells were injected subcutaneously in NSG mice as described previously 27. PDXs of 53M and 67 were established by grafting of patient‐derived tumor pieces into NSG mice subcutaneously 27.
Organotypic cultures
Organotypic cultures were established as previously described 25, 26. In short, cancer cells and PS‐1 cells were placed in a 1:2 ratio on top of gels 63 consisting of 52.5% rat tail Collagen I, 17.5% matrigel, 10% fetal calf serum, 10% fully supplemented DMEM, and 10% 10X DMEM. The following day, gels were placed onto a metal grid and provided with fully supplemented DMEM from below, thereby creating a nutrient gradient and consecutive air–liquid interface. DMEM was refreshed twice a week, and gels were collected for immunohistochemical (IHC) analysis after three weeks. After overnight fixation in 4% paraformaldehyde, gels were dehydrated and embedded in paraffin.
Immunohistochemical staining
Four‐μm‐thick paraffin sections of PDAC patient, xenograft, or organotypic culture tissue were cut using a microtome and subsequently deparaffinized in xylene and dehydrated in a series of ethanol. Hematoxylin and eosin (H&E) and picrosirius red (PSR) stainings were performed according to standard protocol. For IHC staining, dehydrated sections were incubated for 15 min at RT in PBS containing 0.3% hydrogen peroxide (VWR International, Radnor, PA) to block endogenous peroxidase. Heat‐induced epitope retrieval was performed in sodium citrate buffer solution (Lab Vision™ PT Module™, Thermo Scientific, Waltham, MA) at pH 6 for α‐SMA, cytokeratin 19 (CK19), E‐cadherin, and epithelial cell adhesion molecule (EpCAM) staining and in Tris‐EDTA buffer solution (Lab Vision™ PT Module™) at pH 9 for CSF‐1 staining, all for 20 min at 98°C. The primary antibody directed against α‐SMA (ab5694, Abcam, 1:1,000), CK19 (MU246‐UC, BioGenex, Fremont, CA, 1:500), E‐cadherin (EP700Y, Abcam, 1:100), EpCAM (ab32392, Abcam, 1:100), or CSF‐1 (EP1179Y, Ab52864, Abcam, 1:100) was in normal antibody diluent (KliniPath, Radnor, PA) and incubated overnight at 4°C. Subsequently, BrightVision+ post antibody block was incubated for 20 min at RT and secondary antibody BrightVision Poly‐HRP‐anti Ms/Rb IgG (Immunologic, VWR International) was incubated for 30 min at RT. Staining was developed with Bright‐DAB (Immunologic), counterstained with hematoxylin (KliniPath), and dehydrated in a series of ethanol. Sections were mounted in Pertex (HistoLab, Askim, Sweden). For PSR quantification of primary PDAC tumors, of each patient an axial slice containing the entire tumor and duodenum was embedded, stained, and digitized with an Olympus dotSlide virtual slide microscope (Olympus, Tokyo, Japan) using a 10× magnification. The tumor area in these large sections was marked by a pathologist. For PSR and α‐SMA quantification of PDX tumors and the PDAC validation cohort, at least three large representative regions of interest (ROIs) were imaged and quantified of each tumor. For CSF‐1 quantification, in at least three ROIs of each tumor the CSF‐1 positivity was quantified in tumor cells only by drawing separate ROIs around each carcinoma duct. The color deconvolution plugin was used for separation of either the RGB channel for PSR quantification or hematoxylin and DAB channel for CSF‐1 and α‐SMA. Subsequently, the same threshold was set for all images, and the amount of positive staining was analyzed as percentage of area. All quantification was performed using ImageJ software 64.
In vitro treatments and chemicals
Conditioned medium (CM) of PDAC cell lines and PS‐1 cells was collected after three days, spun down at 700 g for 5 min, and stored at 4°C. PS‐1 cells were incubated with CM of PDAC cell lines, PS‐1 cells, or DMEM for three days. For filter experiments, DMEM and CM of PDAC and PS‐1 cells was filtered over Amicon® Ultra‐15 30K and 50K centrifugal filter devices (Merck Millipore, Burlington, MA) according to manufacturer's instructions. For PS‐1 deactivation experiments, PS‐1 cells were treated with 1 or 10 μM all‐trans retinoic acid (ATRA, Sigma). DMEM containing ATRA was refreshed daily for 10 subsequent days. Control condition received DMEM containing 0.1% DMSO. For stimulation of PS‐1 cells with CSF‐1, 250 ng/ml recombinant human CSF‐1 (r‐CSF‐1, Prospec, Rehovot, Israel) was added to CM or DMEM. In addition, CSF‐1 receptor inhibitor (a‐CSF1R, BLZ‐945, AdipoGen Life Sciences) was added to cultures at 10 μM. Conditioned medium (CM) of M2 macrophages was established using the following protocol. THP‐1 cells were treated with 150 nM phorbol 12‐myristate 13‐acetate (PMA) for 24 h in RPMI 1640 (Gibco, Thermo Fischer Scientific, Waltham, MA) medium. Next, adherent activated THP‐1 cells were washed with fresh medium to remove PMA after which cells were cultured in fresh medium for another 24 h after which the medium was refreshed once more. For M2 generation, macrophages were further treated with recombinant IL‐4 and IL‐13 (both Peprotech, Rocky Hill, NJ), 20 ng/ml each, for 72 h, and media were renewed for collection. Macrophage‐conditioned media were collected after 48 h. After collection, media were centrifuged at 350 g for 4 min, filtered with 0.2 μm syringe filters (Corning, New York), and stored at 4°C.
Proliferation and cell cycle analysis
PS‐1 cells were incubated with CM of PDAC cell lines, PS‐1 cells, or DMEM for three days. Cells were harvested and analyzed. For absolute calibrated cell count, samples were measured with CountBright absolute counting beads (Invitrogen) according to manufacturer's instructions. For cell cycle analysis, proliferating cells were stained using the Click‐iT™ Plus EdU Alexa Fluor™ 488 Flow Cytometry Assay Kit (Thermo Fisher Scientific) according to manufacturer's instruction. Cell cycle was assessed by adding 7‐AAD to these samples. For both analyses, events were acquired on a FACS Canto II (BD, Franklin Lakes, NJ). Data were analyzed using FlowJo 10 (Tree Star, Ashland, OR).
Quantitative real‐time PCR
RNA was isolated according to manufacturer's instructions (Macherey‐Nagel, Bioke, Duren, Germany). Subsequently, cDNA was synthesized with Superscript II (Invitrogen, Carlsbad, CA) and SYBR green (Roche, Basel, Switzerland) was used to perform quantitative real‐time PCR (qRT–PCR) on a Lightcycler 480II (Roche) according to manufacturer's instructions. The data were normalized using the delta delta Ct analysis, in which the housekeeping gene was subtracted followed by subtraction of the control‐treated sample and log2 transformation. The following primers were used in this study: hCOL1A1, Fwd: CACACGTCTCGGTCATGGTA, Rv: AAGAGGAAGGCCAAGTCGAG, hACTA2, Fwd: CCAGAGCCATTGTCACACAC, Rv: CAGCCAAGCACTGTCAGG, hMKI67 Fwd: ACGCCTGGTTACTATCAAAAGG, Rv: CAGACCCATTTACTTGTGTTGGA, hSPARC Fwd: GAAAGAAGATCCAGGCCCTC, Rv: CTTCAGACTGCCCGGAGA, hFN1, Fwd: ACCTCGGTGTTGTAAGGTGG, Rv: CCATAAAGGGCAACCAAGAG, hFAP, Fwd: TCAGTGTGAGTGCTCTCATTGTAT, Rv: GCTGTGCTTGCCTTATTGGT, hB2M, Fwd: GTCTTTCAGCAAGGACTGGTC, Rv: CTTCAAACCTCCATGATGC, hRPLPO, Fwd: CCATTCTATCATCAACGGGTACAA, Rv: TCAGCAAGTGGGAAGGTGTAATC, hRPS18, Fwd: AGTTCCAGCATATTTTGCGAG, Rv: CTCTTGGTGAGGTCAATGTC.
Flow cytometry
Flow cytometry stainings were performed as previously described 65, using anti‐E‐cadherin (EP700Y, Abcam, 1:500), anti‐CDH2 (Ab8978, Abcam, 1:100), anti‐VIM (HPA027524, Sigma, 1:100), and anti‐ZEB1 (TA503933/2G7, OriGene). For intracellular staining of CDH2, VIM, and ZEB1, cells were fixed and permeabilized using Cytofix/Cytoperm (BD). Propidium iodide was used to exclude dead cells, and isotype control was subtracted to calculate geometric mean fluorescence intensity (gMFI). Events were acquired on a FACS Canto II (BD, Franklin Lakes, NJ). Data were analyzed using FlowJo 10 (Tree Star, Ashland, OR).
Forward‐phase protein array (FPPA)
Conditioned medium was prepared similarly as described above, except cell protein lysates were generated for normalization. For the protein array, 64 antibodies (Table EV1) were diluted in PBS supplemented with 0.05% Tween‐20 to a final concentration of 200 μg/ml in 384‐well plates. Arrays are printed using an Aushon 2470 microarrayer fitted with 185 μM pins, and antibodies were immobilised on Oncyte supernova nitrocellulose slides (Grace Biolabs, Bend, OR). Four replicate spots were printed for each antibody. After printing, slides were left in the humidified printing chamber for 1 h prior to storage at 4°C. Slides were blocked for 1 h in Superblock T20 (Thermo Fisher) at room temperature with gentle agitation. Block was removed, and samples were incubated with the slides overnight at 4°C with gentle agitation. Slides were washed 3 × 5 min in PBS supplemented with 0.1% Tween 20 (PBS‐T) and re‐blocked for 10 min with SuperG block (Grace Biolabs) at room temperature with gentle agitation. Slides were washed with PBS‐T 3 × 5 min, clamped into a ProPlate 64‐well chamber (Grace Biolabs), and 50 μl of detection Ab was added (diluted at 1:500 from stock in PBS‐T supplemented with 5% BSA and 10% SuperG block). Slides were incubated for 30 min at room temperature, antibodies and clamp were removed, and slides were washed 3× in PBS‐T. Slides were blocked with SuperG for 10 min, washed 3× in PBS‐T, and then incubated with streptavidin IR800CW dye (Cell Signalling Technologies) (1:500 in PBS‐T, 5%BSA, 10% SuperG block) for 30 min in the dark with gentle agitation. Slides were washed with PBS‐T followed by 3 washes in PBS. Slides were dried by spinning at 200 g for 5 min and imaged using an Innopsys 710AL slide scanner. Images were quantified using Mapix software (Innopsys), and median of replicate spots was calculated. Median secreted protein levels were corrected for control (PS‐1 CM). Next, we determined the fold change between groups (Capan2 and AsPC‐1 versus MIA PaCa‐2 and PANC‐1) to identify differentially secreted proteins. Proteins were eligible if they were consistent in secretion levels in the cell lines per group, if they were between 30 and 100 kDa in size to reflect cytokines, and if their gene expression correlated with their protein expression.
Gene expression analysis
Gene expression of COL1A1 through ‐11A1, TGFB1‐3, and PDGFA‐D in PDAC cell lines was determined in two publicly available datasets: Broad Institute (GSE36133) 24 and Maupin et al (GSE21654) 23. Gene expression of cyto‐ and chemokines as assessed in the FPPA was determined in four publicly available datasets: Broad Institute (GSE36133) 24, Maupin et al (GSE21654) 23, Wagner et al (GSE8332) 30, and Wappett (GSE57083) using the web‐based genomics platform R2 (R2: Genomics Analysis and Visualization Platform, http://r2.amc.nl). The average expression of genes corresponding to proteins was determined per cell line, across databases. Janky et al (GSE62165) 18 was used to determine gene expression of CSF‐1, CCL5, COL1A1, or ACTA2 in PDAC patient samples. As CSF‐1 is a well‐known macrophage‐related protein, sub‐groups of low macrophage content and high macrophage content were identified in which median of CD68 gene expression levels was used as cut‐off.
Western blot
PS‐1 cells were starved and subsequently stimulated with 500 ng/ml rh‐CSF‐1 for 0, 5, 10, 15, 30, and 840 (overnight) minutes. Sample were washed with cold PBS and directly lysed in the plate using 2× Laemmli sample buffer (Bio‐Rad) with 10% β‐mercaptoethanol and 1% protease/phosphatase inhibitor cocktail (Cell Signaling). Samples were heated for 5 min at 95°C. Protein levels were determined with protein quantification assay which was compatible with Laemmli buffer (Macherey‐Nagel, Duren, Germany). Samples were run on 4–20% polyacrylamide precast SDS–PAGE gels (Bio‐Rad) and transferred to PVDF membranes. Samples were blocked with 5% BSA (Lonza) in PBS with 0.1% Tween‐20 (TBS‐T) and incubated overnight at 4°C with primary antibodies: phospho‐p44/42 MAPK (Erk1/2) (9101, Cell Signaling), phospho‐AKT (4060/D9E, Cell Signaling), phospho‐p70 S6 Kinase (Thr389) (2211S; Cell Signaling), phospho‐Src Family (6943; Cell Signaling), phospho‐STAT3 (9131; Cell Signaling), and α‐Tubulin (sc‐23948; Santa Cruz). All used 1:1,000. Goat anti‐rabbit horseradish peroxidase (HRP)‐conjugated secondary antibody (Cell Signaling, #7074) was used at 1:5,000 and incubated for 2 h at room temperature. Proteins were imaged using a FujiFilm LAS 4000 imager, using Pierce ECL Western Blotting Substrate (Pierce, Thermo Scientific).
Lentiviral CSF1 silencing
The following hairpins against hCSF‐1 in pLKO.1‐puro were obtained from the Sigma Mission library TRC1.5: shCSF1 #560 (position CSF1 gene: TRCN0000058562:1598‐1619(21), sequence: CCTTTGACTGACACAGGCCAT), shCSF1 #561 (position CSF1 gene: TRCN0000058559:360‐381(21), sequence: CGTGCCAAATTACATTTGAGT), and shCSF1 #562 (position CSF1 gene: TRCN0000058561:576‐597(21), sequence: GCGTCCGAACTTTCTATGAGA). A non‐targeting shRNA was used as a control (pLKO1 shc002). Lentivirus was produced by transfecting HEK293T cells by use of helper plasmids pMD2.G, pMDLg/pRRE and pRSV‐Rev, using Lipofectamine 2000 (Thermo Fisher). Supernatant was harvested at 24 h and 48 h after transfection and filtered through a 0.45‐μm filter (Millipore, Germany). 60% confluent PANC‐1 cells were transduced with the harvested virus and 8 μg/ml polybrene (Sigma) overnight. Cells were selected for stable shRNA expression with 2 μg/ml puromycin (Sigma) until untransduced control cells were dead. Subsequently, RNA was isolated and knockdown of CSF1 was analyzed in these samples with qPCR.
CSF1 overexpression
Overexpression of hCSF‐1 was accomplished by cloning human CSF1 (transcript variant 1, NM_000757.5, OHu11756) into pLeGO‐IV2 (#27344; Addgene, Cambridge, MA 60), provided by Genscript. Lentivirus production was performed as stated above. 60% confluent Capan‐2 cells were transduced with the harvested virus in the presence of 8 μg/ml polybrene (Sigma) overnight. After 7 days, cells were selected by sorting on a BD FACSAria for Venus expression (GFP).
Statistical analyses
Two‐sided unpaired t‐test was used to determine significance. A P‐value of < 0.05 was considered statistically significant. Error bars in bar graphs indicate the standard deviation. The in silico and IHC correlations were determined using linear regression analysis. All statistical analyses were performed using GraphPad Prism 7.03.
Author contributions
Conception and design of the study: AS, MGM, APZ, RK, MFB, and HWML. Generation, collection, assembly, analysis and/or interpretation of data: AS, MGM, APZ, RK, BS, SGG, HMK, CW, JHJ, CT, MGB, ORB, MJV, JV, FD, GT, JWW, JPM, HWML, and MFB. Drafting or revision of the manuscript: AS, MGM, APZ, CT, MFB, and HWML. Approval of the final version of the manuscript: AS, MGM, APZ, RK, BS, SGG, HMK, CW, JHJ, CT, MGB, ORB, MJV, JV, FD, GT, JWW, JPM, HWML, and MFB.
Conflict of interest
HWML has acted as a consultant for BMS, Eli Lilly and Company, MSD, Nordic Pharma Group/Taiho, and Servier, and has received unrestricted research grants from Amgen, Bayer Schering Pharma AG, BMS, Celgene, Eli Lilly and Company, GlaxoSmithKline Pharmaceuticals, MSD, Nordic Pharma Group, Philips, and Roche Pharmaceuticals. MFB has received research funding from Celgene and acted as a consultant to Servier. JWW has received research funding from Celgene and Novartis. HMK has received research funding from Celgene. MGHB has received research funding form Ethicon, Medtronic, and Mylan. None of these companies were involved in the design, conduct, or analysis of this study or drafting of the manuscript and decision to publish.
Supporting information
Expanded View Figures PDF
Table EV1
Review Process File
Source Data for Figure 4
Acknowledgements
We want to thank Tom van Leusden and Helene Damhofer for their help with establishment of the primary PDAC cells and xenografts. We want to thank Prabhu Arumugam and Veronique Veenstra for help with organotypic co‐cultures. We want to thank Tessa Le Large for providing us with culture medium of various PDAC cell lines. We want to thank Solange Lopes Cardozo for help with setting up the patient validation cohort and cutting paraffin sections. This work was supported by KWF Dutch Cancer Society project grants UVA 2013‐5932, UVA 2012‐5607, and a Rene Vogels travel stipend.
EMBO Reports (2020) 21: e48780
See also: https://doi.org/10.15252/embr.202050468 (May 2020)
References
- 1. Ryan DP, Hong TS, Bardeesy N (2014) Pancreatic adenocarcinoma. N Engl J Med 371: 2140 [DOI] [PubMed] [Google Scholar]
- 2. Ren B, Cui M, Yang G, Wang H, Feng M, You L, Zhao Y (2018) Tumor microenvironment participates in metastasis of pancreatic cancer. Mol Cancer 17: 108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bijlsma MF, van Laarhoven HWM (2015) The conflicting roles of tumor stroma in pancreatic cancer and their contribution to the failure of clinical trials: a systematic review and critical appraisal. Cancer Metastasis Rev 34: 97–114 [DOI] [PubMed] [Google Scholar]
- 4. Lafaro KJ, Melstrom LG (2019) The paradoxical web of pancreatic cancer tumor microenvironment. Am J Pathol 189: 44–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Öhlund D, Elyada E, Tuveson D (2014) Fibroblast heterogeneity in the cancer wound. J Exp Med 211: 1503–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McCarroll JA, Naim S, Sharbeen G, Russia N, Lee J, Kavallaris M, Goldstein D, Phillips PA (2014) Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Front Physiol 5: 141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D et al (2009) Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324: 1457–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Erkan M, Reiser‐Erkan C, Michalski CW, Deucker S, Sauliunaite D, Streit S, Esposito I, Friess H, Kleeff J (2009) Cancer‐stellate cell interactions perpetuate the hypoxia‐fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia 11: 497–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kikuta K, Masamune A, Watanabe T, Ariga H, Itoh H, Hamada S, Satoh K, Egawa S, Unno M, Shimosegawa T (2010) Pancreatic stellate cells promote epithelial‐mesenchymal transition in pancreatic cancer cells. Biochem Biophys Res Commun 403: 380–384 [DOI] [PubMed] [Google Scholar]
- 10. Gaggioli C, Hooper S, Hidalgo‐Carcedo C, Grosse R, Marshall JF, Harrington K, Sahai E (2007) Fibroblast‐led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol 9: 1392–1400 [DOI] [PubMed] [Google Scholar]
- 11. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW et al (2014) Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25: 735–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Özdemir BC, Pentcheva‐Hoang T, Carstens JL, Zheng X, Wu C‐C, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV et al (2014) Depletion of carcinoma‐associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25: 719–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Neesse A, Algül H, Tuveson DA, Gress TM (2015) Stromal biology and therapy in pancreatic cancer: a changing paradigm. Gut 64: 1476–1484 [DOI] [PubMed] [Google Scholar]
- 14. Öhlund D, Handly‐Santana A, Biffi G, Elyada E, Almeida AS, Ponz‐Sarvise M, Corbo V, Oni TE, Hearn SA, Lee EJ et al (2017) Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 214: 579–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, Preall J, Tuveson DA (2019) IL1‐induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov 9: 282–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bailey P, Chang DK, Nones K, Johns AL, Patch A‐M, Gingras M‐C, Miller DK, Christ AN, Bruxner TJC, Quinn MC et al (2016) Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531: 47–52 [DOI] [PubMed] [Google Scholar]
- 17. Raphael BJ, Hruban RH, Aguirre AJ, Moffitt RA, Yeh JJ, Stewart C, Robertson AG, Cherniack AD, Gupta M, Getz G et al (2017) Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 32: 185–203.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Janky R, Binda MM, Allemeersch J, Van den broeck A, Govaere O, Swinnen JV, Roskams T, Aerts S, Topal B (2016) Prognostic relevance of molecular subtypes and master regulators in pancreatic ductal adenocarcinoma. BMC Cancer 16: 632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SGH, Hoadley KA, Rashid NU, Williams LA, Eaton SC, Chung AH et al (2015) Virtual microdissection identifies distinct tumor‐ and stroma‐specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet 47: 1168–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L et al (2011) Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med 17: 500–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Puleo F, Nicolle R, Blum Y, Cros J, Marisa L, Demetter P, Quertinmont E, Svrcek M, Elarouci N, Iovanna J et al (2018) Stratification of pancreatic ductal adenocarcinomas based on tumor and microenvironment features. Gastroenterology 155: 1999–2013.e3 [DOI] [PubMed] [Google Scholar]
- 22. Neuzillet C, Tijeras‐Raballand A, Ragulan C, Cros J, Patil Y, Martinet M, Erkan M, Kleeff J, Wilson J, Apte M et al (2019) Inter‐ and intra‐tumoural heterogeneity in cancer‐associated fibroblasts of human pancreatic ductal adenocarcinoma. J Pathol 248: 51–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maupin KA, Sinha A, Eugster E, Miller J, Ross J, Paulino V, Keshamouni VG, Tran N, Berens M, Webb C et al (2010) Glycogene expression alterations associated with pancreatic cancer epithelial‐mesenchymal transition in complementary model systems. PLoS One 5: e13002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D et al (2012) The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483: 603–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Froeling FEM, Mirza TA, Feakins RM, Seedhar A, Elia G, Hart IR, Kocher HM (2009) Organotypic culture model of pancreatic cancer demonstrates that stromal cells modulate E‐cadherin, beta‐catenin, and Ezrin expression in tumor cells. Am J Pathol 175: 636–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Coetzee A, Grose R, Kocher H (2019) Pancreatic cancer organotypic models. In Current Topics in Microbiology and Immunology. Berlin, Heidelberg: Springer; [DOI] [PubMed] [Google Scholar]
- 27. Damhofer H, Ebbing EA, Steins A, Welling L, Tol JA, Krishnadath KK, van Leusden T, van de Vijver MJ, Besselink MG, Busch OR et al (2015) Establishment of patient‐derived xenograft models and cell lines for malignancies of the upper gastrointestinal tract. J Transl Med 13: 115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dijk F, Veenstra VL, Soer EC, Dings MPG, Zhao L, Halfwerk JB, Hooijer GK, Damhofer H, Marzano M, Steins A et al (2019) Unsupervised class discovery in pancreatic ductal adenocarcinoma reveals cell‐intrinsic mesenchymal features and high concordance between existing classification systems. Sci Rep 10: 337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Froeling FEM, Feig C, Chelala C, Dobson R, Mein CE, Tuveson DA, Clevers H, Hart IR, Kocher HM (2011) Retinoic acid‐induced pancreatic stellate cell quiescence reduces paracrine wnt–β‐catenin signaling to slow tumor progression. Gastroenterology 141: 1486–1497.e14 [DOI] [PubMed] [Google Scholar]
- 30. Wagner KW, Punnoose EA, Januario T, Lawrence DA, Pitti RM, Lancaster K, Lee D, von Goetz M, Yee SF, Totpal K et al (2007) Death‐receptor O‐glycosylation controls tumor‐cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med 13: 1070–1077 [DOI] [PubMed] [Google Scholar]
- 31. Lankadasari MB, Mukhopadhyay P, Mohammed S, Harikumar KB (2019) TAMing pancreatic cancer: combat with a double edged sword. Mol Cancer 18: 48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marks DC, Csar XF, Wilson NJ, Novak U, Ward AC, Kanagasundarum V, Hoffmann BW, Hamilton JA (1999) Expression of a Y559F mutant CSF‐1 receptor in M1 myeloid cells: a role for Src kinases in CSF‐1 receptor‐mediated differentiation. Mol Cell Biol Res Commun 1: 144–152 [DOI] [PubMed] [Google Scholar]
- 33. Dey A, She H, Kim L, Boruch A, Guris DL, Carlberg K, Sebti SM, Woodley DT, Imamoto A, Li W (2000) Colony‐stimulating factor‐1 receptor utilizes multiple signaling pathways to induce cyclin D2 expression. Mol Biol Cell 11: 3835–3848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stanley ER, Chitu V (2014) CSF‐1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol 6: a021857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morandi A, Barbetti V, Riverso M, Dello Sbarba P, Rovida E (2011) The colony‐stimulating factor‐1 (CSF‐1) receptor sustains ERK1/2 activation and proliferation in breast cancer cell lines. PLoS One 6: e27450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thomas D, Radhakrishnan P (2019) Tumor‐stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol Cancer 18: 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stark GR, Darnell JE Jr (2012) The JAK‐STAT pathway at twenty. Immunity 36: 503–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sumita N, Bito T, Nakajima K, Nishigori C (2006) Stat3 activation is required for cell proliferation and tumorigenesis but not for cell viability in cutaneous squamous cell carcinoma cell lines. Exp Dermatol 15: 291–299 [DOI] [PubMed] [Google Scholar]
- 39. Terman JR, Kashina A (2013) Post‐translational modification and regulation of actin. Curr Opin Cell Biol 25: 30–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hirshman CA, Zhu D, Pertel T, Panettieri RA, Emala CW (2005) Isoproterenol induces actin depolymerization in human airway smooth muscle cells via activation of an Src kinase and GS . Am J Physiol Cell Mol Physiol 288: L924–L931 [DOI] [PubMed] [Google Scholar]
- 41. Wrobel CN, Debnath J, Lin E, Beausoleil S, Roussel MF, Brugge JS (2004) Autocrine CSF‐1R activation promotes Src‐dependent disruption of mammary epithelial architecture. J Cell Biol 165: 263–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, Stylianopoulos T, Mousa AS, Han X, Adstamongkonkul P et al (2013) Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun 4: 2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Olivares O, Mayers JR, Gouirand V, Torrence ME, Gicquel T, Borge L, Lac S, Roques J, Lavaut M‐N, Berthezène P et al (2017) Collagen‐derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat Commun 8: 16031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hamada S, Masamune A (2018) Elucidating the link between collagen and pancreatic cancer: what's next? Expert Rev Gastroenterol Hepatol 12: 315–317 [DOI] [PubMed] [Google Scholar]
- 45. Han H, Von Hoff D. Targeting Stromal Collagen in Pancreatic Cancer. https://grantome.com/grant/NIH/R01-CA169281-01A1
- 46. van Mackelenbergh MG, Stroes CI, Spijker R, van Eijck CHJ, Wilmink JW, Bijlsma MF, van Laarhoven HWM (2019) Clinical trials targeting the stroma in pancreatic cancer: a systematic review and meta‐analysis. Cancers (Basel) 11: 588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hingorani SR, Zheng L, Bullock AJ, Seery TE, Harris WP, Sigal DS, Braiteh F, Ritch PS, Zalupski MM, Bahary N et al (2018) HALO 202: randomized phase II study of PEGPH20 plus Nab‐Paclitaxel/Gemcitabine versus Nab‐Paclitaxel/gemcitabine in patients with untreated, metastatic pancreatic ductal adenocarcinoma. J Clin Oncol 36: 359–366 [DOI] [PubMed] [Google Scholar]
- 48. Ramanathan RK, McDonough SL, Philip PA, Hingorani SR, Lacy J, Kortmansky JS, Thumar J, Chiorean EG, Shields AF, Behl D et al (2019) Phase IB/II randomized study of FOLFIRINOX plus pegylated recombinant human hyaluronidase versus FOLFIRINOX alone in patients with metastatic pancreatic adenocarcinoma: SWOG S1313. J Clin Oncol 37: 1062–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Froeling FEM, Kocher HM (2015) Homeostatic restoration of desmoplastic stroma rather than its ablation slows pancreatic cancer progression. Gastroenterology 148: 849–850 [DOI] [PubMed] [Google Scholar]
- 50. Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, Collisson EA, Connor F, Van Dyke T, Kozlov S et al (2014) Vitamin D receptor‐mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 159: 80–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang W‐Q, Liu L, Xu H‐X, Luo G‐P, Chen T, Wu C‐T, Xu Y‐F, Xu J, Liu C, Zhang B et al (2013) Intratumoral α‐SMA enhances the prognostic potency of CD34 associated with maintenance of microvessel integrity in hepatocellular carcinoma and pancreatic cancer. PLoS One 8: e71189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li NF, Kocher HM, Salako MA, Obermueller E, Sandle J, Balkwill F (2009) A novel function of colony‐stimulating factor 1 receptor in hTERT immortalization of human epithelial cells. Oncogene 28: 773–780 [DOI] [PubMed] [Google Scholar]
- 53. Rettenmier CW, Roussel MF, Ashmun RA, Ralph P, Price K, Sherr CJ (1987) Synthesis of membrane‐bound colony‐stimulating factor 1 (CSF‐1) and downmodulation of CSF‐1 receptors in NIH 3T3 cells transformed by cotransfection of the human CSF‐1 and c‐fms (CSF‐1 receptor) genes. Mol Cell Biol 7: 2378–2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Manos MM (1988) Expression and processing of a recombinant human macrophage colony‐stimulating factor in mouse cells. Mol Cell Biol 8: 5035–5039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pixley FJ, Stanley ER (2004) CSF‐1 regulation of the wandering macrophage: complexity in action. Trends Cell Biol 14: 628–638 [DOI] [PubMed] [Google Scholar]
- 56. Candido JB, Morton JP, Bailey P, Campbell AD, Karim SA, Jamieson T, Lapienyte L, Gopinathan A, Clark W, McGhee EJ et al (2018) CSF1R+ macrophages sustain pancreatic tumor growth through T cell suppression and maintenance of key gene programs that define the squamous subtype. Cell Rep 23: 1448–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mroczko B, Szmitkowski M, Wereszczynska‐Siemiatkowska U, GrazynaJurkowska G (2004) Stem cell factor and macrophage‐colony stimulating factor in patients with pancreatic cancer. Clin Chem Lab Med 42: 256–260 [DOI] [PubMed] [Google Scholar]
- 58. Vasiliades G, Kopanakis N, Vasiloglou M, Zografos G, Margaris H, Masselou K, Kokosi E, Liakakos T (2012) Role of the hematopoietic cytokines SCF, IL‐3, GM‐CSF and M‐CSF in the diagnosis of pancreatic and ampullary cancer. Int J Biol Markers 27: 186–194 [DOI] [PubMed] [Google Scholar]
- 59. Van Tienhoven G, Versteijne E, Suker M, Groothuis KBC, Busch OR, Bonsing BA, de Hingh IHJT, Festen S, Patijn GA, de Vos‐Geelen J et al (2018) Preoperative chemoradiotherapy versus immediate surgery for resectable and borderline resectable pancreatic cancer (PREOPANC‐1): a randomized, controlled, multicenter phase III trial. J Clin Oncol 36: LBA4002 [Google Scholar]
- 60. Weber K, Mock U, Petrowitz B, Bartsch U, Fehse B (2010) Lentiviral gene ontology (LeGO) vectors equipped with novel drug‐selectable fluorescent proteins: new building blocks for cell marking and multi‐gene analysis. Gene Ther 17: 511–520 [DOI] [PubMed] [Google Scholar]
- 61. van der Heijden M, Miedema DM, Waclaw B, Veenstra VL, Lecca MC, Nijman LE, van Dijk E, van Neerven SM, Lodestijn SC, Lenos KJ et al (2019) Spatiotemporal regulation of clonogenicity in colorectal cancer xenografts. Proc Natl Acad Sci USA 116: 6140–6145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ebbing EA, van der Zalm AP, Steins A, Creemers A, Hermsen S, Rentenaar R, Klein M, Waasdorp C, Hooijer GKJ, Meijer SL et al (2019) Stromal‐derived interleukin 6 drives epithelial‐to‐mesenchymal transition and therapy resistance in esophageal adenocarcinoma. Proc Natl Acad Sci USA 116: 2237–2242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kadaba R, Birke H, Wang J, Hooper S, Andl CD, Di Maggio F, Soylu E, Ghallab M, Bor D, Froeling FE et al (2013) Imbalance of desmoplastic stromal cell numbers drives aggressive cancer processes. J Pathol 230: 107–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Steins A, Ebbing EA, Creemers A, van der Zalm AP, Jibodh RA, Waasdorp C, Meijer SL, van Delden OM, Krishnadath KK, Hulshof MCCM et al (2019) Chemoradiation induces epithelial‐to‐mesenchymal transition in esophageal adenocarcinoma. Int J Cancer 145: 2792–2803 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Review Process File
Source Data for Figure 4