

Abstract
Triple-negative breast cancer (TNBC) has a greater risk of recurrence and metastasis along with a worse prognosis compared with other subtypes of breast cancer. Studies have revealed that mitogenic estrogen signaling is involved in the malignant proliferation of TNBC cells through a novel variant of the estrogen receptor, estrogen receptor α-36 (ER-α36). The results of the present study demonstrated that knockdown of ER-α36 expression in TNBC cells using short hairpin RNA inhibited rapid estrogen signaling bypass activation of the PI3K/AKT signaling pathway. Moreover, the ER-α36 modulator icaritin inhibited the proliferation of TNBC cells both in vitro and in vivo. Here, it was revealed that the combination of icaritin and cetuximab, a therapeutic epidermal growth factor receptor (EGFR) neutralizing antibody, induced apoptosis and inhibited cell proliferation synergistically in TNBC cells. The results of the present study improved the understanding of the underlying mechanisms of TNBC progression and supported the therapeutic potential of combined treatment targeting the ER-α36 and EGFR.
Keywords: estrogen receptor α-36, epidermal growth factor receptor, icaritin, cetuximab, triple-negative breast cancer
Introduction
Breast cancer (BC) was one of the most common types of cancer in US women in 2017 (1). Among the different subtypes, triple-negative breast cancer (TNBC) is defined by the absence of expression of the estrogen receptor (ER), progesterone receptor and the human epidermal growth factor receptor-2 or gene amplification (2,3). These biologic characteristics confer TNBC with greater aggressiveness and relapse risk along with a worse prognosis compared with other subtypes of BC (4,5). Limited options for systemic treatment exist for BC other than chemotherapy (6). BC heterogeneity has limited the successful development of targeted therapy (7). Currently, there are no approved targeted therapies for TNBC (8).
The epidermal growth factor receptor (EGFR) is essential for ductal morphogenesis during the development of normal mammary glands (9) and its upregulation in BC has been well documented (10). Previously, researchers report that the EGFR is commonly upregulated in TNBC compared with other BC subtypes and is associated with poor prognosis (11–13). EGFR inhibition is a promising approach for TNBC; however, minimal benefits have been observed by targeting TNBC in clinical studies (14,15). The molecular mechanisms for the insensitivity of EGFR targeted therapy in patients with TNBC remain unclear (16).
Previously, Wang et al (17) reported a novel ER variant with a molecular weight of 36 kDa, ER-α36, which is located mainly in the plasma membrane and cytoplasm. ER-α36 differs from the estrogen receptor α-66 (ER-α66) as it lacks both transcriptional activation domains [Activation factor (AF)-1 and AF-2], but has the DNA-binding domain and partial ligand-binding domains. ER-α36 possesses a unique 27 amino acid domain that replaces the last 138 amino acids encoded by exons 7 and 8 of the ER-α66 gene. ER-α36 lacks intrinsic transcription ability, but mediates non-genomic estrogen signaling. ER-a36 is generated from a promoter located in the first intron of the ER-α66 gene, indicating that ER-α36 expression is regulated independently from ER-α66. Τhis is consistent with the findings that ER-α36 is expressed in cancer tissue specimens from patients with ER-negative BC and established ER-negative BC cells that lack ER-α66 expression (18,19). It has been suggested that ER-α36 may mediate rapid estrogen signaling, which serves a role in anti-estrogen drug resistance in ER-positive BC and in chemotherapy resistance in ER-negative BC (20). ER-α36 mediates rapid estrogen and antiestrogen signaling and stimulates cell proliferation through the activation of the mitogen-activated protein kinase (MAPK/ERK) and the PI3K/AKT signaling pathways (21).
Icaritin is a prenylflavonoid derivative from the genus Epimedium that has been used in traditional Chinese medicine for centuries (22). Studies have demonstrated that icaritin can be used against different types of cancer. Icatrin can inhibit the proliferation and enhance the radio-sensitivity of BC cells (23); induce apoptosis of human endometrial cancer cells (24); and exhibit potent proliferation inhibition in chronic myeloid leukemia and suppress the growth of renal carcinoma cells (25). Recently, Wang et al (26) demonstrated that icaritin can decrease the expression of the ER-α36 protein in TNBC cells. Thus, it was speculated that the combined application of icaritin and the EGFR inhibitor for patients with TNBC may achieve improved results compared with the individual use of either drug.
In the present study, the function of the ER-α36 in EGFR targeted therapy-resistant TNBC was investigated. Furthermore, the efficiency of combination therapy with ER-α36 molecular inhibitor icaritin and EGFR inhibitor cetuximab for TNBCs was also evaluated.
Materials and methods
Ethical approval
The study protocol was approved by the Animal Care and Use Committee of Third Military Medical University (Army Medical University, Chongqing, China).
Chemicals and antibodies
E2β was purchased from Merck KGaA. The polyclonal anti-ER-α36 antibody was generated and characterized as described previously (14). Antibodies against EGFR (cat. no. 4267), ER-α66 (cat. no. 13258), glyceraldehyde 3-phosphate dehydrogenase (cat. no. 2118), AKT (cat. no. 9272), GAPDH (cat. no. 2118) and phospho-Akt (Ser473; cat. no. 4060) were all obtained from Cell Signaling Technology, Inc. Icaritin was purchased from Shenogen Pharma Group, Ltd., and cetuximab was obtained from Merck KGaA.
Culture and treatment of cells
MCF-7, MDA-MB-231 and MDA-MB-436 cell lines were purchased from American Type Culture Collection. The MDA-MB-231 cell line is a well known cell line of highly aggressive, invasive and poorly differentiated TNBC established in 1978 (27,28). The MDA-MB-436 cell line is also well known and possesses BRCA1 mutations (29). These cell lines were chosen as they are well studied, their behavior is highly predictable. The cells were maintained in DMEM containing 10% fetal calf serum and 1% penicillin/streptomycin (DMEM and fetal calf serum were purchased from HyClone; GE Healthcare Life Sciences and penicillin/streptomycin were purchased from Thermo Fisher Scientific, Inc.) at 37°C in an incubator containing 5% CO2. Prior to treatment with E2β and icaritin, cells were transferred to phenol red-free medium containing 2.5% charcoal-stripped fetal calf serum (HyClone; GE Healthcare Life Sciences) and maintained for 24 h.
Establishment of stable cell lines
MDA-MB-231 and MDA-MB-436 cell lines with the ER-α36 expression knockdown using the short-hairpin (sh) RNA method were established as described previously (30). The ER-α36 shRNA plasmid, vehicle plasmid (pRNAT-U6.1/Neo) and anti-ER-α36 antibody were provided by Dr. Zhao-yi Wang (Department of Medical Microbiology and Immunology, Creighton University Medical School). Transfection of the plasmids were performed after cell confluency reached 60% within 24 h of seeding. Transfection reagent Lipofectamine® 3000 (Invitrogen; Thermo Fisher Scientific, Inc.) was used for plasmid transfection according to the manufacturer's instructions. A total of 10 µg plasmid/1×106 cells was incubated for 12 h at 37°C in a humidified atmosphere with 5% CO2. At 48 h post-transfection, the appropriate antibiotic (neomycin; Sigma-Aldrich; Merck KGaA) was used to screen the transfected cell lines for 3 weeks, and >20 clones of selected cells were pooled and termed MDA-MB-231/V and MDA-MB-231/Sh36 or MDA-MB-436/V and MDA-MB-436/Sh36, respectively. The efficiency of ER-α36 shRNA plasmid transduction was determined by western blotting using anti-ER-α36 antibody (1:1,000).
Cell proliferation assay
Cells were seeded in 60-mm petri dishes at a final concentration of 5×104 cells/dish. After 24 h, the indicated concentrations of cetuximab (1, 5, 10, 50 and 100 µg/ml), icaritin (1, 2.5, 5, 7.5 and 10 µM), cetuximab (100 µg/ml) + icaritin (1, 2.5, 5, 7.5 and 10 µM) or control DMSO were added. After 7 days of culture, cell numbers were determined using the Countess II Automated Cell Counter (Thermo Fisher Scientific, Inc). All cells were maintained at 37°C and 5% CO2 in a humidified incubator.
Western blotting
Cells were washed twice with cold phosphate-buffered saline (PBS) and extracted on ice with RIPA buffer (Beyotime Institute of Biotechnology) containing 1% phenylmethane sulfonyl fluoride and 1% phosphatase inhibitor cocktail solution (Beyotime Institute of Biotechnology). Protein concentrations were quantified using a Bicinchoninic Acid Protein Assay kit (Bio-Rad Laboratories, Inc.). Cell lysates were boiled for 5 min in sodium dodecyl sulfate gel-loading buffer and stored at −20°C for western blotting. Cell lysates containing 50 µg protein were separated using 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride (PVDF) membranes (EMD Millipore). PVDF membranes were blocked for 1 h at room temperature with 5% non-fat milk, and incubated with primary antibody diluted (1:1,000) in 5% non-fat milk overnight at 4°C. Next, the membranes were incubated with horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology, Inc.; 1:4,000) at room temperature for 1 h and developed using an ECL Western Blotting Analysis System (GE Healthcare). GAPDH was used as the control. The density of the immunoreactive bands was quantified using Image J V1.8 (National Institutes of Health).
Flow cytometry
Cells (2×105/well) in 6-well plates were treated with the indicated concentrations of cetuximab, icaritin, cetuximab + icaritin or control DMSO were added for 24 h, collected and washed twice in ice-cold PBS. The apoptosis assay was conducted using an Annexin V-FITC apoptosis detection Kit (Beyotime Institute of Biotechnology) according to the manufacturer's instructions and a BD Accuri™ C6 Flow Cytometer (Becton, Dickinson and Company) was used for fluorescence detection. The results were analyzed using FlowJo 7.6 software (Becton, Dickinson and Company).
Construction of an orthotopic mouse model of BC
A suspension of MDA-MB-231 and MDA-MB-436 cells in a PBS-Matrigel (v/v, 1:1) solution was implanted in the mammary fat pads of female NOD/SCID mice (n=40; age, 4–6 weeks; and weight: 18–20 g) obtained from the Animal Center of the Third Military Medical University. Each mouse was inoculated with 1×106 tumor cells. The tumor volume was calculated as length × width2/2. After the tumors had reached 6–8 mm in diameter, mice were grouped randomly (5 per group) and injected (i.v.) with control (0.9% NaCl), cetuximab (2 mg/kg/week), icaritin (50 mg/kg/week) or cetuximab + icaritin (equivalent dose/week). According to the guidelines of IACUC, the mice were euthanized within 48 h when the diameter of the xenografts reached 1.5 cm.
Statistical analyses
All assays were repeated at least 3 times. Data were described as the mean ± standard deviation (SD) or standard error of mean (SEM) as indicated. Two-sided paired Student's t-tests were used to compare the differences between two groups. One-way ANOVA was used for the comparison of multiple groups, followed by the Bonferroni's post-hoc test. Statistical analyses were performed using SPSS v.19 (IBM Corp). P<0.05 was considered to indicate a statistically significant difference.
Results
High expression of ER-α36 activates the PI3K/AKT signaling pathway downstream of the estrogen receptor
MDA-MB-231 and MDA-MB-436 cell lines expressed high levels of ER-α36 and EGFR compared with MCF7 of ER-positive cell, but undetectable levels of the full length ER-α (Fig. 1A). Following treatment with increasing concentrations of E2β in MDA-MB-231 and MDA-MB-436 cells, western blotting was conducted with a phosphorylation-specific anti-AKT antibody. Basal AKT phosphorylation was notably increased in MDA-MB-231 and MDA-MB-436 cells, particularly at E2β 1 µM concentration (Fig. 1B). To determine if ER-α36 mediated the activation of mitogenic estrogen signaling in these ΤΝΒC cell lines, a stable knockdown of ER-α36 cells by shRNA in MDA-MB-231 and MDA-MB-436 cells was performed. Western blotting demonstrated that ER-α36 expression was down-regulated by ~80% in shRNA-transfected cells compared with control cells (Fig. 1C). E2β treatment failed to induce AKT phosphorylation in the MDA-MB-231 cell line following ER-α36 knockdown. Similar results were observed in the MDA-MB-436 ER-α36-knockdown cell line (Fig. 1D). These results suggested that estrogen may activate the downstream PI3K/AKT signaling pathway through ER-α36.
Figure 1.

High expression of ER-α36 mediates estrogen signaling via the PI3K/AKT signaling pathway in TNBC cells. (A) Western blots displaying the expression of ER-α66, ER-α36 and EGFR in an ER-positive breast-cancer cell line (MCF7) and TNBC cell lines (MDA-MB-231 and MDA-MB-436). (B) MDA-MB-231 and MDA-MB-436 cells were treated with different concentrations (M) of estrogen for 30 min, and AKT phosphorylation levels were investigated by western blotting. (C) Western blots representing ER-α36 expression in variants of MDA-MB-231 and MDA-MB-436 cells; parental cells, MDA-MB-231/P and MDA-MB-436/P; control cells transfected with the empty vector, MDA-MB-231/V and MDA-MB-436/V; and ER-α36 expression knockdown cells, MDA-MB-231/sh36 and MDA-MB-436/sh36. (D) Western blots representing the effects of E2β (1 µM) on the phosphorylation and expression of AKT in variants of MDA-MB-231 and MDA-MB-436 cells, as well as the fold-change of p-AKT/AKT. GAPDH was used as a loading control. Data are presented as the mean ± SEM from three independent experiments. **P<0.01 and ***P<0.001. ER-α36, estrogen receptor α-36; ER-α66, estrogen receptor α-66; EGFR, epidermal growth factor receptor; TNBC, triple negative breast cancer; E2β, 17β-estradiol; p, phosphorylated.
Icaritin downregulates the expression of ER-α36 and inhibits E2β-stimulated AKT phosphorylation
The structure of icaritin was shown in Fig. 2A. Western blotting revealed that icaritin treatment potently reduced ER-α36 expression in both TNBC cell lines (Fig. 2B). The EGFR inhibitor cetuximab was used to treat MDA-MB-231 and MDA-MB-436 cells for 30 min prior to stimulation with EGF and E2β. EGF-stimulated AKT phosphorylation was inhibited by the EGFR inhibitor cetuximab, however, cetuximab failed to influence E2β-stimulated AKT phosphorylation in the two TNBC cancer cell lines (Fig. 2C). In icaritin-treated E2β-stimulated TNBC cells, AKT phosphorylation was inhibited (Fig. 2C and D). These results suggested that E-α36-mediated activation of the estrogen signaling pathway may be associated with EGFR-targeted treatment failure in TNBC cells.
Figure 2.

ICT down-regulates ER-α36 expression and inhibits E2β-stimulated AKT phosphorylation. (A) Chemical structure of ICT. (B) Western blots of ER-α36 expression in MDA-MB-231 and MDA-MB-436 cells treated with different concentrations (M) of icaritin for 12 h. (C) Western blots demonstrating the effects of E2β (1 µM) or EGF (50 ng/ml) on the phosphorylation and expression of AKT in MDA-MB-231 and MDA-MB-436 cells treated with 10 µM of ICT or 100 µg/ml of CTB for 12 h, and (D) fold change of p-AKT/AKT. GAPDH was used as a loading control. Data presented as mean ± SEM obtained from three independent experiments. **P<0.01 and ***P<0.001. CTB, cetuximab; ICT, icaritin; ER-α36, estrogen receptor α-36; EGF, epidermal growth factor; E2β, 17β-estradiol; p, phosphorylated.
Cetuximab plus icaritin inhibits TNBC cell proliferation and induces apoptosis
To determine whether the effects of the combined application of cetuximab and icaritin on the proliferation of TNBC cells were stronger compared with cetuximab or icaritin treatment alone, different concentrations of cetuximab were tested in MDA-MB-231 cells. The quantity of MDA-MB-231 cells did not decrease significantly, even in the highest dose group of 100 µg/ml cetuximab (Fig. 3A). Next, MDA-MB-231 cell were treated with different concentrations of icaritin, and the mean percentages of cells were 1 µM, 99.09±4.80%; 2.5 µM, 93.99±6.62%; 5 µM, 83.52±6.77%; 7.5 µM, 66.4%±5.658 and 10 µM, 41.35±7.00% (Fig. 3A). To identify the combined effects, MDA-MB-231 cells were treated with cetuximab + icaritin; the mean percentages of cells were as follows: 100 µg/ml cetuximab + 1 µM icaritin, 94.45±6.58%; ,100 µg/ml cetuximab + 2.5 µM icaritin, 81.84±4.55%; 100 µg/ml cetuximab + 5 µM icaritin, 66.01±8.75%; 100 µg/ml cetuximab + 7.5 µM icaritin, 47.32±5.82%; and 100 µg/ml cetuximab + 10 µM icaritin, 22.99±4.05% (Fig. 3A). The combined administration of icaritin and cetuximab was more effective in inhibiting the proliferation of MDA-MB-231 cells at 100 µg/ml cetuximab + 2.5 µM icaritin compared with icaritin or cetuximab alone (Fig. 3A). Similarly, the combination strategy significantly reduced the mean percentages of cells compared with single drug treatment in MDA-MB-436 cells (Fig. 3A). These results suggested that once the EGFR and ER signaling pathways were suppressed simultaneously, the proliferation of TNBC cells was inhibited more potently.
Figure 3.
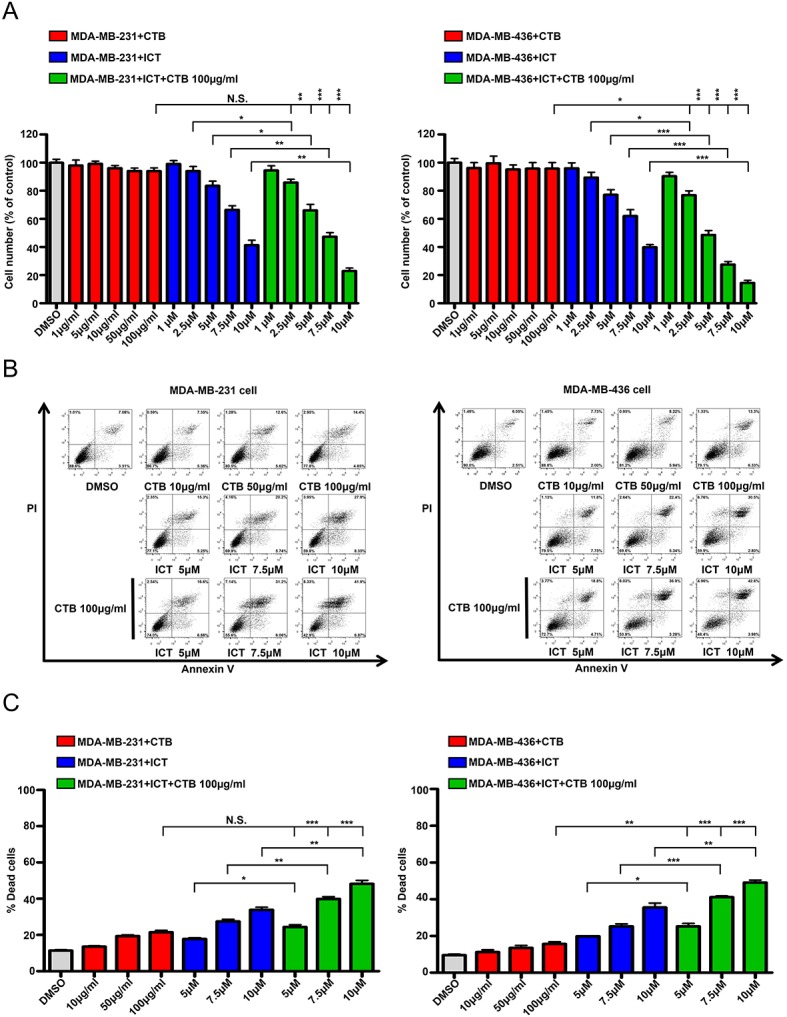
ICT and CTB co-treatment inhibits the proliferation and induce the apoptosis of TNBC cells. (A) Cell numbers (% of control) of MDA-MB-231 and MDA-MB-436 cells compared between CTB (1, 5, 10, 50 and 100 µg/ml), ICT (1, 2.5, 5, 7.5 and 10 µM) and CTB (100 µg/ml) + ICT (1, 2.5, 5, 7.5 and 10 µM) treatment for 7 days. (B and C) Apoptosis assay performed using MDA-MB-231 and MDA-MB-436 cells treated with CTB (10, 50 and 100 µg/ml), ICT (5, 7.5 and 10 µM) and CTB (100 µg/ml) + ICT (5, 7.5 and 10 µM) treatment for 24 h. Data presented as mean ± SD obtained from three independent experiments. *P<0.05, **P<0.01 and ***P<0.001. CTB, cetuximab; ICT, icaritin; TNBC, triple-negative breast cancer.
To ascertain whether this inhibition was caused by apoptosis, the Annexin V/PI double labeling apoptosis assay was performed. The percentages of apoptotic cells in the combination groups were as follows: 100 µg/ml cetuximab + 5 µM icaritin, 24.35±2.14%; 100 µg/ml cetuximab + 7.5 µM icaritin, 39.85±2.26%; and 100 µg/ml cetuximab + 10 µM icaritin, 48.19±3.34%, which were higher compared with the cetuximab alone treatment group (21.47±1.81%) or icaritin gradient treatment group (5 µM icaritin, 17.80±1.15%; 7.5 µM icaritin, 27.45±2.06%; 10 µM icaritin, 33.85±2.53%) (Fig. 3B and C). Similar results were also obtained using the MDA-MB-436 cell line (Fig. 3B and C). Together, these results demonstrated that the combination of cetuximab and icaritin may more effectively promote the apoptosis of TNBC cells compared with either drug used alone. (Fig. 3B and C).
Therapeutic effects of the combined treatment with cetuximab and icaritin on TNBC cells in vivo
To evaluate the effects of cetuximab monotherapy, icaritin monotherapy and combined treatment on TNBC cells in MDA-MB-231 and MDA-MB-436 ×enografts, human BC cell xenografts were created in immunocompromised NOD/SCID mice using MDA-MB-231 or MDA-MB-436 cells. The mice were divided into 4 groups: Control, cetuximab, icaritin and cetuximab + icaritin (Fig. 4A and B). Cetuximab monotherapy was ineffective in MDA-MB-231 ×enografts [tumor doubling time (TDT)=12±2 days, N.S.] compared with the control (TDT=12±3 days, P=0.556) and icaritin monotherapy (TDT=21±3 days, P<0.001; Fig. 4A) groups. The combined therapy induced a significant reduction in the tumor growth of MDA-MB-231 ×enografts compared with cetuximab (P<0.001) or icaritin (P<0.001) monotherapy. Similar results could be observed in the xenografts derived from the MDA-MB-436 cell line (Fig. 4B). These results indicated that the combination of cetuximab and icaritin exhibited greater therapeutic effects compared with those elicited by cetuximab monotherapy or icaritin monotherapy.
Figure 4.

Xenograft mouse models derived from MDA-MB-231 and MDA-MB-436 cell lines treated with cetuximab monotherapy, icaritin monotherapy or cetuximab + icaritin. After the xenografts reached 6–8 mm in diameter, the mice were grouped randomly (5 mice/group) and injected (i.v.) with control, NaCl, 0.9%; cetuximab, 2 mg/kg/week; icaritin, 50 mg/kg/week or cetuximab + icaritin, equivalent dose/week to treat (A) MDA-MB-231 and (B) MDA-MB-436 tumors. Tumor diameter was measured every 3 days. The xenografts and growth curves of tumors are displayed.
Discussion
The EGFR regulates the development of epithelial tissue and maintains homeostasis. The EGFR is a driver of tumorigenesis in lung cancer, BC and glioblastoma (31). Inappropriate activation of the EGFR in cancer results mainly from amplifications and point mutations at the genomic locus (32). Experimental and clinical studies (11,33) have suggested that EGFR expression in TNBC is higher compared with other subtypes of BC, and that EGFR expression is associated with a poor prognosis, the 5-year disease free survival (DFS) for EGFR-positive and EGFR-negative patients were 69.0% and 83.8%, respectively. DFS was significantly poorer for the EGFR-positive patients (HR=2.11, P=0.011) (34). Thus, EGFR inhibition is a promising approach against TNBC. Although, a variety of EGFR inhibitors have been developed, clinical studies have reported that the use of cetuximab alone in the treatment of patients with TNBC did not achieve the expected results, and most TNBC patients exhibit sustained activation of the PI3K/AKT signaling pathway downstream of the EGFR, suggesting that most had alternate mechanisms for pathway activation (35).
Previously, Zhang et al (36) reported that E2β-stimulated proliferation of ER-negative BC cells is through a novel variant of the ER, ER-α36. It has also been demonstrated that E2β induced the physical interaction between ER-α36 and Src, and consequently, the auto-phosphorylation of Src-Y416 in ER-negative BC cells (37). In the present study, ER-α36 was upregulated in TNBC cell lines, and it was demonstrated that E2β serves an important role in activating the downstream PI3K/AKT signaling pathway by binding to ER-α36. These results are consistent with those of Tsai et al (38) who reported that E2β could induce PI3K/AKT phosphorylation in MDA-MB-231 ER-negative cells. Friedl et al (39) reported that the malignant growth of MD-MB-231 cells was stimulated by estrogen in immunodeficient mice. Therefore, the role of estrogen may be beyond classical activation of ER signaling. These data suggest that non-genomic and mitogenic estrogen signaling is retained in TNBC cells (39). Therefore, knowing the specific signaling pathway is important for therapeutic strategies targeting ER-α36. Τhe present study demonstrated that icaritin effectively downregulated the expression of ER-α36, inhibited TNBC cell proliferation and induced apoptosis. However, the present study did not demonstrate that the effects of icaritin on the proliferation and apoptosis of TNBC cells were directly due to ER-α36 inhibition, future studies are required to ascertain this.
The present study demonstrated that EGF and estrogen activated the AKT signaling pathway. In vitro, MDA-MB-231 and MDA-MB-436 cells starved for 24 h in low-concentration serum and a phenol red-free environment, which has a weak estrogen-like effect, were used to investigate estrogen activation of the AKT signaling pathway. Cetuximab alone inhibited the activation of the AKT signaling pathway induced by EGF. In mice, the presence of endogenous estrogen activated the AKT signaling pathway, therefore, cetuximab alone was not sufficient to inhibit the activation of the AKT signaling pathway and tumor growth. This result was consistent with the hypothesis of an AKT bypass activation mechanism by the ER-α36 mediated rapid estrogen signaling pathway in TNBC cells.
In summary, the existence of ER-α36-mediated rapid estrogen signaling bypass activation AKT signaling pathway was demonstrated in TNBC cells. This ER-α36 mediated rapid estrogen signaling pathway is one of the mechanisms for the resistance of TNBC cells to EGFR-targeted therapy. It was also revealed that the combination of the ER-α36 molecular inhibitor icaritin and the EGFR inhibitor cetuximab may more effectively inhibit the proliferation and promote the apoptosis of TNBC cells compared with either individual drug. The current data may help to develop novel therapeutic strategies against TNBC.
Acknowledgements
The authors would like to thank Dr Zhao-yi Wang (Department of Medical Microbiology and Immunology, Creighton University Medical School) for donating the plasmid and antibody.
Funding
This study was supported by grants from the National Key Research and Development Program of China (grant no. 2016YFA0202104), the National Natural Science Foundation of China (grant no. 81602730) and the Key Clinical Research Program of Southwest Hospital (grant nos. SWH2016ZDCX1005, SWH2017ZDCX1003 and SWH2019TD-01).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request
Authors' contributions
LY and XZL acquired, analyzed and interpreted data and drafted the manuscript; XWQ, ZYY and RLC acquired data; SCY, LC and HJC analyzed and interpreted data, critically revised the manuscript for intellectual content, obtained funding and supervised the study; all authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
The study protocol was approved by the Animal Care and Use Committee of the Third Military Medical University (Army Medical University, Chongqing, China).
Patient consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
References
- 1.DeSantis CE, Ma J, Goding Sauer A, Newman LA, Jemal A. Breast cancer statistics, 2017, racial disparity in mortality by state. CA Cancer J Clin. 2017;67:439–448. doi: 10.3322/caac.21412. [DOI] [PubMed] [Google Scholar]
- 2.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363:1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 4.Haffty BG, Yang Q, Reiss M, Kearney T, Higgins SA, Weidhaas J, Harris L, Hait W, Toppmeyer D. Locoregional relapse and distant metastasis in conservatively managed triple negative early-stage breast cancer. J Clin Oncol. 2006;24:5652–5657. doi: 10.1200/JCO.2006.06.5664. [DOI] [PubMed] [Google Scholar]
- 5.Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin Cancer Res. 2007;13:4429–4434. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- 6.Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F, Ollila DW, Sartor CI, Graham ML, Perou CM. The triple negative paradox: Primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res. 2007;13:2329–2334. doi: 10.1158/1078-0432.CCR-06-1109. [DOI] [PubMed] [Google Scholar]
- 7.Hudis CA, Gianni L. Triple-negative breast cancer: An unmet medical need. Oncologist. 2011;16(Suppl 1):S1–S11. doi: 10.1634/theoncologist.2011-S1-01. [DOI] [PubMed] [Google Scholar]
- 8.Chang-Qing Y, Jie L, Shi-Qi Z, Kun Z, Zi-Qian G, Ran X, Hui-Meng L, Ren-Bin Z, Gang Z, Da-Chuan Y, Chen-Yan Z. Recent treatment progress of triple negative breast cancer. Prog Biophys Mol Biol. 2019 Nov 21; doi: 10.1016/j.pbiomolbio.2019.11.007. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 9.Troyer KL, Lee DC. Regulation of mouse mammary gland development and tumorigenesis by the ERBB signaling network. J Mammary Gland Biol Neoplasia. 2001;6:7–21. doi: 10.1023/A:1009560330359. [DOI] [PubMed] [Google Scholar]
- 10.Kumar R, Wang RA. Protein kinases in mammary gland development and cancer. Microsc Res Tech. 2002;59:49–57. doi: 10.1002/jemt.10176. [DOI] [PubMed] [Google Scholar]
- 11.Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z, Hernandez-Boussard T, Livasy C, Cowan D, Dressler L, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004;10:5367–5374. doi: 10.1158/1078-0432.CCR-04-0220. [DOI] [PubMed] [Google Scholar]
- 12.Corkery B, Crown J, Clynes M, O'Donovan N. Epidermal growth factor receptor as a potential therapeutic target in triple-negative breast cancer. Ann Oncol. 2009;20:862–867. doi: 10.1093/annonc/mdn710. [DOI] [PubMed] [Google Scholar]
- 13.Park HS, Jang MH, Kim EJ, Kim HJ, Lee HJ, Kim YJ, Kim JH, Kang E, Kim SW, Kim IA, Park SY. High EGFR gene copy number predicts poor outcome in triple-negative breast cancer. Mod Pathol. 2014;27:1212–1222. doi: 10.1038/modpathol.2013.251. [DOI] [PubMed] [Google Scholar]
- 14.Masuda H, Zhang D, Bartholomeusz C, Doihara H, Hortobagyi GN, Ueno NT. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res Treat. 2012;136:331–345. doi: 10.1007/s10549-012-2289-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gelmon K, Dent R, Mackey JR, Laing K, McLeod D, Verma S. Targeting triple-negative breast cancer: Optimising therapeutic outcomes. Ann Oncol. 2012;23:2223–2234. doi: 10.1093/annonc/mds067. [DOI] [PubMed] [Google Scholar]
- 16.Nakai K, Hung MC, Yamaguchi H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am J Cancer Res. 2016;6:1609–1623. [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. Identification, cloning, and expression of human estrogen receptor-alpha36, a novel variant of human estrogen receptor-alpha66. Biochem Biophys Res Commun. 2005;336:1023–1027. doi: 10.1016/j.bbrc.2005.08.226. [DOI] [PubMed] [Google Scholar]
- 18.Lee LM, Cao J, Deng H, Chen P, Gatalica Z, Wang ZY. ER-alpha36, a novel variant of ER-alpha, is expressed in ER-positive and -negative human breast carcinomas. Anticancer Res. 2008;28:479–483. [PMC free article] [PubMed] [Google Scholar]
- 19.Shi L, Dong B, Li Z, Lu Y, Ouyang T, Li J, Wang T, Fan Z, Fan T, Lin B, et al. Expression of ER-{alpha}36, a novel variant of estrogen receptor {alpha}, and resistance to tamoxifen treatment in breast cancer. J Clin Oncol. 2009;27:3423–3429. doi: 10.1200/JCO.2008.17.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang ZY, Yin L. Estrogen receptor alpha-36 (ER-α36): A new player in human breast cancer. Mol Cell Endocrinol. 2015;418:193–206. doi: 10.1016/j.mce.2015.04.017. [DOI] [PubMed] [Google Scholar]
- 21.Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. A variant of estrogen receptor-{alpha}, hER-{alpha}36: Transduction of estrogen- and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc Natl Acad Sci USA. 2006;103:9063–9068. doi: 10.1073/pnas.0603339103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang XJ, Xi YM, Li ZJ. Icaritin: A novel natural candidate for hematological malignancies therapy. Biomed Res Int. 2019;2019:4860268. doi: 10.1155/2019/4860268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong J, Zhang Z, Lv W, Zhang M, Chen C, Yang S, Li S, Zhang L, Han D, Zhang W. Icaritin synergistically enhances the radiosensitivity of 4T1 breast cancer cells. PLoS One. 2013;8:e71347. doi: 10.1371/journal.pone.0071347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li S, Priceman SJ, Xin H, Zhang W, Deng J, Liu Y, Huang J, Zhu W, Chen M, Hu W, et al. Icaritin inhibits JAK/STAT3 signaling and growth of renal cell carcinoma. PLoS One. 2013;8:e81657. doi: 10.1371/journal.pone.0081657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu JF, Li ZJ, Zhang GS, Meng K, Kuang WY, Li J, Zhou XF, Li RJ, Peng HL, Dai CW, et al. Icaritin shows potent anti-leukemia activity on chronic myeloid leukemia in vitro and in vivo by regulating MAPK/ERK/JNK and JAK2/STAT3/AKT signalings. PLoS One. 2011;6:e23720. doi: 10.1371/journal.pone.0023720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X, Zheng N, Dong J, Liu L, Huang J. Estrogen receptor-α36 is involved in icaritin induced growth inhibition of triple-negative breast cancer cells. J Steroid Biochem Mol Biol. 2017;171:318–327. doi: 10.1016/j.jsbmb.2017.05.009. [DOI] [PubMed] [Google Scholar]
- 27.Brinkley BR, Beall PT, Wible LJ, Mace ML, Turner DS, Cailleau RM. Variations in cell form and cytoskeleton in human breast carcinoma cells in vitro. Cancer Res. 1980;40:3118–3129. [PubMed] [Google Scholar]
- 28.Cailleau R, Young R, Olive M, Reeves WJ., Jr Breast tumor cell lines from pleural effusions. J Natl Cancer Inst. 1974;53:661–674. doi: 10.1093/jnci/53.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chavez KJ, Garimella SV, Lipkowitz S. Triple negative breast cancer cell lines: One tool in the search for better treatment of triple negative breast cancer. Breast Dis. 2010;32:35–48. doi: 10.3233/BD-2010-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang L, Wang ZY. Breast cancer cell growth inhibition by phenethyl isothiocyanate is associated with down-regulation of oestrogen receptor-alpha36. J Cell Mol Med. 2010;14:1485–1493. doi: 10.1111/j.1582-4934.2009.00877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 32.Sigismund S, Avanzato D, Lanzetti L. Emerging functions of the EGFR in cancer. Mol Oncol. 2018;12:3–20. doi: 10.1002/1878-0261.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 34.Liu D, He J, Yuan Z, Wang S, Peng R, Shi Y, Teng X, Qin T. EGFR expression correlates with decreased disease-free survival in triple-negative breast cancer: A retrospective analysis based on a tissue microarray. Med Oncol. 2012;29:401–405. doi: 10.1007/s12032-011-9827-x. [DOI] [PubMed] [Google Scholar]
- 35.Carey LA, Rugo HS, Marcom PK, Mayer EL, Esteva FJ, Ma CX, Liu MC, Storniolo AM, Rimawi MF, Forero-Torres A, et al. TBCRC 001: Randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J Clin Oncol. 2012;30:2615–2623. doi: 10.1200/JCO.2010.34.5579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X, Ding L, Kang L, Wang ZY. Estrogen receptor-alpha 36 mediates mitogenic antiestrogen signaling in ER-negative breast cancer cells. PLoS One. 2012;7:e30174. doi: 10.1371/journal.pone.0030174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang XT, Ding L, Kang LG, Wang ZY. Involvement of ER-α36, Src, EGFR and STAT5 in the biphasic estrogen signaling of ER-negative breast cancer cells. Oncol Rep. 2012;27:2057–2065. doi: 10.3892/or.2012.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsai EM, Wang SC, Lee JN, Hung MC. Akt activation by estrogen in estrogen receptor-negative breast cancer cells. Cancer Res. 2001;61:8390–8392. [PubMed] [Google Scholar]
- 39.Friedl A, Jordan VC. Oestradiol stimulates growth of oestrogen receptor-negative MDA-MB-231 breast cancer cells in immunodeficient mice by reducing cell loss. Eur J Cancer. 1994;30A:1559–1564. doi: 10.1016/0959-8049(94)00293-E. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request