

Abstract
The tumor suppressor Smad4, a key mediator of the TGF‐β/BMP pathways, is essential for development and tissue homeostasis. Phosphorylation of Smad4 in its linker region catalyzed by the mitogen‐activated protein kinase (MAPK) plays a pivotal role in regulating its transcriptional activity and stability. In contrast, roles of Smad4 dephosphorylation as a control mechanism of TGF‐β/BMP signaling and the phosphatases responsible for its dephosphorylation remain so far elusive. Here, we identify Wip1 as a Smad4 phosphatase. Wip1 selectively binds and dephosphorylates Smad4 at Thr277, a key MAPK phosphorylation site, thereby regulating its nuclear accumulation and half‐life. In Xenopus embryos, Wip1 limits mesoderm formation and favors neural induction by inhibiting TGF‐β/BMP signals. Wip1 restrains TGF‐β‐induced growth arrest, migration, and invasion in human cells and enhances the tumorigenicity of cancer cells by repressing the antimitogenic activity of Smad4. We propose that Wip1‐dependent dephosphorylation of Smad4 is critical for the regulation of TGF‐β signaling.
Keywords: phosphatase, Smad4, TGF‐β, Wip1, Xenopus
Subject Categories: Development & Differentiation; Post-translational Modifications, Proteolysis & Proteomics; Signal Transduction
Phosphorylation of Smad4, a central mediator of TGF‐β signaling, plays a pivotal role in controlling its activity and stability. Here, Wip1‐mediated dephosphorylation of Smad4 in its linker region is identified as a novel regulatory mechanism of TGF‐β signaling.

Introduction
The transforming growth factor‐β (TGF‐β) superfamily of secreted signaling molecules regulates a broad range of biological processes in metazoans, including embryonic development, inflammation, immune regulation, and cancer 1, 2. TGF‐β family members signal by binding to a heteromeric receptor complex consisting of type I and type II serine/threonine kinase receptors. Upon binding TGF‐β ligands, type II receptors phosphorylate and activate type I receptors, which in turn phosphorylate Smad transcription factors at their distal C‐terminus. These receptor‐regulated Smads (R‐Smads) include Smad1, Smad5, and Smad8 in the BMP pathway and Smad2 and Smad3 in the TGF‐β/activin/nodal pathway. This C‐terminal phosphorylation of R‐Smads requires arginine methylation of Smad6 or Smad7 by PRMT1, which leads to the dissociation of these inhibitory Smads from the type I receptor, thereby allowing derepression of R‐Smad phosphorylation in response to TGF‐β or BMP 3, 4. Phosphorylated R‐Smads associate with the common mediator Smad4 and then translocate into the nucleus, where R‐Smad‐Smad4 complexes bind other DNA‐binding factors for target gene selection and transcriptional regulation.
The phosphorylation status of Smads, determined by dynamic interplay between protein kinases and phosphatases, plays key roles in controlling their functions 5. Receptor‐mediated phosphorylation of R‐Smads at the C‐terminal SSXS motif is a prerequisite for their transcriptional activities in the nucleus. Protein phosphatase PPM1A dephosphorylates the C‐terminal tail motif of the TGF‐β‐specific Smad2 and Smad3, as well as of the BMP‐specific Smad1 and Smad5, limiting the duration of their activities and thereby attenuating TGF‐β‐ and BMP‐regulated gene responses 6, 7. Besides, the FGF/MAPK pathway phosphorylates Smad1, Smad2, and Smad3 in the linker region between the MH1 and MH2 domains. This linker phosphorylation prevents nuclear accumulation of R‐Smads and Smad‐dependent transcriptional responses induced by agonists such as TGF‐β and BMP 8, 9. In contrast, agonist‐induced linker phosphorylation of R‐Smads, mediated by cyclin‐dependent kinases CDK8 and CDK9 in the nucleus, promotes Smad transcriptional activity, concurrently marking the Smads for proteasome‐dependent degradation 10.
Unlike R‐Smads, Smad4 undergoes no C‐terminal tail phosphorylation, though it is phosphorylated in the linker region. Extracellular signal‐regulated kinase (ERK) has been shown to phosphorylate Smad4 in the linker at Thr277 (equivalent to mouse Thr276) 11, 12. This phosphorylation contributes to TGF‐β‐induced nuclear accumulation and peak transcriptional activity of Smad4 by facilitating the interaction between Smad4 and FAM/USP9x, a deubiquitinating enzyme 13. FAM competitively prevents ectodermin/TIF1γ, a monoubiquitin ligase from binding and monoubiquitinating Smad4, maintaining Smad4 nuclear retention, and stabilizing the R‐Smad‐Smad4 complex in the nucleus. Thus, the phosphorylation status of Smad4 linker region appears to regulate the monoubiquitination/deubiquitination cycle of Smad4 required for TGF‐β signaling. Thr277 phosphorylation also functions to prime inhibitory GSK3‐mediated phosphorylations at the threonine residues near the MAPK/ERK sites in the linker, which leads to the E3‐ligase β‐TrCP‐mediated polyubiquitination and subsequent degradation of Smad4 12. A recent study has revealed that PRMT1 methylation of an arginine residue within the GSK3 site in Smad4 is essential for its phosphorylation by GSK3 14. In TGF‐β signaling, transcriptional dynamics has been shown to be associated with the timing of Smad4 nuclear localization rather than with the activities of the R‐Smads 15. After entering the nucleus with the R‐Smads, Smad4 mediates temporal adaptation of the signaling pathways that terminates transcription. Given the importance of Smad4 phosphorylation at Thr277 for its nuclear retention, dephosphorylation as well as phosphorylation of Smad4 at this residue might play a crucial role in control of transcriptional dynamics of TGF‐β signaling. However, phosphatase(s) involved in dephosphorylation of Smad4 at the MAPK/ERK site remains unknown.
Here, we identified wild‐type p53‐inducible protein 1 (Wip1) as a phosphatase responsible for the dephosphorylation of Smad4 at Thr277. Wip1, encoded by the gene PPM1D, is a member of the PP2C family of Ser/Thr protein phosphatases and a major homeostatic regulator of the DNA damage response 16, 17. Wip1 has been implicated in oncogenic transformation, but Wip1‐null mice display tumor‐resistant phenotypes 18, 19. We found that gain of function and loss of function of Wip1 inhibit and enhance TGF‐β‐induced gene responses, respectively. Wip1 interacts with and dephosphorylates Smad4 at the MAPK site in the linker region, thereby controlling the nuclear accumulation and stability of Smad4. This function of Wip1 is critical for the control of germ‐layer specification in early development and of proliferation, migration, and tumorigenicity of cancer cells.
Results
Gain‐of‐Wip1 function prevents TGF‐β‐induced gene responses
Wip1 regulates several signaling networks in the context of different conditions, such as oncogenesis, DNA damage response, and aging 20. This critical role of Wip1 as a regulator of homeostasis prompted us to unravel its biological activities in vertebrate early development. We employed stem cell‐like animal cap tissue of Xenopus early embryo and analyzed the effects of increased levels of Wip1 on the expression of target genes triggered by key signaling ligands. Interestingly, co‐expression of wild‐type (wt) Wip1 repressed the expression of the dorsal mesodermal marker Goosecoid (Gsc), the pan‐mesodermal marker Xbra, and the ventral mesodermal marker XWnt8 induced ectopically in Xenopus Nodal‐1 (Xnr1)‐stimulated animal caps (Fig 1A). BMP4 induction of the expression of ventral target genes, such as Xbra, Xhox3, and Xmsx1, was also inhibited by co‐injection of wt Wip1 (Fig 1B). Besides, overexpression of wt Wip1 promoted the expression of the neural markers NCAM and Otx2 at the expense of the epidermal marker E. keratin (Fig 1C), indicative of Wip1 inhibition of BMP signaling in animal caps. These effects of Wip1 on the gene responses required its phosphatase activity as evidenced by the result that a catalytically inactive Wip1 mutant, Wip1 (D277A), had no effect on the expression of markers induced by Xnr1 or BMP4 (Fig 1A–C). We also analyzed the effects of overexpression of Wip1 on the activity of TGF‐β signal‐responsive promoters in cultured cells. Reporter assays revealed that wt Wip1, but not Wip1 (D277A), could inhibit significantly the activity of activin/nodal response element (ARE3) and of BMP response element (BRE), which was induced by ALK4* (active form of activin/nodal receptor) and BMPR1* (active form of BMP receptor), respectively (Fig 1D and E). Together, these results indicate the negative role of Wip1 as a phosphatase in the regulation of the gene responses induced by TGF‐β signals.
Figure 1. Overexpression of Wip1 inhibits TGF‐β signaling.
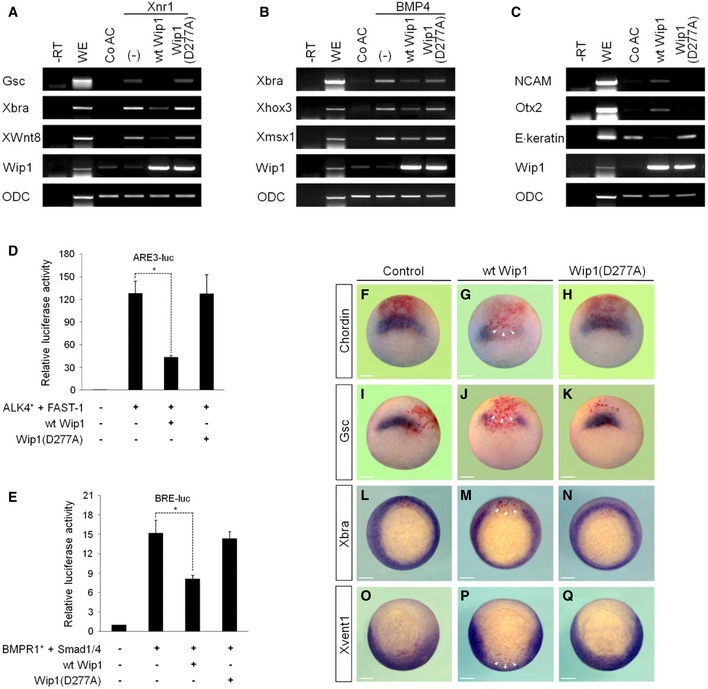
-
A–CWip1 represses the gene responses induced by nodal or BMP signaling in animal caps as analyzed by RT–PCR. ODC serves as a loading control. –RT, control in the absence of reverse transcriptase. WE, whole embryo. Co AC, uninjected control animal caps. (–), no injection of wild‐type (wt) Wip1 or Wip1 (D277A) mRNA. The amount of mRNA injected: wt Wip1 (1 ng), Wip1 (D277A) (1 ng), Xnr1 (50 pg), and BMP4 (100 pg).
-
D, Ewt Wip1, but not Wip1 (D277A), down‐regulates the activity of activin‐ or BMP‐responsive promoters in HEK293T cells. FAST1, Smad1, and Smad4 were co‐transfected as indicated for efficient activation of the promoters. Data are expressed as the mean ± SEM (n = 3 biological replicates). *P < 0.05 by unpaired Student's t‐test.
-
F–QDorsal (F‐N) or ventral (O‐Q) injection of wt Wip1 abrogates the in vivo expression of mesodermal markers at stage 10.25 as analyzed by in situ hybridization. Embryos are shown in vegetal views with dorsal to the top. Arrowheads denote the absence of expression of mesodermal markers in the injected region of embryos. Control, an embryo injected with LacZ only. The amount of mRNA injected: wt Wip1 (1 ng), Wip1 (D277A) (1 ng), and LacZ (100 pg). Scale bar, 150 μm.
Source data are available online for this figure.
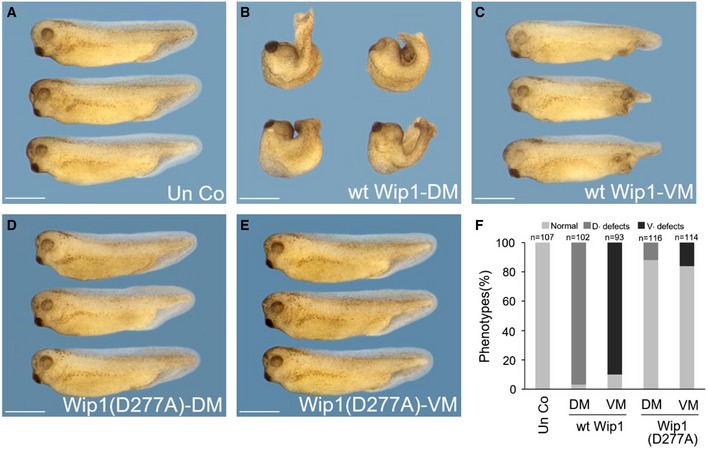
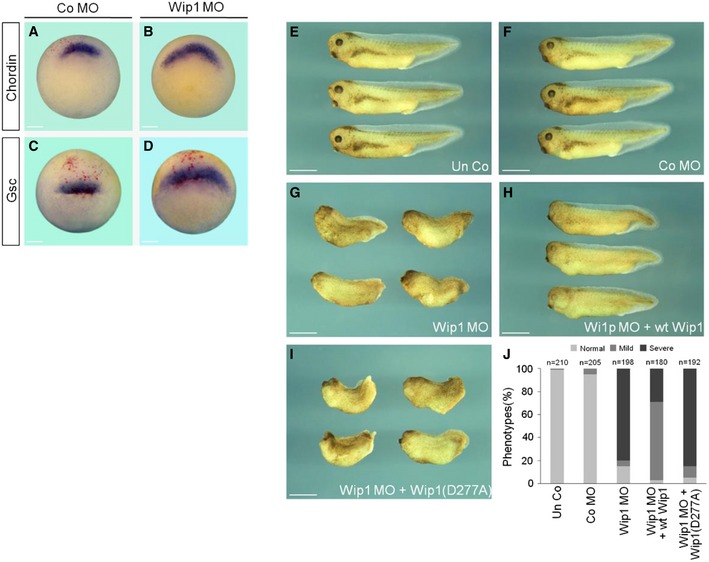
These in vitro results led us to speculate that the gain‐of‐Wip1 function would interfere with mesoderm formation in vivo in Xenopus embryos, which is regulated by activin/nodal as well as BMP signaling 21. As expected, forced expression of wt Wip1, but not Wip1 (D277A), impeded strongly the expression of activin/nodal‐responsive genes, including Gsc (86%, n = 35 for wt Wip1; 4%, n = 25 for Wip1(D277A)), Chordin (83%, n = 30 for wt Wip1; 4%, n = 25 for Wip1(D277A)), and Xbra (98%, n = 60 for wt Wip1; 2%, n = 53 for Wip1(D277A)), and of BMP4‐responsive gene Xvent1 (96%, n = 51 for wt Wip1; 2%, n = 43 for Wip1(D277A)), in the areas stained positively for a lineage tracer, LacZ, in gastrulae (Fig 1F–Q). In line with these inhibitory effects on the expression of early markers, overexpression of Wip1 had powerful long‐term phenotypic effects. Dorsal injection of wt Wip1 caused loss of eyes, microcephaly, and gastrulation defects, such as spina bifida and shortened and dorsally kinked body axis (Fig EV1B and F), indicative of a defective dorsal mesoderm formation, but Wip1 (D277A) had fewer phenotypic effects (Fig EV1D and F). Ventral injection of wt Wip1 generated defective and shortened tail structures, suggesting defects in ventral mesoderm formation, but not Wip1 (D277A) mutant (Fig EV1C, E and F). These data suggest that the increased phosphatase activity of Wip1 down‐regulates Xenopus mesoderm differentiation which depends on activin/nodal and/or BMP signals.
Figure EV1. Morphological phenotypes of embryos overexpressing Wip1 .
-
A–EFour‐cell stage embryos were injected in the dorsal or ventral marginal region of two blastomeres with wt Wip1 (1 ng) or Wip1 (D277A) (1 ng) mRNA and cultured to tadpole stages. Embryos are shown in lateral views with anterior to the left. Scale bar, 1 mm.
-
FQuantification of the phenotypes shown in (A–E). D. defects (dorsal defects) include shortened and kinked body axis, spina bifida, and gastrulation defects. V. defects (ventral defects) denote malformed posterior structures. DM, dorsal marginal zone; VM, ventral marginal zone.
Knockdown of Wip1 enhances TGF‐β signaling
To assess the developmental relevance of Wip1, we achieved its loss of function using two anti‐sense morpholino oligonucleotides (MO1 and MO2). These MOs were designed to target simultaneously to the sequences upstream of or around translation initiation sites of Wip1 mRNA, blocking its translation. MO1 and MO2 each, but not control (Co) MO, blocked the production of Wip1 protein without affecting actin level (Appendix Fig S1A and B), confirming the specificity and efficacy of both MOs. In further experiments, the two MOs were mixed at an equimolar ratio and injected (the mixed MOs will be designated as Wip1 MO hereafter). siRNA‐mediated knockdown of Wip1 was also carried out for its loss of function in cultured human cells. Two Wip1 siRNAs each, but not Co siRNA, silenced efficiently Wip1 expression in HEK293T cells (Appendix Fig S1C).
Prior to the loss‐of‐Wip1 function analysis in Xenopus embryo, we examined the spatio‐temporal expression pattern of its transcripts during early development. RT–PCR analysis revealed both maternal transcription and zygotic transcription of Wip1 during early embryogenesis (Appendix Fig S2A). Of note, its maternal messages exhibited markedly higher levels than its zygotic ones. Spatially, strong expression of Wip1 was detectable in the animal and marginal regions of embryo from the cleavage stages to the early gastrula stages (Appendix Fig S2B and C). At the tadpole stages, its specific expression was found in the eyes, branchial arches, otic vesicles, and pronephros (Appendix Fig S2D).
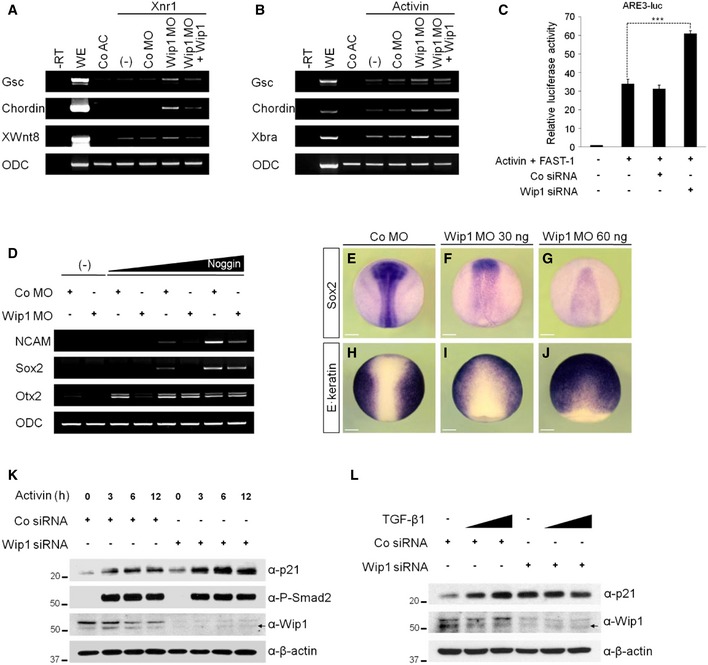
As overexpression of Wip1 down‐regulated TGF‐β signaling, we expected that its knockdown would enhance signaling responses triggered by TGF‐β ligands. Depletion of Wip1 augmented the ectopic expression of target genes, including Gsc, Chordin, XWnt8, and Xbra, induced by stimulation with Xnr1 or activin (Fig 2A and B). These enhancing effects of Wip1 MO were reversed by co‐expression of Wip1 mRNA immune to MO inhibition, supporting the specificity of Wip1 MO. siRNA‐mediated silencing of Wip1 also sensitized the response to activin in HEK293T cells, further up‐regulating the activity of the reporter whose expression is under the control of activin response element (ARE3) (Fig 2C). However, depletion of Wip1 had no effect on its activity in unstimulated cells (Appendix Fig S3).
Figure 2. Depletion of Wip1 up‐regulates TGF‐β signaling.
-
A, BActivin/nodal‐induced gene responses are enhanced further in the absence of Wip1 as analyzed by RT–PCR. Animal caps in (B) were treated with activin protein (5 ng/ml). (–), no injection of Co MO, Wip1 MO, and/or Wip1 mRNA. The dose of reagent injected: Xnr1 (5 pg), Co MO (60 ng), Wip1 MO (60 ng), and Wip1 (1 ng).
-
CKnockdown of Wip1 up‐regulates the activity of activin/nodal‐responsive promoter. HEK293T cells transfected with Co siRNA (50 nM) or Wip1 siRNA (50 nM) were stimulated with activin protein (10 ng/ml) for 10 h and then subjected to luciferase assays. Data are expressed as the mean ± SEM (n = 3 biological replicates). ***P < 0.001 by unpaired Student's t‐test.
-
D–JDepletion of Wip1 promotes epidermal differentiation at the expense of neural fate. (–), no injection of Noggin mRNA. Embryos in (E–J) are shown in dorsal views with anterior to the top. The amount of MO and mRNA injected: Co MO (60 ng), Wip1 MO (60 ng for D, G, and J; 30 ng for F and I), and Noggin (5, 10, 20 pg). Scale bar, 150 μm.
-
K, LActivin or TGF‐β induction of p21 is up‐regulated upon Wip1 knockdown. HEK293T cells were transfected with Co siRNA or Wip1 siRNA and 48 h after siRNA transfection were stimulated with activin (10 ng/ml) for 3–12 h as indicated or with increasing concentration of TGF‐β1 (10, 20 ng/ml) for 3 h. t = 0 in (K) is 48 h after siRNA transfection. P‐Smad2 serves as an indicator of active activin/nodal signaling. Arrows indicate non‐specific bands.
Source data are available online for this figure.
Since Wip1 favored neural differentiation by down‐regulating BMP signaling (Fig 1C), we checked whether Wip1 knockdown would conversely promote epidermal development at the expense of neural fate. Ectopic expressions of neural genes NCAM, Sox2, and Otx2 were triggered differentially in animal cap explants by the extracellular BMP antagonist, Noggin, in a dose‐dependent manner (Fig 2D). These inductions of the neural markers were down‐regulated by co‐injection of Wip1 MO, suggesting that knockdown of Wip1 leads to unrestrained intracellular BMP signaling, which may attenuate the cell response to the extracellular neural inducer. To substantiate this conclusion, we also examined the formation of neural and epidermal tissues in Wip1‐depleted whole embryo at the mid‐neurula stages. Wip1 morphants exhibited reduction or absence of neural tissue as evidenced by the decreased expression of neural marker Sox2 (95%, n = 40; Fig 2E–G). In contrast, knockdown of Wip1 caused expansion of epidermal marker E. keratin into the presumptive neural plate at the same stages (97%, n = 35; Fig 2H–J). As E. keratin is a target gene of BMP signaling, depletion of Wip1 appears to promote epidermal differentiation by enhancing the signaling activity of BMP pathway.
To test whether knockdown of Wip1 would also promote TGF‐β responses, we monitored the cellular levels of p21, a cyclin‐dependent kinase (CDK) inhibitor and a key mediator of TGF‐β‐mediated growth suppression 22. As shown in Fig 2K and L, silencing of Wip1 up‐regulated the basal levels as well as the induced levels of p21 as response to activin or TGF‐β1. Together, these results indicate that the loss‐of‐Wip1 function leads to the enhanced sensitivity to TGF‐β signals in mammalian cells as observed in Wip1‐depleted Xenopus embryonic cells.
Wip1 restrains TGF‐β‐induced cytostasis and cell migration
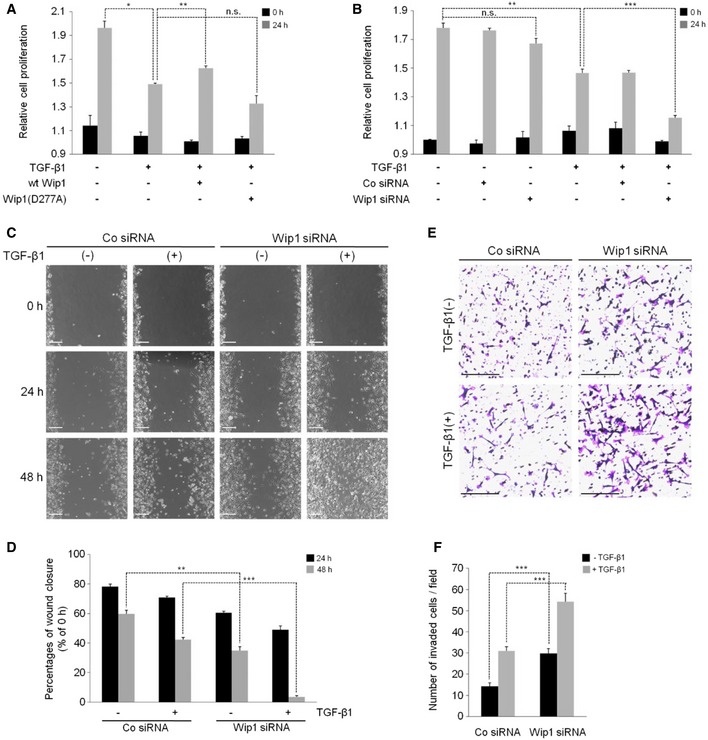
Given its negative effects on the gene responses induced by TGF‐β/activin/nodal and BMP signaling, Wip1 might limit TGF‐β‐induced biological effects, such as growth arrest and promotion of migratory/invasive behaviors of cancer cells 23. To test this hypothesis, we first conducted Cell Counting Kit‐8 (CCK‐8) assays to measure the proliferation of cells transfected with wt Wip1 or Wip1 (D277A) mutant. As shown in Fig 3A, forced expression of wt Wip1, but not Wip1 (D277A), prevented growth arrest of HEK293T cells induced by TGF‐β. Conversely, depletion of Wip1 had the opposite effect on TGF‐β‐induced cytostasis as evidenced by significantly enhanced antiproliferative effect of TGF‐β ligand in Wip1‐silenced cells (Fig 3B).
Figure 3. Silencing of Wip1 promotes the biological effects of TGF‐β.
-
A, BEffects of the gain of function or loss of function of Wip1 on TGF‐β‐induced growth arrest. HEK293T cells were transfected or not with wt Wip1, Wip1 (D277A), Co siRNA, or Wip1 siRNA as indicated and subjected to CCK‐8 assays before (0 h) and after stimulation with TGF‐β1 (40 ng/ml) (24 h). Data are presented as the mean ± SEM (n = 3 biological replicates). *P < 0.05, **P < 0.01, ***P < 0.001 by unpaired Student's t‐test. n.s., not significant.
-
C–FDepletion of Wip1 augments TGF‐β‐induced migration and invasion of MDA‐MB231 breast cancer cells. MDA‐MB231 cells transfected with Co siRNA or Wip1 siRNA were scratched with a 20‐μl pipette tip and then incubated in the absence or presence of TGF‐β1 for the indicated times (C, D) or subjected to transwell assay with or without TGF‐β1 for 20 h (E, F). Data in (D, F) were quantified from three biological replicates and are represented as mean ± SEM. **P < 0.01, ***P < 0.001 by unpaired Student's t‐test. Scale bar, 200 μm.
We next determined whether Wip1 has a repressive effect on TGF‐β‐induced invasion and migration of MDA‐MB231 breast cancer cells. Wip1 has been found to be amplified and overexpressed in various primary human tumor types including breast cancer 16. These metastatic cancer cells increase their motility as response to TGF‐β signaling, which can be quantified by in vitro transwell migration assays and wound healing assays. Depletion of Wip1 significantly augmented the migration and invasion of these breast cancer cells both in the absence and in the presence of TGF‐β signal (Fig 3C and E; quantification in Fig 3D and F). Collectively, these results indicate that Wip1 plays critical roles in controlling TGF‐β‐induced biological effects.
Wip1 regulates germ‐layer specification in Xenopus embryos
In Xenopus, mesoderm is induced and patterned in the marginal zone of embryo by a gradient of nodal signal emanating from its vegetal hemisphere 21. Given that Wip1 is expressed in the marginal region of embryo and has a regulatory role in activin/nodal signaling, it might be responsible for appropriate induction and patterning of mesoderm in Xenopus embryo. To test this, we examined the effects of Wip1 MO on the expression of mesodermal markers in the marginal zone of gastrula stage embryos. As shown in Fig 4A–D, depletion of Wip1 up‐regulated and expanded the expression of dorsal mesodermal markers, Gsc and Chordin, toward the lateral region of embryos (75%, n = 28 for Chordin; 46%, n = 28 for Gsc), compared with that in Co MO‐injected embryos. In addition, Wip1‐depleted embryos displayed several severe defects, including microcephaly, no eyes, shortened and kinked body axis, and exo‐gastrulation, along the antero‐posterior body axis (Fig 4E–G; quantification in 4J). Of note, these malformed phenotypes could be rescued efficiently by co‐expression of a MO‐insensitive wt Wip1 mRNA, but not by Wip1 (D277A) mutant mRNA (Fig 4H and I; quantification in 4J), corroborating the specific effects of Wip1 MO on the embryonic phenotypes. Those defects in Wip1 morphants are reminiscent of those in embryos exposed to enhanced levels of activin/nodal signal 24, 25. Therefore, together with the molecular effects, these phenotypes of Wip1 morphants suggest that knockdown of Wip1 expands mesoderm by up‐regulating activin/nodal signal, interfering with the correct patterning of whole embryo. As shown above, Wip1 is also critical for the decision between epidermal cell fate and neural cell fate during ectodermal patterning. Therefore, we conclude that Wip1 functions as a phosphatase to regulate germ‐layer specification in Xenopus early embryos.
Figure 4. Mesoderm expansion in Wip1‐depleted embryos.
-
A–DKnockdown of Wip1 expands the expression of dorsal mesodermal markers. Embryos injected dorsally with Co MO (40 ng) or Wip1 MO (40 ng) were subjected to in situ hybridization at stage 10.25. Embryos are shown in dorso‐vegetal views with dorsal to the top. Scale bar, 150 μm.
-
E–IMorphological phenotypes of Wip1 morphants. Embryos were injected radially in the marginal zone with Co MO (80 ng), Wip1 MO (80 ng), wt Wip1 (1 ng), and Wip1 (D277A) mRNA as indicated and cultured to tadpole stages. Embryos are shown in lateral views with anterior to the left. Un Co, uninjected control. Scale bar, 1 mm.
-
JQuantification of the phenotypes shown in (E–I). Severe defects include microcephaly, shortened and kinked body axis, and no eye. Mild defects indicate normal body axis with malformed eyes.
Wip1 physically associates with Smad4
While Wip1 is transcriptionally up‐regulated after DNA damage in a p53‐dependent manner, it can contribute to p53 deactivation directly or indirectly, thereby controlling cell cycle checkpoints 16. In addition, p53 regulates a subset of gene responses to TGF‐β/activin signal through the association with Smad complexes 26. Given these, it is possible that the inhibitory effects of Wip1 on TGF‐β signaling may be mediated by its down‐regulation of p53 activity. To test this possibility, we checked whether Wip1 could repress gene induction in response to p53 overexpression. Co‐expression of Wip1 did not inhibit the ectopic expression of mesodermal genes induced by p53 in animal cap cells (Appendix Fig S4). Furthermore, knockdown of Wip1 had no effect on p53‐induced gene responses. These results indicate that Wip1 does not target p53 in regulation of TGF‐β signaling.
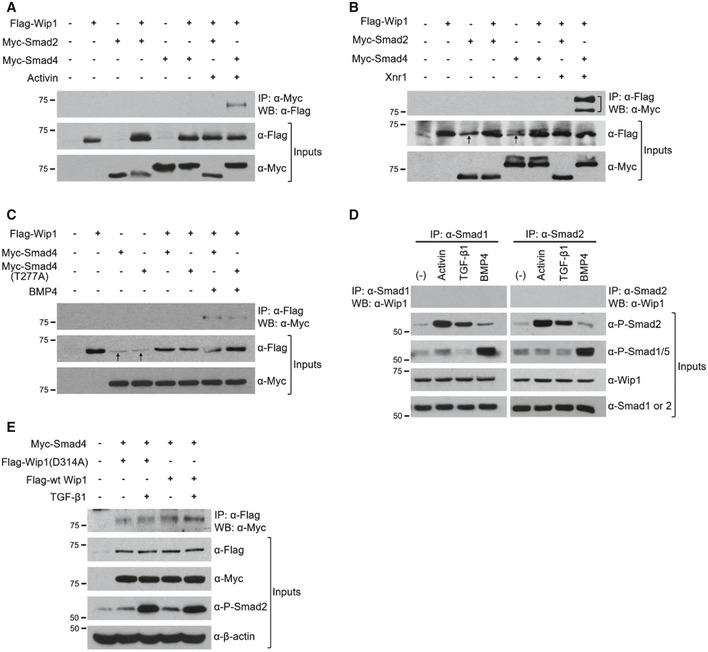
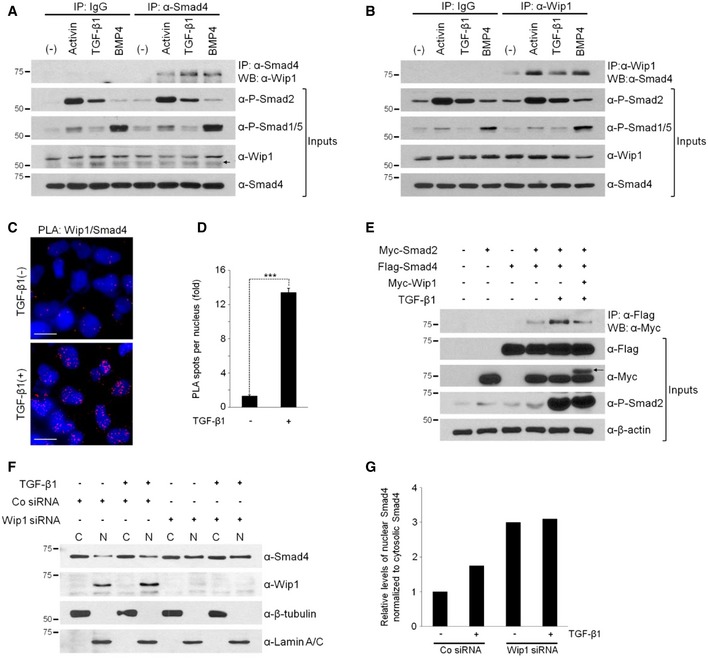
Wip1 regulates negatively TGF‐β/activin/nodal as well as BMP signaling pathways. These two branches of TGF‐β signaling are mediated not only by distinct effectors, Smad2/3 and Smad1/5/8, respectively, but also by a common effector, Smad4 2. Thus, we postulated that the mechanism underlying Wip1 interference with both pathways might involve down‐regulation of a common mediator Smad4. To test this assumption, we first examined whether Wip1 would interact physically with the Smad proteins. Notably, co‐immunoprecipitation experiments with epitope‐tagged proteins produced from Xenopus embryos revealed that Wip1 binds to Smad4 in a signal‐dependent manner (Fig EV2A–C). To determine whether this interaction might occur at physiological levels of protein expression, we treated HEK293T cells with TGF‐β, activin, or BMP4 and then immunopurified endogenous Smad4 from their lysates and probed for its association with endogenous Wip1. As shown in Fig 5A, Wip1 and Smad4 formed a complex in vivo in a signal‐dependent fashion. The same endogenous association was confirmed in reverse, using anti‐Wip1 antibody for immunoprecipitation (Fig 5B). In contrast, immunoprecipitation of Smad1 or Smad2 from the lysates followed by monitoring for co‐purified Wip1 detected no interaction between the proteins (Fig EV2D). Proximity ligation assays also demonstrated that endogenous Wip1 and Smad4 dynamically form a complex in the nucleus in a ligand‐dependent manner (Fig 5C; quantification in 5D). Furthermore, the catalytically inactive mutant of Wip1 still interacted with Smad4, though its binding appears to be weaker and inefficient, compared with that of wt Wip1 (Fig EV2E). This suggests the necessity of the phosphatase activity of Wip1 for its effective association with Smad4.
Figure EV2. Wip1 binds to Smad4 in a signal‐dependent manner.
-
A–CAnimal caps from embryos injected with Flag‐Wip1 (1 ng), Myc‐Smad4 (1 ng), Myc‐Smad4 (T277A) (1 ng), Myc‐Smad2 (1 ng), Xnr1 (100 pg), and BMP4 (200 pg) as indicated were cultured in the presence (A) or absence (B, C) of activin protein (10 ng/ml) and harvested for co‐immunoprecipitation analysis. Note that Smad4 (T277A) mutant interacts with Wip1, albeit more weakly than wt Smad4, as shown in (C). A bracket in (B) denotes Myc‐Smad4. Anti‐Flag antibody often produced non‐specific bands at the size of Flag‐Wip1 protein as indicated by arrows in (B, C).
-
DNo interaction between Wip1 and Smad1 or Smad2. The cell lysates were subjected to immunoprecipitation with anti‐Smad1 or anti‐Smad2 antibody and subsequent Western blotting with anti‐Wip1 antibody.
-
EPhosphatase‐inactive Wip1 mutant weakly binds to Smad4. HEK293T cells transfected with Myc‐Smad4, Flag‐wt Wip1, and phosphatase‐dead Flag‐Wip1 (D314A) as indicated were treated or not with TGF‐β1 (20 ng/ml) for 1 h and subjected to immunoprecipitation followed by Western blotting.
Source data are available online for this figure.
Figure 5. Wip1 interacts with Smad4 in vivo .
-
A, BWip1 and Smad4 form an endogenous protein complex. Cell lysates of HEK293T cells, which were stimulated or not with activin (10 ng/ml), TGF‐β1 (40 ng/ml), or BMP4 (20 ng/ml) for 1 h, were immunoprecipitated with anti‐Smad4 antibody followed by Western blotting with anti‐Wip1 antibody (A) and vice versa (B). (–), no treatment with protein ligand. IgG, control IgG. An arrow in (A) indicates non‐specific bands.
-
CUpon ligand stimulation, Wip1 forms a complex with Smad4 in the nucleus. HeLa cells were treated or not with TGF‐β1 (40 ng/ml) for 2 h, fixed, and then subjected to proximity ligation assays with primary antibodies against Wip1 and Smad4. Blue: DAPI; red: PLA signal. Scale bar, 20 μm.
-
DQuantitative analysis of Wip1/Smad4 interactions shown in (C). Data are represented as the mean ± SEM (n = 3 biological replicates). Twenty cells were analyzed per sample. ***P < 0.001 by Student's t‐test.
-
EWip1 interferes with the ability of Smad4 to bind to Smad2. HEK293T cells co‐transfected with the indicated constructs were treated or not with TGF‐β1 for 1 h and then harvested. An arrow denotes Myc‐tagged Wip1 protein.
-
FWip1 has a negative effect on nuclear accumulation of Smad4. HEK293T cells were transfected with Co siRNA or Wip1 siRNA, treated with TGF‐β1 (40 ng/ml, 1 h) 48 h after siRNA transfection and subjected to nuclear/cytoplasmic fractionation and Western blotting analysis. β‐tubulin and lamin A/C serve as cytoplasmic and nuclear markers, respectively. C, cytoplasm; N, nucleus. The experiment was repeated two times with similar results.
-
GQuantification of the levels of nuclear Smad4 normalized to those of cytoplasmic Smad4 shown in (F).
Source data are available online for this figure.
Upon ligand stimulation, the activated R‐Smads associate with Smad4, then translocating into the nucleus to regulate transcription of target genes, though they exhibit no interaction with Wip1 as shown above. Thus, we examined the effect of Wip1 on the association between Smad4 and R‐Smads. Treatment with TGF‐β1 enhanced the formation of a complex between Smad2 and Smad4, which could be prevented by co‐expression of Wip1 (Fig 5E), suggesting a competition between Wip1 and R‐Smads in binding to Smad4.
To investigate whether Smad4 might be indeed a Wip1‐regulated target, we further examined the effects of Wip1 knockdown on the nuclear versus cytoplasmic localization of Smad4. As reported previously 27, endogenous Wip1 was mostly localized in the nucleus of cultured cells regardless of TGF‐β stimulation (Fig 5F). In contrast, Smad4 was distributed in both cytoplasm and nucleus of Co siRNA‐treated control cells, and its nuclear accumulation was promoted by treatment with TGF‐β (Fig 5F; quantification in 5G). Notably, silencing of Wip1 increased markedly the basal levels of nuclear Smad4 to the extent that its nuclear levels were not enhanced further as a response to TGF‐β (Fig 5F; quantification in 5G). Since Wip1 acts as a phosphatase controlling TGF‐β signaling, Wip1 might regulate the phosphorylation status of Smad4, modulating its nuclear retention and/or export.
Wip1 dephosphorylates Smad4 in its linker region
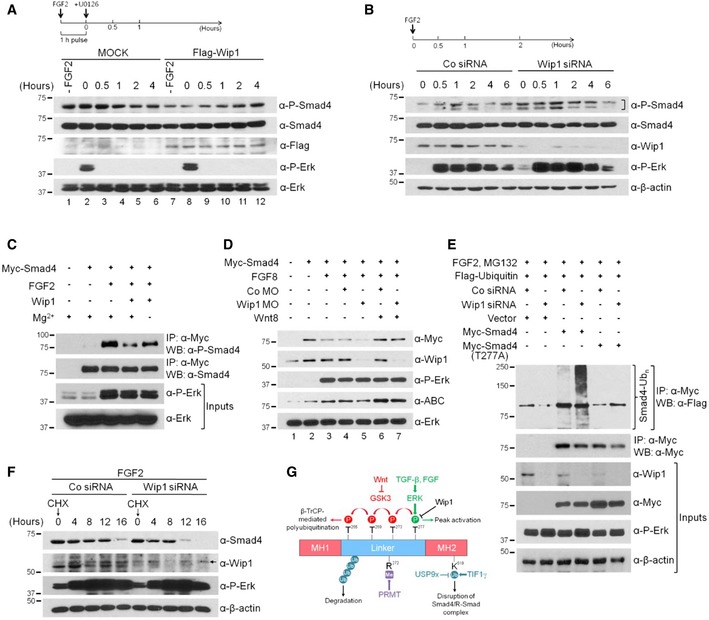
Smad4 has been known to be constitutively phosphorylated in cells 8, though most of the phosphorylation sites in Smad4 remain unknown. However, stimulation with FGF or TGF‐β signal has been found to induce Smad4 phosphorylation by extracellular signal‐regulated kinase (ERK) in the linker region at Thr277 12, 13. This phosphorylation is essential for TGF‐β‐induced nuclear accumulation and peak transcriptional activity of Smad4. As such, it is tempting to speculate that Wip1 might dephosphorylate Smad4 at this site, regulating its nuclear accumulation and transcriptional activity. To test this hypothesis, we first performed a pulse‐chase analysis to examine the effects of the gain‐of‐Wip1 function on the levels of Smad4 phosphorylation. For this experiment, cells were treated with 1‐h pulse of FGF2 ligand and subsequently with U0126, a MEK inhibitor as shown in Fig 6A. This prevents Smad4 phosphorylation by the residual active ERK, allowing for the measurement of the changes in the levels of its phosphorylation induced only for the brief interval. Smad4 phosphorylated at Thr277 was detectable at basal levels in unstimulated HEK293T cells (lane 1). Treatment with FGF2 pulse led to its elevated levels, which were gradually decreased over a period of 2 h (Fig 6A, lanes 2‐5; quantification in Fig EV3A). Of note, forced expression of Wip1 lowered the levels of phosphorylated Smad4 in FGF2‐pulsed as well as unstimulated cells, compared with those in MOCK‐transfected control cells (Fig 6A, lanes 7–11; quantification in Fig EV3A). In contrast, the total levels of Smad4 remained constant throughout the time course of the experiments, indicative of the immediate effects of Wip1 on Smad4 phosphorylation. Moreover, the phosphatase‐dead mutant of Wip1 had no effect on the levels of phosphorylated Smad4 (Fig EV4A). Conversely, depletion of Wip1 enhanced markedly not only the basal levels but also the FGF2‐induced levels of phosphorylated Smad4, compared with those in Co siRNA‐treated cells (Fig 6B; quantification in Fig EV3B). As a test for the specificity of Wip1 toward Smad4, we also examined the effects of Wip1 on the phosphorylation status of the R‐Smads, Smad1 and Smad2. Co‐expression of Wip1 or Wip1 (D277A) did not affect the levels of C‐terminally phosphorylated Smad1 and Smad2 induced in response to BMP4 and Xnr1, respectively (Fig EV4B and C). In addition, knockdown of Wip1 did not alter activin‐induced phosphorylation of Smad2 (Fig 2K). Together, these results indicate the specific effect of Wip1 on the phosphorylation status of Smad4.
Figure 6. Wip1 is a Smad4 phosphatase.
-
AOverexpression of Wip1 down‐regulates Smad4 phosphorylation at Thr277. MOCK‐ or Flag‐Wip1‐transfected HEK293T cells were treated with 1‐h pulse of FGF2 (10 ng/ml), followed by addition of U0126 (40 μM) and harvesting for Western blotting at the indicated times. –FGF2, no treatment with FGF2.
-
BTime course of Smad4 phosphorylation at Thr277 induced by FGF2 in control and Wip1‐depleted HEK293T cells, showing that knockdown of Wip1 up‐regulates Smad4 phosphorylation at the MAPK site. A bracket denotes phospho‐Smad4.
-
CWip1 directly dephosphorylates Smad4 at Thr277 as assayed by in vitro phosphatase assay. Myc‐tagged Smad4 was expressed in HEK293T cells, which were then treated or not with FGF2 (40 ng/ml, 2 h), and the Myc‐Smad4 was immunoprecipitated using anti‐Myc antibody and subsequently incubated with or without recombinant human Wip1 protein (0.5 μg) in the absence or presence of Mg2+ as indicated.
-
DFGF‐induced destabilization of Smad4 is enhanced further in Wip1‐depleted animal cap cells as analyzed by Western blotting. The amount of reagent injected: Myc‐Smad4 (400 pg), FGF8 (100 pg), Wnt8 (400 pg), Co MO (60 ng), and Wip1 MO (60 ng).
-
EPolyubiquitination of wt Smad4 is promoted in Wip1‐silenced cells. HEK293T cells were transfected with the indicated combinations of Flag‐ubiquitin, Co siRNA, Wip1 siRNA, Myc‐Smad4, and Myc‐Smad4 (T277A) and then treated with FGF2 (10 ng/ml, 4 h) in the presence of the proteasome inhibitor MG132, and Myc‐Smad4 and its phosphorylation‐resistant mutant were immunoprecipitated with anti‐Myc antibody. Ubiquitin‐conjugated Smad4 (Smad4‐Ubn) was detected by Western blotting with anti‐Flag antibody.
-
FWip1 prolongs the half‐life of Smad4. HEK293T cells were transfected with Co siRNA or Wip1 siRNA and then treated with FGF2 along with cycloheximide (CHX, 100 ng/ml) for the indicated times and harvested for Western blotting. An arrow indicates non‐specific bands.
-
GDiagram of the posttranslational Smad4 modifications by modifying enzymes and their downstream functions. P, phosphorylation; Ub, monoubiquitination; Me, methylation.
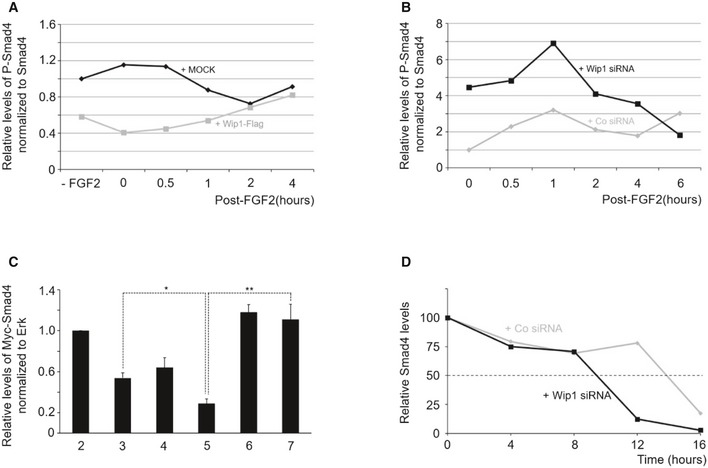
Figure EV3. Quantification of the results shown in Fig 6A, B, D and F.
-
AQuantification of the levels of phosphorylated Smad4 (normalized to Smad4) in Fig 6A.
-
BQuantification of Western blotting results shown in Fig 6B.
-
CQuantification of the levels of Myc‐Smad4 (normalized to ERK) in Fig 6D. Data are expressed as the mean ± SEM (n = 3 biological replicates). *P < 0.05, **P < 0.01 by unpaired Student's t‐test.
-
DQuantification of Western blotting results shown in Fig 6F.
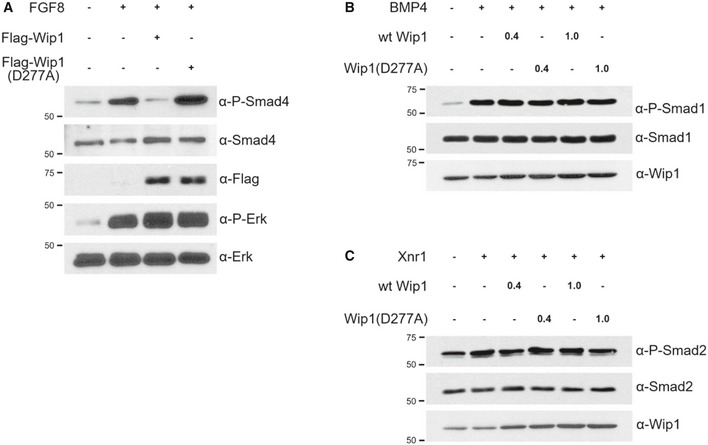
Figure EV4. Wip1 specifically dephosphorylates Smad4 in its linker region but not Smad1 and Smad2 at their C‐termini.
-
A–CAnimal caps from embryos injected with wt Wip1 (0.4, 1 ng), Wip1 (D277A) (0.4, 1 ng), Flag‐Wip1 (1 ng), Flag‐Wip1 (D277A) (1 ng), Xnr1 (100 pg), BMP4 (200 pg), and FGF8b (100 pg) as indicated were harvested at stage 15 (A) or 10.25 (B, C) for Western blotting analysis. Total Smad1, Smad2, and Erk serve as loading controls.
Source data are available online for this figure.
As Wip1 could affect the phosphorylation status of Smad4 via another phosphatase, we next carried out an in vitro phosphatase assay to check the direct involvement of Wip1 in dephosphorylating Smad4 at Thr277. Phospho‐Smad4 immunoprecipitated from the cell lysates could be effectively dephosphorylated by recombinant Wip1 protein (Fig 6C). Wip1 activity required Mg2+, as Wip1 failed to dephosphorylate the phospho‐Smad4 in the absence of this metal ion. These results suggest that phospho‐Smad4 at Thr277 is a direct target of Wip1.
The ERK phosphorylation of Smad4 at Thr277 acts to prime inhibitory GSK3 phosphorylation of linker region near the MAPK/ERK site, which leads to ubiquitination‐dependent degradation of Smad4 12. Wnt signal prevents this turnover of Smad4 by inhibiting its phosphorylation by GSK3, potentiating TGF‐β signaling in the presence of FGF. In light of these findings, we investigated whether knockdown of Wip1 might affect the stability of Smad4. Co‐expression of FGF8 lowered the levels of Myc‐Smad4 protein in animal caps (Fig 6D, lanes 2 and 3; quantification in Fig EV3C), presumably by inducing its phosphorylation by ERK and GSK3. FGF8‐reduced levels of Myc‐Smad4 were down‐regulated further by co‐injection of Wip1 MO (lane 5). Notably, co‐injection of Wnt8 reversed efficiently the decrease in Myc‐Smad4 levels caused by FGF8 alone or with Wip1 MO (lanes 6 and 7), supporting that reduction in Myc‐Smad4 levels was mediated by sequential ERK and GSK3 phosphorylation of its linker region. Consistent with these, Smad4 polyubiquitination was augmented by depletion of Wip1 in FGF2‐stimulated cells, compared with that in Co siRNA‐treated cells, and required an intact phosphorylation site (Thr277) for MAPK (Fig 6E). Cycloheximide chase experiments also revealed that the half‐life of Smad4 can be shortened in the absence of Wip1 (Fig 6F; quantification in EV3D). Taken together, these results indicate that Wip1 targets Smad4 to dephosphorylate Thr277 in its linker region, thereby controlling the stability of Smad4 (Fig 6G).
Wip1 down‐regulation of TGF‐β signaling is mediated by its dephosphorylation of Smad4 at Thr277
As Smad4 phosphorylation at Thr277 is required for its peak activity, we tested whether Wip1 dephosphorylation of Smad4 at this site might be relevant to its inhibition of TGF‐β signaling. Overexpression of Wip1 in the marginal zone of embryo repressed the in vivo expression of mesodermal markers, Chordin and Xbra (Fig 7A–D; quantification in 7K). These inhibitory effects of Wip1 were rescued by co‐expression of wt Smad4 or Smad4 (T277E) mutant in which a phospho‐mimetic amino acid is introduced at the MAPK site, but not by an ERK phosphorylation‐resistant mutant, Smad4 (T277A) (Fig 7E–J; quantification in 7K). Moreover, Wip1‐induced ectopic expression of neural markers, NCAM and Otx2, in animal caps was down‐regulated by co‐injection of wt Smad4 or Smad4 (T277E), but not by Smad4 (T277A) (Fig 7L). These results suggest that inhibition of activin/nodal and BMP signaling by increased Wip1 levels can be counterbalanced by co‐expression of Smad4 with a phosphorylatable or phospho‐mimetic amino acid at the MAPK site.
Figure 7. Wip1 inhibits TGF‐β signaling by dephosphorylating Smad4 in its linker region.
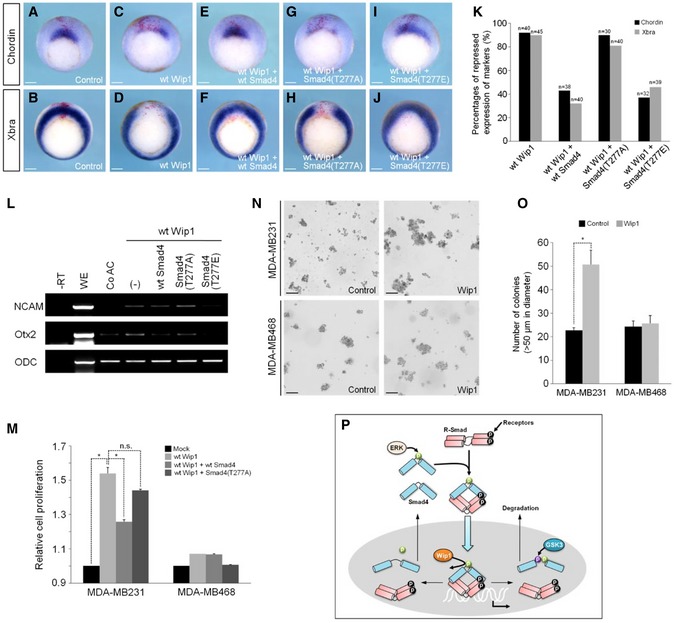
-
A–JEmbryos injected dorsally with wt Wip1 (1 ng), wt Smad4 (1 ng), Smad4 (T277A) (1 ng), and Smad4 (T277E) (1 ng) as denoted were subjected to in situ hybridization against Chordin or Xbra at stage 10.25. Control, control embryo injected with LacZ mRNA only. Scale bar, 150 μm.
-
KQuantification of the results from in situ hybridization analysis in (A–J).
-
LWip1‐mediated neural induction is inhibited by Smad4 with a phosphorylatable or phospho‐mimetic amino acid at the MAPK site. (–), no co‐injection of wt or mutated Smad4.
-
MMDA‐MB231 and MDA‐MB468 cells were transfected with wt Wip1 alone or with wt Smad4 or Smad4 (T277A) and then subjected to CCK‐8 assays to measure cell growth. Data are presented as the mean ± SEM (n = 3 biological replicates). *P < 0.05 by unpaired Student's t‐test.
-
NAnchorage‐independent colony‐formation assays were performed in MDA‐MB231 and MDA‐MB468 cells transfected with Wip1 or negative control constructs. Scale bar, 100 μm.
-
OQuantification of the assays shown in (N). The number of colonies larger than 50 μm in diameter was scored. Data are expressed as the mean ± SEM (n = 3 biological replicates). *P < 0.05 by unpaired Student's t‐test.
-
PProposed model of the mechanism by which Wip1 controls the transcriptional activity and stability of Smad4. See the discussion for details.
Source data are available online for this figure.
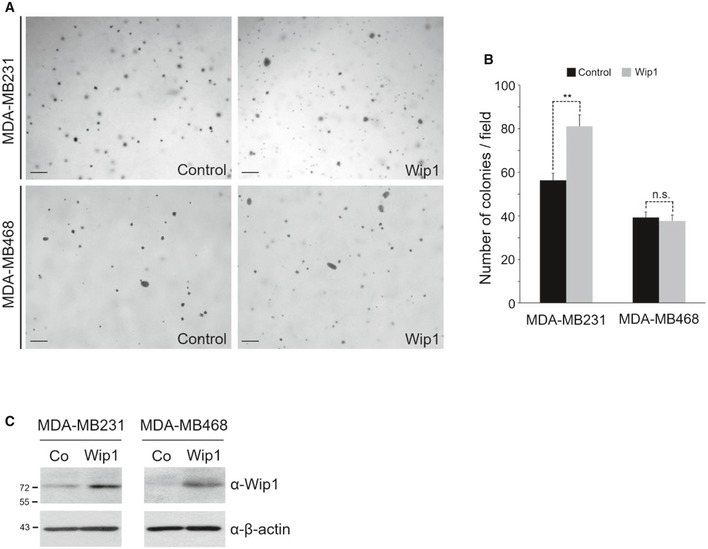
We next sought to determine whether Wip1 might also target Smad4 to control cell proliferation. For this, we used the MDA‐MB231 breast cancer cell line, which retains low levels of wt Smad4 and exhibits attenuated TGF‐β responsiveness 28 and as a negative control, the MDA‐MB468 cells lacking endogenous Smad4. As shown in Fig 7M, transfection of wt Wip1 augmented markedly cell proliferation in MDA‐MB231 cells, but not in Smad4‐null MDA‐MB468 cells, indicating that this promotion of cell growth is mediated by targeting Smad4. In support of this, MDA‐MB231 cells overexpressing Wip1 were growth‐arrested significantly upon co‐transfection of wt Smad4, but not of non‐phosphorylatable Smad4 (T227A) mutant. We further assessed the effect of Wip1 on the tumorigenic properties of breast cancer cells. Transient or stable overexpression of Wip1 significantly enhanced the anchorage‐independent growth of MDA‐MB231 cells, but not of MDA‐MB468 cells, as determined by the increase in colony number and colony size on soft agar (Figs 7N; quantification in 7O, and EV5A; quantification in EV5B). Collectively, these results suggest that Wip1 regulates the proliferation and tumorigenicity of cancer cells by targeting Smad4 for the dephosphorylation of its linker region.
Figure EV5. Overexpression of Wip1 promotes the anchorage‐independent growth of MDA‐MB231 cells, but not of MDA‐MB468 cells.
-
ASoft agar colony‐formation assays were performed in MDA‐MB231 and MDA‐MB468 cells stably overexpressing Wip1 or negative control constructs. Scale bar, 200 μm.
-
BQuantitative analysis of colony formation shown in (A). Data are expressed as the mean ± SEM (n = 3 biological replicates). **P < 0.01 by unpaired Student's t‐test. n.s., not significant.
-
CWestern blotting analysis of Wip1 expression in MDA‐MB231 and MDA‐MB468 cells stably overexpressing Wip1. Co, Vector control stable cell line; Wip1, Wip1 stable cell line.
Source data are available online for this figure.
Discussion
Reversible phosphorylation of Smad4 in its linker region
While Smad4 lacks the C‐terminal SSXS motif found in R‐Smads, undergoing no receptor‐mediated phosphorylation, it is phosphorylated by MAP kinases in its linker region. Specifically, ERK phosphorylation of Smad4 at residue Thr277 in the linker region is required for its nuclear accumulation and maximal transcriptional activity 11, 12. The phosphorylation status of this residue also plays a pivotal role in control of Smad4 stability. Given the significance of Smad4 phosphorylation at Thr277, dephosphorylation of Smad4 at the MAPK site would be crucial for regulating cellular responses to TGF‐β signals. However, the potential of Smad4 dephosphorylation as a regulatory mechanism of TGF‐β signaling and the phosphatase implicated in this dephosphorylation remain unknown. The present work demonstrated that Wip1 functions as a Smad4 phosphatase to control the phosphorylation status of Thr277, modulating the duration and activity of TGF‐β signaling. Thus, we suggest that reversible phosphorylation of Smad4 at Thr277, mediated by ERK and Wip1, is essential for dynamic cell responses to TGF‐β signaling.
A model for the regulation of TGF‐β signaling by ERK and Wip1
ERK phosphorylation of Smad4 at Thr277 has been shown to be induced by FGF or TGF‐β signals in different cell types 11, 13. This phosphorylation of Smad4 acts to prime it for inhibitory GSK3 phosphorylation of the linker region, leading to ubiquitin ligase‐driven, proteasomal degradation of Smad4 12. In this respect, Thr277 phosphorylation of Smad4 is similar to linker phosphorylation of R‐Smads by antagonists such as FGF and EGF via MAPK 9. On the other hand, TGF‐β/ERK‐mediated phosphorylation of Smad4 at the MAPK site also appears to be similar to agonist‐induced linker phosphorylation of R‐Smads mediated by cyclin‐dependent kinase, CDK8/9 10. While linker phosphorylation of Smad4 triggered by TGF‐β stimulation also involves a kinase distinct from ERK 29, it remains to be investigated whether it might be CDK8/9. Antagonist‐induced linker phosphorylation of R‐Smads is mediated by cytoplasmic MAPK, whereas agonist‐induced one involves nuclear cyclin‐dependent kinases 10, 30. Cytoplasmic as well as nuclear activities of EKRs have been demonstrated to control of transcription 31. However, since linker phosphorylation of Smad4 is relevant to a cycle of its monoubiquitination/deubiquitination 13, it would be reasonable to postulate that FGF or TGF‐β‐induced phosphorylation of Smad4 at Thr277 is mediated by cytoplasmic ERK. Cytoplasmic FAM/USP9x‐mediated deubiquitination of Smad4 is essential for TGF‐β signaling, but its monoubiquitination by nuclear ectodermin/TIF1γ acts to disrupt the R‐Smad‐Smad4 complex and to assist nuclear export of Smad4 to terminate signaling 32. ERK phosphorylation of Smad4 at Thr277 promotes Smad4‐FAM/USP9x interaction and R‐Smad‐Smad4 complex formation 13. As such, it is plausible that cytoplasmic MAPK might phosphorylate the linker region of monoubiquitinated Smad4, which is exported from the nucleus, thereby facilitating its interaction with and subsequent deubiquitination by cytoplasmic FAM/USP9x (Fig 7P). Deubiquitinated Smad4, which is still phosphorylated at Thr277, would form a stable complex with receptor‐activated R‐Smads. This R‐Smad‐Smad4 complex translocates into the nucleus, where it controls transcription of target genes in cooperation with other DNA‐binding cofactors. First, nuclear Wip1 might dephosphorylate Smad4 at Thr277 in order to recycle Smad4. Intact catalytic site in Wip1 is important for its efficient association with Smad4 (Fig EV2). In addition, Wip1 acts on Smad4, resulting in dissociation of Smad4 and R‐Smads (Fig 5). Monoubiquitination of Smad4 causes disassembly of the R‐Smad‐Smad4 complex and subsequent nuclear export of monoubiquitinated Smad4 32. Thus, Wip1 dephosphorylation of Smad4 would promote its association with and monoubiquitination by ectodermin/TIF1γ in the nucleus. In support of the proposed effect of Wip1 on the subcellular distribution of Smad4, depletion of Wip1 enhances the nuclear retention of Smad4 (Fig 5) and the phospho‐resistant mutant, Smad4 (T277A), has been shown to accumulate less efficiently in the nucleus after stimulation by TGF‐β 11. R‐Smad, disengaged from Smad4, might be dephosphorylated by Smad phosphatase, such as PPM1A, at the C‐terminal tail, and also shuttles back to the cytoplasm to be reactivated by receptors 7, 33.
Even in the absence of TGF‐β signaling, Smad4, but not R‐Smads, undergoes continuous nucleocytoplasmic shuttling, which is dictated by the relative strengths of its nuclear localization signal (NLS) and nuclear export signal (NES) 34. Notably, mutation of the NES, which renders Smad4 constitutively nuclear, does not promote basal transcription in unstimulated cells. Furthermore, mouse embryo homozygous for constitutively nuclear Smad4 mutant alleles develops normally 35. In contrast, knockdown of Wip1, which also prolongs the nuclear retention of Smad4, enhances target gene transcription in the absence as well as in the presence of TGF‐β signals (Fig 2). It has been demonstrated that the timing of the transcriptional response in TGF‐β signaling coincides with the duration of Smad4 nuclear localization rather than with R‐Smad activity 15; that is, intrinsic temporal control of transcription is reflected only in Smad4. Thus, we propose that Wip1‐mediated dephosphorylation of Smad4 is primarily responsible for the determination of transcriptional dynamics. Subsequently, among the steps presumed to follow Wip1 dephosphorylation of Smad4, ectodermin/TIF1γ‐mediated monoubiquitination of Smad4, which disrupt the R‐Smad‐Smad4 complex, would be critical for termination of TGF‐β signaling, but not Smad4 nuclear export.
Control of transcriptional dynamics and Smad4 nuclear export as a response to TGF‐β signals depends on new protein synthesis 15. Given its key role in turning off TGF‐β signaling, it is tempting to speculate that modulation of Wip1 expression may be a pivotal regulatory mechanism of transcriptional dynamics. Of note, the phosphatase activity of Wip1 has not been shown to be modulated by posttranslational modification; the major regulatory mechanism of Wip1 activity involves control of its expression levels 17. Wip1 functions as a homeostatic regulator in double‐strand break (DSB) repair signaling to dephosphorylate and inhibit several key DNA damage‐responsive proteins. In the early stages of DNA damage response, low levels of Wip1 protein are maintained by miR‐16‐mediated inhibition of its production and/or its phosphorylation by HIPK2, which leads to its proteasomal degradation 36, 37. At the late stages, increased levels of Wip1 are induced by reduction in miR‐16 expression and escape from HIPK2‐driven its degradation. This temporal control of Wip1 expression ensures a completion of the early DNA damage response, preventing a premature termination of DSB repair signaling. We observed that TGF‐β signals have no effect on transcription of Wip1 (Appendix Fig S5). Recently, DNA damage has been shown to stabilize the TGF‐β type II receptor via ATM and c‐Cbl, thereby promoting TGF‐β signaling 38. As TGF‐β signals induce ERK phosphorylation of Smad4 at Thr277, this DNA damage up‐regulation of TGF‐β signaling might lead to the linker phosphorylation of Smad4. DNA damage also induces sequential activation of ATM, CHK2, and p53, which leads to activation of Wip1 transcription 20. The increased levels of Wip1 down‐regulate ATM, CHK2, and p53 in a negative feedback loop. We speculate that DNA damage‐induced Wip1 expression would down‐regulate TGF‐β signaling by dephosphorylating Smad4 as part of a negative feedback loop. Together, these findings suggest that stress signals such as DNA damage may also regulate Smad4 phosphorylation at Thr277 and TGF‐β signaling through a delayed induction of Wip1 expression. It will be of great interest to investigate whether and how temporal control of Wip1 expression would also contribute to the dynamics of the transcriptional response to TGF‐β signals under normal physiological conditions. In addition, this potential mechanism would be crucial for regulation of Smad4 stability as phosphorylation of Smad4 at Thr277 leads to its phosphorylation by GSK3 and subsequent degradation driven by an E3‐ligase. PRMT1 methylation of an arginine within the GSK3 site in Smad4 is essential for its phosphorylation by GSK3 14. TGF‐β and Wnt signals have been shown to facilitate PRMT1 methylation of Smad6/7 and Smad4 at their arginine residues, respectively 3, 4, 14. Wip1 depletion alone accelerates Smad4 degradation via its phosphorylation by GSK3 in FGF‐stimulated cells (Fig 6). This suggests the possibility that the activity of a methyltransferase is affected by FGF signal and/or Wip1 expression level, though its basal activity may already be high enough to modify Smad4. Smad4 is recycled for the next rounds of signaling or enters the degradative pathway, depending on the levels of Wip1 expression as shown in this study. Thus, it appears that the stability of Smad4 at the crossroad of Wnt, TGF‐β, and FGF signaling pathways is regulated by the status of its phosphorylation and methylation in the linker, which is determined by the integration of these signals as well as the levels of Wip1 expression.
Wip1‐dependent control of cell proliferation and tumor formation
PPM1D, the gene encoding Wip1, has been found to be aberrantly amplified and even mutated in numerous human cancer types 16. Wip1 exhibits oncogenic properties by negatively controlling several tumor suppressor pathways 19. Wip1 inhibits p53 activity by dephosphorylating p53 itself at Ser15 or the upstream kinases that phosphorylate p53, including ATM, p38MAPK, Chk1, and Chk2. In addition, Wip1 dephosphorylates Mdm2, an E3 ubiquitin ligase at Ser395, resulting in Mdm2 stabilization and subsequent Mdm2‐mediated degradation of p53. Wip1‐null mice have been shown to be resistant to tumorigenesis in several mouse models of cancer 39. In a mouse model of APCMin‐driven colorectal cancer, Wip1 deficiency represses polyp formation by enhancing p53‐dependent apoptosis of intestinal stem cells, thereby increasing the survival of mice 18. As Wip1‐null cells have already elevated level and/or activity of p53, constitutive activation of Wnt signaling on APC mutation, which also leads to p53 activation, would induce efficiently apoptosis of intestinal stem cells of Wip1‐null/APCMin mice but not wild‐type APCMin mice. Based on this speculation, it has been proposed that loss of Wip1 reduces the threshold for apoptosis of cancer‐initiating stem cells, inhibiting APCMin‐induced polyposis 18.
The TGF‐β/Smad pathway is involved in tumor suppression or progression, depending on the context in colorectal cancer development 1, 23. TGF‐β signal triggers apoptosis in the context of pre‐malignant cells, such as intestinal stem cells of APCMin mice. In contrast, genetic ablation of TGF‐β type II receptor (TGF‐βRII) or Smad4 potentiates carcinoma progression in APC‐deficient mice. Besides, two‐point mutations in Smad4 (Pro130Ser and Asn351His), which are commonly found in human pancreatic and colorectal cancers, have been shown to promote GSK3 phosphorylation of its linker region, accelerating its degradation 29. It is worthwhile noting that p53 cooperates with Smads for the full transcriptional activation of a significant subset of TGF‐β target genes 26. Thus, it is possible that Wip1 might target Smad4 as well as p53 in intestinal stem cells. In Wip1‐null/APCMin cells, dephosphorylation of Smad4 at Thr277 would not occur due to the loss of Wip1. In this condition, the activity and duration of Smad4‐mediated signaling might be highly up‐regulated compared with those in wild‐type APCMin mice. Inhibition of GSK3 activity in Wip1‐null/APCMin cells would allow Smad4 to reach its maximal activity by impeding GSK3 phosphorylation of Smad4 in the linker region and its subsequent degradation. Consistently, apoptosis of intestinal stem cells in Wip1‐null/APCMin mice has been enhanced further by treatment of GSK3 inhibitors 18. As such, it is plausible that the combinatorial control of Smads and p53, both of which are highly activated in the absence of Wip1, might efficiently contribute to TGF‐β‐mediated apoptosis in pre‐malignant Wip1‐null/APCMin cells. During intestinal homeostasis, BMP signaling restricts the self‐renewal of intestinal stem cells by inhibiting Wnt‐β‐catenin signaling via the PTEN‐PI3K pathway or through Smad‐mediated direct transcriptional suppression of stem cell signature genes 40, 41. Wip1 has been shown to be expressed in intestinal stem cells of mice 18. Overexpression of Wip1 in intestinal stem cells would inhibit the BMP/Smad pathway as shown in our data, promoting the expression of stem cell signature genes, their Wnt‐independent self‐renewal, and subsequent polyp formation. In line with this, Wip1 expression is highly increased in intestinal polyp of APCMin mice, and Wip1 deficiency has no effect on β‐catenin‐dependent gene transcription program 18. Further studies are warranted to investigate whether Wip1 might function as a mediator of the cross‐talk between BMP and Wnt signaling to regulate intestinal homeostasis.
Role of Wip1 in germ‐layer specification
As a maternal factor, Wip1 is expressed in the marginal as well as in the animal regions of embryo during germ‐layer specification. Since Wip1 has inhibitory effects on TGF‐β signals, it would function to limit the mesoderm‐inducing activin/nodal signal from the vegetal pole to ensure that cells in the marginal zone of embryo adopt a mesodermal fate, not an endodermal one. In addition, Wip1 would down‐regulate activin/nodal as well as BMP signals in the animal pole region to ensure that ectodermal cells adopt the default neural fate but not an epidermal or mesodermal fate. Therefore, our results suggest that Wip1 plays critical roles in determination of cellular fates along the animal–vegetal axis.
The elevated levels of Wip1 inhibit mesoderm formation and induce neural fate in naïve ectodermal cells, which can be reverted by co‐expression of wild‐type Smad4 or its phospho‐mimetic mutant, Smad4 (T277E), but not by phospho‐resistant mutant, Smad4 (T277A). This finding demonstrates that Smad4 phosphorylation at Thr277 is necessary for mesoderm formation and epidermal differentiation. Consistently, Smad4 MO‐mediated inhibition of the expression of mesodermal marker was not restored efficiently by co‐expression of phospho‐resistant mutant, Smad4 (T277V) 12. By keeping this phosphorylation of Smad4 under control, Wip1 appears to lower its activity in favor of mesodermal differentiation and the default neurulation. It has been shown that during blastula stages, activated MAPK is detected in the animal cap and marginal zone of the embryo, with its level in the animal region being lower than that in the marginal zone 42, 43. Given this, the interplay between MAPK and Wip1 would generate a gradient of Smad4 phosphorylation at Thr277, contributing to correct specification of embryonic mesoderm and ectoderm.
In summary, Wip1 functions as a Smad4 phosphatase to dephosphorylate it at Thr277 in the linker region, controlling its activity, stability, and nuclear accumulation. By negative regulation of TGF‐β signaling, Wip1 contributes to germ‐layer specification in the vertebrate embryo and to the control of growth and migration of mammalian cells. This work provides insight into the mechanism by which the phosphorylation status of Smad4 regulates the activity and dynamics of TGF‐β signaling. Since dysregulated TGF‐β signaling leads to oncogenesis, the mechanistic link between Wip1 and TGF‐β signaling suggests the potential of Wip1 as a therapeutic target for cancer treatment, which merits further study.
Materials and Methods
Cell culture and transfections
MDA‐MB231, MDA‐MB468, HEK293T, and HeLa cells were cultured in complete Dulbecco's modified Eagle's medium (DMEM, GE Healthcare) supplemented with 10% fetal bovine serum (FBS, Corning), 100 units/ml penicillin, and 100 mg/ml streptomycin at 37°C in a 5% CO2 atmosphere. Cells were transfected using Lipofectamine 2000 (Invitrogen) for DNA constructs or Lipofectamine RNAiMax (Invitrogen) for siRNAs and then subjected to subsequent experiments after 24–48 h.
Establishment of cell lines stably overexpressing Wip1
For cell lines stably overexpressing Wip1, MDA‐MB231 and MDA‐MB468 cells were transduced with a lentivirus carrying the Wip1 gene and selected in the presence of puromycin (1 μg/ml) for 1 week to eliminate the untransfected cells. After selection, puromycin‐resistant cells were pooled and screened by Western blotting (Fig EV5C).
Embryo manipulation
This study was compliant with all relevant ethical regulations regarding animal research. All animal experiments were approved by the Institutional Animal Care and Use Committee at the Asan Institute for Life Sciences, Asan Medical Center. Male and female Xenopus laevis frogs were purchased from NASCO & Korean Xenopus Resource Center for Research. Eggs were obtained from female frogs primed with 800 units of human chorionic gonadotropin (HCG, Daesung Microbiological Labs.). They were in vitro fertilized using macerated testis, dejellied in 2% cysteine (Sigma) solution (pH 7.8), cultured in 0.33× Modified Ringer (MR) until stage 8, and then transferred to 0.1× MR with gentamicin (Gibco). Staging of embryos was according to Nieuwkoop and Faber's normal table of development 44. Unless otherwise specified, embryos were microinjected at four‐cell stage in the animal pole region in 0.33× MR containing 4% Ficoll 400 (GE Healthcare) using a Pico‐Injector (Harvard Apparatus). Injected embryos were cultured in 0.33× MR with 4% Ficoll 400 until stage 8.5–9 and then transferred to 0.1× MR without Ficoll 400 for subsequent culture. For animal assays, animal cap explants were dissected at stages 8, cultured to stage 10.25 (Figs 1A and B, and 2A and B, and EV4B and C), 10.5 (Fig EV3A–C, Appendix Fig S1A and B, and S4), 15 (Figs 6D and EV4A), or 18 (Figs 1C, 2D and 7L) in 1× MR containing 10 μg/ml of bovine serum albumin (BSA, Roche) and 50 μg/ml of gentamicin, and then subjected to further analysis.
Plasmids
The entire coding region of Xenopus Wip1 was amplified from a stage 10 cDNA library by using the primer combinations 5′‐GGAATTCCATGGAGACTCCATTCTCCCTGCG‐3′ (forward) and 5′‐GCTC TAGAGCTTAACAGACACATATTGTTTTTCTGTGCTG‐3′ (reverse) and subcloned into the EcoRI and XbaI sites of the pCS2+ vector to generate the Wip1‐CS2+ construct. The Wip1‐T construct was produced by inserting the coding region of Xenopus Wip1 into the pGEM‐T easy vector (Promega). To obtain the phosphatase‐inactive version of Xenopus Wip1, the single amino acid change from D to A at position 277 was achieved by site‐directed mutagenesis using Pfu DNA polymerase (Agilent) and the primers, 5′‐CATCATTATAGGCAGTGCTGGTCTCTGGAACATGG‐3′ (forward) and 5′‐CCATGTTCCAGAGACCAGCACTGCCTATAATGATG‐3′ (reverse). This mutant was cloned into the EcoRI and XbaI sites of the pCS2+ vector to generate the Wip1 (D277A)‐CS2+ construct. To produce the Flag‐tagged Xenopus Wip1 construct (Flag‐Wip1‐CS2+), a DNA fragment was amplified by PCR using the primers, 5′‐GGAATTCCATGGATTACAAGGATGACGACGATAAGTCGGTAGAGACTCCATTCTCCCTGCGGGTGA‐3′ (forward, harboring the Flag epitope sequence) and 5′‐GCTCTAGAGCTTAACAGACACATATTGTTTTTCTGTGCTG‐3′ (reverse) and inserted into the EcoRI and XbaI sites of the pCS2+ vector. The Wip1‐Myc‐CS2+ construct, which contains the target sequence of Wip1 morpholino oligos, was generated by subcloning into the BamHI and ClaI sites of the pCS2‐MT vector, a DNA fragment from a PCR with the primers, 5′‐CGGATCCGAAAGAAGGGGTGCAATGGAGACTCCATTCTCCCT‐3′ (forward) and 5′‐CATCGATGACAGACACATATTGTTTTTCTGTGCTGCTG‐3′ (reverse). The phospho‐resistant Smad4 construct, Myc‐Smad4 (T277A)‐CS2+, and the phospho‐mimetic Smad4 construct, Myc‐Smad4 (T277E)‐CS2+, were generated by site‐directed mutagenesis using Myc‐human Smad4‐CS2+ as a PCR template and the primers 5′‐AGGACTGCAC CATACGCACCTAATTTGCCTCAC‐3′ (forward, for Smad4 (T277A)) or 5′‐AGGACTGCACCATACGAACCTAATTTGCCTCAC‐3′ (forward, for Smad4 (T277E)) and 5′‐GTGAGGCAAATTAGGTGCGTATGGTGCAGTCCT‐3′ (reverse, for both). Other plasmids are generous gifts as specified: Xenopus FGF8b, Xenopus BMP4, Myc‐Xenopus Smad1, Myc‐Xenopus Smad2, and Myc‐human Smad4 (J.K. Han); Flag‐human Wip1 and Flag‐human Wip1 (D314A) (J. Choi); Noggin, LacZ (R. Harland); Xenopus FAST‐1, constitutively active human Alk4 (Alk4 (T206D)), constitutively active Xenopus BMPR1 (BMPR1 (Q228D)) and ARE3‐Luc (C.Y. Yeo); Xenopus Wnt8 (R.T. Moon); Xnr1 (C.V.E. Wright); and BRE‐Luc (J.B. Kim).
Morpholino oligos, siRNAs, and recombinant proteins
The anti‐sense morpholino oligos (MOs) were purchased from Gene Tools. The MO oligo sequences were as follows: Wip1 MO1, 5′‐GGAGTCTCCATTGCACCCCTTCTTT‐3′; MO2, 5′‐ATGGAATCTC CATTGCACCCCTTCT‐3′. For complete knockdown of Wip1, equal amounts of the MO1 and MO2 were mixed and microinjected. Control (Co) MO was a standard morpholino oligo from Gene Tools whose sequence was 5′‐CCTCTTACCTCAGTTACAATTTATA‐3′. SMARTpool siRNA targeting human Wip1 (Cat. No., L‐004554‐00) was purchased from Dharmacon. Another synthetic Wip1 siRNA (Cat. No., S16127) and silencer negative control siRNA #1 (AM4635) were obtained from Thermo Scientific, Inc. Recombinant activin A (Cat. No., 338‐AC), FGF2 (Cat. No., 233‐FB), TGF‐β1 (Cat. No., 240‐B‐010), and BMP4 (Cat. No., 314‐BP‐010) proteins were purchased from R&D systems.
mRNA synthesis
Capped synthetic sense mRNAs were generated by using the Ambion mMessage mMachine SP6 (AM1340) and T7 (AM1344) kits (Thermo Scientific, Inc.). The expression constructs Wip1‐CS2+, Wip1 (D277A)‐CS2+, Flag‐Wip1‐CS2+, Wip1‐Myc‐CS2+, Myc‐Smad2‐CS2+, Myc‐Smad4‐CS2+, Myc‐Smad4 (T277A)‐CS2+, Myc‐Smad4 (T277E)‐CS2+, XWnt8‐CS2+, FGF8b‐CS2+, and Noggin‐CS2+ were linearized with NotI, and their respective mRNAs were synthesized using SP6 RNA polymerase. Xnr1‐CS2+ was linearized with Asp718, and its mRNA was synthesized using SP6 RNA polymerase. BMP4‐T7TS construct was cut with EcoRI, and its mRNA was synthesized using T7 RNA polymerase.
Reverse transcription–polymerase chain reaction (RT–PCR)
RT–PCR analysis was carried out as described 24. Briefly, total RNA was extracted from whole embryos or dissected animal cap explants with TRIzol Reagent (Life Technologies) and treated with RNase‐free DNase Ι (Roche) to remove genomic DNA. Approximately 5 μg of total RNA was reverse‐transcribed by using random hexamers and M‐MLV reverse transcriptase (Promega). PCRs were performed in a standard 50 μl of PCR with Taq polymerase (LaboPass). The following primers were used: Wip1, (forward) 5′‐GGGAGGACGAGCTGCCTTGG‐3′, (reverse) 5′‐ACTGGCTGTGGTTCCGGACG‐3′; Xmsx1, (forward) 5′‐ACTGGTGTGAAGCCGTCCCT‐3′, (reverse) 5′‐TTCTCTCGGGACTCTCAGGC‐3′; NCAM, (forward) 5′‐CACAGTTCCACCAAATGC‐3′, (reverse) 5′‐GGAATCAAGCGGTACAGA‐3′; and ODC, (forward) 5′‐GTCAATGATGGAGTGTATGGATC‐3′, (reverse) 5′‐TCCATTCCGCTCTCCTGAGCAC‐3′. Other primers are described in De Robertis’ homepage (http://www.hhmi.ucla.edu/derobertis/index.html).
Immunoprecipitation and Western blot analysis
For co‐immunoprecipitations of ectopically expressed proteins in Xenopus embryos (Fig EV2A‐C), animal cap explants were excised from injected whole embryos at stage 9 and cultured to early gastrula stages with or without activin for co‐IP. The animal caps were homogenized in Triton X‐100 lysis buffer (50 mM Tris (pH 7.6), 1% Triton X‐100, 50 mM NaCl, 1 mM EDTA, 1 mM sodium vanadate, 10 mM NaF, 1 mM PMSF, 20 μg/ml aprotinin, 20 μg/ml leupeptin). For co‐immunoprecipitations of proteins in cell lines (Figs 5A, B and E, and EV2D and E), cultured cells were treated with or without activin, TGF‐β1, or BMP4 for 1 h and then lysed in Triton X‐100 lysis buffer. Proteins were collected from lysates using Protein G Mag Sepharose (GE Healthcare)‐bound antibodies for overnight at 4°C on a rotating platform. Purified rabbit IgG or mouse IgG was used as negative controls. Immunoprecipitates and protein lysates were resolved by 6–12% SDS–PAGE. Western blot analysis was performed according to standard protocol. Antibodies used for IPs, PLA, and Western blot analysis are as follows: anti‐Smad1 (for IP, Santa Cruz, sc‐7965), anti‐Smad1 (Cell Signaling, #6944), anti‐P‐Smad1/5 (Cell Signaling, #9516), anti‐Smad2 (for IP, Santa Cruz, sc‐101153), anti‐Smad2 (Cell Signaling, #5339), anti‐P‐Smad2 (Cell Signaling, #3108), anti‐Smad4 (for IP and PLA, Santa Cruz, sc‐7966), anti‐Smad4 (Cell Signaling, #9515), anti‐P‐Smad4 (T277) (Abnova, PAB1965), anti‐Wip1 (for IP, Santa Cruz, sc‐20712), anti‐Wip1 (for PLA, Cell Signaling, #11901S), anti‐Wip1 (Bethyl Laboratories, A300‐664A), anti‐p21 (Cell Signaling, #2947), anti‐Lamin A/C (Abcam, ab108595), anti‐P‐ERK (Cell Signaling, #4377), anti‐ERK (Cell Signaling, #4695), anti‐ABC (Cell Signaling, #4270), anti‐Myc (Santa Cruz, sc‐40), anti‐Flag (Sigma, F1804), and anti‐β‐actin (Santa Cruz, sc‐47778)) antibodies. The intensities of Western blot bands were measured using ImageJ software.
Cell Counting Kit‐8 (CCK‐8) assay
Cell proliferation was measured by CCK‐8 assay. Briefly, transfected cells were seeded in a 96‐well plate at a cell density of 5.0 × 103 cells per well and incubated for 12 h. Subsequently, the cells were cultured in DMEM supplemented with fetal bovine serum (10%) in the presence or absence of TGF‐β1 (40 ng/ml) for 24 h. 10 μl of CCK‐8 solution (Dojindo Molecular Technologies, Inc.) was added to each well of the plate, and then, the plate was incubated for 3 h in the incubator. The total number of cells in each well was determined by measuring the absorbance at 450 nm using a microplate reader.
In vitro phosphatase assay
To obtain phosphorylated Smad4 protein, HEK293T cells were transfected with the expression plasmid encoding Myc‐tagged wt Smad4 and then cultured in the presence of FGF2 (40 ng/ml) for 2 h prior to harvest. Phosphorylated Myc‐Smad4 was immunoprecipitated using anti‐Myc antibody, and the protein bound to the beads was resuspended in phosphatase buffer (50 mM Tris–HCl (pH 7.5), 0.1 mM EGTA, 0.02% 2‐mercaptoethanol) with or without 30 mM MgCl2 and used as a substrate in the in vitro phosphatase assay. Phosphatase reactions were initiated by adding recombinant GST‐Wip1 (Abnova, 0.5 μg/reaction) to phospho‐Smad4 and incubated for 20 min at 30°C. Subsequently, the eluted proteins were subjected to SDS–PAGE followed by Western blotting using anti‐phospho‐Smad4 or anti‐Smad4 antibodies.
Polyubiquitination assays
Ubiquitination assays were carried out as described in Ref. 12. HEK293T cells were transfected with siRNAs and 48 h later, with the indicated DNA constructs. 24 h posttransfection, the cells were treated with FGF2 and MG132 (20 μM) and allowed to incubate for 4 h. Total cell lysates were prepared by sonication in RIPA lysis buffer. Prior to immunoprecipitation, lysates were boiled in 1% SDS at 95°C for 10 min to break protein–protein interactions and then diluted 10‐fold with RIPA buffer. The diluted samples were subjected to an anti‐Myc immunoprecipitation followed by immunoblotting with anti‐Flag antibody to detect Flag‐ubiquitin conjugated to Smad4.
Proximity ligation assays (PLA)
PLA was performed using Duolink In Situ Orange Starter Kit Mouse/Rabbit (Sigma‐Aldrich). Briefly, HeLa cells on coated coverslips were grown to 60–70% confluency and then treated or not with TGF‐β1 for 2 h. Subsequently, cells were fixed in 4% paraformaldehyde in PBS for 10 min and permeabilized in 0.4% Triton X‐100 in PBS for 10 min. After three 5‐min washes with PBS, cells were treated with blocking solution for 1 h at 37°C and then incubated with specific combination of primary antibodies [mouse monoclonal Smad4 (1:100) and rabbit polyclonal Wip1 (1:100)] at 4°C overnight in a humidity chamber. The PLA reaction was carried out using the two PLA probes (Duolink PLA anti‐mouse minus and anti‐rabbit plus) diluted 1:5 in Duolink antibody diluent and incubated for 1 h at 37°C. Subsequent ligation, amplification, and final washes were performed using supplied reagents according to the manufacturer's instructions. Coverslips were mounted on slides with Duolink PLA mounting media with DAPI. Images were acquired on a Nikon Eclipse Ti fluorescent microscope.
Soft agar colony‐formation assays
Complete medium containing 0.5% agar was laid down in a 6‐well plate. 5 × 103 cells (control, Wip1‐overexpressed MDA‐MB231 and MDA‐MB468 cells) were suspended in complete medium with 0.3% agar, and then, the agar‐cell mixture was plated on top of a bottom layer. At 15 days of culture at 37°C, the colonies were stained with 0.005% (w/v) Crystal Violet. Total colonies or colonies larger than 50 μm in diameter were counted.
Wound healing assays
MDA‐MB231 cells were seeded in 6‐well plates and transfected with siRNA. 48 h after siRNA transfection, 80–90% confluent cell monolayers were scratched with a 20‐μl pipette tip to obtain two parallel wounds in each well, washed with medium and replenished with fresh medium with or without TGF‐β1 (40 ng/ml), and photographed. Migration was followed until cells were grown to 24 or 48 h after the scratch, fixed in 4% paraformaldehyde, and photographed. Experiments were repeated three times independently. The gap distance was quantitatively evaluated using Image J software.
Transwell invasion assays
Transwell invasion assays were carried out in Matrigel‐coated 24‐well PC inserts (SPL 8.0 μm pore size). MDA‐MB231 cells were transfected with siRNA, and 48 h posttransfection, cells (5 × 104) were resuspended in pre‐warmed serum‐free medium, plated in transwell inserts, and then treated or not with TGF‐β1 (40 ng/ml) for 20 h. Non‐invaded cells in the upper part of transwell were removed with a cotton swab. Migrated cells were fixed in 4% paraformaldehyde for 10 min and after PBS washes, stained with 0.5% Crystal Violet for 1 min. The dried transwell membranes were photographed, and the number of cells in different fields of view was counted.
Cytoplasmic and nuclear fractionation
Cell pellets were suspended in F1 buffer (20 mM Tris–HCl (pH 7.6), 50 mM 2‐mercaptoethanol, 0.1 mM EDTA, 2 mM MgCl2, 1 mM PMSF) and then incubated on ice for 10 min. After addition of NP‐40 to a final concentration of 1%, the cell suspension was homogenized by pipetting and subsequently centrifuged at 600 g for 5 min at 4°C. The supernatant (cytoplasmic fraction) was collected and stored at −80°C. The nuclear pellet was washed three times in F1 buffer containing 1% NP‐40 and then lysed in nuclear extraction (NE) buffer (20 mM HEPES (pH 7.4), 0.4 M NaCl, 25% glycerol, 1 mM EDTA, 1 mM PMSF, 0.5 mM DTT) by repeated cycles of freezing in liquid nitrogen followed by thawing at 37°C. The supernatant containing soluble nuclear proteins was obtained by centrifugation at 20,000 g for 20 min.
In situ hybridization
Whole‐mount in situ hybridization was performed with digoxigenin (DIG)‐labeled probes as described 24. Anti‐sense RNA in situ probes were in vitro synthesized using the plasmids containing cDNAs, linearizing restriction enzymes, and SP6 or T7 RNA polymerase as specified: Wip1, pGEM‐T‐Wip1 (NcoI, SP6); Chordin, pGEM‐T‐Chordin (SpeI, T7); Sox2, pGEM‐T‐Sox2 (BamHI, SP6); E. keratin, pGEM‐T‐E. keratin (NcoI, SP6); Xbra, pXbra (XhoI, SP6); Goosecoid, pG500 (BamHI, T3); and Xvent1, pBluescript‐Xvent1 (SalI, T7). Hybridization was detected with an alkaline phosphatase (AP)‐conjugated anti‐digoxigenin antibody and visualized using BM purple as a substrate (Roche).
Reporter gene assay
Cultured cells were homogenized in 1× Passive Lysis Buffer (Promega). Reporter activities were measured using Dual‐Luciferase Reporter Assay System (Promega) according to the manufacture's protocol. All values were normalized for transfection efficiency against the activity of pRL‐TK Renilla luciferase. All experiments were performed in triplicate and repeated three times.
Statistical analysis
Experimental data were analyzed using t‐test in Microsoft Excel software. A P‐value of < 0.05 was considered as significant.
Author contributions
S‐CC conceived the study and supervised the project. D‐SP, E‐YK, TL, G‐HY, and KK designed and performed the experiments. S‐CC, E‐JC, PCWL, E‐YK, and D‐SP analyzed and discussed the data. S‐CC and D‐SP wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
This work was supported by Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Science, ICT, & Future Planning (2016R1A2B4013355 and 2019R1H1A2080117).
EMBO Reports (2020) 21: e48693
See also: https://doi.org/10.15252/embr.202050246 (May 2020)
References
- 1. Massague J (2012) TGFbeta signalling in context. Nat Rev Mol Cell Biol 13: 616–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wakefield LM, Hill CS (2013) Beyond TGFbeta: roles of other TGFbeta superfamily members in cancer. Nat Rev Cancer 13: 328–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xu J, Wang AH, Oses‐Prieto J, Makhijani K, Katsuno Y, Pei M, Yan L, Zheng YG, Burlingame A, Bruckner K et al (2013) Arginine methylation initiates BMP‐induced Smad signaling. Mol Cell 51: 5–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Katsuno Y, Qin J, Oses‐Prieto J, Wang H, Jackson‐Weaver O, Zhang T, Lamouille S, Wu J, Burlingame A, Xu J et al (2018) Arginine methylation of SMAD7 by PRMT1 in TGF‐beta‐induced epithelial‐mesenchymal transition and epithelial stem‐cell generation. J Biol Chem 293: 13059–13072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wrighton KH, Lin X, Feng XH (2009) Phospho‐control of TGF‐beta superfamily signaling. Cell Res 19: 8–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Duan X, Liang YY, Feng XH, Lin X (2006) Protein serine/threonine phosphatase PPM1A dephosphorylates Smad1 in the bone morphogenetic protein signaling pathway. J Biol Chem 281: 36526–36532 [DOI] [PubMed] [Google Scholar]
- 7. Lin X, Duan X, Liang YY, Su Y, Wrighton KH, Long J, Hu M, Davis CM, Wang J, Brunicardi FC et al (2006) PPM1A functions as a Smad phosphatase to terminate TGFbeta signaling. Cell 125: 915–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xu P, Lin X, Feng XH (2016) Posttranslational regulation of Smads. Cold Spring Harb Perspect Biol 8: a022087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fuentealba LC, Eivers E, Ikeda A, Hurtado C, Kuroda H, Pera EM, De Robertis EM (2007) Integrating patterning signals: Wnt/GSK3 regulates the duration of the BMP/Smad1 signal. Cell 131: 980–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alarcon C, Zaromytidou AI, Xi Q, Gao S, Yu J, Fujisawa S, Barlas A, Miller AN, Manova‐Todorova K, Macias MJ et al (2009) Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF‐beta pathways. Cell 139: 757–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roelen BA, Cohen OS, Raychowdhury MK, Chadee DN, Zhang Y, Kyriakis JM, Alessandrini AA, Lin HY (2003) Phosphorylation of threonine 276 in Smad4 is involved in transforming growth factor‐beta‐induced nuclear accumulation. Am J Physiol Cell Physiol 285: C823–C830 [DOI] [PubMed] [Google Scholar]
- 12. Demagny H, Araki T, De Robertis EM (2014) The tumor suppressor Smad4/DPC4 is regulated by phosphorylations that integrate FGF, Wnt, and TGF‐beta signaling. Cell Rep 9: 688–700 [DOI] [PubMed] [Google Scholar]
- 13. Wu Y, Yu X, Yi X, Wu K, Dwabe S, Atefi M, Elshimali Y, Kemp KT II, Bhat K, Haro J et al (2017) Aberrant phosphorylation of SMAD4 Thr277‐mediated USP9x‐SMAD4 interaction by free fatty acids promotes breast cancer metastasis. Cancer Res 77: 1383–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Albrecht LV, Ploper D, Tejeda‐Munoz N, De Robertis EM (2018) Arginine methylation is required for canonical Wnt signaling and endolysosomal trafficking. Proc Natl Acad Sci USA 115: E5317–E5325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Warmflash A, Zhang Q, Sorre B, Vonica A, Siggia ED, Brivanlou AH (2012), Vol. 109, pp E1947–E1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhu YH, Bulavin DV (2012) Wip1‐dependent signaling pathways in health and diseases. Prog Mol Biol Transl Sci 106: 307–325 [DOI] [PubMed] [Google Scholar]
- 17. Lowe J, Cha H, Lee MO, Mazur SJ, Appella E, Fornace AJ Jr (2012) Regulation of the Wip1 phosphatase and its effects on the stress response. Front Biosci 17: 1480–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Demidov ON, Timofeev O, Lwin HN, Kek C, Appella E, Bulavin DV (2007) Wip1 phosphatase regulates p53‐dependent apoptosis of stem cells and tumorigenesis in the mouse intestine. Cell Stem Cell 1: 180–190 [DOI] [PubMed] [Google Scholar]
- 19. Lu X, Nguyen TA, Moon SH, Darlington Y, Sommer M, Donehower LA (2008) The type 2C phosphatase Wip1: an oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metastasis Rev 27: 123–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Le Guezennec X, Bulavin DV (2010) WIP1 phosphatase at the crossroads of cancer and aging. Trends Biochem Sci 35: 109–114 [DOI] [PubMed] [Google Scholar]
- 21. Agius E, Oelgeschlager M, Wessely O, Kemp C, De Robertis EM (2000) Endodermal Nodal‐related signals and mesoderm induction in Xenopus . Development 127: 1173–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Datto MB, Li Y, Panus JF, Howe DJ, Xiong Y, Wang XF (1995) Transforming growth factor beta induces the cyclin‐dependent kinase inhibitor p21 through a p53‐independent mechanism. Proc Natl Acad Sci USA 92: 5545–5549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meulmeester E, Ten Dijke P (2011) The dynamic roles of TGF‐beta in cancer. J Pathol 223: 205–218 [DOI] [PubMed] [Google Scholar]
- 24. Yun CH, Choi SC, Park E, Kim SJ, Chung AS, Lee HK, Lee HJ, Han JK (2007) Negative regulation of Activin/Nodal signaling by SRF during Xenopus gastrulation. Development 134: 769–777 [DOI] [PubMed] [Google Scholar]
- 25. Branford WW, Yost HJ (2002) Lefty‐dependent inhibition of Nodal‐ and Wnt‐responsive organizer gene expression is essential for normal gastrulation. Curr Biol 12: 2136–2141 [DOI] [PubMed] [Google Scholar]
- 26. Cordenonsi M, Dupont S, Maretto S, Insinga A, Imbriano C, Piccolo S (2003) Links between tumor suppressors: p53 is required for TGF‐beta gene responses by cooperating with Smads. Cell 113: 301–314 [DOI] [PubMed] [Google Scholar]
- 27. Moorhead GB, Trinkle‐Mulcahy L, Ulke‐Lemee A (2007) Emerging roles of nuclear protein phosphatases. Nat Rev Mol Cell Biol 8: 234–244 [DOI] [PubMed] [Google Scholar]
- 28. Chen CR, Kang Y, Massague J (2001) Defective repression of c‐myc in breast cancer cells: a loss at the core of the transforming growth factor beta growth arrest program. Proc Natl Acad Sci USA 98: 992–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Demagny H, De Robertis EM (2016) Point mutations in the tumor suppressor Smad4/DPC4 enhance its phosphorylation by GSK3 and reversibly inactivate TGF‐beta signaling. Mol Cell Oncol 3: e1025181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sapkota G, Alarcon C, Spagnoli FM, Brivanlou AH, Massague J (2007) Balancing BMP signaling through integrated inputs into the Smad1 linker. Mol Cell 25: 441–454 [DOI] [PubMed] [Google Scholar]
- 31. Plotnikov A, Zehorai E, Procaccia S, Seger R (2011) The MAPK cascades: signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim Biophys Acta 1813: 1619–1633 [DOI] [PubMed] [Google Scholar]
- 32. Dupont S, Mamidi A, Cordenonsi M, Montagner M, Zacchigna L, Adorno M, Martello G, Stinchfield MJ, Soligo S, Morsut L et al (2009) FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell 136: 123–135 [DOI] [PubMed] [Google Scholar]
- 33. Schmierer B, Tournier AL, Bates PA, Hill CS (2008) Mathematical modeling identifies Smad nucleocytoplasmic shuttling as a dynamic signal‐interpreting system. Proc Natl Acad Sci USA 105: 6608–6613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pierreux CE, Nicolas FJ, Hill CS (2000) Transforming growth factor beta‐independent shuttling of Smad4 between the cytoplasm and nucleus. Mol Cell Biol 20: 9041–9054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Biondi CA, Das D, Howell M, Islam A, Bikoff EK, Hill CS, Robertson EJ (2007) Mice develop normally in the absence of Smad4 nucleocytoplasmic shuttling. Biochem J 404: 235–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Choi DW, Na W, Kabir MH, Yi E, Kwon S, Yeom J, Ahn JW, Choi HH, Lee Y, Seo KW et al (2013) WIP1, a homeostatic regulator of the DNA damage response, is targeted by HIPK2 for phosphorylation and degradation. Mol Cell 51: 374–385 [DOI] [PubMed] [Google Scholar]
- 37. Zhang X, Wan G, Mlotshwa S, Vance V, Berger FG, Chen H, Lu X (2010) Oncogenic Wip1 phosphatase is inhibited by miR‐16 in the DNA damage signaling pathway. Cancer Res 70: 7176–7186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li Y, Liu Y, Chiang YJ, Huang F, Li Y, Li X, Ning Y, Zhang W, Deng H, Chen YG (2019) DNA damage activates TGF‐beta signaling via ATM‐c‐Cbl‐mediated stabilization of the type II receptor TbetaRII. Cell Rep 28: 735–745 e734 [DOI] [PubMed] [Google Scholar]
- 39. Nannenga B, Lu X, Dumble M, Van Maanen M, Nguyen TA, Sutton R, Kumar TR, Donehower LA (2006) Augmented cancer resistance and DNA damage response phenotypes in PPM1D null mice. Mol Carcinog 45: 594–604 [DOI] [PubMed] [Google Scholar]
- 40. He XC, Zhang J, Tong WG, Tawfik O, Ross J, Scoville DH, Tian Q, Zeng X, He X, Wiedemann LM et al (2004) BMP signaling inhibits intestinal stem cell self‐renewal through suppression of Wnt‐beta‐catenin signaling. Nat Genet 36: 1117–1121 [DOI] [PubMed] [Google Scholar]
- 41. Qi Z, Li Y, Zhao B, Xu C, Liu Y, Li H, Zhang B, Wang X, Yang X, Xie W et al (2017) BMP restricts stemness of intestinal Lgr5(+) stem cells by directly suppressing their signature genes. Nat Commun 8: 13824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Curran KL, Grainger RM (2000) Expression of activated MAP kinase in Xenopus laevis embryos: evaluating the roles of FGF and other signaling pathways in early induction and patterning. Dev Biol 228: 41–56 [DOI] [PubMed] [Google Scholar]
- 43. LaBonne C, Whitman M (1997) Localization of MAP kinase activity in early Xenopus embryos: implications for endogenous FGF signaling. Dev Biol 183: 9–20 [DOI] [PubMed] [Google Scholar]
- 44. Nieuwkoop PD, Faber J (1994) Normal table of Xenopus laevis (Daudin): a systematical and chronological survey of the development from the fertilized egg till the end of metamorphosispp Garland Pub.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7