

Abstract
RNA processing occurs co‐transcriptionally through the dynamic recruitment of RNA processing factors to RNA polymerase II (RNAPII). However, transcriptome‐wide identification of protein–RNA interactions specifically assembled on transcribing RNAPII is challenging. Here, we develop the targeted RNA immunoprecipitation sequencing (tRIP‐seq) method that detects protein–RNA interaction sites in thousands of cells. The high sensitivity of tRIP‐seq enables identification of protein–RNA interactions at functional subcellular levels. Application of tRIP‐seq to the FUS‐RNA complex in the RNAPII machinery reveals that FUS binds upstream of alternative polyadenylation (APA) sites of nascent RNA bound to RNAPII, which retards RNAPII and suppresses the recognition of the polyadenylation signal by CPSF. Further tRIP‐seq analyses demonstrate that the repression of APA is achieved by a complex composed of FUS and U1 snRNP on RNAPII, but not by either one alone. Moreover, our analysis reveals that FUS mutations in familial amyotrophic lateral sclerosis (ALS) that impair the FUS‐U1 snRNP interaction aberrantly activate the APA sites. tRIP‐seq provides new insights into the regulatory mechanism of co‐transcriptional RNA processing by RNA processing factors.
Keywords: CLIP, FUS, RNA polymerase II, tRIP, U1 snRNP
Subject Categories: Methods & Resources, RNA Biology
The targeted RNA immunoprecipitation, tRIP, method enables identification of protein‐RNA interactions and provides insight into FUS and U1 snRNP‐mediated control of alternative polyadenylation (APA) during co‐transcriptional RNA processing.
Introduction
RNA processing is a finely tuned mechanism involving the production of mature mRNA 1. Following the initiation of transcription by RNA polymerase II (RNAPII), processing of nascent RNA, including 5′ end capping, splicing, and 3′ end processing, occurs co‐transcriptionally on the transcribing RNAPII 2. Each step in the processing is tightly regulated and processed by a distinct complex comprising multiple proteins and RNA, such as the 5′ capping complex, the spliceosome, and the 3′ end processing machinery. These complexes are recruited to the RNAPII machinery through the dynamic regulation of the phosphorylation pattern of C‐terminal domain (CTD) of RNAPII 3, 4.
On RNAPII, a diverse array of RNA‐binding proteins (RBPs) bind to distinct sites of nascent RNA in a competitive or cooperative manner to regulate the co‐transcriptional RNA processing. A given RBP often operates across a wide range of machineries and participates in various types of RNA processing 1, making it difficult to identify protein–RNA interaction sites specifically assembled on transcribing RNAPII. For example, FUS, EWS, TAF15, and TDP‐43 are RBPs causally associated with amyotrophic lateral sclerosis (ALS) 5. These RBPs are localized in the spliceosome 6 and in the microRNA‐processing machinery 7 to regulate alternative splicing 5, 8, 9 and miRNA production 9, 10, 11, respectively. TDP‐43 and FUS also participate in mRNA transport and local translation in the cytoplasm 9. Interactome analysis using mass spectrometry has revealed robust associations of FUS, EWS, and TAF15 with U1 snRNP 12, an essential spliceosomal complex. Furthermore, FUS, EWS, and TAF15 also directly interact with RNAPII 12, 13, 14, 15. Recent advances in cross‐linking immunoprecipitation (CLIP) methodology have helped disclose endogenous protein–RNA interaction sites in living cells. However, most CLIP methods require tens of millions of cells, which hinders identification of protein–RNA interactions in a specific RNA machinery within a specific subcellular fraction.
U1 snRNP is composed of U1 snRNA, Sm proteins, and three U1‐specific proteins, i.e., U1A, U1C, and U1‐70K 16. U1 snRNP recognizes the 5′ splice site through base‐pairing interactions with the 5′ end of the U1 snRNA and the 5′ splice site 17. Interestingly, lack of U1 snRNP rapidly eliminates upstream antisense RNAs arising from bidirectional transcription 18. In addition, Dreyfuss and colleagues demonstrated that U1 snRNP globally suppresses alternative polyadenylation (APA) to prevent shortening of mRNA length 19, 20. The suppression of APA by U1 snRNP promotes the production of full‐length mRNAs, which is more prevalent in longer genes 21. A recent study identified that U1 snRNP additionally associates with the 3′ end processing factors to suppress APA 22. Although binding of U1 snRNA to nascent RNA is required for the suppression of APA, specific positions of base pairings that suppress APA are not well dissected 23. In addition, molecule(s) involved in the specific base pairing remain mostly unelucidated.
An ALS‐associated RBP, FUS, is involved in a variety of RNA metabolic processes, such as transcription, splicing, polyadenylation, and transport of RNA 5. FUS is directly associated with U1 snRNP 24, 25 as well as with CTD of the largest RNAPII subunit 13. FUS‐binding sites are enriched around alternative splice (AS) sites 26 and APA sites 27 to regulate the respective RNA processing events. Our previous CLIP‐seq analysis using whole cells identified that binding of FUS downstream and upstream to APA sites enhances and represses APA, respectively 27. Furthermore, minigene analysis revealed that the downstream FUS‐binding recruits CPSF160 to nascent RNA to promote APA. However, three questions remain unsolved: (i) Which subcellular/subnuclear fraction is involved in the APA regulation, (ii) How polyadenylation is repressed when FUS binds upstream to APA, and (iii) Why FUS and U1 snRNP exhibit a similar APA‐repressing activity?
In an effort to investigate transcriptome‐wide protein–RNA interactions in a specific machinery within a specific subcellular fraction, we developed the targeted RNA immunoprecipitation sequencing (tRIP‐seq) method, which detected the UV‐crosslinked protein–RNA interaction sites from thousands of cells. In addition, the high sensitivity of tRIP‐seq enabled identification of RBP‐binding sites of serially immunoprecipitated protein–protein–RNA complex isolated from a subnuclear fraction. Application of tRIP‐seq to RNAPII and FUS revealed that FUS binds upstream to APA sites of nascent RNA under transcription by RNAPII in the chromatin fraction. In addition, tRIP‐seq of U1 snRNP showed the recruitment of U1 snRNP upstream to similar APA sites of nascent RNA to suppress APA. Integrated analysis of tRIP‐seq and polyA‐seq revealed that depletion of either FUS or U1 snRNP disrupts the binding of its counterpart to nascent RNA and that each depletion activates APA to a similar extent. Our analysis demonstrated that both FUS and U1 snRNP are indispensable constituents of the RNAPII machinery to suppress APA in co‐transcriptional RNA processing.
Results
Development of tRIP‐seq
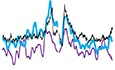
During the examination of enzymes that degrade RNA, we noticed that the nucleolytic activity of the terminator 5′‐phosphate‐dependent exonuclease (TEX) is blocked by insertion of a biotinylated nucleotide (Appendix Fig S1A). TEX is a processive 5′‐3′ exonuclease, which specifically degrades single‐stranded RNA/DNA with a 5′ monophosphate end. We found that TEX treatment degraded an RNA/DNA hybrid probe of 32 nucleotides (nt) carrying the 5′ monophosphate end (Appendix Fig S1A, lanes 3 and 4). Attachment of biotin in the middle of the probe blocked TEX‐dependent degradation of the probe at the biotinylated nucleotide (Appendix Fig S1A, lanes 1 and 2). We expected that tethering of a protein on RNA would protect the protein‐bearing RNA from TEX‐dependent digestion, whereas protein‐free RNA would be efficiently eliminated (Fig 1A).
Figure 1. Identification of protein–RNA interactions by tRIP‐seq.
- Schematic representation of terminator 5′‐phosphate‐dependent exonuclease (TEX) treatment of immunoprecipitated RNA. TEX digestion stops at the antibody‐tethered nucleotide, while TEX eliminates the non‐specific RNA and linkers remaining on the beads. Green circle represents m6A nucleotide.
- Distribution of RBFOX2‐eCLIP reads and RBFOX2‐tRIP reads on chr10: 75,544,000–75,635,000 (GRCh37/hg19).
- Distributions of FUS‐HITS‐CLIP reads, FUS‐tRIP reads, and ContAb‐tRIP reads on chr15: 26,165,000–28,520,000 (GRCm38/mm10). The number of N2A cells is indicated.
- Correlation of read densities constituting MACS‐defined peaks between RBFOX2‐tRIP and RBFOX2‐eCLIP. Scatter plot indicates RPM of tRIP‐seq and eCLIP constituting each peak.
- Correlation in read densities constituting MACS‐defined peaks between FUS‐tRIP and FUS‐HITS‐CLIP. Scatter plot indicates RPM of tRIP‐seq and HITS‐CLIP constituting each peak.
- The RBFOX2 motif identified by MEME in the peaks of RBFOX2‐tRIP.
- The FUS motif identified by MEME in the peaks of FUS‐tRIP of 1 × 108 cells.
- Distributions of FUS‐tRIP reads (upper panel) and ContAb‐tRIP reads (lower panel) mapped to the relative positions of all coding genes in mouse. The ngs.plot tool 58 was used to calculate the average RPM for a gene structure. The average RPM at each position was normalized based on the total RPM mapped to each gene. The standard error of normalized RPM is shown as a semi‐transparent shade around the average curve. TSS, transcriptional start site; TTS, transcriptional termination site.
We applied TEX treatment to RBP‐tethered RNA fragments, which were immunoprecipitated with a specific antibody against the RBP (tRIP; Fig EV1, see Materials and Methods). An RBP was UV‐crosslinked to RNA and immunoprecipitated by a specific antibody. The antibody–protein–RNA complex was tethered on Protein G beads, fragmented by endonucleases, and treated by TEX. We confirmed that TEX treatment efficiently eliminated free linkers and non‐specific RNA remaining on beads (Appendix Fig S1B), suggesting that the contamination of non‐specific RNA was low in tRIP. In addition, the lack of UV crosslinking (Appendix Fig S1C) as well as that of an antibody specific to an RBP (Appendix Fig S1D) efficiently eliminated RNA, suggesting that the UV crosslinking of a protein on an RNA prevents the TEX‐dependent elimination of the RNA, as expected.
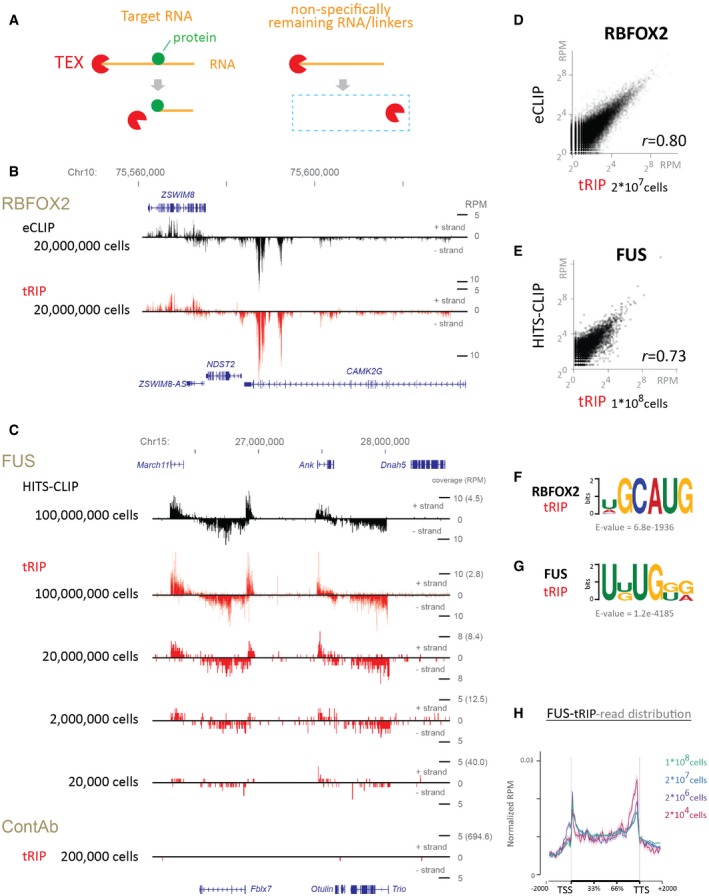
Figure EV1. Schematic representation of the tRIP workflow.
RNA‐binding protein (RBP)–RNA complex is immunoprecipitated with an antibody against the RBP. Then, the immunoprecipitated RNA is partially digested with RNase on beads. After stringent washes, a 64‐nt linker is ligated to the 3′ end of the RNA fragment, and immunoprecipitants are treated with TEX to eliminate non‐specifically remaining RNA and free linkers. The TEX digests immunoprecipitated RNA up to the RBP‐tethered site. The immunoprecipitants are then treated with proteinase K, followed by column purification of RNA. After the first‐strand synthesis with reverse transcription, a polyA tail is added to the 3′ end of the first‐strand cDNA. After the second‐strand synthesis with the tagging primer, the double‐strand cDNAs are PCR‐amplified and subjected to high‐throughput sequencing analysis.
We applied tRIP to well‐characterized RBPs, RBFOX2, and FUS, using 2 × 107 HEK293T cells and 1 × 108 N2A cells, respectively, and performed high‐throughput sequencing analysis (tRIP‐seq). The mapped tRIP reads of RBFOX2 and FUS were compared with those of eCLIP of RBFOX2 (Gene Expression Omnibus accession number, GSM2055434) 28 and HITS‐CLIP of FUS (DDBJ Sequence Read Archive accession number, DRR014227) 27, respectively. We used the same cell lines and the same cell numbers for the respective comparisons.
We observed that the reads were similarly distributed on representative regions in tRIP‐seq and eCLIP of RBFOX2 (Fig 1B), and in tRIP‐seq and HITS‐CLIP of FUS (Fig 1C). The correlation coefficient (r) between the number of reads comprising a MACS‐defined tRIP‐seq peak and that of an eCLIP peak was 0.80 for RBFOX2 (Fig 1D). Similarly, the correlation coefficient (r) between tRIP‐seq and HITS‐CLIP was 0.73 for FUS (Fig 1E). Motif analysis of RBFOX2‐tRIP revealed the enrichment of a previously reported motif of GCAUG 29 (Fig 1F). The RBFOX2 motif was markedly concentrated at the 5′ ends of RBFOX2‐tRIP reads (Appendix Fig S1E), as in iCLIP 30. We previously reported that FUS‐binding motifs are highly degenerative with GU enrichment 31, and motif analysis of FUS‐tRIP indeed showed a GU‐rich motif (Fig 1G). Thus, tRIP‐seq was able to detect RBP‐binding sites, as in CLIP.
Efficient identification of protein–RNA interaction sites by tRIP‐seq
CLIP requires SDS–PAGE and membrane transfer followed by the excision and purification of a specific protein–RNA complex, whereas tRIP does not require these steps (Appendix Table S1). In tRIP, a cDNA library is generated by a single RNA purification step. In addition, the library preparation is performed in a single tube, similar to the single‐cell RNA‐seq analysis 32, to minimize the loss of RNA. These modifications simplify the protocol and increase sensitivity. The tRIP method requires only 2 days to generate a cDNA library with a single RNA purification step (Appendix Table S1). In addition, to make cDNA libraries, eCLIP of RBFOX2 required 16 PCR cycles 28, whereas five independent replicates of tRIP required on average 10.2 PCR cycles from the same number of HEK 293T cells (Appendix Fig S1F). Similarly, HITS‐CLIP of FUS required 21 PCR cycles, whereas tRIP required 12 PCR cycles from the same number of N2A cells (Appendix Fig S1F). The reduced number of PCR cycles was expected to increase the number of unique reads and reduce the amount of input RNA. Although decreasing the number of cells lowered the number of uniquely mapped reads (Fig 1C, Appendix Fig S1G), tRIP reads were similarly mapped to each gene, irrespective of the number of cells (Fig 1H). Accumulations of FUS around transcriptional start sites (TSS) and transcriptional termination sites (TTS) were observed, as previously reported 13, 26. When immunoprecipitation was performed using control antibody from 2 × 105 N2A cells (ContAb‐tRIP), no distinct peaks were observed (Fig 1C).
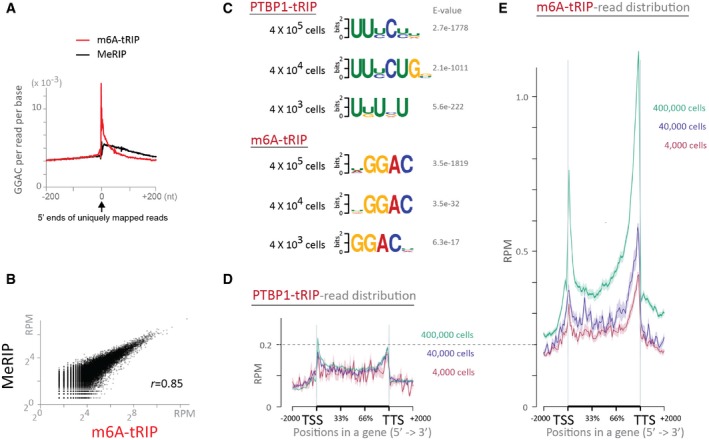
We also performed tRIP‐seqs of polypyrimidine tract‐binding protein (PTBP1), a ubiquitously expressed RBP, using 4 × 105, 4 × 104, and 4 × 103 C2C12 cells. In addition, we performed tRIP‐seqs of N6‐methyladenosine (m6A) using same numbers of C2C12 cells without UV crosslinking. Similar to the UV‐crosslinked protein on RNA, binding of anti‐m6A antibody at an m6A nucleotide prevented the TEX‐dependent RNA digestion, which was indicated by the prominent concentration of the m6A consensus motif of GGAC 33 at the 5′ ends of the m6A‐tRIP reads (Fig EV2A). Positions of m6A‐tRIP peaks were similar to those of methylated RNA immunoprecipitation (MeRIP) peaks with anti‐m6A antibody 33 (Fig EV2B). Motif analysis detected the CU‐rich PTBP1 motifs in PTBP1‐tRIPs and m6A motif of GCAG in m6A‐tRIPs (Fig EV2C). The PTBP1‐tRIP reads were enriched at both the 5′ and 3′ ends of a gene (Fig EV2D). In contrast, the m6A‐tRIP reads were enriched more at the 3′ end of a gene than at the 5′ end of the gene (Fig EV2E), as previously reported 34. Taken together, tRIP‐seq has high sensitivity with low background, which enables detection of PTBP1‐binding sites and m6A‐modification sites using thousands of cells. Although less RNA input resulted in detection of fewer protein‐binding sites, spatial profiles of detected sites remained essentially unchanged.
Figure EV2. Read distributions of m6A‐tRIP and PTBP1‐tRIP .
-
AThe enrichment of m6A motif (GGAC) around the 5′ ends of mapped reads obtained from m6A‐tRIP and m6A‐MeRIP (RNA‐seq of conventional RNA immunoprecipitation using At‐m6A antibody).
-
BCorrelation of read densities constituting MACS‐defined peaks between m6A‐MeRIP and m6A‐tRIP. Scatter plot indicates reads per million mapped reads (RPM) of tRIP and MeRIP constituting each peak. Pearson's correlation coefficient (r) is indicated.
-
CThe CU‐rich PTBP1 motifs (upper panels) and the m6A motifs (lower panels) identified by MEME in the peaks of PTBP1‐tRIPs and m6A‐tRIPs, respectively, performed using indicated number of C2C12 cells.
-
D, EDistributions of PTBP1‐tRIP reads (D) and m6A‐tRIP reads (E) mapped to the relative positions of all mouse coding genes. The ngs.plot tool 58 was used to calculate average RPM on a gene structure. Shown are tRIP‐seqs of indicated number of C2C12 cells. The standard error of average RPM is shown as a semi‐transparent shade around the average curve.
Identification of protein–nascent RNA interaction sites in the RNAPII machinery by tRIP‐seq
We next employed tRIP‐seq to explore RBP–nascent RNA interactions that are co‐transcriptionally formed on RNAPII. Following UV crosslinking of protein–RNA interactions, the chromatin fraction was isolated from the cells, and the RNAPII machinery was immunoprecipitated using an antibody against RNAPII β subunit (Fig 2A). We observed that various RBPs, including FUS, the components of U1 snRNP, and the 3′ end processing factors, were co‐immunoprecipitated with RNAPII (Fig 2B) from the UV‐crosslinked lysate (Fig 2B, lanes 1 and 3) in an RNA‐dependent manner (Fig 2B, lanes 1 and 2), demonstrating the role of these factors on the processing of nascent RNA under transcription, such as co‐transcriptional splicing 35 and 3′ end formation 36. These results indicated that protein–RNA–RNAPII complexes were isolated as expected.
Figure 2. Identification of protein–RNA interactions in the RNAPII machinery by tRIP‐seq.
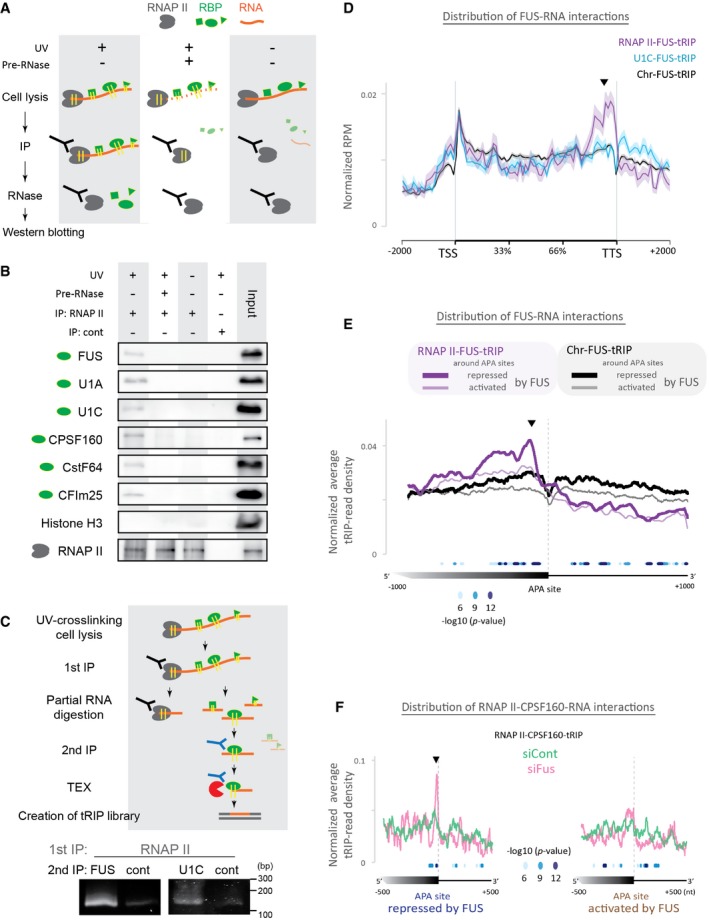
- Schematic representation of immunoprecipitation of protein–RNA complexes engaged to RNAPII shown in (B). With UV crosslinking of protein–RNA interactions (UV+) or without (UV−), the chromatin fraction is isolated from the cells and treated with (Pre‐RNase+) or without RNase A (Pre‐RNase−). RNAPII, shown in gray, is 1st immunoprecipitated. The immunoprecipitants are treated with RNase A to release a protein, shown in green, from the protein–RNA–RNAPII complex.
- RNAPII‐RBP‐RNA complex was immunoprecipitated by anti‐RNAPII antibody or control antibody (cont) and subjected to immunoblotting. The input lanes contained 0.4% of lysates used in the immunoprecipitation experiments.
- Generation of tRIP‐seq libraries of RNAPII‐RBP‐RNA complexes. After UV crosslinking, protein–RNA complexes were immunoprecipitated with anti‐RNAPII (1st IP). RNA of the isolated RNAPII‐RBP‐RNA complexes was partially digested with RNase on beads, and RBP‐RNA complexes dissociated from the bead were further immunoprecipitated with anti‐FUS or anti‐U1C antibody bound to the other beads (2nd IP). The immunoprecipitated FUS‐RNA or U1C‐RNA complex was treated with TEX and then subjected to tRIP‐seq library generation. The bottom panels show PCR‐amplification of tRIP‐seq libraries generated from RNAPII‐FUS‐RNA complex (left panel) and RNAPII‐U1C‐RNA complex (right panel).
- Read distributions of Chr‐FUS‐tRIP, U1C‐FUS‐tRIP, and RNAPII‐FUS‐tRIP mapped to the relative positions of all coding genes in mouse. The ngs.plot tool 58 was used to calculate the average RPM for a gene structure. The average RPM at each position was normalized for the total RPM mapped to each gene. The standard error of normalized RPM is shown as a semi‐transparent shade around the average curve. Arrowhead indicates a peak before the transcription termination site (TTS) in RNAPII‐FUS‐tRIP.
- Read distributions of RNAPII‐FUS‐tRIP (purple lines) and Chr‐FUS‐tRIP (black lines) around the APA sites repressed (bold lines) or activated (thin lines) by FUS. The P‐values for the differences between RNAPII‐FUS‐tRIP and Chr‐FUS‐tRIP around FUS‐repressed APA sites are indicated by circles. An arrowhead indicates a peak upstream to APA sites in RNAPII‐FUS‐tRIP.
- Read distributions of RNAPII‐CPSF160‐tRIPs generated from Fus‐silenced cells (siFus, pink line) and those of control siRNA‐treated cells (siCont, green line). The P‐values for the differences between siFus and siCont are indicated by circles. An arrowhead indicates a peak before APA sites in RNAPII‐CPSF160‐tRIP of siFus‐treated cells.
We next exploited downscaling of the required amount of protein–RNA complex in tRIP to construct a tRIP library from the immunoprecipitated RNAPII complex (Fig 2C). First, the RNAPII complex was partially digested with RNase. Then, the released protein–RNA complex was immunoprecipitated with FUS or U1C. Finally, the immunoprecipitated protein–RNA complex was treated with TEX to construct a tRIP library. We confirmed that tRIP libraries were generated only in the presence of a specific antibody against FUS or U1C (Fig 2C). Thus, tRIP enabled serial immunoprecipitations of RNAPII followed by another (FUS or U1C).
Following this procedure, we performed tRIP‐seq of FUS in the RNAPII machinery (RNAPII‐FUS‐tRIP). We similarly performed tRIP‐seq of FUS in the U1 snRNP machinery, in which a tRIP library was generated by serial immunoprecipitations by U1C and FUS (U1C‐FUS‐tRIP; Appendix Fig S2AB). For comparison, we also performed tRIP‐seq of FUS in the chromatin fraction (Chr‐FUS‐tRIP). We found that reads of Chr‐FUS‐tRIP and U1C‐FUS‐tRIP were similarly distributed on a gene, whereas reads of RNAPII‐FUS‐tRIP made a cluster near the 3′ ends of genes (Fig 2D), indicating that RNAPII‐associated FUS has unique RNA‐binding sites. As FUS is involved in the regulation of APA 27, we next analyzed the read distributions of these tRIP‐seqs around APA sites. We reanalyzed previously reported polyA‐seq of N2A cells (DDBJ accession number, DRA002447) 27 and found that Fus silencing upregulated 26,764 APA sites (FUS‐repressed APA sites) and downregulated 46,581 APA sites (FUS‐activated APA sites) by more than twofold. We found that RNAPII‐FUS‐tRIP reads were enriched upstream to the FUS‐repressed APA sites compared to Chr‐FUS‐tRIP reads (purple lines in Fig 2E and Appendix Fig S2C), suggesting that FUS is recruited upstream to APA sites to repress APA in the RNAPII machinery. In contrast, RNAPII‐FUS‐tRIP reads were rapidly diminished downstream to APA sites compared to Chr‐FUS‐tRIP reads (purple lines in Fig 2E and Appendix Fig S2C). This is likely because the downstream FUS‐RNA interactions are formed after the dissociation of RNA from RNAPII by the cleavage at polyadenylation sites during 3′ end processing.
We next dissected how the upstream binding of FUS represses APA using three additional tRIP‐seq experiments in the presence and absence of FUS. First, we observed that RNAPII‐tRIP showed accumulation of RNAPII upstream to the FUS‐repressed APA sites, which was obscured by Fus silencing (Fig EV3A). Second, we performed tRIP‐seq of CPSF160 in the RNAPII machinery (RNAPII‐CPSF160‐tRIP, Fig 2F). CPSF160 is a core component of the cleavage and polyadenylation specificity factor (CPSF) 37, and makes a complex with FUS 25, 27. RNAPII‐CPSF160‐tRIP showed that Fus silencing induced marked binding of CPSF160 at ~20 nucleotides (nt) upstream to the FUS‐repressed APA sites (Fig 2F, pink line), where polyadenylation (polyA) signals were located (Fig EV3B, blue line). Third, tRIP‐seq of CPSF160 in the chromatin fraction (Chr‐CPSF160‐tRIP) similarly showed that Fus silencing induced the binding of CPSF160 upstream to the FUS‐repressed APA sites (Fig EV3C, pink line). Interestingly, the peaks of FUS, RNAPII, and CPSF160 were ~20 nucleotides (nt) apart (Fig EV3B), which may represent a temporal profile of binding of these proteins in the course of transcription.
Figure EV3. The recruitment of FUS upstream to polyA sites slows down transcription and inhibits the recognition of polyA signal by CPSF .
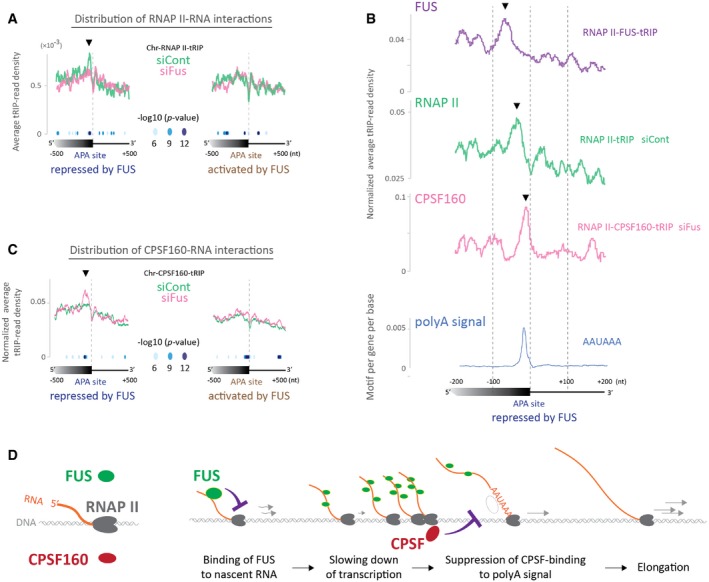
- Read distributions of Chr‐RNAP II‐tRIPs of Fus‐silenced cells (siFus, pink line) and control siRNA‐treated cells (siCont, green line) around APA sites repressed (left graph) or activated (right graph) by FUS. The P‐values for the differences between siFus and siCont are indicated by circles. An arrowhead indicates a peak upstream to the repressed APA sites detected in Chr‐RNAP II‐tRIP of siCont‐treated cells.
- Expanded view of read distributions of RNAP II‐FUS‐tRIP (Fig 2E, bold purple line), RNAP II‐tRIP of siCont‐treated cells (Fig EV3A, green line), and RNAP II‐CPSF160‐tRIP of Fus‐silenced cells (Fig 2F, pink line) around FUS‐repressed APA sites. The bottom graph shows frequency of the AAUAAAA polyadenylation signal (blue line). Arrowheads indicate noticeable peaks detected in the respective tRIP‐seqs.
- Read distributions of Chr‐CPSF160‐tRIPs of Fus‐silenced cells (siFus, pink line) and control siRNA‐treated cells (siCont, green line) around APA sites repressed (left graph) or activated (right graph) by FUS. The P‐values for the differences between siFus and siCont are indicated by circles. An arrowhead indicates a peak upstream to the repressed APA sites detected in Chr‐CPSF160‐tRIP of Fus‐silenced cells.
- A proposed model for FUS‐dependent repression of APA. The recruitment of FUS upstream to polyA sites slows down transcription by RNAP II and inhibits the recognition of polyA signal by CPSF, which suppresses subsequent APA and keeps elongation by RNAP II.
In summary, tRIP‐seq improved the resolution of FUS‐RNA interactions to a co‐transcriptional level and disclosed that recruitment of FUS upstream to APA induces accumulation of RNAPII and inhibits the recognition of a polyA signal by CPSF, which suppresses activation of APA to make a longer transcript (Fig EV3D).
Identification of FUS‐dependent enhancement of U1 snRNP‐RNA interactions around alternative splice (AS) sites
FUS interacts with U1 snRNP 24 and regulates AS 25, 26. Therefore, we looked into the effect of FUS on U1 snRNP‐RNA interactions around AS sites. Chr‐FUS‐tRIP reads were enriched around AS sites, as previously reported with FUS‐CLIP 26 (Fig 3A). U1C‐FUS‐tRIP reads (Fig 3B, blue line), U1A‐FUS‐tRIP reads (Appendix Fig S3A, blue line), and RNAPII‐FUS‐tRIP reads (Fig 3B, purple line) were similarly enriched around AS sites. Thus, U1C, U1A, and RNAPII accompany FUS around AS sites.
Figure 3. Identification of protein–RNA interactions in the RNAPII machinery by tRIP‐seq to investigate FUS‐dependent enhancement of U1 snRNP‐RNA interactions around alternative splice sites.
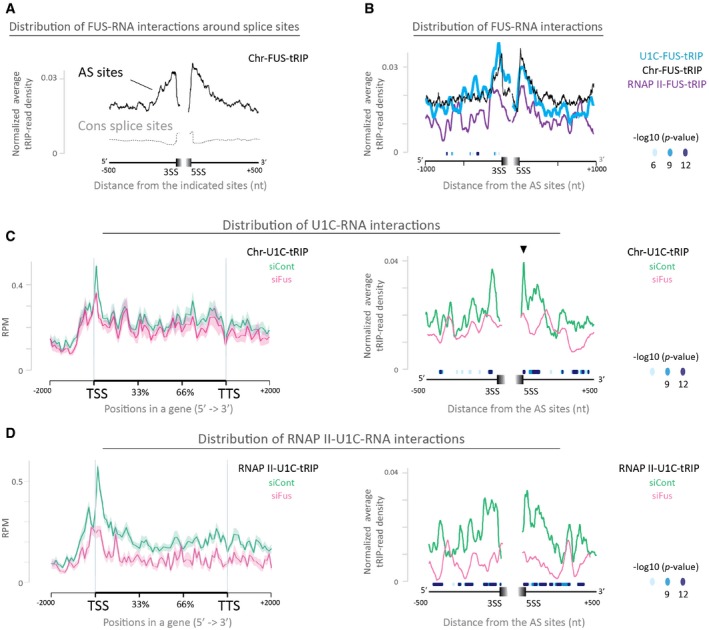
- Read distributions of Chr‐FUS‐tRIP mapped around constitutive splice sites (CS sites, gray dotted line) and alternative splice sites (AS sites, black solid line). Positions of CS sites and AS sites are according to the ENSEMBL annotations on GRCm38/mm10. 3SS, 3′ splice site; 5SS, 5′ splice site.
- Read distributions of RNAPII‐FUS‐tRIP (RNAPII‐FUS, purple line), U1C‐FUS‐tRIP (U1C‐FUS, blue line), and Chr‐FUS‐tRIP (Chr‐FUS, black line) around AS sites. The P‐values for the differences between RNAPII‐FUS and Chr‐FUS are indicated by circles.
- Read distributions of Chr‐U1C‐tRIPs generated from Fus‐silenced cells (siFus, pink line) and those of control siRNA‐treated cells (siCont, green line). Arrowhead indicates a peak at the 5′ splice site, which disappears upon Fus silencing.
- Read distributions of RNAPII‐U1C‐tRIPs generated from Fus‐silenced cells (siFus, pink line) and control siRNA‐treated cells (siCont, green line).
To examine whether FUS affects the U1 snRNP‐RNA interaction, we treated N2A cells with siRNA specific to Fus (siFus) or control siRNA (siCont), as previously reported 27, and performed tRIP‐seq of U1C in the chromatin fraction (Chr‐U1C‐tRIP). We similarly performed tRIP‐seqs of U1A (Chr‐U1A‐tRIP) and U1‐70K (Chr‐U1‐70K‐tRIP) in the chromatin fraction of N2A cells with or without Fus silencing. We observed that substantial numbers of tRIP reads of U1C, U1A, and U1‐70K were aligned to U1 snRNA in these tRIP‐seqs (Appendix Fig S3B), representing direct interactions between U1 snRNA and these proteins 16. Consistent with a previous report showing transcriptome‐wide U1 snRNA‐RNA interactions 38, Chr‐U1C/U1A/U1‐70K‐tRIP reads were enriched around transcriptional start sites (TSSs), as well as around the 5′ and 3′ splice sites (green lines in Fig 3C and Appendix Fig S3C and D). In addition, in contrast to broad peaks at the 5′ splice site of Chr‐U1A‐tRIP reads (Appendix Fig S3C) and Chr‐U1‐70K‐tRIP reads (Appendix Fig S3D), Chr‐U1C‐tRIP reads made a narrow peak at the 5′ splice sites (Fig 3C, arrow head in the right panel), representing the direct association of the 5′ splice site with U1C, but not with U1A or U1‐70K 39.
Fus silencing inhibited the enrichment of Chr‐U1C‐tRIP reads around AS sites (Fig 3C, pink line in the right panel), as well as the enrichment of RNAPII‐U1C‐tRIP reads around AS sites (Fig 3D, pink line in the right panel). These results suggest the involvement of FUS in the recognition of AS sites by U1 snRNP. We observed that Fus silencing had no essential effect on the overall read distributions of Chr‐U1C‐tRIP (Fig 3C, pink line in the left panel). In contrast, Fus silencing decreased the overall read distributions of RNAPII‐U1C‐tRIP (Fig 3D, pink line in the left panel), which is consistent with a previous in vitro report showing that FUS enhances the interaction between U1 snRNP and RNAPII 40. Our results indicate that FUS is likely to recruit U1 snRNP to and around the 5′ and 3′ splice sites in the RNAPII machinery.
FUS‐U1 snRNP complex on RNAPII suppresses APA
U1 snRNP globally suppresses APA to prevent shortening of mRNA length 19, 20. To investigate the involvement of FUS‐U1 snRNP interaction in the suppression of APA, we performed polyA‐seq 41 under Fus silencing with siRNA and/or U1 snRNP depletion with antisense morpholino oligonucleotide (U1AS) 20. We confirmed that U1 snRNP globally represses proximal APA sites (Appendix Fig S4A), as previously reported 19, 20. Comparison of polyA‐seq of U1AS‐treated and Fus‐silenced cells revealed that U1 snRNP and FUS similarly repress APA sites (Fig 4A). Furthermore, Fus silencing had no additional effect on the repression of APA, when combined with U1AS treatment (Fig 4B). These results suggest that U1 snRNP and FUS cooperatively repress target APA sites.
Figure 4. FUS‐U1 snRNP complex on RNAPII suppresses APA .
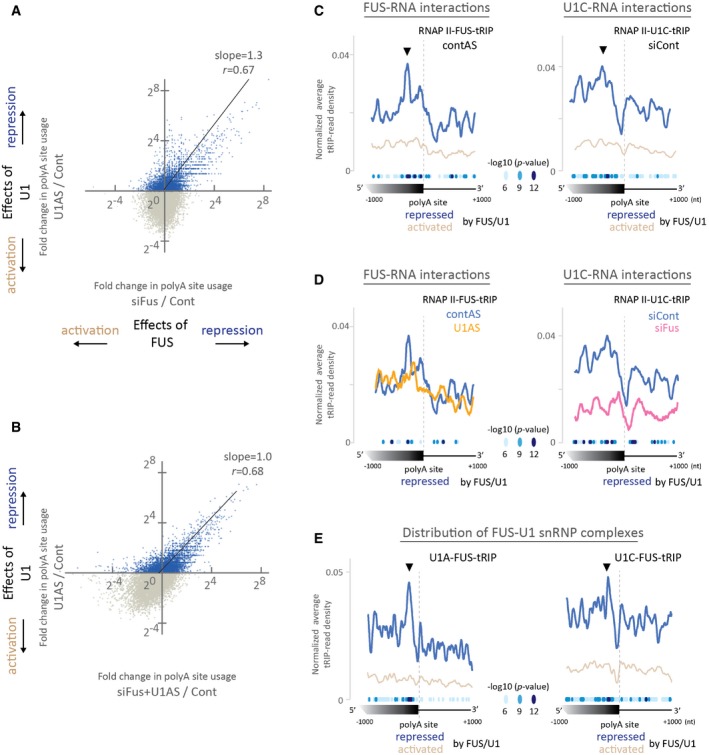
-
A, BPolyA‐seq was performed using N2A cells to examine the effect of downregulation of FUS and U1 snRNP on polyA site usage. Cells were treated with siRNA against Fus (siFus) and/or U1 antisense morpholino (U1AS). Control cells (Cont) were treated with both control siRNA and control antisense morpholino. (A) Correlation of changes in polyA site usage between U1 inhibition and Fus silencing. Fold change in the RPM of a polyA site between siFus and Cont (siFus/Cont) was plotted against that between U1AS and Cont (U1AS/Cont). Pearson's correlation coefficient (r) and the slope of the regression line are shown. (B) Correlation of changes in polyA site usage between U1 inhibition alone and the combination of Fus silencing and U1 inhibition. Fold change in the RPM of a polyA site between siFus + U1AS and Cont (siFus + U1AS/Cont) was plotted against that between U1AS and Cont (U1AS/Cont). Pearson's correlation coefficient (r) and the slope of the regression line are shown. A slope of 1.0 indicates that Fus silencing has no additive effect on U1 inhibition.
-
CRead distributions of RNAPII‐FUS‐tRIP (left) and RNAPII‐U1C‐tRIP (right) around APA sites repressed [the first quadrant in (A)] or activated [the third quadrant in (A)] by both FUS and U1 snRNP. The P‐values for the differences between the repressed and the activated APA sites are indicated by circles. Arrowheads indicate noticeable peaks of RNAPII‐FUS and RNAPII‐U1C upstream to the repressed APA sites.
-
DEffects of Fus silencing (pink line) and U1 inhibition (yellow line) on read distributions of RNAPII‐FUS‐tRIP (left) and RNAPII‐U1C‐tRIP (right) around APA sites repressed by both FUS and U1 snRNP [the first quadrant in (A)]. The P‐values for the differences between contAS and U1AS (left panel), as well as those between siCont and siFus (right panel), are indicated by circles.
-
ERead distributions of U1A‐FUS‐tRIP (left) and U1C‐FUS‐tRIP (right) around APA sites repressed [the first quadrant in (A)] or activated [the third quadrant in (A)] by both FUS and U1 snRNP. The P‐values for the differences between repressed and activated APA sites are indicated by circles. Arrowheads indicate noticeable peaks of U1A/U1C‐FUS upstream to the repressed APA sites.
We performed tRIP‐seq of FUS in the RNAPII machinery (RNAPII‐FUS‐tRIP) in U1AS‐ and control AS‐treated cells. In control cells, we observed that RNAPII‐FUS‐tRIP reads were enriched several hundred nucleotides upstream to APA sites that were repressed by both FUS and U1 snRNP (FUS/U1‐repressed APA sites) (Fig 4C, left panel). In accordance with RNAPII‐FUS‐tRIP reads, we observed that, in control cells, RNAPII‐U1C‐tRIP reads were also enriched several hundred nucleotides upstream to FUS/U1‐repressed APA sites (Fig 4C, right panel). We confirmed similar enrichment of Chr‐U1A‐tRIP, Chr‐U1‐70K‐tRIP, and Chr‐U1C‐tRIP reads at the FUS‐repressed APA sites (Appendix Fig S4B). As predicted, the upstream regions, where RNAPII‐FUS‐tRIP reads (Fig 4C, left panel) and RNAPII‐U1C‐tRIP reads (Fig 4C, right panel) were clustered, were enriched with motifs for FUS and U1 snRNP, respectively (Appendix Fig S4C). Thus, FUS and U1 snRNP are recruited to the overlapping upstream regions to suppress APA in the RNAPII machinery.
We also observed that Fus silencing eliminated the enrichment of RNAPII‐U1C‐tRIP reads upstream to the FUS/U1‐repressed APA sites (Fig 4D, right panel). Conversely, U1AS reduced the enrichment of RNAPII‐FUS‐tRIP reads upstream to the FUS/U1‐repressed APA sites (Fig 4D, left panel). Furthermore, enrichments of U1A‐FUS‐tRIP reads and U1C‐FUS‐tRIP reads were similarly observed upstream to the FUS/U1‐repressed APA sites, suggesting that FUS is recruited to the upstream regions along with U1 snRNP (Fig 4E). These results indicate that both FUS and U1 snRNP make a complex and bind to the upstream regions to suppress APA in the RNAPII machinery.
Taken together, our analysis revealed that FUS and U1 snRNP repress APA sites by interacting with each other. In the RNAPII machinery, both FUS and U1 snRNP bind to overlapping or neighboring sites, which are several hundred nucleotides upstream to the target APA sites. Depletion of either FUS or U1 snRNP disrupts the binding of its counterpart to nascent RNA, and activates APA to a similar extent. In addition, depletion of both FUS and U1 snRNP has no additional effect compared with depletion of U1 snRNP alone. Thus, the APA‐suppressing RNAPII machinery needs to carry both FUS and U1 snRNP, but not either one alone (Fig 5).
Figure 5. A proposed model for the repression of APA by a complex composed of FUS and U1 snRNP .
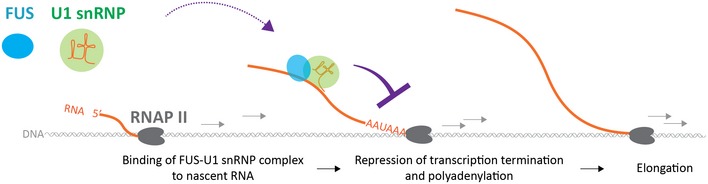
FUS interacts with U1 snRNP, and binds upstream to APA to repress transcription termination and polyadenylation, which allows further elongation of nascent RNA.
ALS mutations in FUS aberrantly activate APA sites repressed by FUS‐U1 snRNP complex
Amyotrophic lateral sclerosis mutations in FUS cluster in the C‐terminus of FUS, where nuclear localized signal (NLS) is located 42. The NLS mutations of FUS reduce the interaction of FUS with U1 snRNP, in addition to inducing cytoplasmic mislocalization of FUS 25. To examine whether the reduced association between mutant FUS and wild‐type U1 snRNP perturbs APA, we introduced a dominant NLS‐truncation mutation, R495X, into N2A cells by CRISPR/Cas9 system. We obtained one N2A cell line carrying a heterozygous R495X mutation (R495X_N2A, Appendix Fig S5A). Parental N2A cells were used as a wild‐type control (WT_N2A). We observed mislocalization of FUS to the cytoplasm (Fig 6A) and reduced interaction between FUS and U1 snRNP (Fig 6B) in R495X_N2A cells, as expected. In addition, U1 snRNA was upregulated in the cytoplasm in R495X_N2A cells, as previously reported 43.
Figure 6. The ALS mutation in FUS, R495X, aberrantly activates APA sites, which are normally repressed by FUS‐U1 snRNP complex.
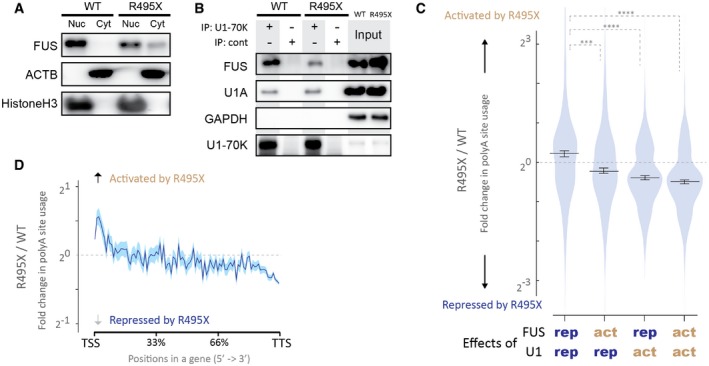
- Localization of FUS in R495X_N2A cells (R495X) and wild‐type N2A cells (WT). Cells were fractionated to separate nucleus and cytoplasm. Equal proportion of cell extracts from nuclear and cytoplasmic fractions were immunoblotted with indicated antibodies.
- Interactions between FUS and U1 snRNP in the R495X_N2A cells (R495X) and wild‐type N2A cells (WT). Total cell lysates were immunoprecipitated with anti‐U1‐70K antibody or a control antibody (cont) and immunoblotted with indicated antibodies.
- Effects of the R495X mutation on four categories of polyA sites. Four categories of polyA sites are comprised of (1) FUS‐repressed (rep)/U1‐repressed sites, (2) FUS‐activated (act)/U1‐repressed sites, (3) FUS‐repressed/U1‐activated sites, and (4) FUS‐activated/U1‐activated sites, according to Fig 4A. Fold change of each polyA site usage by the R495X mutation (R495X/WT) is shown in violin plots. Bars indicate the median and 95% confidential intervals for each category. ***P < 0.001, ****P < 0.0001, based on the Steel–Dwass test for comparing all 6 pairs of categories.
- Change in polyA site usage by the R495X mutation. Fold change of polyA site usage by the R495X mutation (R495X/WT) on the relative positions of all mouse coding genes is plotted. The average is shown in blue line. The standard error of the mean is shown in semi‐transparent blue shade.
We performed polyA‐seq analysis of R495X_N2A and WT_N2A cells. Our analysis detected 32,467 polyA sites that were used in both cells, of which 904 and 2134 sites were upregulated and downregulated more than fourfold, respectively, in R495X_N2A cells (Appendix Fig S5C, left panel). Gene ontology analysis showed that these misregulated polyA sites were enriched in the genes involved in neuronal functions (Appendix Table S2). Comparison with the polyA‐seq of Fus‐silenced and U1AS‐treated N2A cells (Fig 4A) revealed that APA sites repressed by both FUS and U1 snRNP were significantly activated in R495X_N2A cells (Fig 6C, lane 1). In addition, APA sites proximal to TSS were preferentially activated in R495X_N2A cells (Fig 6D), as was observed in U1‐depleted cells (Appendix Fig S4A). These results suggest that the reduced interaction between the mutant FUS and wild‐type U1 snRNP (Fig 6B) compromised the suppression of APA (Fig 5).
We next investigated the effect of another mutation of FUS, P525L, which also disrupts the interaction between FUS and U1 snRNP 25. We reanalyzed RNA‐seq of human motor neurons derived from iPS cells carrying P525L (GEO accession number, GSE94888) 44, using the QAPA tool 45, which infers APA from conventional RNA‐seq data. The analysis detected 19,836 polyA sites, of which 528 sites were affected more than fourfold by the mutation (Appendix Fig S5C, right panel). Gene ontology analysis showed that the affected polyA sites were enriched in the genes associated with the term “transmission of nerve impulse” (P = 0.0003). The preferential activation of proximal APA sites was observed in P525L‐mutant cells, as in R495X_N2A cells (Appendix Fig S5D, left panel). In contrast, the activation of proximal APA was not observed in SOD1‐mutated motor neurons cells derived from ALS‐iPS cells (GEO accession number, GSE95089) (Appendix Fig S5D, right panel). In summary, our analysis revealed that FUS mutations lead to misregulation of APA especially in genes involved in neuronal functions. FUS mutations disrupt the interaction between FUS and U1 snRNP and activate the APA sites, which are normally repressed by FUS‐U1 snRNP complex.
Discussion
RNA processing is spatiotemporally regulated by complex interactions between proteins and RNA. Detection of protein–RNA interaction is one of the key strategies to understand regulation of RNA processing. In the present study, we showed that a newly developed tRIP‐seq method markedly improves the detection efficiency of protein–RNA interactions, which enables the identification of protein–RNA interactions in the specific RNA machinery. We performed 17 kinds of tRIP‐seq analyses (Appendix Table S4) to dissect features of tRIP‐seq and to characterize specific interactions of nascent RNA with FUS (Fig 2D and E), CPSF160 (Fig 2F), and U1C (Figs 3D and 4C and D) in the RNAPII machinery among the numerous protein–RNA interactions inside the cells. CLIP and its variations have been widely used to identify direct endogenous protein–RNA interactions 46. CLIP‐seq technologies produce robust transcriptome‐wide maps of protein–RNA interactions at high resolution. As CLIP requires a large amount of input materials due to loss of materials during multiple purification steps of a trace amount of immunoprecipitated RNA fragments, CLIP experiments have been performed using whole cell extracts or large amounts of subcellular fractions 46. In tRIP, RNA fragments are treated with TEX, which efficiently eliminates linkers and non‐specific RNA with no need for SDS–PAGE and membrane transfer used in CLIP. In addition, a cDNA library is generated by a single RNA purification step. These modifications reduce the number of experimental steps and hands‐on time (Appendix Table S1), and increase sensitivity.
Our tRIP‐seq analysis revealed that U1 snRNP binds several hundred nucleotides upstream to APA sites to suppress APA (Fig 4C, Appendix Fig S4B). The enrichments of U1C‐RNA interactions around APA sites were also observed in the RNA‐seq of RNA fragments co‐immunoprecipitated with formaldehyde‐crosslinked U1C 22. In this experiment, the reads of RNA‐seq were broadly distributed around APA sites (± 1 kb), probably due to large sizes of RNA fragments (100–500 nt). In tRIP‐seq, most of fragments were < 50 nt and the protein–RNA interaction sites were highly concentrated at the 5′ ends of the fragments (Appendix Fig S1E), which enhanced the resolution of the analysis and enabled the identification of the U1 snRNP‐RNA interactions concentrated several hundred nt upstream to APA sites.
RNA processing is coupled with transcription through the dynamic recruitments of RNA processing factors to the RNAPII machinery 3. In the present study, we performed tRIP‐seq analysis of protein–RNA complexes in the RNAPII machinery, which revealed direct interaction maps between the recruited proteins and nascent RNA during co‐transcriptional RNA processing. RNAPII‐FUS‐tRIP revealed that FUS–nascent RNA interactions are enriched upstream of FUS‐repressed APA sites (Fig 2E) and around AS sites (Fig 3B) in the RNAPII machinery. RNAPII‐CPSF160‐tRIP in the presence or absence of FUS showed that FUS suppresses the enrichment of CPSF160–nascent RNA interactions upstream to APA sites in the RNAPII machinery (Fig 2F). RNAPII‐U1C‐tRIP in the presence or absence of FUS revealed that FUS is essential for the U1C–nascent RNA interactions around APA sites (Fig 4D) and AS sites (Fig 3D) in the RNAPII machinery. These results suggest a role for FUS in the position‐specific assembly of CPSF160 and U1C on nascent RNA during co‐transcriptional RNA processing. FUS forms droplets through phase separation in cells, which can directly interact with CTD of RNAPII 47. Interestingly, CTD of RNAPII itself forms droplets through phase separation 48. The association of these two phase‐separated molecules may contribute to the unique features of FUS in the RNAPII machinery.
Our analysis revealed that U1 snRNP makes a complex with FUS and binds upstream to APA sites of nascent RNA to suppress APA (Fig 5). FUS directly binds to RNAPII 13 and enhances the interaction between U1 snRNP and RNAPII in vitro 40. FUS is involved in the dynamic regulation of CTD phosphorylation 13 and local transcription activity of RNAPII 27. Furthermore, we here proved that FUS regulates specific recruitments of U1 snRNP and CPSF160 to their respective target sites of nascent RNA engaged to RNAPII (Figs 2F and 4D). These results suggest that the suppression of APA by the U1 snRNP is substantially dependent on the function of FUS in the RNAPII machinery. In accordance with our observations, a large complex (~1.8 MDa) comprising FUS, CPSF160, and U1 snRNP was identified in HeLa cells 25. A recent study also identified the interaction of U1 snRNP with 3′ end processing machinery including CPSF, CstF, and CFIm, as well as FUS 22. These observations indicate that finely tuned interactions among FUS, U1 snRNP and 3′ end machinery govern the suppression of APA.
FUS is causally associated with the onset of ALS 49, and ALS‐related mutations in FUS reduce its interaction with U1‐snRNP 25. Our analysis revealed that FUS mutations lead to widespread misregulation of APA, including aberrant activation of FUS/U1‐repressd APA sites (Fig 6C). Interestingly, these misregulated APA sites were enriched in genes involved in neuronal functions. The affected FUS‐U1 snRNP‐RNA interactions may be associated with the neurodegeneration in ALS through aberration of APA regulation.
Materials and Methods
Antibodies
Anti‐PTBP1 (N‐20), anti‐FUS (4H11), anti‐RNAPII (N20), anti‐CPSF160 (G‐10), anti‐CFIm25 (2203C3), control rabbit IgG, and control mouse IgG were purchased from Santa Cruz Biotechnology. Anti‐RBFOX2 (A300‐864A), anti‐CstF64 (A301‐092A), and anti‐CFIm59 (A301‐359A) were purchased from Bethyl Laboratories. Anti‐U1A (PA5‐27474) was purchased from Thermo Fisher Scientific. Anti‐U1‐70k (H111) was purchased from Synaptic Systems. Anti‐Rpb1 CTD (4H8) and anti‐Rpb1 NTD (D8L4Y) were purchased from Cell Signaling Technology. Anti‐U1 snRNP C (U1C) (4H12) was purchased from Bio Academia.
Cell culture
HEK293T cells and C2C12 cells were grown in DMEM with 10% fetal bovine serum at 37°C in 5% CO2. N2A mouse neuroblastoma cells were grown in MEM with 10% fetal bovine serum.
RNAi and transfection
The sequence of the siRNA against mouse Fus was as previously described 27. N2A cells were transfected with siRNA using Lipofectamine RNAiMAX (Life Technologies) according to the manufacturer's instructions. We purchased the AllStar negative control siRNA (1027281) from Qiagen.
Transfection with antisense morpholino oligonucleotides
The sequences of the U1 and control antisense morpholinos (Gene Tools) were as previously described 19, 20. N2A cells were transfected using the NEPA21 electroporation system (NEPAGENE). The NEPA21 electroporator was operated with poring pulses of voltage, 125 V; pulse length, 1.5 ms; pulse interval, 50 ms; number of pulses, 2; decay rate, 10%; and polarity +/−, and with transfer pulses of voltage, 20 V; pulse length, 50 ms; pulse interval, 50 ms; number of pulses, 5; decay rate 40%; and polarity +/−. Two million N2A cells were transfected with 5 nmol of U1AS or a control AS for 8 h, prior to the analysis.
Western blotting
Western blotting was performed as previously described 27. Cytoplasmic and nuclear lysates were harvested as previously described 50.
Construction of the 3′ linker for tRIP‐seq
The 3′ linker sequence was as follows: 5′‐phosphate‐AGATCGGAAGAGCACACGTCTGAACTCCAGTCACATCTCGTATGCCGTCTTCTGCTTG‐3′‐biotin, where represents 6‐nt Illumina TruSeq LT indices. The 5′‐phosphorylated linker was converted to the 5′‐adenylated form using the 5′ DNA adenylation kit (NEB) according to the manufacturer's instructions, followed by column purification with Oligo Clean & Concentrator (Zymo Research).
Primer sequences for generating tRIP‐seq library
Sequences used for the tRIP‐seq library construction were as follows: 1st‐RT‐primer, 5′‐CAAGCAGAAGACGGCAT‐3′; 2nd‐str‐primer, 5′‐CGACACGTCGCGTTTTTTTTTTVN‐3′; PCR‐P5‐full, 5′‐AATGATACGGCGACCACCGAGATCTACACGCACGCTGTCCCGACACGTCGCGTTTT‐3′; PCR‐P7‐full, 5′‐CAAGCAGAAGACGGCATACGAGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT‐3′, where represents 6‐nt Illumina TruSeq LT indices (001, CGTGAT; 002, ACATCG; 003, GCCTAA; 004, TGGTCA; 005, CACTGT; 006, ATTGGC; 007, GATCTG; 008, TCAAGT; 009, CTGATC; 010, AAGCTA; 011, GTAGCC; 012, TACAAG; 013, TTGACT; 014, GGAACT; 015, TGACAT; 016, GGACGG; 018, GCGGAC; 019, TTTCAC; 020, GGCCAC; 021, CGAAAC; and 022, CGTACG).
Sequencing primers for Illumina high‐throughput sequencer were as follows: sequencing primer, 5′‐GCACGCTGTCCCGACACGTCGCGTTTTTTTTTT‐3′; and Index1 Seq Primer, 5′‐GATCGGAAGAGCACACGTCTGAACTCCAGTCAC‐3′. Note that tRIP‐seq uses custom sequencing primers.
Purification of RNA bound to proteins
The following protocol is for the preparation of FUS‐tRIP library using 2 × 107 N2A cells. Constituents of reagents for each tRIP are summarized in Appendix Table S3. N2A cells were rinsed with ice‐cold PBS, were collected by cell scraping, and were counted. Then, cells were UV‐irradiated with 254 nm UV‐C at 400 mJ/cm2 prior to the harvest of cells. Pelleted cells were lysed in 1 ml of lysis buffer (50 mM Tris–HCl pH 7.4, 100 mM NaCl, 1 mM MgCl2, 0.1 mM CaCl2, 1% NP‐40, 0.5% sodium deoxycholate, and 0.1% SDS) and incubated on ice for 5 min. After sonication (MySonic, Power 30, 2 cycles of 10‐s burst and 30‐s rest), the lysates were treated with 60 μl of RNase‐free DNase I (Qiagen) for 10 min at 37°C, followed by addition of 10 μl 0.5 M EDTA. After centrifugation at 19,000 g for 5 min at 4°C, 10 μg of specific antibody and 100 μl of Dynabeads Protein G were added to the supernatants and incubated for 5 h at 4°C. Immunoprecipitants were washed twice with wash buffer (1 PBS, 0.1% SDS, 0.5% deoxycholate, and 0.5% NP‐40) and twice with 1× RNase III buffer (10 mM Tris–HCl pH 7.9, 50 mM NaCl, 10 mM MgCl2, and 1 mM DTT). After suspension in 200 μl of 1× RNase III buffer with 0.2 units of RNase III (New England Biolabs), the immunoprecipitants were incubated for 4 min at 37°C. RNase III cleaves single‐stranded RNA at low salt concentrations 51, although its primary target is double‐stranded RNA. Additionally, both Benzonase and RNase III produce RNA fragments with the 5′ monophosphate and the 3′ OH on their ends, which eliminates the need for dephosphorylation of the 3′ end with phosphatases before 3′ linker ligation and also for phosphorylation of the 5′ end with kinase before TEX treatment. Following the partial digestion of RNA on beads with RNase III, the immunoprecipitants were quickly rinsed with wash buffer, stringently washed twice with high salt buffer (50 mM Tris–HCl pH 7.4, 1 M NaCl, 1% NP‐40, 0.1% SDS, and 0.5% deoxycholate) for 10 min at RT, and washed twice with PNK buffer (50 mM Tris–HCl pH 7.4, 10 mM MgCl2, and 0.5% NP‐40) for 5 min at RT. Ligations were performed on beads overnight at 15°C with 100 units of T4 RNA ligase 2 truncated KQ (New England Biolabs) in 30 μl of 1× supplied reaction buffer containing 10% PEG 8000. The next day, the beads were washed twice with wash buffer and once with PNK buffer, and incubated with 50 units of 5′ deadenylase (New England Biolabs) for 45 min at 30°C in 20 μl of 1× supplied Buffer 1. After washing once with wash buffer and twice with PNK buffer, the beads were incubated with 0.5 units of terminator exonuclease (Epicenter) for 60 min at 30°C in 20 μl of 1× supplied Buffer A (TEX treatment). Following sequential washes once with wash buffer, twice with high salt buffer, and once with wash buffer, the beads were treated with 5 mg/ml proteinase K in 50 μl of 1× PK buffer (100 mM Tris–HCl pH 7.5, 100 mM NaCl, and 20 mM EDTA) for 40 min at 37°C. RNA fragments eluted in the supernatants were purified with Quick‐RNA MicroPrep (Zymo Research) and were extracted with 6 μl H2O.
Library construction for tRIP‐seq
We generated cDNA library by modifying a protocol for single‐cell RNA‐seq 32. The eluate (total ~5.8 μl) was transferred to a 0.2‐ml PCR tube containing 0.4 μl each of 0.5 pmol/μl 1st‐RT‐primer and 10 mM dNTP mix, and denaturation and priming were performed at 70°C for 5 min and 25°C for 30 s. Subsequently, 0.8 μl of 10 × PCR buffer (TaKaRa), 0.4 μl of SuperScript IV (Thermo Fisher Scientific), and 0.4 μl of 100 mM DTT were added to the tube on ice. The reverse transcription was performed at 42°C for 20 min and 50°C for 20 min, and the reactions were heat‐inactivated at 70°C for 10 min. Following addition of 1.2 μl of exonuclease I (TaKaRa), 0.4 μl of 10× supplied exonuclease buffer, and 0.4 μl H2O, primer digestion was performed at 37°C for 30 min, and the reactions were heat‐inactivated at 80°C for 20 min. Then, 6.5 μl of polyA‐tailing mix [0.8 μl of 10× PCR buffer, 0.2 μl of 100 mM dATP, 0.2 μl of RNase H (TaKaRa), 0.6 μl of terminal transferase (New England Biolabs), and 4.7 μl H2O] was added to the tube on ice, and the polyA‐tailing reaction was performed at 37°C for 50 s, followed by heat inactivation at 65°C for 10 min. Then, 28 μl of 2 × MightyAmp Buffer v2 (TaKaRa a), 0.7 μl of 10 μM 2nd‐str‐primer, 2 μl of MightyAmp DNA Polymerase (TaKaRa), and 8.8 μl H2O were added to the tube, and the second‐strand synthesis was performed at 98°C for 130 s, 40°C for 1 min, and 68°C for 1 min. After addition of 25 μl of 2× MightyAmp Buffer v2 (TaKaRa), 1 μl of 50 μM PCR‐P5‐full primer, 1 μl of 50 μM PCR‐P7‐full primer, and 23 μl H2O to the tube, 7.5 μl of the PCR mix (1st PCR mix) was transferred to a well of a 96‐well plate containing 0.5 μl of 1/20 diluted EvaGreen (Biotium). The 1st PCR mix was quantified using LightCycler 480 (Roche) with the following program: 2 cycles of 98°C for 10 s, 52°C for 1 min, and 68°C for 1 min, and then 35 cycles of 98°C for 10 s, 65°C for 15 s, and 68°C for 30 s. The rest of the PCR mix was amplified with the same program as for the 1st PCR mix except that the number of PCR cycles was set to 3 cycles less than the threshold count (Ct) obtained with the 1st PCR mix. Final libraries were purified using the PCR purification column (Qiagen) or AMPure XP beads (Beckman Coulter). One μl of the library was applied to a HS‐DNA chip on the Bioanalyzer (Agilent Technologies) to estimate sizes of the library.
High‐throughput sequencing and data processing for tRIP‐seq
Samples were sequenced on the Illumina HiSeq 4000 with 50 bp single read (BGI), NextSeq500 with 75 bp single read (Kazusa DNA Research Institute), or MiSeq with 150 bp single read (Nagoya University). Mapping of sequenced reads were performed as previously described with some modifications 28. Briefly, after standard HiSeq demultiplexing, reads were adapter‐trimmed and reads less than 18 bp were discarded using cutadapt (v1.10) 52 with parameters of “—match‐read‐wildcards –times 1 –e 0.1 –O 1 –quality‐cutoff 6 –m 18″. Mapping was first performed against the human or mouse repetitive elements in RepBase 53 with STAR (v2.5.2b) 54. Repeat‐mapped reads were segregated for separate analysis, and all others were then mapped against the masked human genome (hg19) or the masked mouse genome (mm10) with STAR (v 2.5.2b). Ensemble hard masked genome files (Homo_sapiens.GRCh37.dna_rm.toplevel.fa and Mus_musculus.GRCm38.dna_rm.toplevel.fa, respectively) were used. Multiply mapped reads were filtered out. Duplicates of reads uniquely mapped to the human or mouse genome were removed by Picard (REMOVE_DUPLICATES=true VALIDATION_STRINGENCY=LENIENT) (https://broadinstitute.github.io/picard/). To identify tRIP‐tag peaks, we used MACS (v1.4.2) 55 with the following parameters “–f BAM —nomodel —shiftsize 25”. Information contents 56 were calculated using seqLogo R package (http://works.bepress.com/bembom/11/). Motifs in 10 nucleotides around the start position of tRIP‐tag in peaks were searched by MEME‐ChIP (v 4.11.2) 57 with the following parameters “–rna –meme‐minw 4 –meme‐maxw6 –dreme‐m 5”.
The ngs.plot tool (v2.61) 58 was used with default settings to plot average reads per million mapped reads (RPM) on a gene structure.
Normalized average tRIP read density was calculated as follows. tRIP read density around an APA site or an AS site was computed from coverage of mapped reads at each nucleotide position divided by total coverage of mapped reads within the window spanned 2000 nt centered around the analyzed site. Note that the average distance between adjacent APA sites is 56,438 nt (median, 5,065 nt) and that between adjacent AS sites is 35,677 nt (median, 1,961 nt). Then, the average of tRIP read density was calculated for each nucleotide position in all sites considered and was multiplied by ratio of total coverage of reads within the window to total million coverage of all mapped reads of a respective tRIP‐seq library to represent differential read alignment depths around the analyzed sites.
The statistical analyses of the differences in tRIP‐read distributions were carried out using Welch's test according to the previous report 59.
tRIP‐seq analysis of protein–RNA interactions in RNAPII machinery
The following protocol is for preparation of RNAPII‐FUS‐tRIP library. Constituents of reagents for each tRIP are summarized in Appendix Table S3. N2A cells (18 million cells) were UV‐crosslinked as described above. The cells were lysed with 1 ml of Buffer A (10 mM HEPES‐KOH pH 7.8, 10 mM KCl, 0.1 mM EDTA, and 0.1% NP‐40) and were centrifuged at 2,000 g for 1 min at 4°C, to discard cytoplasmic fractions and collect nuclear fractions. Then, chromatin fractions were further extracted from nuclear pellets as described elsewhere 27. Briefly, nuclear pellets were resuspended in 1 ml of NUC buffer (20 mM HEPES pH 7.6, 150 mM NaCl, 2 mM EDTA, and 1 mM DTT) and mixed with 1 ml of 2× NUN buffer (50 mM HEPES pH 7.6, 2 M urea, 2% NP‐40, 600 mM NaCl, and 1 mM DTT). After incubation on ice for 20 min, chromatin fractions were collected with centrifugation at 19,000 g for 5 min at 4°C and lysed with 1 ml of lysis buffer. Following sonication (MySonic, Power 30, 4 cycles of 10‐s burst and 30‐s rest), the extracted chromatin lysates were incubated with 30 μl of DNase I for 5 min at 37°C and immunoprecipitated with 5 μg of anti‐RNAPII antibody and 50 μl of Dynabeads Protein G overnight at 4°C. The immunoprecipitants were stringently washed once with wash buffer, twice with high salt buffer, and twice with 1× RNase III buffer. After suspension in 200 μl of 1× RNase III buffer with 1 unit of RNase III, the immunoprecipitants were incubated at 37°C for 4 min. Then, the beads were isolated for generation of Chr‐RNAPII‐tRIP library, and the supernatants were collected for generation of RNAPII‐FUS‐tRIP library. The beads were stringently washed once with wash buffer, twice with high salt buffer, and twice with PNK buffer, followed by 3′ linker ligation overnight at 15°C to generate Chr‐RNAPII‐tRIP library. The supernatants were added with 500 μl wash buffer, 500 μl high salt buffer, and 50 mM EDTA (final conc.) to terminate RNase III reaction. The supernatants were then cleared of remaining beads and antibody by incubating with 5 μl of untreated Dynabeads Protein G for 30 min twice. Then, the supernatants were immunoprecipitated with 0.5 μg of anti‐FUS antibody and 5 μl of Dynabeads Protein G for 6 h at 4°C. The immunoprecipitants were stringently washed once with wash buffer, twice with high salt buffer, and twice with PNK buffer, followed by 3′ linker ligation overnight at 15°C. RNAPII‐FUS‐tRIP library was constructed as described above.
PolyA‐seq analysis
N2A cells were transfected with siRNA against Fus or control siRNA for 36 hrs. Then, the transfected cells were further transfected by electroporation with control or antisense morpholino against U1 snRNA. After 8 h, total RNA was harvested from the cells using Quick‐RNA MicroPrep (Zymo Research).
For the construction of the polyA‐seq 41 libraries, we used QuantSeq 3′ mRNA‐Seq Library Prep Kits (Lexogen) according to the manufacturer's instructions (QuantSeq REV kit). High‐throughput 150‐bp single‐end sequencing was performed with the MiSeq sequencer (Nagoya University) using the Custom Read1 sequencing primer (Lexogen).
Mapping of sequenced reads was performed as follows. After standard HiSeq demultiplexing, reads were adapter‐trimmed and reads < 18 bp were discarded using cutadapt (v1.10) 52 with parameters of “—match‐read‐wildcards –times 1 –e 0.1 ‐O 1 –quality‐cutoff 6 –m 18″. Mapping was performed against the mouse genome (GRCm38/mm10) with STAR (v 2.5.2b). Multiply mapped reads were filtered out. The reads flanking downstream 10 bp genomic regions harboring 7 or more A nucleotides were discarded to remove internal polyA priming reads. After the remaining reads were clustered into 25‐nt bins, the clusters locating in Ensemble protein‐coding genes were selected. The position within a particular cluster that gave the largest read coverage was defined as the polyA site of that cluster and used for analysis. The polyA site usage was defined as the RPM of polyA‐seq reads in the cluster. Among 32,157 detected polyA sites, 5,799 sites were detected only in U1‐depleted or Fus‐silenced cells but not in control cells (RPM = 0). In Figs 4C–E and Appendix Fig S4B and C, 4,704 and 12,878 polyA sites that were upregulated (fold change > 1, repressed by FUS/U1) and downregulated (fold change < 1, activated by FUS/U1), respectively, by U1 inhibition and also by Fus silencing, were used for analysis. In Fig 4A and B, fold change in polyA site usage was calculated as follows, to minimize the effects of extreme values and errors produced by faintly detected polyA sites. We limited the analysis for 10,986 polyA sites, of which RPM is more than 2 in Fus‐silenced cells and U1‐depleted cells. Then, 2 was added to all RPM values to avoid division by zero, which occurs in the calculation of the sites not detected in control cells. Since mean RPMs of the analyzed polyA sites are 69.9 ± 1.5 (mean ± SEM), the addition of 2 is expected to have minimal effects on the fold‐change values of most of polyA sites. In Fig 6C, the analysis was performed using 7,134 polyA sites detected in all four polyA‐seq analyses: polyA‐seqs of siFus‐treated cells, U1AS‐treated cells, R495X_N2A cells, and WT_N2A cells. Among the detected APA sites, 1,646 sites were repressed both FUS and U1 snRNP, 1,490 sites were repressed by U1 snRNP but not by FUS, 1,580 sites were repressed by FUS but not by U1 snRNP, and 2,427 sites were upregulated by both FUS and U1 snRNP, at a fold‐change cutoff 1.0.
In Figs 2E and F, and EV3A–C, and Appendix Fig S2C, we analyzed read distributions of tRIP‐seqs around polyA sites detected in the previously performed polyA‐seq of N2A cells (DRA002447) 27. Mapping and filtering of polyA sites were according to the previous report 27. We limited our analysis to the upregulated 26,764 polyA sites and downregulated 46,581 polyA sites more than twofold by Fus silencing that were covered with at least two reads of the polyA‐seq.
The QAPA tool 45 was used with default settings to extract and quantify the usage of polyA sites from RNA‐seq data.
Immunoprecipitation
Cells were lysed in 1 ml of lysis buffer and incubated on ice for 20 min. Then, the lysates were treated with 30 μl of RNase‐free DNase I (Qiagen) for 10 min at 37°C. After centrifugation at 19,000 g for 5 min at 4°C, 5 μg of At‐U1 70K antibody and 25 μl of Dynabeads Protein G were added to the supernatants and incubated overnight at 4°C. Immunoprecipitants were washed three times with lysis buffer and analyzed by Western blotting.
Generation of the N2A cell line carrying a R495X mutation (R495X_N2A) by CRISPR/Cas9 system
To generate the N2A cell line carrying a R495X mutation (R495X_N2A), we used the CRISPR‐Cas9 system in combination with a single‐stranded oligonucleotide (ssODN) as homologous template containing the R495X mutation. Two synonymous mutations were also inserted to avoid repeated cutting by the CRISPR‐Cas9 and to add a restriction enzyme recognition site for Not I. ssODN was ordered from Thermo Fisher. The CRISPR/Cas9 target sequence was 5′‐GGCTTCAGAGGGGGCCGGGG‐3′, which was designed with the software (http://www.genome-engineering.org). The single guide RNA (sgRNAs) was generated by in vitro transcription system as follows. The template for in vitro transcription of sgRNA was amplified by PCR using the pX330 vector (Addgene) as template DNA with the primers: forward primer, 5′‐TAATACGACTCACTATAGGG‐[20‐bp sgRNA target sequence]‐GTTTTAGAGCTAGAAATA‐3′, and reverse primer, 5′‐AAAAGCACCGACTCGGTGCC‐3′. The underlined sequence in the forward primer indicates the T7 promoter region. The segment of tracer RNA was obtained by PCR using the pX330 vector as a template. The amplified PCR fragment was purified with AMPure XP beads (Beckman Coulter) and was in vitro‐transcribed using RiboMAX Large Scale RNA Production Systems‐T7 (Promega). The sgRNA was purified with Quick‐RNA MicroPrep Kit (ZYMO RESEARCH).
N2A cells were transfected with ssODN using Fugene 6 (Promega), followed by transfection of sgRNA and Cas9 enzyme (Integrated DNA Technologies) using Lipofectamine CRISPRMAX (Thermo Fisher), according to the manufacturer's instructions. Next day, cells were seeded at 0.5 cells/well and 0.2 cells/well in 96‐well plates. After 14 days, all single‐cell clones were screened with Not I restriction enzyme digestion of PCR amplicons. PCR was performed using a pair of primers, 5′‐TCACGGGAGGGAAGGTAGAA‐3′ and 5′‐TCCATGAGCGATCCTGAATTG‐3′. The amplicons were also sequenced to confirm the insertion of R495X mutation.
Author contributions
AM designed and executed experiments, analyzed the data, and wrote the manuscript. TK and JT designed and performed the computational analyses. BO and MI helped with the experiments. KO designed the experiments and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figure PDF
Review Process File
Acknowledgements
We thank Dr. Daisuke Kaida at Toyama University for productive discussions. The authors wish to acknowledge Division for Medical Research Engineering, Nagoya University Graduate School of Medicine for technical support of the sequencing analysis using MiSeq. This work was supported by Grants‐in‐Aids from the Japan Society for the Promotion of Science [JP18K06058, JP17K07094, JP18K06483, JP16H06279 (PAGS), and JP18K14684]; the Ministry of Health, Labour, and Welfare of Japan (H29‐Nanchi‐Ippan‐030); the Japan Agency for Medical Research and Development (JP19gm1010002, JP19ek0109230, JP19ek0109281, and JP19bm0804005); the Hori Sciences And Arts Foundation; the Naito Foundation; and the Intramural Research Grant for Neurological and Psychiatric Disorders of NCNP (29‐4).
EMBO Reports (2020) 21: e49890
Data availability
All data are presented in the manuscript, and raw files will be made available upon request. All tRIP‐seq and polyA‐seq data generated for this study have been deposited in the DDBJ Sequence Read Archive (https://www.ddbj.nig.ac.jp/dra/index-e.html). The details of data and accession numbers are listed in Appendix Table S4. tRIP‐seq data: DRA005742 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA005742), DRA005743 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=RA005743), DRA005746 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA005746), DRA005757 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA005757), DRA008137 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA008137), DRA008138 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA008138), DRA008140 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA008140), DRA008141 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA008141), DRA008142 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA008142), DRA008251 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA008251), and DRA008252 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA008252). PolyA‐Seq data: DRA008136 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA008136), and DRA009662 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA009662).
References
- 1. Gerstberger S, Hafner M, Tuschl T (2014) A census of human RNA‐binding proteins. Nat Rev Genet 15: 829–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bentley DL (2014) Coupling mRNA processing with transcription in time and space. Nat Rev Genet 15: 163–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harlen KM, Churchman LS (2017) The code and beyond: transcription regulation by the RNA polymerase II carboxy‐terminal domain. Nat Rev Mol Cell Biol 18: 263–273 [DOI] [PubMed] [Google Scholar]
- 4. Nojima T, Rebelo K, Gomes T, Grosso AR, Proudfoot NJ, Carmo‐Fonseca M (2018) RNA polymerase II phosphorylated on CTD serine 5 interacts with the spliceosome during Co‐transcriptional splicing. Mol Cell 72: 369–379.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Masuda A, Takeda J, Ohno K (2016) FUS‐mediated regulation of alternative RNA processing in neurons: insights from global transcriptome analysis. Wiley Interdiscip Rev RNA 7: 330–340 [DOI] [PubMed] [Google Scholar]
- 6. Zhou Z, Licklider LJ, Gygi SP, Reed R (2002) Comprehensive proteomic analysis of the human spliceosome. Nature 419: 182–185 [DOI] [PubMed] [Google Scholar]
- 7. Rappsilber J, Ryder U, Lamond AI, Mann M (2002) Large‐scale proteomic analysis of the human spliceosome. Genome Res 12: 1231–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tan AY, Manley JL (2009) The TET family of proteins: functions and roles in disease. J Mol Cell Biol 1: 82–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ratti A, Buratti E (2016) Physiological functions and pathobiology of TDP‐43 and FUS/TLS proteins. J Neurochem 138(Suppl 1): 95–111 [DOI] [PubMed] [Google Scholar]
- 10. Ballarino M, Jobert L, Dembele D, de la Grange P, Auboeuf D, Tora L (2013) TAF15 is important for cellular proliferation and regulates the expression of a subset of cell cycle genes through miRNAs. Oncogene 32: 4646–4655 [DOI] [PubMed] [Google Scholar]
- 11. Sohn EJ, Park J, Kang SI, Wu YP (2012) Accumulation of pre‐let‐7g and downregulation of mature let‐7g with the depletion of EWS. Biochem Biophys Res Commun 426: 89–93 [DOI] [PubMed] [Google Scholar]
- 12. Chi B, O'Connell JD, Yamazaki T, Gangopadhyay J, Gygi SP, Reed R (2018) Interactome analyses revealed that the U1 snRNP machinery overlaps extensively with the RNAP II machinery and contains multiple ALS/SMA‐causative proteins. Sci Rep 8: 8755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schwartz JC, Ebmeier CC, Podell ER, Heimiller J, Taatjes DJ, Cech TR (2012) FUS binds the CTD of RNA polymerase II and regulates its phosphorylation at Ser2. Genes Dev 26: 2690–2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bertolotti A, Melot T, Acker J, Vigneron M, Delattre O, Tora L (1998) EWS, but not EWS‐FLI‐1, is associated with both TFIID and RNA polymerase II: interactions between two members of the TET family, EWS and hTAFII68, and subunits of TFIID and RNA polymerase II complexes. Mol Cell Biol 18: 1489–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kwon I, Kato M, Xiang S, Wu L, Theodoropoulos P, Mirzaei H, Han T, Xie S, Corden JL, McKnight SL (2013) Phosphorylation‐regulated binding of RNA polymerase II to fibrous polymers of low‐complexity domains. Cell 155: 1049–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pomeranz Krummel DA, Oubridge C, Leung AK, Li J, Nagai K (2009) Crystal structure of human spliceosomal U1 snRNP at 5.5 A resolution. Nature 458: 475–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wahl MC, Will CL, Luhrmann R (2009) The spliceosome: design principles of a dynamic RNP machine. Cell 136: 701–718 [DOI] [PubMed] [Google Scholar]
- 18. Almada AE, Wu X, Kriz AJ, Burge CB, Sharp PA (2013) Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature 499: 360–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kaida D, Berg MG, Younis I, Kasim M, Singh LN, Wan L, Dreyfuss G (2010) U1 snRNP protects pre‐mRNAs from premature cleavage and polyadenylation. Nature 468: 664–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berg MG, Singh LN, Younis I, Liu Q, Pinto AM, Kaida D, Zhang Z, Cho S, Sherrill‐Mix S, Wan L et al (2012) U1 snRNP determines mRNA length and regulates isoform expression. Cell 150: 53–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oh JM, Di C, Venters CC, Guo J, Arai C, So BR, Pinto AM, Zhang Z, Wan L, Younis I et al (2017) U1 snRNP telescripting regulates a size‐function‐stratified human genome. Nat Struct Mol Biol 24: 993–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. So BR, Di C, Cai Z, Venters CC, Guo J, Oh JM, Arai C, Dreyfuss G (2019) A complex of U1 snRNP with cleavage and polyadenylation factors controls telescripting, regulating mRNA transcription in human cells. Mol Cell 76: 590–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Venters CC, Oh JM, Di C, So BR, Dreyfuss G (2019) U1 snRNP telescripting: suppression of premature transcription termination in introns as a new layer of gene regulation. Cold Spring Harb Perspect Biol 11: a032235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamazaki T, Chen S, Yu Y, Yan B, Haertlein TC, Carrasco MA, Tapia JC, Zhai B, Das R, Lalancette‐Hebert M et al (2012) FUS‐SMN protein interactions link the motor neuron diseases ALS and SMA. Cell Rep 2: 799–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sun S, Ling SC, Qiu J, Albuquerque CP, Zhou Y, Tokunaga S, Li H, Qiu H, Bui A, Yeo GW et al (2015) ALS‐causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1‐snRNP. Nat Commun 6: 6171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ishigaki S, Masuda A, Fujioka Y, Iguchi Y, Katsuno M, Shibata A, Urano F, Sobue G, Ohno K (2012) Position‐dependent FUS‐RNA interactions regulate alternative splicing events and transcriptions. Sci Rep 2: 529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Masuda A, Takeda J, Okuno T, Okamoto T, Ohkawara B, Ito M, Ishigaki S, Sobue G, Ohno K (2015) Position‐specific binding of FUS to nascent RNA regulates mRNA length. Genes Dev 29: 1045–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin‐Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K et al (2016) Robust transcriptome‐wide discovery of RNA‐binding protein binding sites with enhanced CLIP (eCLIP). Nat Methods 13: 508–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yeo GW, Coufal NG, Liang TY, Peng GE, Fu XD, Gage FH (2009) An RNA code for the FOX2 splicing regulator revealed by mapping RNA‐protein interactions in stem cells. Nat Struct Mol Biol 16: 130–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huppertz I, Attig J, D'Ambrogio A, Easton LE, Sibley CR, Sugimoto Y, Tajnik M, Konig J, Ule J (2014) iCLIP: protein‐RNA interactions at nucleotide resolution. Methods 65: 274–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Takeda JI, Masuda A, Ohno K (2017) Six GU‐rich (6GUR) FUS‐binding motifs detected by normalization of CLIP‐seq by Nascent‐seq. Gene 618: 57–64 [DOI] [PubMed] [Google Scholar]
- 32. Sasagawa Y, Nikaido I, Hayashi T, Danno H, Uno KD, Imai T, Ueda HR (2013) Quartz‐Seq: a highly reproducible and sensitive single‐cell RNA sequencing method, reveals non‐genetic gene‐expression heterogeneity. Genome Biol 14: R31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR (2012) Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149: 1635–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhao BS, Roundtree IA, He C (2017) Post‐transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol 18: 31–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Naftelberg S, Schor IE, Ast G, Kornblihtt AR (2015) Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu Rev Biochem 84: 165–198 [DOI] [PubMed] [Google Scholar]
- 36. Porrua O, Libri D (2015) Transcription termination and the control of the transcriptome: why, where and how to stop. Nat Rev Mol Cell Biol 16: 190–202 [DOI] [PubMed] [Google Scholar]
- 37. Schonemann L, Kuhn U, Martin G, Schafer P, Gruber AR, Keller W, Zavolan M, Wahle E (2014) Reconstitution of CPSF active in polyadenylation: recognition of the polyadenylation signal by WDR33. Genes Dev 28: 2381–2393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Engreitz JM, Sirokman K, McDonel P, Shishkin AA, Surka C, Russell P, Grossman SR, Chow AY, Guttman M, Lander ES (2014) RNA‐RNA interactions enable specific targeting of noncoding RNAs to nascent Pre‐mRNAs and chromatin sites. Cell 159: 188–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kondo Y, Oubridge C, van Roon AM, Nagai K (2015) Crystal structure of human U1 snRNP, a small nuclear ribonucleoprotein particle, reveals the mechanism of 5′ splice site recognition. eLife 4: e04986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yu Y, Reed R (2015) FUS functions in coupling transcription to splicing by mediating an interaction between RNAP II and U1 snRNP. Proc Natl Acad Sci USA 112: 8608–8613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Derti A, Garrett‐Engele P, Macisaac KD, Stevens RC, Sriram S, Chen R, Rohl CA, Johnson JM, Babak T (2012) A quantitative atlas of polyadenylation in five mammals. Genome Res 22: 1173–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kapeli K, Martinez FJ, Yeo GW (2017) Genetic mutations in RNA‐binding proteins and their roles in ALS. Hum Genet 136: 1193–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yu Y, Chi B, Xia W, Gangopadhyay J, Yamazaki T, Winkelbauer‐Hurt ME, Yin S, Eliasse Y, Adams E, Shaw CE et al (2015) U1 snRNP is mislocalized in ALS patient fibroblasts bearing NLS mutations in FUS and is required for motor neuron outgrowth in zebrafish. Nucleic Acids Res 43: 3208–3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. De Santis R, Santini L, Colantoni A, Peruzzi G, de Turris V, Alfano V, Bozzoni I, Rosa A (2017) FUS mutant human motoneurons display altered transcriptome and microrna pathways with implications for ALS pathogenesis. Stem Cell Reports 9: 1450–1462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ha KCH, Blencowe BJ, Morris Q (2018) QAPA: a new method for the systematic analysis of alternative polyadenylation from RNA‐seq data. Genome Biol 19: 45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee FCY, Ule J (2018) Advances in CLIP technologies for studies of protein‐RNA interactions. Mol Cell 69: 354–369 [DOI] [PubMed] [Google Scholar]
- 47. Burke KA, Janke AM, Rhine CL, Fawzi NL (2015) Residue‐by‐residue view of in vitro fus granules that bind the C‐terminal domain of RNA polymerase II. Mol Cell 60: 231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Boehning M, Dugast‐Darzacq C, Rankovic M, Hansen AS, Yu T, Marie‐Nelly H, McSwiggen DT, Kokic G, Dailey GM, Cramer P et al (2018) RNA polymerase II clustering through carboxy‐terminal domain phase separation. Nat Struct Mol Biol 25: 833–840 [DOI] [PubMed] [Google Scholar]
- 49. Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323: 1205–1208 [DOI] [PubMed] [Google Scholar]
- 50. Masuda A, Andersen HS, Doktor TK, Okamoto T, Ito M, Andresen BS, Ohno K (2012) CUGBP1 and MBNL1 preferentially bind to 3′ UTRs and facilitate mRNA decay. Sci Rep 2: 209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dunn JJ (1976) RNase III cleavage of single‐stranded RNA. Effect of ionic strength on the fideltiy of cleavage. J Biol Chem 251: 3807–3814 [PubMed] [Google Scholar]
- 52. Martin M (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet J 17: 10–12 [Google Scholar]
- 53. Bao W, Kojima KK, Kohany O (2015) Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6: 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schneider TD, Stephens RM (1990) Sequence logos: a new way to display consensus sequences. Nucleic Acids Res 18: 6097–6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Machanick P, Bailey TL (2011) MEME‐ChIP: motif analysis of large DNA datasets. Bioinformatics 27: 1696–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shen L, Shao N, Liu X, Nestler E (2014) ngs.plot: quick mining and visualization of next‐generation sequencing data by integrating genomic databases. BMC Genom 15: 284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fagerland MW, Sandvik L (2009) Performance of five‐two‐sample location tests for skewed distributions with unequal variances. Contemp Clin Trials 30: 490–496 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figure PDF
Review Process File
Data Availability Statement
All data are presented in the manuscript, and raw files will be made available upon request. All tRIP‐seq and polyA‐seq data generated for this study have been deposited in the DDBJ Sequence Read Archive (https://www.ddbj.nig.ac.jp/dra/index-e.html). The details of data and accession numbers are listed in Appendix Table S4. tRIP‐seq data: DRA005742 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA005742), DRA005743 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=RA005743), DRA005746 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA005746), DRA005757 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA005757), DRA008137 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA008137), DRA008138 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA008138), DRA008140 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA008140), DRA008141 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA008141), DRA008142 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA008142), DRA008251 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA008251), and DRA008252 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA008252). PolyA‐Seq data: DRA008136 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA008136), and DRA009662 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA009662).