

Abstract
Objective:
Alpha-synuclein (α-syn) is a major component of Lewy bodies, which are the pathological hallmark in Parkinson’s disease, and its genetic mutations cause familial forms of Parkinson’s disease. Patients with α-syn G51D mutation exhibit severe clinical symptoms. However, in vitro studies showed low propensity for α-syn with the G51D mutation. We studied the mechanisms associated with severe neurotoxicity of α-syn G51D mutation using a murine model generated by G51D α-syn fibril injection into the brain.
Methods:
Structural analysis of wild-type and G51D α-syn-fibrils were performed using Fourier transform infrared spectroscopy. The ability of α-syn fibrils forming aggregates was first assessed in in vitro mammalian cells. An in vivo mouse model with an intranigral injection of α-syn fibrils was then used to evaluate the propagation pattern of α-syn and related cellular changes.
Results:
We found that G51D α-syn fibrils have higher β-sheet contents than wild-type α-syn fibrils. The addition of G51D α-syn fibrils to mammalian cells overexpressing α-syn resulted in the formation of phosphorylated α-syn inclusions at a higher rate. Similarly, an injection of G51D α-syn fibrils into the substantia nigra of a mouse brain induced more widespread phosphorylated α-syn pathology. Notably, the mice injected with G51D α-syn fibrils exhibited progressive nigral neuronal loss accompanied with mitochondrial abnormalities and motor impairment.
Conclusion:
Our findings indicate that the structural difference of G51D α-syn fibrils plays an important role in the rapidly developed and more severe neurotoxicity of G51D mutation-linked Parkinson’s disease.
Keywords: Parkinson’s disease, alpha-synuclein, Lewy body, propagation, animal model
Parkinson’s disease (PD) is characterized primarily by motor features (bradykinesia, tremor, rigidity, and postural instability), and its neuropathological hallmarks are degeneration of the dopaminergic neurons in the substantia nigra pars compacta (SNc) and α-synuclein (α-syn) accumulation forming Lewy bodies (LB) and Lewy neurites (LN) in neuronal perikarya and processes, respectively.1 α-syn pathology is thought to play a key role in PD and other neurodegenerative diseases, such as multiple system atrophy and dementia with Lewy bodies, which are characterized by abnormal accumulation of α-syn aggregates in neurons, nerve fibers, or glial cells and are collectively called α-synucleinopathies.2 α-syn is a natively unfolded protein mainly expressed in presynaptic terminals3 where it is involved in exocytosis, neurotransmission, and synaptic plasticity.4–7 Aggregation of α-syn is a nucleation-dependent process whereby the initially formed aggregate can act as a nucleus that induces new aggregate assembly.8–10 Studies of α-syn fibril injections into the brain of various animal species have shown the propagation of α-syn pathology,11–13 which may underlie the widespread neurodegeneration present in PD from the brainstem to the cortex. The recent propagation hypothesis posits that the pathological transcellular spreading of α-syn in PD is mediated by a templated seeding in the central nervous system.14–16 However, the accumulation and propagation that occurs with wild and mutated types of α-syn may result in different patterns of α-syn pathology.
Several mutations of α-syn (A30P, E46K, H50Q, G51D, A53T, A53E and whole-gene multiplication) have been identified in familial PD.17,18 Patients carrying the G51D mutation exhibit a more severe clinical form, including earlier onset, pyramidal signs, and significant psychiatric and autonomic disturbances.19–21 Postmortem brain examination of patients carrying the G51D mutation revealed atypical neurodegenerative changes, such as atrophy of the frontal and temporal lobes and neuronal loss in hippocampus CA2/3 regions. Furthermore, G51D pathology includes oligodendroglial inclusions similar to those observed in multiple system atrophy.19,21 The distinctive phenotype associated with the G51D mutation regarding both clinical expression and brain pathology underscores the role of the α-syn structure in PD pathogenesis.
The aggregation ability of G51D-mutated α-syn in vitro is equal to or weaker than that of wild-type (WT) α-syn,21–24 which is at odds with its increased cytotoxicity.23,25 We studied the structural profile of G51D-mutated α-syn using protein secondary structure analysis and characterized the pathology associated with this mutation by examining cellular changes and propagation following the intranigral injection of α-syn fibrils in mice. Our studies showed that G51D-mutated α-syn fibrils have a distinctive β-sheet rich structure, which causes severe pathology, including nigral cell death in mice.
Materials and Methods
Generation of Recombinant α-Syn Fibrils
Human α-syn gene was amplified from complementary DNA (cDNA) of human brain (Cap site cDNA dT: Nippon Gene, Tokyo, Japan) as previously described.26 The site-directed mutagenesis of WT α-syn was performed to generate a point mutation of G51D α-syn using the following primers: forward, 5′-ccaaggagggagtggtgcatgatgtggcaa-3′; reverse, 5′-ttgccacatcatgcaccactccctccttgg-3′. Whole construction and mutation were confirmed by sequencing. Human WT or G51D α-syn was purified from Escherichia coli as previously described.27
Forced amyloid fibrillation was induced with the HANdai Amyloid Burst Inducer (HANABI, Elekon Science Co. Ltd. and Corona Electric Co., Osaka, Japan) system and thioflavin T (ThT) assay. In the HANABI system, a microplate reader was combined with a water bath-type ultrasonicator,28 and to obtain the preformed fibril of WT or G51D α-syn, we used reaction mixtures composed of 5 mg/ml of α-syn monomer in 150 mM Sodium Chloride (NaCl), 50 mM Tris hydrochloride (Tris-HCL), and Potential of Hydrogen (pH) 7.4. The reaction mixtures in the1.5-mL tube were ultrasonicated from 3 directions (i.e., 2 sides and the bottom) for 3 minutes and then incubated under quiescence for 7 minutes. This process was repeated for 48 hours at 37°C. To monitor the kinetics of fibril formation, ThT was added to the reaction mixtures at a final concentration of 10 μM and assayed in a 96 well plate by a significant enhancement in ThT fluorescence. The excitation and emission wavelengths were 455 and 485 nm, respectively, which were set with band path filters. HANABI automatically measured the fluorescence of samples every 10 minutes. Fibrils were diluted 10-fold and immediately placed on 400-mesh carbon-coated copper grid (Nissin EM, Tokyo, Japan) for transmission electron microscopy. Adsorbed fibrils on the grid were negatively stained with a 2% (w/v) uranyl acetate solution. Electron micrographs were acquired using a H-7650 transmission electron microscopy (Hitachi, Tokyo, Japan) at 80 kV. The end-point products of WT or G51D α-syn fibrils were lyophilized and resuspended by 150 mM NaCl, 50 mM Tris HCl, and pH 7.4 in double-distilled water for Fourier transform infrared (FT-IR) spectroscopy. A 5-μl sample was placed on the sample cell, and the spectra were detected by JASCO 4000 FT-IR spectrometers (JASCO Co, Tokyo, Japan). Spectra were recorded at a resolution of 4 cm−1 and accumulated 64 times at a wave number range from 1400 to 1900 cm−1.
In Vitro Analyses of α-Syn Fibrils
Human neuroblastoma cell line SH-SY5Y (American type culture collection (ATCC) CRL-2266) cells were cultured in Dulbecco’s modified eagle’s medium (Sigma-Aldrich, MO, USA) and supplemented with 10% fetal bovine serum (Thermo Fisher, Carlsbad, CA) at 37°C. Cells were routinely subcultured when confluent. Human WT α-syn was subcloned into the NheI and NotI site of pcDNA vector. SH-SY5Y cells were transfected with the pcDNA vector (Thermo Fisher Scientific) in opti-MEM using the Lipofectamine 2000 reagent (Invitrogen, CA, USA). Stable transfected clones were selected with G418 (500 μg/ml), and the resistant clone was picked and cultured. Human WT α-syn expression was examined by Western blot analysis using the human specific antibody syn211 (Invitrogen). In this study, human WT α-syn stably expressing SH-SY5Y cells were not neuronally differentiated.
The cells were grown to 80% to 90% confluence in culture dishes and transfected with α-syn fibrils using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. At 48 hours after the addition of α-syn fibrils, human WT α-syn stable-expressing SH-SY5Y cells were fixed with 4% paraformaldehyde for 30 minutes at room temperature. After washing in phosphate-buffered saline (PBS), the cells were incubated in PBS with 10% blockace (Yukizirushi, Tokyo, Japan) for 1 hour and subsequently with primary anti-phosphorylated α-syn (p-α-syn) (1:1000; Abcam, Cambridge, UK)29 for 4°C overnight. After several washes in PBS, the cells were incubated with Cy3-conjugated anti-rabbit antibody (Jackson ImmunoResearch, PA, USA) for 1 hour at room temperature. After several washes in PBS, the cells were counterstained with 4’,6-diamidino-2-phenylindole mounting medium (Vector Laboratories, CA, USA) and were observed using confocal scanning microscopy (FV1200; Olympus, Tokyo, Japan). A percentage of p-α-syn-positive cells of total cells was quantified by counting cells from 6 random 20× magnification fields. Cell death was assessed by assaying lactate dehydrogenase (LDH) activity using a cytotoxicity detection kit (Roche, Indianapolis, IN) 48 hours after α-syn fibril transfection. The collected culture medium was centrifuged at 300g for 5 minutes before assaying according to the manufacturer’s instruction. Released LDH reduced the tetrazolium salt to a red-colored formazan salt. The amount of formazan, which correlated directly with the amount of LDH activity, was quantified by absorbance at the wavelength of 490 nm.
Animals
Studies were conducted in male C57BL/6 mice (8 weeks old; weighing 20–25 g, Charles River Laboratories, Yokohama, Japan). Animals were kept 4 per cage under 12-hour light/dark cycles and standard housing condition with ad libitum access to food and water for 1 week before the experiment. All animal experiments were conducted according to the Osaka University Medical School Guideline for the Care and Use of Laboratory Animals. Animal surgeries and experimental procedures were approved by the Osaka University Medical School Animal Care and Use Committee.
Stereotaxic Injection of α-Syn Fibril into the Substantia Nigra of Mice
Mice were anesthetized for unilateral injections of WT α-syn fibrils or G51D α-syn fibrils (20 μg at 5 μg/μL) into the SNc using a Hamilton microsyringe (Hamilton Co, NV, USA) under stereotaxic surgery (1.3 mm lateral, −2.8 mm posterior from the bregma, 4.3 mm below the dural surface). At 12 or 24 weeks postinjection, fibrilinoculated mice were deeply anesthetized and perfused transcardially with 4% paraformaldehyde in PBS. The brains were removed, postfixed overnight in 4% paraformaldehyde in PBS, and immersed in PBS containing 30% sucrose until sinking.
Immunohistochemistry and Mapping of P-α-Syn Pathology
Immunohistochemistry was performed on 20 μm serial sections cut using a cryostat (CM1850; Leica Microsystems, Wetzlar, Germany). The primary antibodies and working dilutions used for immunohistochemistry are listed in Supplementary Table 1. For double immunofluorescence staining, appropriate fluorescent secondary antibodies conjugated to Cy3 and FITC (all were made in donkey, diluted to 1:500, and purchased from Jackson ImmunoResearch) were used. Incubation was done in PBS for 1 hour at Room Temperature, and sections were washed with PBS 3 times, counterstained with 4′,6-diamidino-2-phenylindole mounting medium (Vectashield, Vector Laboratories), and observed by confocal scanning microscopy (FV1200; Olympus). For histological and cell mapping studies, coronal sections were incubated with the biotinylated second antibody (anti-rabbit immunoglobulin G (IgG) antibody, 1:500; Vector Laboratories), and the reaction products were visualized with avidin-biotin-peroxidase complex (Vector Laboratories) using 3′-diaminobenzidine (Sigma Aldrich) as a chromogen. The sections were then counterstained with Mayer’s hematoxylin (MUTO PURE CHEMICALS Co., Ltd. Tokyo, Japan). The p-α-syn immunoreactive inclusions/cells and neurites were mapped at multiple rostrocaudal levels corresponding to 0.02, −1.46, and −2.92 mm relative to Bregma. Images were acquired by BZ-9000 and then manually marked and digitized by Hybrid Cell Count Software (Keyence, Osaka, Japan). Collages were assembled using Adobe Photoshop software (Adobe Systems, Mountain View, CA). We quantified the area of p-α-syn-positive LN-like pathology in the substantia nigra (SN), striatum, and Cortex (Ctx). A total of 6 fields of view (×20) were randomly taken per animal, and automated sections and measurements of the 3,3′-Diaminobenzidine (DAB) stained areas were performed using H3C Hybrid Cell Count Software (Keyence; N = 4). For stereological assessment of the total number of tyrosine hydroxylase (TH) and Nissl double immunopositive neurons, serial nigral sections were prepared as previously reported.30 Every fourth section was stained through the entire extent of the SN. Cells were counted based on the method of Furuya and colleagues.30
Behavioral Assessment
Apomorphine-induced rotation was assessed starting 10 minutes after injection of apomorphine (0.05 mg/kg i.p.; Sigma-Aldrich) and for 5 minutes while using a video recorder. The number of 360° turns (scored as 1 turn) ipsilateral and contralateral to the lesion were counted manually.31
Results
G51D α-Syn Fibrils Have a Rich β-Sheet Structure Compared With WT α-Syn Fibrils
To study the protein structure of G51D α-syn, in vitro aggregation analysis was performed using HANABI, a system that uses ultrasonication force to trigger the fibrillation of proteins, and a thioflavin T (ThT) fluorescence assay for fibril detection.28 The aggregation kinetics of G51D α-syn were not significantly different from that of the WT protein (T[1/2] = 7.776 ± 0.2711 hour and 8.5452 ± 0.4546 hour [P = 0.50377], WT and G51D, respectively); however, the final absolute value of the plateau ThT signal of the G51D mutant was lower, consistent with a previous report22 (Fig. 1A). Transmission electron microscopy revealed no apparent morphological difference between the WT and G51D α-syn fibrils (Fig. 1B). Moreover, the WT and G51D α-syn fibrils showed no difference in proteinase K resistance (Supplementary Fig. S1). The secondary structure content of each type of fibrils was examined using FT-IR, analyzing the amide I region of the FT-IR spectrum (1700–1600 cm−1), which corresponds to the absorption of the carbonyl peptide bond of the main amino acid chain of the protein. This is a sensitive marker of the protein secondary structure. After deconvolution of the FT-IR spectra, we were able to measure the individual secondary structure elements. As β-sheet structure greatly contributes to the properties of protein aggregates, we compared the peaks at 1625 cm−1 that correspond to the presence of amyloid-like intermolecular β-sheet structure.32 Notably, the proportion of β-sheet was significantly higher in G51D α-syn fibrils than WT α-syn fibrils (64.0 ± 0.458% vs. 39.3 ± 1.6%; Fig. 1C,D). The proportion of α-helix of G51D α-syn fibrils slightly decreased when compared with WT α-syn fibrils (WT, 33.7 ± 1.55% vs. G51D, 30.4 ± 0.35%; Fig. 1C,D). Therefore, G51D α-syn fibrils distinctively acquire a rich β-sheet structure when compared with WT α-syn fibrils.
FIG. 1.
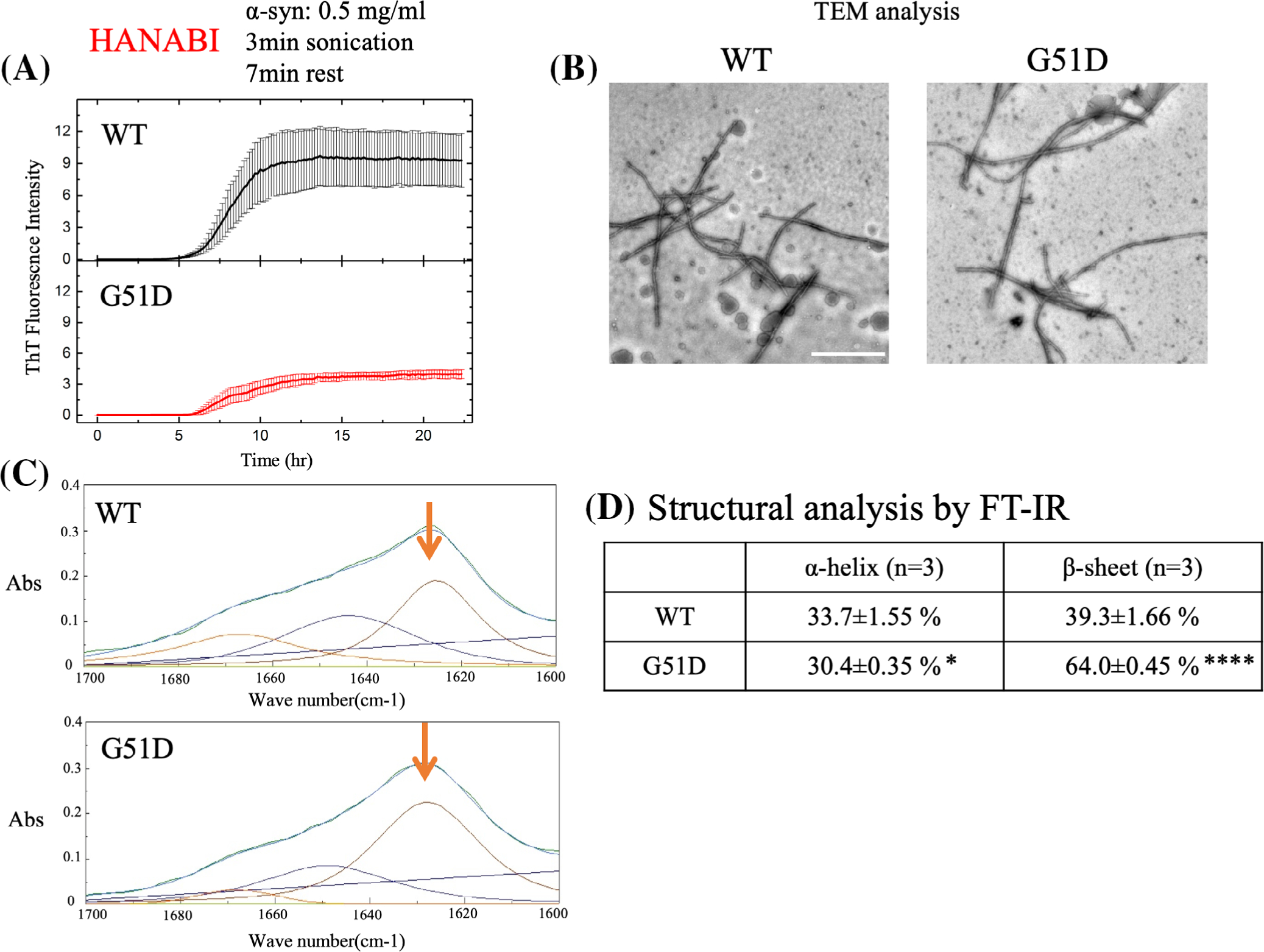
G51D α-syn fibrils have distinct structural characteristics. (A) Average time curve of ThT fluorescence with recombinant human WT α-syn and G51D α-syn fibrils. G51D α-syn fibrils displayed lower ThT fluorescence intensity than WT α-syn fibrils. (B) Electron micrographs showing WT α-syn and G51D α-syn fibrils formed from constructs used in this study. Scale bar: 1 μm. (C) Green line in each panel shows the FT-IR spectrum obtained from WT α-syn and G51D α-syn fibrils. Red line (indicated by arrow) represents contributions of β-sheet structures. Blue line represents the fitted curve. Data were fitted using a Gaussian species model centered at 1628 (β-sheets, red line) cm−1. (D) FT-IR deconvolution. The deconvolution indicates that the β-sheet content of G51D α-syn fibrils (64.0 ± 0.45%) is higher than that of WT α-syn fibrils (39.3 ± 1.6%). Furthermore, the α-helix content (30.4 ± 0.35%) of G51D α-syn fibrils is lower than that of WT α-syn fibrils (33.7 ± 1.55 %). Data are shown as mean ± standard deviation of 3 independent experiments (*P < 0.05, ****P < 0.0001; Student’s t test). α-syn, alpha-synuclein; abs, Absorbance 280nm; FT-IR, Fourier transform infrared; TEM, transmission electron microscopy; ThT, thioflavin T; WT, wild-type.
G51D α-Syn Fibrils Induce Formation of Phosphorylated α-Syn Inclusions in Mammalian Cells
We assessed whether G51D α-syn fibrils induces aggregate formation in mammalian cells. Each type of α-syn fibril was transfected into SH-SY5Y cells with stable overexpression of human α-syn and immunostained using the p-α-syn antibody 48 hours after the addition of fibrils was compared (Fig. 2A–C). WT α-syn fibrils increased the p-α-syn-positive cells when compared with controls, but G51D α-syn fibrils showed a further increase in p-α-syn-positive cells in a concentration-dependent manner (control 0.16%, 400 nM; WT 1.49%, 800 nM; WT 3.23%, 400 nM; G51D 3.3%, 800 nM; G51D 7.85%; Fig. 2D). The intracellular p-α-syn inclusions colocalized with ubiquitin binding protein p62, but not ubiquitin (Supplementary Fig. S2). Neither WT nor G51D α-syn fibrils had any effect on cell survival (Fig. 2E).22
FIG. 2.
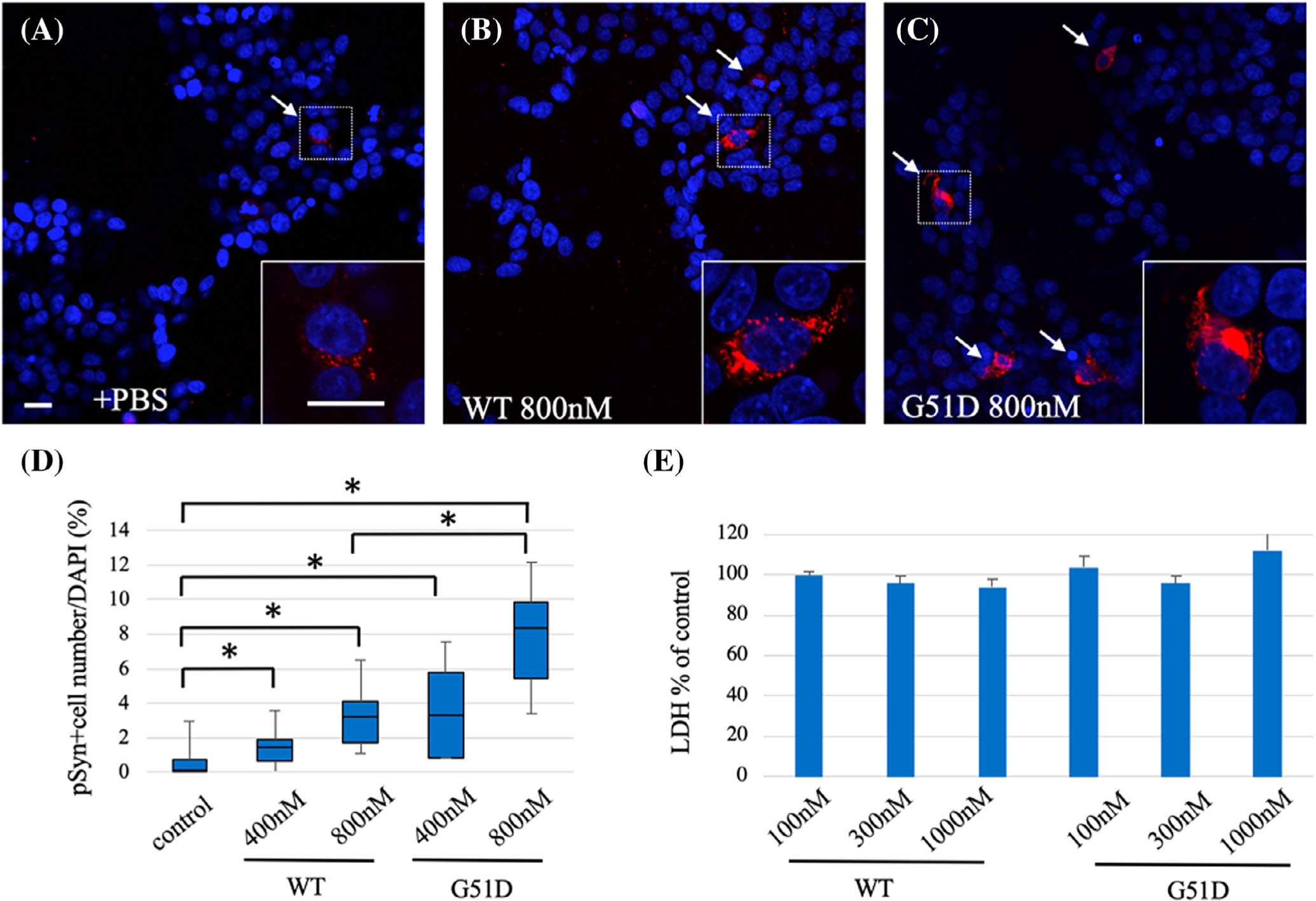
G51D α-syn fibrils promote formation of phosphorylated α-syn inclusion in mammalian cells. Representative images of dual staining of p-α-syn (red) and nuclear stain DAPI (blue) of human WT α-syn stably overexpressing SH-SY5Y cells (SH-SY5Y WT α-syn) treated with (A) PBS, (B) WT, or (C) G51D α-syn fibrils. Bottom right panels depict high-power photomicrographs of areas indicated by the squares, respectively. (D) Quantification of percentage of p-α-syn-positive cells of total cell from 6 random 20× magnification microscopic fields of view showed a higher percentage of p-α-syn-positive cells of total cell in G51D α-syn fibril-treated cells compared with WT α-syn-fibrils treated cells. (N = 4 independent experiments.) *P < 0.05, Wilcoxon signed-rank test. (E) LDH assay revealed addition of α-syn fibrils (100–1000 nM) did not induce LDH release in SH-SY5Y WT α-syn cells at 24 hours. Results are shown as mean ± SEM. Scale bar: 10 μm. α-syn, alpha-synuclein; DAPI, 4’,6-diamidino-2-phenylindole; LDH, lactate dehydrogenase; p-α-syn, phosphorylated α-syn; PBS, phosphate-buffered saline; WT, wild-type; SEM, standard error of mean.
G51D α-Syn Fibril-Injected Mice Exhibit Abundant P-α-Syn-Positive Lewy-Like Pathology
To evaluate the propagation of pathology in the mouse brain, p-α-syn immunostaining was analyzed at 12 and 24 weeks after intranigral α-syn fibril injection. The distribution of p-α-syn pathology (Fig. 3: a–i, ipsilateral; a′–i′, contralateral) in the brain regions was different between the WT α-syn (Fig. 3B,C) and G51D α-syn (Fig. 3D,E) fibril-injected mice. In the WT α-syn fibril-injected mice, LN-like pathology was observed in all regions on the ipsilateral side at 12 weeks, whereas LB-like inclusion was detected in amygdala (Fig. 3B: d) and SN (Fig. 3B: h). A few LN-like pathologies were detectable on ipsilateral hippocampal CA3, but not on contralateral side (Fig. 3B: f′). After 24 weeks, LB-like inclusion can be detected even in the periaqueductal gray (Fig. 3C: i). In G51D α-syn fibril-injected mice, both LN-like and LB-like pathologies were observed in all regions on the ipsilateral side starting at 12 weeks (Fig. 3D,E). Also important, LB-like inclusion can be detected in the contralateral sensory cortex, dorsomedial hypothalamus nucleus, SNc, and periaqueductal gray starting at 12 weeks (Fig. 3D: a′, e′, h′, i′), and it is further extended to the globus pallidus, amygdala, and ectorhinal cortex at 24 weeks (Fig. 3E: a′, b′, d′, e′, g′, h′, i′). In summary, G51D α-syn fibril-injected mice clearly displayed more widespread p-α-syn pathology when compared with WT α-syn fibril-injected mice (Fig. 3F), indicating that G51D α-syn fibrils have a higher seeding ability to the spread of α-syn pathology in vivo. We quantified the area of p-α-syn-positive LN-like pathology in the SN, striatum, and Ctx. As a result of quantification, G51D fibrils significantly induced LN-like pathology when compared with WT fibrils (P < 0.05; Fig. 3G).
FIG. 3.
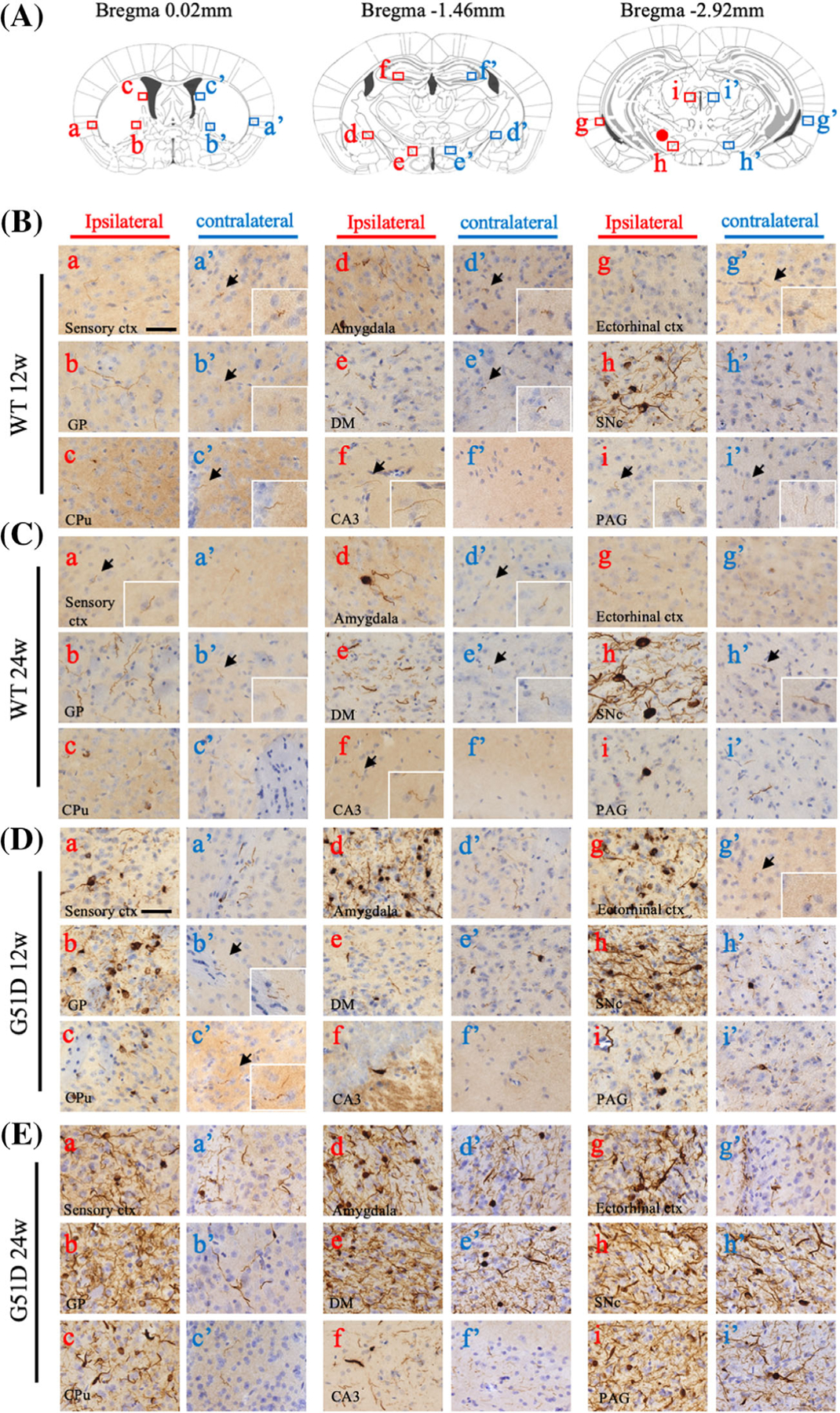
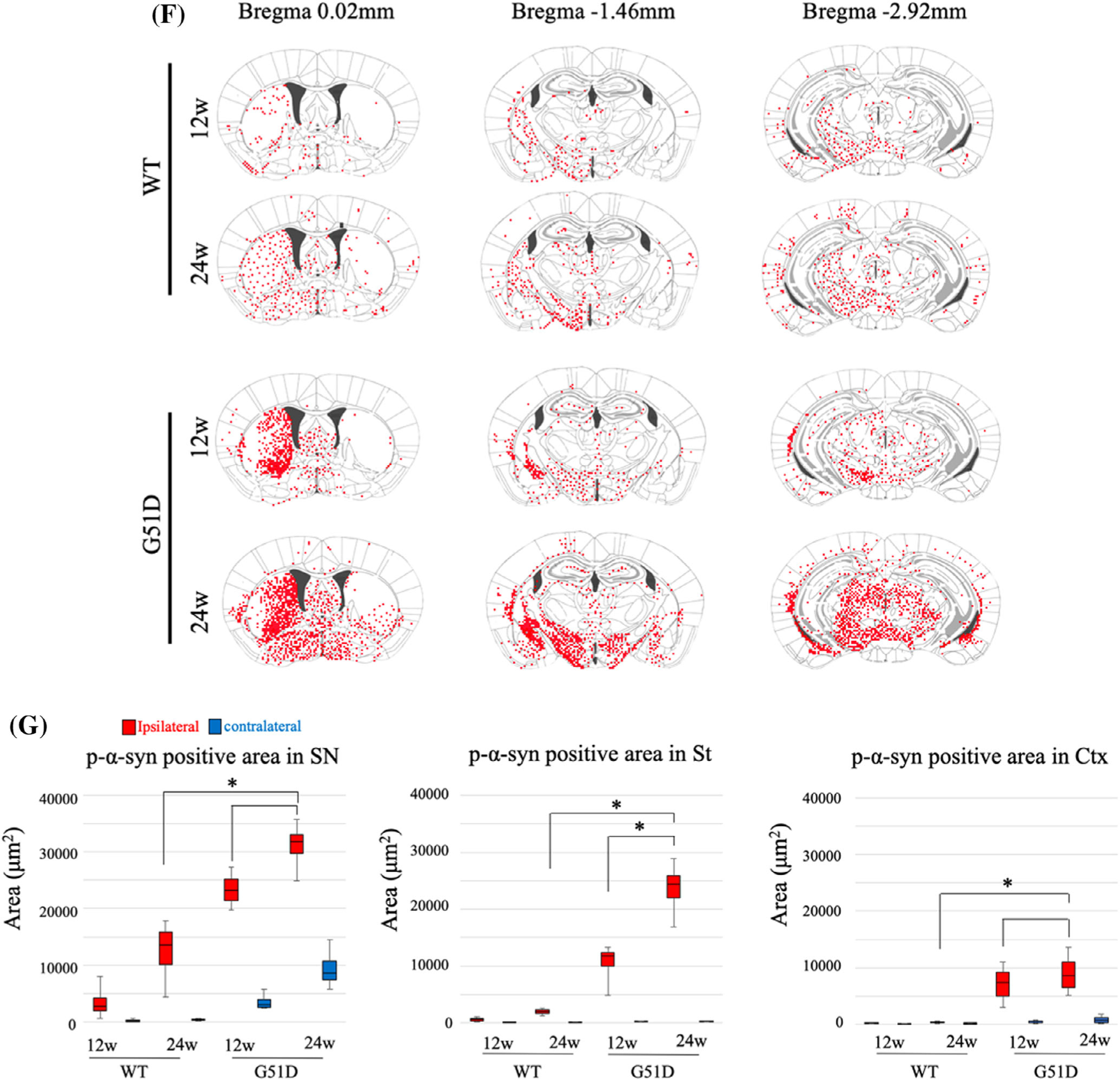
G51D α-syn fibrils induce robust p-α-syn-positive Lewy-like pathology. (A–E) Coronal mouse brain sections, collected at 12 and 24 weeks after intranigral α-syn fibrils injection, were stained for p-α-syn-positive cells. The distribution of p-α-syn pathology (a–i: ipsilateral, a′–i′: contralateral) in each brain region of WT α-syn or G51D α-syn fibril-injected mice at 12 and 24 weeks are shown. (B–E) Insets show the area indicated by arrow at high magnification. (B–C) In WT α-syn fibril-injected mice, Lewy neurite-like pathology can be observed in all regions on the ipsilateral side at 12 weeks, whereas Lewy body-like inclusion can be detected in amygdala (B: d) and SN (B: h). A few Lewy neurite-like pathologies were detectable in the contralateral side but not in CA3 (B: f′). After 24 weeks, Lewy body-like inclusion can be detected even in PAG (C: i). (D–E) In G51D α-syn fibril-injected mice, both Lewy neurite-like and Lewy body-like pathologies were observed in all regions on the ipsilateral side at 12 and 24 weeks. Lewy body-like inclusion can be detected in the contralateral sensory ctx, DMH, SN, and PAG at 12 weeks (a′, e′, h′, i′) and further extended to the GP, amydala, and ectorhinal ctx at 24 weeks (a′, b′, d′, e′, g′, h′, i′). Sensory ctx (a, a′), GP (b, b′), CPu (c, c′), amygdala (d, d′), DM (e, e′), CA3 (f, f′), ectorhinal ctx (g, g′), SNc (h, h′), and PAG (i, i′). (F) Distribution of p-α-syn-positive pathology in α-syn fibril-injected mouse brains. Red dots indicate Lewy body-like and Lewy neurite-like pathology. (G) Quantification of p-α-syn-positive Lewy body-like and Lewy neurite-like pathologies in the contralateral and ipsilateral of the SN, St, and cortex of α-syn fibril-injected mice. *P < 0.05, Wilcoxon signed-rank test. Scale bar: 50 μm. α-syn, alpha-synuclein; CA3, field CA3 hippocampus; CPu, caudate putamen; cxt, cortex; DM, dorsomedial hypothalamus nucleus; GP, globus pallidus; PAG, periaqueductal gray; p-α-syn, phosphorylated α-syn; SNc, substantia nigra pars compacta; St, striatum; WT, wild-type.
We assessed whether endogenous mouse α-syn is involved in WT and G51D α-syn fibril-induced p-α-syn pathology using anti-mouse-specific antibodies for α-syn immunostaining. Exogenous human α-syn fibrils induce the formation of p-α-syn-positive aggregates from the endogenous mouse α-syn. This mechanism may play a key role in the propagation of α-syn pathology.33 Inclusions of mouse α-syn were observed in the SN of both WT and G51D α-syn fibril-injected mice at both 12 and 24 weeks (Supplementary Fig. S3).
G51D α-Syn Fibril-Injected Mice Showed P-α-Syn Inclusions in Dopaminergic Neurons and Oligodendrocytes
Following the intranigral injection of both WT and G51D α-syn fibrils, p-α-syn-positive Lewy-like pathology developed in dopaminergic neurons of the SN. Characteristically, injection of WT α-syn fibrils caused formation of LB-like inclusion bodies widely distributed in the cytoplasm. Similarly, G51D α-syn fibril-injected mice caused the formation of LB-like inclusion bodies that are widely distributed in the cytoplasm. Moreover, LB inclusion-like spherical masses were detected abundantly in G51D α-syn fibril-injected mice. The 2-dimensional structure of each type of α-syn fibril likely underlies the pattern of intracellular protein aggregation (Fig. 4A).
FIG. 4.

G51D fibrils induce Lewy body-like inclusion in dopaminergic neurons and also induce p-α-syn aggregation not only in neuronal cells but also in oligodendrocytes. (A) Double staining of TH and p-α-syn of WT α-syn fibril-injected mice is shown. WT α-syn fibril-injected mice showed intracytoplasmic p-α-syn inclusion. G51D α-syn fibrils-injected mice also showed intracytoplasmic p-α-syn inclusion in TH-positive cells, similar to WT α-syn fibrils-injected mice (upper panel). Lewy body-like inclusions were also observed (lower panel). (B) The p-α-syn-positive inclusion colocalized with microglial cell marker Iba1. (C) Colocalization of p-α-syn-positive inclusion with oligodendrocyte marker olig2 was detected in G51D α-syn fibrils, but not WT α-syn fibril-injected mice. (D) The p-α-syn-positive inclusion did not colocalize with astrocytes marker GFAP in both the WT α-syn fibril-injected and G51D α-syn fibril-injected mice. Scale bar: 20 μm. α-syn, alpha-synuclein; GFAP, Glial fibrillary acidic protein; Iba1, microglia; olig2, oligodendrocytes; p-α-syn, phosphorylated α-syn; TH, tyrosine hydroxylase; WT, wild-type.
To determine the specific cell types that form the p-α-syn aggregates, we used markers for microglia, oligodendrocytes, astrocytes, and neurons for double immunofluorescence staining with p-α-syn (Fig. 4B–D; Supplementary Figs. S4, S5). The p-α-syn inclusions were predominantly formed in the neurons, and only a small proportion of p-α-syn inclusions was formed in the microglia in both WT and G51D α-syn fibril-injected mice (Fig. 4B). Notably, p-α-syn was also detected in oligodendrocytes following G51D, but not WT α-syn fibril injection, recapitulating the pathological characteristics reported in patients carrying G51D mutation (Fig. 4C).19 No α-syn aggregates were detected in the astrocytes of either WT or G51D α-syn fibril-injected mice (Fig. 4D).
The p-α-syn inclusion bodies have been found to colocalize with various proteins, and postmortem studies have shown that G51D α-syn colocalizes particularly with phosphorylated tau.19,38,39 We assessed the colocalization of α-syn with the autophagy-related markers p62 and ubiquitin and with phosphorylated tau (AT8). In both WT and G51D α-syn fibril-injected mice, p-α-syn-positive inclusions were found to colocalize with p62 and ubiquitin (Supplementary Fig. S6A). We observed AT8-positive LN only in G51D α-syn fibril-injected mice, but not WT α-syn fibril-injected mice, similar to the findings in human brains (Supplementary Fig. S6B). α-syn aggregates have been associated with mitochondrial changes in PD pathology.34 The staining pattern of double fluorescent staining of p-α-syn and the mitochondrial marker Tom 20 was different between WT and G51D α-syn fibril-injected mice (Supplementary Fig. S6C). By electron microscopy of neurons in the SNc of G51D α-syn fibril-injected mice, cytosolic aggregates with a similar ultrastructural profile of those in the α-syn-expressed Human Embryonic Kidney cells 293 (HEK 293)35 were often located adjacent to mitochondria (Supplementary Fig. S6E,F). Furthermore, neurons with aggregates possessed deformed mitochondria with enlarged cristae and/or small caves (Supplementary Fig. S6F,G). Immunostaining with lysosomal and autophagy markers (Lysosomal-associated membrane protein 2 (LAMP2) and Microtubule-associated proteins 1A/1B light chain 3B (LC3)) revealed that p-α-syn inclusions induced no remarkable changes in the morphology and cellular distribution of lysosomes and autophagy-related structures (Supplementary Fig. S7). These results indicated that mitochondria are particularly affected by the α-syn pathology developed following injection of G51D fibrils in mice.
G51D α-Syn Fibril Injection Causes Nigrostriatal Dopaminergic Degeneration
As mentioned previously, our mouse model showed Lewy pathology and propagation. The presence of Lewy pathology in TH-positive neurons prompted us to investigate the impact on neuronal survival. Significantly, G51D α-syn fibril-injected mice showed a reduction of approximately 60% in the number of TH-positive cells in SNc compared to the contralateral side at 24 weeks after injection, whereas no dopamine cell loss was detected in the SN of WT fibril-injected mice (Fig. 5A,E). TH-positive staining of terminals in the striatum was also reduced (Fig. 5D). The number of TH-positive neurons in the SN of G51D fibril-injected mice tended to decrease at 12 weeks (Fig. 5E; P > 0.05). Conversely, the intrastriatal injection of G51D α-syn fibrils did not cause dopaminergic neuronal loss in the SN even at week 24 (Supplementary Fig. S8). Because the reduction of TH-positive cells was observed in the injection side of G51D fibril-injected mice, we performed an apomorphine-induced rotation test to evaluate the functional imbalance in the nigrostriatal dopaminergic system between both hemispheres.36,37 The unilateral 6-hydroxydopamine lesion model was used as a positive control. The G51D α-syn fibril-injected mice showed a stronger rotational response when compared with normal control and WT fibril-injected mice, indicating motor dysfunction as a result of dopamine cell loss (Fig. 5F). These results show that G51D α-syn fibrils, as opposed to WT α-syn fibrils, can induce slowly progressive cell death.
FIG. 5.

G51D α-syn pathology induces progressive dopaminergic neuron degeneration. substantia nigra sections of (A) WT α-syn fibril-injected and (C) G51D α-syn fibril-injected mice immunostained for TH and counterstained with Nissl. (B, D) Striatum sections were immunostained for TH and nuclear staining. G51D α-syn fibril-injected mice showed degeneration of TH-positive fibers in the ipsilateral striatum at 24 weeks. (C, D) Arrows show degeneration of dopaminergic neuron and fiber. (E) Number of dopaminergic cell bodies and (F) total neurons in the substantia nigra. At 24 weeks, G51D α-syn fibril injection caused a significant degeneration of the nigrostriatal dopaminergic neurons. (N = 8 mice per group. *P < 0.05, **P < 0.01, ***P < 0.001, Wilcoxon signed-rank test.) (G) Apomorphine-induced turning behavior was evaluated at 24 weeks after α-syn fibril injection. 6-hydroxydopamine (6-OHDA)-injected mice serve as positive control. Each group comprises 6 mice. *P < 0.05, Wilcoxon signed-rank test. Scale bars: (A, C) 200 μm, (B, D) 500 μm. α-syn, alpha-synuclein; TH, tyrosine hydroxylase; WT, wild-type.
Discussion
In this study, we report that the injection of G51D α-syn fibrils into the SN of WT mice leads to the development of widespread α-syn pathology and significant progressive neuronal death, both of which are the pathological characteristic features of PD, whereas no neuronal death was observed in WT α-syn fibril-injected mice. Structural analyses revealed higher β-sheet contents than WT α-syn fibrils. Therefore, our study provides the basis to establish the G51D mutation-linked mouse model of PD, which recapitulates the severe pathology of human PD, and underscores the important role of α-syn fibril structures in its pathogenetic mechanisms.
We consider that lower ThT intensity and higher β-sheet structure of G51D α-syn fibrils were related to its structural difference to WT α-syn fibrils. Indeed, Sidhu and colleagues40 recently demonstrated the existence of ThT differential binding affinity as a result of structural polymorphism.
The present data demonstrate the accelerated seeding ability of G51D α-syn fibrils in vitro and in vivo. Our cell culture experiments revealed that the addition of G51D α-syn fibrils more strongly induces the formation of p-α-syn-positive aggregates in α-syn-expressing SH-SY5Y cells than WT α-syn fibrils (Fig. 2A–D). We further showed by injecting α-syn fibrils into the mouse brain that G51D α-syn fibrils exhibit more widespread α-syn pathology than WT α-syn fibrils (Fig. 3).
Consistent with our study, Rutherford and colleagues41 have reported a similar study using G51D α-syn fibrils, but we showed a more robust pathology when compared with their findings. As fibrils seed aggregation in a dose-dependent manner, the dose of 4 μg used by Rutherford and colleagues may be low. Furthermore, Iba and colleagues42 found that the injection of tau fibrils into the striatum rather than the hippocampus induced extensive tau pathology, suggesting that the lesion may not spread in the hippocampus. Differences in the α-syn fibril strains have been shown to induce different propagation potency in mouse models.9 Because G51D α-syn fibrils have a higher β-sheet-rich structure, we propose that the structural differences of G51D α-syn fibrils may be key to the propagation mechanisms in vivo.
Most important, we demonstrated that G51D α-syn fibrils caused about 60% loss of dopaminergic neurons in the SN and the reduction of TH-positive fibers in the striatum at 24 weeks after injection, whereas WT α-syn fibrils did not cause significant neurotoxicity (Fig. 4). We further showed that G51D α-syn fibril-injected mice develop motor dysfunction reflecting dopamine neuron loss in the rotation test (Fig. 4F). Of note, the number of dopamine neurons was lower, although not statistically significant, in the G51D α-syn fibril-injected mice even at 12 weeks and significantly decreased at 24 weeks (Fig. 4E), suggesting that the G51D α-syn fibril-injected mice were likely exhibiting slowly progressive neurodegeneration. We previously proposed “the exposed β-sheet hypothesis” in the pathogenesis of neurodegenerative diseases, in which the β-sheet structure exposed in misfolded proteins gains leads to neurotoxicity.43 The present data lead us to conclude that the higher β-sheet contents of G51D α-syn fibril structure enhance neurotoxicity in vivo.
We also showed that p-α-syn inclusions in the G51D α-syn fibril-injected mice were also immunoreactive for p62, ubiquitin, and phosphorylated tau (AT8), which reflect the pathological characteristics of PD patients carrying the G51D mutation.19,21 The interaction between tau pathology and α-syn pathology has been well discussed, and studies have shown that α-syn oligomers induce the conversion of monomeric tau into β-sheet rich toxic tau oligomers.44–46 Phosphorylated tau-positive Lewy pathology was detected in only G51D α-syn fibril-injected mice, but not in WT α-syn fibril-injected mice (Supplementary Fig. S4), suggesting that the G51D mutation underlies the tau conversion ability of α-syn. Notably, p-α-syn-positive inclusion bodies were present in oligodendrocytes in the G51D α-syn fibril-injected mice, but not in the WT α-syn fibril-injected mice. PD patients carrying the G51D mutation also develop glial cytoplasmic inclusions within oligodendrocytes in addition to broad cortical and subcortical neuronal α-syn inclusion bodies,19,21 which is consistent with our experimental findings. Previous studies have shown that α-syn aggregation causes mitochondrial dysfunction and neurodegeneration.47–49 Furthermore, G51D-mutated α-syn exacerbates mitochondrial fragmentation in primary neurons.22 These data suggest that mitochondrial abnormalities caused by G51D α-syn fibrils may slowly underlie progressive neuronal death.
The present data show that the intranigral G51D α-syn fibril-injection model recapitulates many features of α-syn G51D mutation-linked familial PD, such as the development of α-syn pathology, progressive degeneration of dopaminergic neurons in the SN, and accompanying movement disorders. A key distinction in this mutation is the higher β-sheet contents of G51D α-syn fibrils, which is associated with severe neurotoxicity in vivo. The novel mouse model of PD based on α-syn G51D mutation is therefore expected to be a powerful tool to investigate pathological mechanisms and develop new disease-modifying therapies for PD.
Supplementary Material
Funding agencies:
This work was supported by Japan Society for the Promotion of Science (JSPS) KAKENHI Grant JP16k19512 (to H.H.), Grant-in-Aid for Scientific Research Grant JP17K09299 (to K.B.), Grant in-Aid for Scientific Research on Innovative Areas (Brain Protein Aging and Dementia Control; 15H01558) from Ministry of Education, Culture, Sports, Science and Technology (MEXT), and the Brain Mapping by Integrated Neurotechnologies for Disease Studies from the Japan Agency for Medical Research and Development (AMED) Brain Mapping Grant 19dm0207070, P3GM-GRIFIN Grant 19km0405206, SENTAN Grant JP18hm0102037, JP18km0405206, KAKENHI Grant JP18H02741 and JST CREST Grant Number JPMJCR18H4, JPMJCR17H6 (to H.M.). Dr. Papa is funded by NIH grants NS073994, NS045962, RR000165 and OD011132.
Footnotes
Relevant conflicts of interests/financial disclosures: Nothing to report.
Supporting Data
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site.
References
- 1.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature 1997:839–840. [DOI] [PubMed] [Google Scholar]
- 2.Recasens A, Ulusoy A, Kahle PJ, Di Monte DA, Dehay B. In vivo models of alpha-synuclein transmission and propagation. Cell Tissue Res 2017;373:183–193. [DOI] [PubMed] [Google Scholar]
- 3.Iwai AM, Yoshimoto E, Ge M, et al. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995;14:467–475. [DOI] [PubMed] [Google Scholar]
- 4.Abeliovich A, Schmitz Y, Farinas I, et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000;25:239–252. [DOI] [PubMed] [Google Scholar]
- 5.Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 2005;123:383–396. [DOI] [PubMed] [Google Scholar]
- 6.Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010;329:1663–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Logan T, Bendor J, Toupin C, Thorn K, Edwards RH. Alpha-Synuclein promotes dilation of the exocytotic fusion pore. Nat Neurosci 2017;20:681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wood SJ, Wypych J, Steavenson S, Louis JC, Citron M, Biere AL. Alpha-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson’s disease. J Biol Chem 1999;274: 19509–19512. [DOI] [PubMed] [Google Scholar]
- 9.Bousset L, Pieri L, Ruiz-Arlandis G, et al. Structural and functional characterization of two alpha-synuclein strains. Nat Commun 2013; 4:2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peelaerts W, Bousset L, Van der Perren A, et al. Alpha-synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015;522:340–344. [DOI] [PubMed] [Google Scholar]
- 11.Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med 2012;209:975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thakur P, Breger LS, Lundblad M, et al. Modeling Parkinson’s disease pathology by combination of fibril seeds and alpha-synuclein overexpression in the rat brain. Proc Natl Acad Sci U S A 2017;114: E8284–E8293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimozawa A, Ono M, Takahara D, et al. Propagation of pathological alpha-synuclein in marmoset brain. Acta Neuropathol Commun 2017;5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Volpicelli-Daley LA, Luk KC, Patel TP, et al. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011;72:57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelvin C, Luk VK, Carroll J, et al. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012;338:949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tofaris GKG, Goedert M, Spillantini MG. The transcellular propagation and intracellular trafficking of alpha-synuclein. Cold Spring Harb Perspect Med 2017;7(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petrucci S, Ginevrino M, Valente EM. Phenotypic spectrum of alpha-synuclein mutations: new insights from patients and cellular models. Parkinsonism Relat Disord 2016;22(suppl 1):S16–S20. [DOI] [PubMed] [Google Scholar]
- 18.Rosborough K, Patel N, Kalia LV. α-Synuclein and parkinsonism: updates and future perspectives. Curr Neurol Neurosci Rep 2017; 17(4):31. [DOI] [PubMed] [Google Scholar]
- 19.Kiely AP, Asi YT, Kara E, et al. alpha-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol 2013;125:753–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tokutake TI, Yoshimura A, Miyashita N, Kuwano A, Nishizawa R, Ikeuchi T. Clinical and neuroimaging features of patient with early-onset Parkinson’s disease with dementia carrying SNCA p.G51D mutation. Parkinsonism Relat Disord 2014;20:262–264. [DOI] [PubMed] [Google Scholar]
- 21.Lesage S, Anheim M, Letournel F, et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol 2013;73:459–471. [DOI] [PubMed] [Google Scholar]
- 22.Fares MB, Ait-Bouziad N, Dikiy I, et al. The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of alpha-synuclein, and enhances its secretion and nuclear localization in cells. Hum Mol Genet 2014;23: 4491–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rutherford NJ, Moore BD, Golde TE, Giasson BI. Divergent effects of the H50Q and G51D SNCA mutations on the aggregation of alpha-synuclein. J Neurochem 2014;131:859–867. [DOI] [PubMed] [Google Scholar]
- 24.Stefanovic AN, Lindhoud S, Semerdzhiev SA, Claessens MM, Subramaniam V. Oligomers of Parkinson’s disease-related alpha-synuclein mutants have similar structures but distinctive membrane permeabilization properties. Biochemistry 2015;54:3142–3150. [DOI] [PubMed] [Google Scholar]
- 25.Ysselstein D, Joshi M, Mishra V, et al. Effects of impaired membrane interactions on alpha-synuclein aggregation and neurotoxicity. Neurobiol Dis 2015;79:150–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yagi H, Kusaka E, Hongo K, Mizobata T, Kawata Y. Amyloid fibril formation of alpha-synuclein is accelerated by preformed amyloid seeds of other proteins: implications for the mechanism of transmissible conformational diseases. J Biol Chem 2005;280:38609–38616. [DOI] [PubMed] [Google Scholar]
- 27.Araki K, Yagi N, Nakatani R, et al. A small-angle X-ray scattering study of alpha-synuclein from human red blood cells. Sci Rep 2016; 6:30473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Umemoto AY, So H, Goto MY. High-throughput analysis of ultrasonication-forced amyloid fibrillation reveals the mechanism underlying the large fluctuation in the lag time. J Biol Chem 2014; 289:27290–27299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prusiner SB, Woerman AL, Mordes DA, et al. Evidence for a-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A 2015;112: E5308–E5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furuya T, Hayakawa H, Yamada M, et al. Caspase-11 mediates inflammatory dopaminergic cell death in the 1-methyl-4-phenyl-1,-2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. J Neurosci 2004;24:1865–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Francardo V, Recchia A, Popovic N, Andersson D, Nissbrandt H, Cenci MA. Impact of the lesion procedure on the profiles of motor impairment and molecular responsiveness to L-DOPA in the 6-hydroxydopamine mouse model of Parkinson’s disease. Neurobiol Dis 2011;42:327–340. [DOI] [PubMed] [Google Scholar]
- 32.Lazaro DF, Dias MC, Carija A, et al. The effects of the novel A53E alpha-synuclein mutation on its oligomerization and aggregation. Acta Neuropathol Commun 2016;4:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Masuda-Suzukake M, Nonaka T, Hosokawa M, et al. Prion-like spreading of pathological α-synuclein in brain. Brain 2013;136: 1128–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem 2008; 283:9089–9100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paxinou E, Chen Q, Weisse M, et al. Induction of alpha-synuclein aggregation by intracellular nitrative insult. J Neurosci 2001;21: 8053–8061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ungerstedt U, Arbuthnott GW. Quantitative recording of rotational behavior in rats after 6-hydroxy-dopamine lesions of the nigrostriatal dopamine system. Brain Res 1970;24:485–493. [DOI] [PubMed] [Google Scholar]
- 37.Schwarting RK, Huston JP. The unilateral 6-hydroxydopamine lesion model in behavioral brain research. Analysis of functional deficits, recovery and treatments. Prog Neurobiol 1996; 50:275–331. [DOI] [PubMed] [Google Scholar]
- 38.Wakabayashi KT, Mori K, Takahashi FH. The Lewy body in Parkinson’s disease: molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology 2007; 27:494–506. [DOI] [PubMed] [Google Scholar]
- 39.Xu YD, Qing YH. The phosphorylation of alpha-synuclein: development and implication for the mechanism and therapy of the Parkinson’s disease. J Neurochem 2015;135:4–18. [DOI] [PubMed] [Google Scholar]
- 40.Sidhu A, Vaneyck J, Blum C, Segers-Nolten I, Subramaniam V. Polymorph-specific distribution of binding sites determines thioflavin-T fluorescence intensity in α-synuclein fibrils. Amyloid 2018;25:189–196. [DOI] [PubMed] [Google Scholar]
- 41.Rutherford NJ, Dhillon J-KS, Riffe CJ, Howard JK, Brooks M, Giasson BI. Comparison of the in vivo induction and transmission of α-synuclein pathology by mutant α-synuclein fibril seeds in transgenic mice. Hum Mol Genet 2017;26:4906–4915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VMY. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci 2013;33:1024–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagai Y, Popiel HA. Conformational changes and aggregation of expanded polyglutamine proteins as therapeutic targets of the poly-glutamine diseases: exposed beta-sheet hypothesis. Curr Pharm Des 2008;14:3267–3279. [DOI] [PubMed] [Google Scholar]
- 44.Lasagna-Reeves CA, Castillo-Carranza DL, Guerrero-Muoz MJ, Jackson GR, Kayed R. Preparation and characterization of neurotoxic tau oligomers. Biochemistry 2010;49:10039–10041. [DOI] [PubMed] [Google Scholar]
- 45.Sengupta U, Guerrero-Munoz MJ, Castillo-Carranza DL, et al. Pathological interface between oligomeric alpha-synuclein and tau in synucleinopathies. Biol Psychiatry 2015;78:672–683. [DOI] [PubMed] [Google Scholar]
- 46.Castillo-Carranza DL, Guerrero-Munoz MJ, Sengupta U, Gerson JE, Kayed R. Alpha-synuclein oligomers induce a unique toxic tau strain. Biol Psychiatry 2018;84:499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dryanovski DI, Guzman JN, Xie Z, et al. Calcium entry and alpha-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J Neurosci 2013;33:10154–10164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guardia-Laguarta C, Area-Gomez E, Rub C, et al. Alpha-synuclein is localized to mitochondria-associated ER membranes. J Neurosci 2014;34:249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Plotegher N, Gratton E, Bubacco L. Number and brightness analysis of alpha-synuclein oligomerization and the associated mitochondrial morphology alterations in live cells. Biochim Biophys Acta 2014; 1840:2014–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.