

ABSTRACT
Type 2 diabetes mellitus (T2DM) is a multisystemic disease that afflicts more than 415 million people globally—the incidence and prevalence of T2DM continues to rise. It is well‐known that T2DM has detrimental effects on bone quality that increase skeletal fragility, which predisposes subjects to an increased risk of fracture and fracture healing that results in non‐ or malunion. Diabetics have been found to have perturbations in metabolism, hormone production, and calcium homeostasis—particularly PTH expression—that contribute to the increased risk of fracture and decreased fracture healing. Given the perturbations in PTH expression and the establishment of hPTH (1–34) for use in age‐related osteoporosis, it was determined logical to attempt to ameliorate the bone phenotype found in T2DM using hPTH (1–34). Therefore, the present study had two aims: (i) to establish a suitable murine model of the skeletal fragility present in T2DM because no current consensus model exists; and (ii) to determine the effects of hPTH (1–34) on bone fractures in T2DM. The results of the present study suggest that the polygenic mouse of T2DM, TALLYHO/JngJ, most accurately recapitulates the diabetic osteoporotic phenotype seen in humans and that the intermittent systemic administration of hPTH (1–34) increases fracture healing in T2DM murine models by increasing the proliferation of mesenchymal stem cells. © 2020 The Authors. JBMR Plus published by Wiley Periodicals, Inc. on behalf of American Society for Bone and Mineral Research.
Keywords: DIABETIC OSTEOPOROSIS, FRACTURE HEALING, ORTHOPEDIC TRAUMA, PARATHYROID HORMONE, TYPE 2 DIABETES
Introduction
PTH, an 84‐amino acid peptide released by the parathyroid glands, is an important calcium‐regulating hormone that aids in regulating bone remodeling. PTH is released from the parathyroid glands in response to hypocalcemic states and increases serum calcium levels by stimulating the production of 1,25‐dihydroxyvitamin D [1,25(OH)2D], which in turn increases gastrointestinal calcium absorption, renal calcium reabsorption, and the rate of bone resorption by osteoclasts.1 Although the sustained increase in PTH secretion seen in hyperparathyroidism results in bone loss caused by an upregulation of osteoclastic bone resorption, transient rises in PTH levels have been shown to increase bone mass as PTH's anabolic properties outweigh its catabolic activity when used in this modality.2 Indeed, multiple animal studies have shown that brief increases in PTH levels stimulate endochondral bone formation and promote mesenchymal stem cell (MSC) differentiation into osteoblasts.1, 3, 4 Additionally, intermittent s.c. administration of PTH has been shown to increase the load properties and mineral content in fractures.5 Currently, daily s.c. injections of recombinant human PTH amino‐terminal peptide 1–34 [hPTH (1–34)], or the full‐length protein, hPTH (1–84), are used to treat osteoporosis in human patients.6
Multiple studies have shown that patients with type 2 diabetes mellitus (T2DM) are more likely to suffer osteoporotic fractures than nondiabetics.7, 8, 9 Undeniably, such fractures are a debilitating complication of diabetes that impairs quality of life in millions of people globally. This also leads to the expenditure of billions of health care dollars in the United States caused by the direct care and the loss of productivity of patients from complications that follow fractures.10, 11, 12, 13, 14 Unfortunately, the rate of T2DM and its associated fractures continues to rise in the United States. As of 2017, the Centers for Disease Control and Prevention reports that 23.1 million Americans, or 7.2% of the population, have diagnosed diabetes, with only 5% being T1DM.15 When they include estimates for those undiagnosed, this number increases to 30.3 million, or 9.4% of the population. These figures are in addition to the 84.1 million Americans 18 years of age or older who have prediabetes and are at risk of developing the disease in the next 5 years.15
The exact connection between diabetic pathophysiology and an increased risk of bone fracture is unknown. Hyperglycemic states have been shown to cause an increase in urinary calcium loss, resulting in a negative calcium balance that may contribute to the increased rate of bone fractures in diabetic patients.7 Perhaps more significantly, the persistent inflammatory state seen with chronic hyperglycemia increases chondrocyte and osteoblast apoptosis, while simultaneously encouraging osteoclast survival, tipping the scales of bone remodeling in favor of bone loss.16 Similarly, inflammatory markers, including TNFα, inhibit angiogenesis, a vital component of fracture healing and bone health.17 This state of calcium depletion, remodeling inequality, and angiogenic insufficiency may contribute both to the increased fracture risk and the delayed fracture healing seen in diabetic populations.
T2DM has a diverse phenotype stemming from many etiologies in humans because genetics and environment both play a significant role in the development of the disease. Further, T2DM is associated with many comorbidities, particularly obesity, dyslipidemia, and hypertension, a trend that holds true even in adolescent populations.18 As such, there is no single mouse model that fully encompasses all the varied features of clinical T2DM. The present study utilizes two distinct murine models of T2DM. The first murine model, B6.BKS(D)‐Leprdb/J (db/db), has a monogenetic knockout mutation in the leptin receptor gene resulting in hyperphagia, marked obesity, and consequent hyperglycemia. The second murine model, TALLYHO/JngJ, is a polygenetic model of T2DM that accurately reflects the many characteristic phenotypes of human T2DM. Together, these models cover a wide variety of T2DM etiologies and offer insight into the human disease.
With these chosen models of T2DM, the current study investigates: (i) which murine model of skeletal fragility found in T2DM is most representative of the human phenotype; and (ii) the potential use of systemic hPTH (1–34) in diabetic fracture healing. Intermittent systemic treatment of hPTH (1–34) has previously been successful in improving bone quality for osteoporotic human patients.6 Moreover, diabetes is associated with increased urinary calcium excretion, and although the associated low levels of serum calcium usually induce an increase in parathyroid activity, PTH levels in patients with DM have been shown to be 55% of normal.19 Of note, low PTH secretion is itself a cause of poor bone quality in T2DM patients, and is not suggestive of resistance to PTH at the osteoblast level.20 This functional hypoparathyroidism makes PTH an area of interest in diabetic bone research and identifies T2DM patients with fractures as ideal candidates for potential treatment with exogenous PTH.
The data from the present study suggest that the monogenic murine model db/db is the most accurate representation of human diabetic osteoporosis and that the intermittent administration of hPTH (1–34) may improve fracture healing capacity in human patients with T2DM as evidenced by this effect in the chosen murine models.
Materials and Methods
Murine models
All animal experiment protocols were approved by Yale University School of Medicine's Institutional Animal Care and Use Committee. Up to five mice were housed in sterile, ventilated cages and were allowed food and water ad libitum within Yale University School of Medicine's animal facility. Chow was provided by Yale University. Male, 10‐week‐old C57BL/6J (Stock No: 000664), B6.BKS(D)‐Leprdb/J (db/db) (Stock No: 000697), Swiss Webster/J (SWR/J) (Stock No: 000689), and TALLYHO/JngJ (Stock No: 005314) strains were all purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Animals were randomized to treatment and control groups; researchers were not blinded. No important adverse events occurred during animal experiments.
Blood glucose measurements
All blood glucose measurements were obtained using a commercially available glucometer and test strips (One Touch Ultra 2; LifeScan, Inc., Milpitas, CA, USA) after fasting the mice for 16 hours. Blood was obtained from the tail vein following anesthesia.
Surgical method
A ketamine (Zoetis Inc., Kalamazoo, MI, USA) / xylazine solution (Akorn Inc., Lake Forest, IL, USA) was administered i.p. at a dose of 100 mg/kg / 10 mg/kg to induce anesthesia. The depth of anesthesia was assessed by response to tail, pinnae, or pedal pinch. Preemptively, buprenorphine (0.05 mg/kg i.p.) was administered for general pain control, whereas the surgical incision was infiltrated with bupivacaine (AuroMedics Pharma LLC, E. Windsor, NJ, USA) for local pain control (4 mg/kg s.q.). An incision extending from the hip to the knee joint was made on the lateral right hindlimb. The patella was displaced laterally, and a 1‐inch 23‐gauge needle was inserted into the intramedullary space along the entire length of the femur from the femoral condyles to the femoral neck. The lateral thigh muscles were spread apart bluntly to expose the femoral shaft, and a length of Gigli saw wire was passed around the femur with care to protect the soft tissues and neurovascular structures (Fig. 1 D, upper panel). The intramedullary needle was then withdrawn halfway, and the Gigli saw wire was used to create a transverse midshaft fracture in the femur (Fig. 1 D, middle panel). The needle was then readvanced past the created fracture into the proximal femur and the musculature was returned to anatomical position (Fig. 1 D, lower panel). The needle was then clipped at the end of the distal femur, leaving a length of needle in the intramedullary space. The patella was returned to anatomical position; the musculature was opposed using suture; and the skin incision was closed with surgical staples. hPTH (1–34) (40 μg/kg; Alfa Aesar, Haverhill, MA, USA) was injected s.c. immediately following surgery for mice in the treatment group. Analgesic (buprenorphine; Reckitt Benckiser Healthcare, Hull, England) and antibiotic (10 mg/kg enrofloxacin; Bayer, Leverkusen, Germany) treatments were maintained for 72 hours following surgery. X‐ray was used to verify pin placement (Fig. 1 E).
Figure 1.
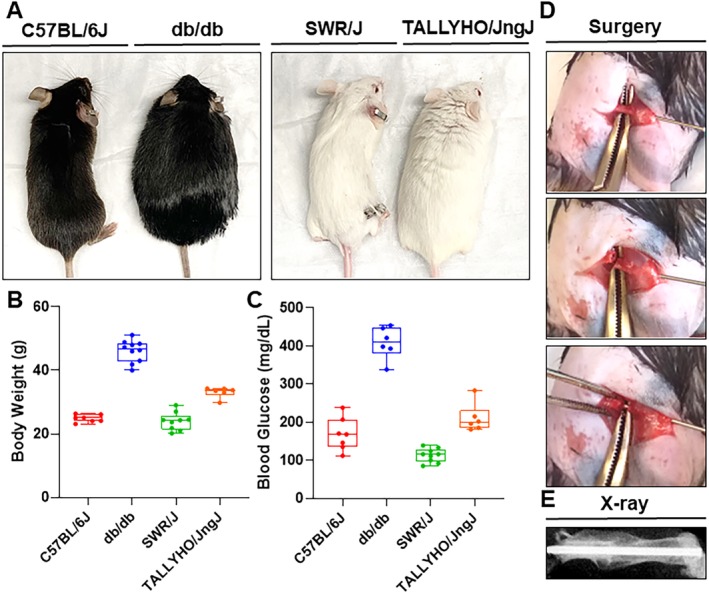
Body mass, blood glucose measurements, and surgical technique in type 2 diabetes mellitus murine models. (A) Gross body weight differences in murine strains. (B) Body weight (g) of each murine strain was quantified using an analytical scale. C57BL/6J models showed an average body weight of 24.8 ± 1.2 g (n = 6), whereas the db/db model average body weight was 46.1 ± 3.5 g (n = 6). The SWR/J model average body weight was 24.0 ± 2.8 g (n = 9), whereas TALLYHO/JngJ models had an average body weight of 33.1 ± 1.7 g (n = 6). (C) Blood glucose measurements (mg/dL) of murine models were taken using a glucometer (One Touch Ultra 2; LifeScan, Inc., Milpitas, CA, USA) after 16 hours of fasting. C57BL/6J mice showed average blood glucose levels of 148.0 ± 43.1 mg/dL (n = 6); db/db mice showed average glucose levels of 408.5 ± 42.5 mg/dL (n = 6). SWR/J showed average glucose levels of 112.9 ± 18.1 mg/dL (n = 9); TALLYHO/JngJ showed average glucose levels of 210.3 ± 37.6 mg/dL (n = 6). Body weights and fasting plasma glucose levels of all mice were measured at 10 weeks of age. (D) Key steps during surgery: placement of Gigli saw wire for fracture induction (upper panel), induction of transverse fracture (middle panel), and fixation of fracture (lower panel). (E) X‐ray image verifying correct pin placement after surgery.
μCT studies
μCT analysis of femurs was performed in 10‐week‐old mice for baseline studies and at 28 days postfracture for fracture healing studies. An 11.5‐mm sample of the femoral shaft was used for mineralized callus μCT analysis on a Scanco μCT 35 system (Scanco Medical, Brüttisellen, Switzerland). A 10‐μm voxel size, 55 KVp, 0.36‐degree rotation step (180‐degree angular range), and a 400‐ms exposure per view were used for the scans that were performed in PBS solution. The Scanco μCT software (HP, DECwindows Motif 1.6) was used for 3D reconstruction and viewing of images. After 3D reconstruction, callus volumes with extracted pre‐existing cortex were segmented using a global threshold of 0.4 g/c. Tissue mineral density (TMD), direct total volume (TV), bone volume (BV), and bone volume fraction (BV/TV) were calculated for each callus. Starting 100 μm from the growth plate, 1.35 mm of the distal part of each femur and a 1.4 mm part of the mid‐diaphysis were used for trabecular and cortical bone (respectively) μCT analysis on the Scanco μCT 35 system, as described above. Relative cortical area (Ct.Ar/Tt.Ar), thickness of the cortex (Ct.Th), and polar moment of inertia were calculated for cortical bone. TMD, apparent density, and directly measured bone volume fraction (BV/TV), surface‐to‐volume ratio (BS/BV), trabecular thickness (Tb.Th), trabecular number (Tb.N), and trabecular separation (Tb.Sp) were calculated for the trabecular bone.
Histology and histomorphometry
Harvested femora were fixed for 1 day in 4% paraformaldehyde and PBS directly after euthanasia. After fixation, the bones were rinsed and decalcified with 10% EDTA (pH 7.2 to 7.4) for 2 weeks on a shaker. The intramedullary pins were removed from the bones before embedding and sectioning. After, the specimens were processed to a thickness of 5 μm. Immunohistochemistry (IHC) was performed using the MACH 4 Universal HRP‐Polymer (Biocare Medical, LLC, Pacheco, CA, USA) to detect anti‐CD51 mouse antibody (MAI‐33578; Abcam, Cambridge, MA, USA) and anti‐PPARɤ rabbit monoclonal antibody (2435; Cell Signaling Technology, Inc., Danvers, MA, USA). Reactions were then visualized with diaminobenzidine (DAB) as substrate. The sections were counterstained with hematoxylin. All staining procedures were followed as instructed by the manufacturer. ImageJ software (NIH, Bethesda, MD, USA; https://imagej.nih.gov/ij/) was used to count the number of positive stained cells. Additionally, multiplex immunohistochemistry (m‐IHC) was performed using fluorescein, cyanine 3, and cyanine 5 dyes (PerkinElmer, Boston, MA, USA) for m‐IHC according to the manufacturer's instructions. Lastly, cartilage was defined as tissue with positive safranin O/fast green staining. Images of histologic sections were captured using a Cytation 5 Imaging Reader (Biotek, Winooski, VT, USA). ImageJ software was used to count the number of pixels for analysis.
Bone mechanical strength testing
After scanning, the specimen was mounted on a three‐point‐bending jig on an electromagnetic loading machine. The loading location was determined at 55% of the femur length from the femoral head. Dynamic mechanical analysis (DMA) was performed by applying nondestructive compressive oscillatory displacement with a mean level of 0.01 mm and amplitude of 0.005 mm at the range of 0.5 to 3 Hz. A displacement transducer with 15‐nm resolution was utilized to control displacement and detect the small magnitude of signal changes. Dynamic complex stiffness (K*) was computed from elastic (storage; K′) and viscous (loss; K′′) stiffness with an equation of K* = K′ + iK′′ using the cyclic force and displacement. Tangent delta (tan δ), which accounts for ability of loading energy dissipation, was computed by K′′/K′. Following the nondestructive DMA testing, static fracture testing was conducted with a displacement rate of 0.5 mm/s up to fracture. Static stiffness was measured using the maximum slope of force‐displacement curve and maximum force was assessed at the fracture point.
Statistical analysis
All analyses were performed using GraphPad Prism (GraphPad Software, Inc., La Jolla, CA, USA). Data were analyzed using one‐way ANOVA and Tukey post hoc correction. Statistical significance was assigned for p ≤ 0.05. All experiments were done in triplicate.
Results
T2DM murine models recapitulate the human T2DM phenotype
Body weight data showed experimental mouse models, db/db and TALLYHO/JngJ, to have increased body mass as compared with their control strains, C57BL/6J and SWR/J (Fig. 1 A,B). In comparison with C57BL/6J mice that have an average body weight of 24.8 ± 1.2 g, db/db mice had an average body weight of 46.1 ± 3.5 g. Also, in comparison with SWR/J, which had an average body mass of 24.0 ± 2.8 g, TALLYHO/JngJ mice had an elevated average weight of 33.1 ± 1.7 g. Moreover, the current study found hyperglycemia in both experimental mouse strains by measuring baseline blood glucose levels in each mouse group after 16 hours of fasting. Both db/db and TALLYHO/JngJ mice had elevated average blood glucose levels of 408.5 ± 42.5 mg/dL and 210.3 ± 37.6 mg/dL as compared with control strains C57BL/6J and SWR/J, which had average blood glucose levels of 148.0 ± 43.1 mg/dL and 112.9 ± 18.1 mg/dL, respectively (Fig. 1 C).
μCT analysis of femora from T2DM murine models
Untreated, harvested femora from 10‐week‐old mice from each strain were analyzed using μCT analysis to determine the characteristics of each strain's cortical Fig. 2 A–D and trabecular Fig. 3 A–E bone at baseline. μCT analysis of C57BL/6J mouse femoral cortical bone showed an average cortical thickness (Ct.Th) of 0.16 ± 0.01 mm, cortical bone mineral density (Ct.BMD) of 1028.7 ± 18.6 mg/cm3, and cortical bone volume fraction (Ct.BV/TV) of 0.91 ± 0.006. On analysis of db/db cortical bone, average Ct.Th was 0.14 ± 0.002 mm, Ct.BMD was 1057.4 ± 9.8 mg/cm3, and Ct.BV/TV was 0.90 ± 0.001. SWR/J cortical bone showed an average Ct.Th of 0.16 ± 0.007 mm, Ct.BMD of 1278.0 ± 5.6 mg/cm3, and Ct.BV/TV of 0.43 ± 0.02. Lastly, analysis of the cortical bone of TALLYHO/JngJ showed an average Ct.Th of 0.22 ± 0.004 mm, Ct.BMD of 1134.7 ± 21.0 mg/cm3, and Ct.BV/TV of 0.94 ± 0.002 Fig. 2 B–D.
Figure 2.
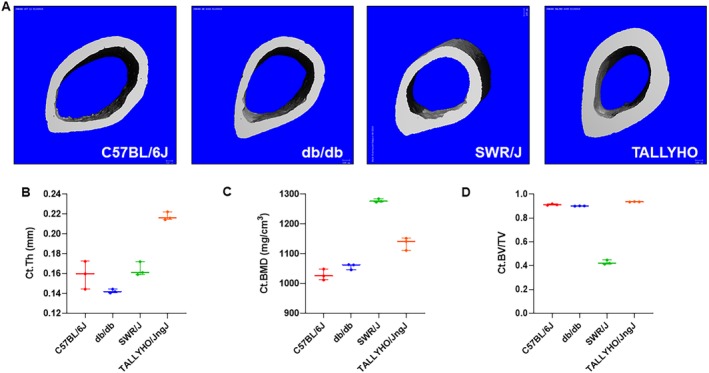
μCT analysis of femoral cortical bone from type 2 diabetes mellitus murine models. (A) Representative images of the cross sections of the mid‐diaphysis of murine femurs. (B–D) Untreated femora from 10‐week‐old mice were analyzed using a Scanco μCT 35 system (Scanco Medical, Brüttisellen, Switzerland). C57BL/6J mice (n = 3) showed an average cortical thickness (Ct.Th) of 0.16 ± 0.01 mm, cortical bone mineral density (Ct.BMD) of 1028.7 ± 18.6 mg/cm3, and cortical bone volume fraction (Ct.BV/TV) of 0.91 ± 0.006. Db/db mice (n = 3) showed an average Ct.Th of 0.14 ± 0.002 mm, Ct.BMD was 1057.4 ± 9.8 mg/cm3, and Ct.BV/TV was 0.90 ± 0.001. SWR/J mice (n = 3) showed an average Ct.Th of 0.16 ± 0.007 mm, Ct.BMD of 1278.0 ± 5.6 mg/cm3, and Ct.BV/TV of 0.43 ± 0.02. TALLYHO/JngJ mice (n = 3) showed an average Ct.Th of 0.22 ± 0.004 mm, Ct.BMD of 1134.7 ± 21.0 mg/cm3, and Ct.BV/TV of 0.94 ± 0.002.
Figure 3.
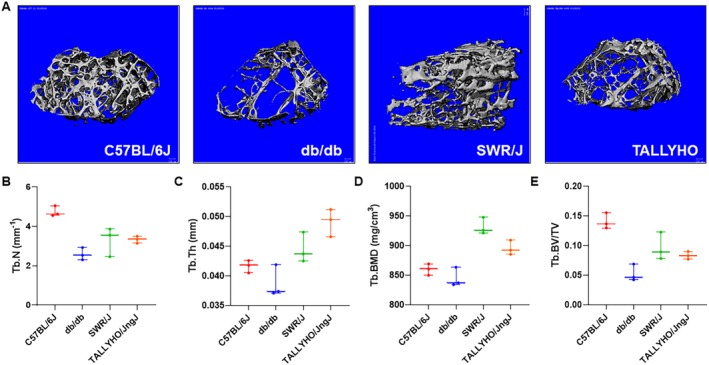
μCT analysis of femoral trabecular bone from type 2 diabetes mellitus murine models. (A) Representative images of the trabecular architecture of murine femurs. (B–E) Untreated femora from 10‐week old mice were analyzed using a Scanco μCT 35 system (Scanco Medical, Brüttisellen, Switzerland). C57BL/67 (n = 3) showed an average trabecular number (Tb.N) of 4.7 ± 0.3 mm−1, trabecular thickness (Tb.Th) of 0.04 ± 0.001 mm, trabecular bone mineral density (Tb.BMD) of 859.9 ± 9.7 mg/cm3, and trabecular bone volume fraction (Tb.BV/TV) of 0.14 ± 0.01. Db/db mice (n = 3) showed an average Tb.N of 2.6 ± 0.3 mm−1, Tb.Th of 0.04 ± 0.003 mm, Tb.BMD of 844.9 ± 16.2 mg/cm3, and Tb.BV/TV of 0.05 ± 0.01. SWR/J mice (n = 3) showed an average Tb.N of 3.3 ± 0.7 mm−1, Tb.Th of 0.04 ± 0.003 mm, Tb.BMD of 931.8 ± 14.1 mg/cm3, and Tb.BV/TV of 0.1 ± 0.02. TALLYHO/JngJ mice (n = 3) showed an average Tb.N of 3.3 ± 0.2 mm−1, Tb.Th of 0.05 ± 0.002 mm, Tb.BMD of 895.7 ± 12.3 mg/cm3, and Tb.BV/TV of 0.08 ± 0.01.
μCT analysis of C57BL/6J mouse femoral trabecular bone showed an average trabecular number (Tb.N) of 4.7 ± 0.3 mm−1, trabecular thickness (Tb.Th) of 0.04 ± 0.001 mm, trabecular bone mineral density (Tb.BMD) of 859.9 ± 9.7 mg/cm3, and trabecular bone volume fraction (Tb.BV/TV) of 0.14 ± 0.01. Analysis of db/db trabecular bone showed an average Tb.N of 2.6 ± 0.3 mm−1, Tb.Th of 0.04 ± 0.003 mm, Tb.BMD of 844.9 ± 16.2 mg/cm3, and Tb.BV/TV of 0.05 ± 0.01. SWR/J trabecular bone analysis showed an average Tb.N of 3.3 ± 0.7 mm−1, Tb.Th of 0.04 ± 0.003 mm, Tb.BMD of 931.8 ± 14.1 mg/cm3, and Tb.BV/TV of 0.1 ± 0.02. TALLYHO/JngJ trabecular bone showed an average Tb.N of 3.3 ± 0.2 mm−1, Tb.Th of 0.05 ± 0.002 mm, Tb.BMD of 895.7 ± 12.3 mg/cm3, and Tb.BV/TV of 0.08 ± 0.01 Fig. 3 B–E.
hPTH (1–34) increases cartilage development and CD51+ MSC proliferation in T2DM fracture calluses
Safranin O staining for cartilage revealed increased cartilaginous formation in fracture calluses in mice treated with hPTH (1–34) Fig. 4 A–B. In db/db mice, the average ratio of cartilage area to callus area was increased to 28.9% ± 11.1% for mice treated with hPTH (1–34) compared with 16.3% ± 2.0% in untreated mice (p = 0.0274); C57BL/6J mice showed an average cartilage‐to‐callus area ratio of 24.9% ± 2.6% Fig. 4 C. In TALLYHO/JngJ mice, this average ratio was 30.1% ± 13.5% in treated mice compared with 14.4% ± 2.4% in untreated mice (p = 0.0471); SWR/J showed an average cartilage‐to‐callus area ratio of 11.6% ± 13.3% Fig. 4 D. Moreover, IHC staining for CD51+ MSC proliferation in reclaimed calluses from T2DM mouse femora was performed 7 days postfracture; quantification of IHC staining showed increased proliferation of CD51+ MSC in both db/db and TALLYHO/JngJ mice treated with hPTH (1–34) Fig. 5 A. In db/db mice, the average ratio of CD51+ cells to total cells was 80.1% ± 11.3% for mice treated with hPTH (1–34) compared with 57.6% ± 18.0% in untreated db/db mice (p = 0.0353); C57BL/6J showed an average ratio of CD51+ cells to total cells of 78.7% ± 7.3 (Fig. 5 B). In TALLYHO/JngJ mice, this average ratio was 78.0% ± 12.0% for treated mice compared with 50.1% ± 28.8% in untreated mice (p = 0.0374); SWR/J showed an average ratio of CD51+ cells to total cells of 86.1% ± 6.8% (Fig. 5 C). Furthermore, m‐IHC showed colocalization of PTHr1 and Ki67 in CD51+ cells (Fig. 6).
Figure 4.
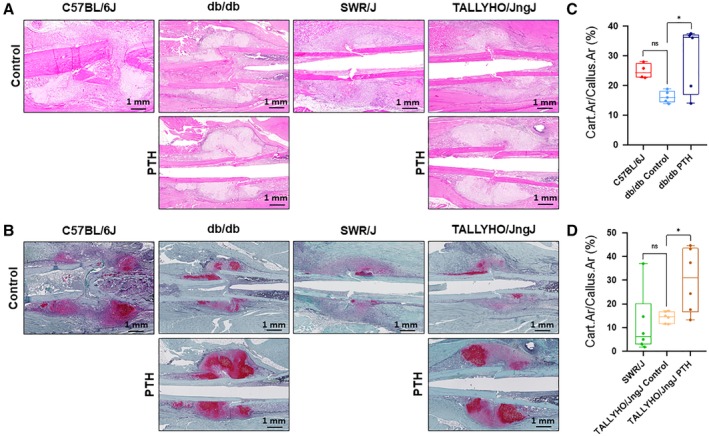
Safranin O staining for cartilage deposition in fracture callus. (A) Representative H&E histology of fracture calluses. (B) Representative safranin O staining of fracture calluses. (C,D) quantification of cartilage area in calluses from 10‐week‐old mice treated with 40 μg / kg hPTH (1–34) or without intervention are shown at day 7 postfracture. In untreated db/db mice, the average cartilage to callus area was 16.3 ± 2.0% (n = 5), whereas in db/db mice treated with hPTH (1–34) the average cartilage‐to‐callus area was 28.9 ± 11.1% (n = 5; p = 0.0274). C57BL/6J mice showed an average cartilage‐to‐callus area of 24.9 ± 2.6% (n = 4). Untreated TALLYHO/JngJ mice showed an average cartilage‐to‐callus area of 14.4 ± 2.4% (n = 6), whereas treated mice showed an increased average area of 30.1 ± 13.5% (n = 6; p = 0.0471). SWR/J mice showed an average cartilage‐to‐callus area of 11.6 ± 13.3% (n = 6). The difference between C57BL/6J and db/db control was not significant (p = 0.157). The difference between SWR/J and TALLYHO/JngJ control was not significant (p = 0.868). Red staining on safranin O denotes cartilage deposition. Measurements were performed using ImageJ software. ns = p ≥ 0.05, *p < 0.05; one‐way ANOVA.
Figure 5.
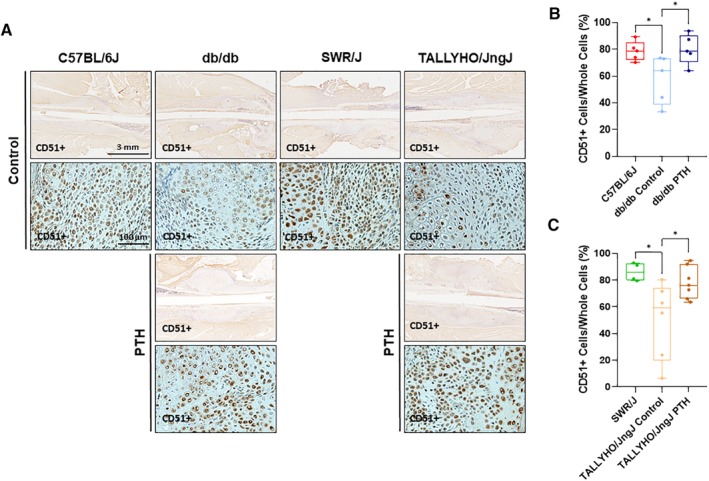
Immunohistochemistry for CD51+ MSC in fracture calluses. (A) Representative images of immunohistochemistry staining for CD51+ cells. (B,C) Quantification of immunohistochemistry analysis of callus formation in 10‐week‐old mice is shown at 7 days postfracture. C57BL/6J mice showed an average CD51+ percentage of 78.7 ± 7.3% (n = 5). Db/db mice treated with hPTH (1–34) showed an increase in average CD51+ cells to 80.1 ± 11.3% (n = 5) as compared with untreated db/db mice, which showed an average of 57.6 ± 18.0% (n = 5; p = 0.0353). SWR/J mice showed an average CD51+ percentage of 86.1 ± 6.8 (n = 4). TALLYHO/JngJ mice treated with hPTH (1–34) showed an increase in average CD51+ cells to 78.0 ± 12.0% (n = 7) as compared with untreated TALLYHO/JngJ mice, which showed an average 50.1 ± 28.8% (n = 6; p = 0.0374). The difference between C57BL/6J and db/db controls was significant (p = 0.0488) as was that between SWR/J and TALLYHO/JngJ controls (p = 0.0214). Yellow‐brown staining suggests a positive CD51 stain, a marker for mesenchymal stem cells. Measurements were performed using ImageJ software. *p < 0.05; one‐way ANOVA.
Figure 6.
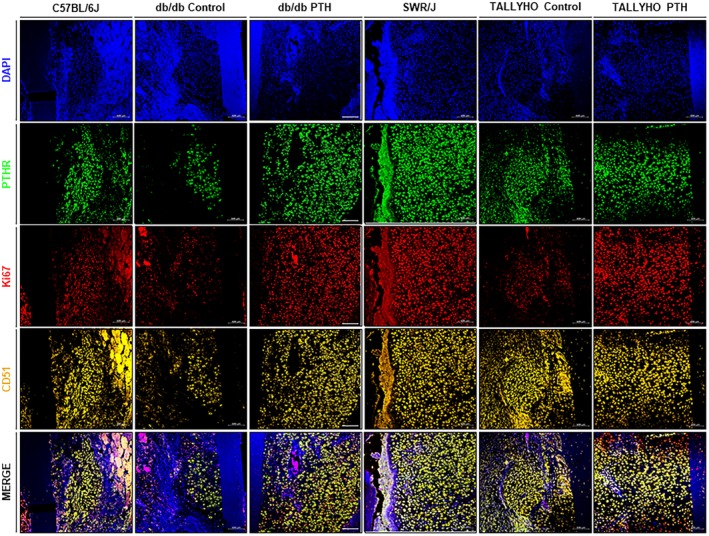
Multiplex immunohistochemistry staining for colocalization of PTHr1 and Ki67 in CD51+ mesenchymal stem cells (MSCs). Representative images of multiplex immunohistochemistry (m‐IHC) staining. Following quantification of IHC staining for CD51+, m‐IHC was performed to colocalize PTHr1 and Ki67. The membrane receptor PTHr1 colocalizes with the MSC membrane receptor CD51+; additionally, the nuclear protein Ki67 localizes to CD51+/PTHr1+ cells, which is demonstrative of accelerated proliferation in these cells. The data suggest that hPTH (1–34) increases MSC proliferation through a direct mechanism after interaction with PTHr1. Blue (4,6‐diamidino‐2‐phenylindole) stains for nuclei; green (fluorescein) stains for PTHr1; red (cyanine 5) stains for Ki67; orange (cyanine 3) stains for CD51.
hPTH (1–34) administration increases bone mineralization in T2DM murine models
μCT analysis of femora reclaimed from db/db and TALLYHO/JngJ mice treated with hPTH (1–34) for 28 days postfracture showed that hPTH (1–34) administration promoted fracture healing in diabetic mice Fig. 7 A. In femora reclaimed from diabetic mice, fracture calluses were appreciably smaller in size and showed more porous hard callus formation in untreated mice compared with calluses from fractured femora in mice treated with hPTH (1–34). Furthermore, db/db and TALLYHO/JngJ mice treated with hPTH (1–34) showed an increased fraction of mineralized bone (BV/TV) in healing calluses (Fig. 7). In db/db mice, average BV/TV was 0.25 ± 0.05 in mice treated with hPTH (1–34) compared with 0.13 ± 0.03 in untreated db/db mice (p = 0.0020); C57BL/6J showed an average BV/TV of 0.24% ± 0.02% (Fig. 7 B). In TALLYHO/JngJ mice, average BV/TV was 0.37% ± 0.09% in treated mice compared with 0.26 ± 0.08% in untreated TALLYHO/JngJ mice (p = 0.0054); SWR/J showed an average BV/TV of 0.33% ± 0.03% (Fig. 7 C).
Figure 7.
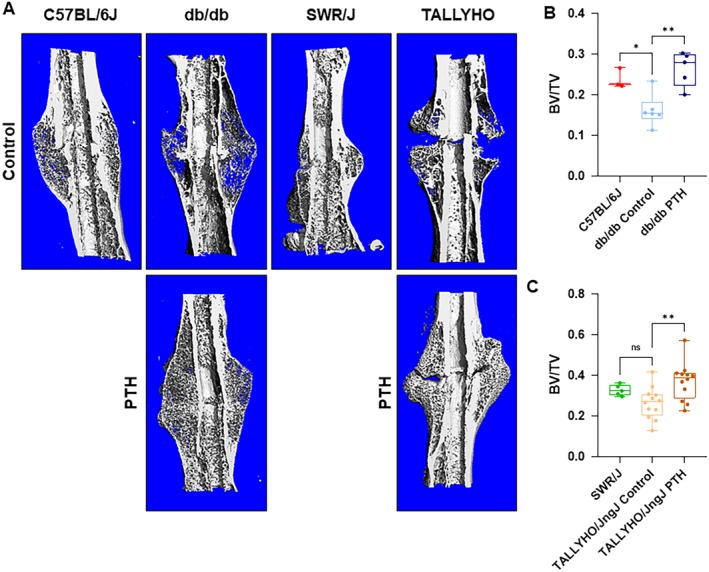
μCT analysis of fracture calluses. (A) Representative images of μCT‐scanned and graphically reconstructed global images of fracture calluses at 28 days postfracture. (B,C) Assessment of the ratio of bone volume to total volume (BV/TV) of healing femora from C57BL/6J (n = 3), db/db (Control n = 6, PTH n = 5), SWR/J (n = 5), and TALLYHO/JngJ mice (Control n = 12, PTH n = 12). Db/db and TALLYHO/JngJ mice treated with hPTH (1–34) showed a significantly increased average fraction of mineralized bone (BV/TV) at 0.25 ± 0.05 (p = 0.0020) and 0.37 ± 0.09 (p = 0.0054), respectively. This is compared with untreated db/db and TALLYHO/JngJ mice groups at 0.13 ± 0.03 and 0.26 ± 0.08, respectively. C57BL/6J and SWR/J mice showed an average fraction of mineralized bone of 0.24 ± 0.02 and 0.33 ± 0.03, respectively. The difference between C57BL/6J and db/db control was significant (p = 0.0326), whereas SWR/J and TALLYHO/JngJ controls were not (p = 0.254). ns = p ≥ 0.05, *p < 0.05, **p < 0.01; one‐way ANOVA.
hPTH (1–34) administration increases force required to induce a repeat fracture
Three‐point mechanical strength testing (MST) of femora reclaimed from db/db and TALLYHO/JngJ treated with hPTH (1–34) showed an increase in the maximal load (Fmax) required to induce a new fracture following initial fracture healing (Fig. 8). Db/db mouse femora treated with hPTH (1–34) required a significantly increased average Fmax of 8.3 ± 2.5 N to induce a fracture as compared with control db/db mice, which required an average of 2.2 ± 1.3 N (p = 0.0034); C57BL/6J required an average of 15.7 ± 3.4 N to induce fracture (Fig. 8 A). hPTH (1–34) treated TALLYHO/JngJ mouse femora required an average Fmax of 8.3 ± 4.1 N as compared with their control counterparts, which required an average of 4.2 ± 2.4 N to induce a fracture (p = 0.0456); SWR/J required an average of 18.0 ± 5.3 N to induce a new fracture (Fig. 8 B).
Figure 8.
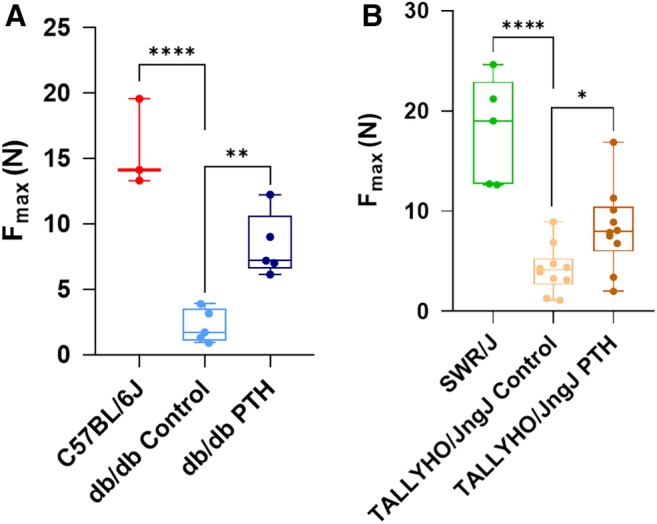
Three‐point mechanical stress testing of healed femora. (A,B) Quantification of mechanical stress testing results of mouse femora harvested 28 days after fracture induction with and without daily injections of hPTH (1–34). Db/db mouse femora treated with hPTH (1–34) required an average Fmax of 8.3 ± 2.5 N (n = 5; p = 0.0034) to induce a fracture as compared with control db/db mice, which required an average of 2.2 ± 1.3 N (n = 5). C57BL/6J mice required an average of 15.7 ± 3.4 N (n = 3) to induce fracture. hPTH (1–34) treated TALLYHO/JngJ mouse femora required an average Fmax of 8.3 ± 4.1 N (n = 10; p = 0.0456) as compared with their control counterparts, which required an average of 4.2 ± 2.4 N (n = 10) to induce a fracture. SWR/J mice required an average of 18.0 ± 5.3 N (n = 5) to induce fracture. The difference between C57BL/6J and db/db controls was significant (p = 0.0001). The difference between SWR/J and TALLYHO/JngJ controls was also significant (p = 0.0001). *p < 0.05, **p < 0.01; ****p < 0.0001; one‐way ANOVA.
Discussion
TALLYHO/JngJ murine models of T2DM best mimic that of the human skeletal phenotype in T2DM
In this study, the effects of hPTH (1–34) on fracture healing in T2DM were tested. Currently, the intermittent systemic administration of hPTH (1–34) is utilized to treat senile osteoporosis,21, 22 osteoporosis secondary to prolonged glucocorticoid administration,23, 24, 25 and bisphosphonate‐associated osteonecrosis of the jaw.26, 27, 28, 29 Based on prior literature establishing that T2DM patients have both an increased risk of fracture30, 31, 32, 33, 34 and decreased PTH levels,19, 35, 36 it was determined practical to utilize hPTH (1–34) in the treatment of bone fractures secondary to T2DM. Therefore, the hypothesis of the present study was that hPTH (1–34) would improve the decreased bone‐quality phenotype and subsequently decrease the risk of bone fractures present in T2DM. To test this hypothesis, the determination of a relevant animal model was necessary. Given the complexities and polygenic etiology of T2DM in humans, there is currently no singular animal model of human T2DM that recapitulates the varied phenotypic presentations of the disease. Moreover, there is no consensus on the best murine model to study skeletal fragility secondary to T2DM. Therefore, two murine models of T2DM were analyzed: a single gene knockout murine model [B6.BKS(D)‐Leprdb/J (db/db)] and a polygenic murine model (TALLYHO/JngJ).
The db/db mouse is produced by a single gene mutation in the leptin receptor on the C57BL/6J background that results in extreme hyperphagia, subsequent morbid obesity, marked hyperglycemia, a compensatory hyperinsulinemia, and the resultant expansion of pancreatic β‐cell mass37; the data of the present study reiterated the obesity and hyperglycemia present in this strain. Furthermore, although db/db mice have elevated body mass and adiposity, which generally serves to increase bone mass,38, 39, 40 db/db mice have decreased bone volume (BV/TV) in both cortical and trabecular bone41—a fact recapitulated by the data of the present study. Additionally, prior studies have utilized histology to establish that db/db mice have decreased Ct.Th, Tb.N, and Tb.Th in long bones42; all of these metrics were verified in the present study. Moreover, Takeshita and colleagues determined that BMD of cortical and trabecular bone is decreased in db/db mice after 12 weeks of age, but not prior to this age range43; however, the mice used in this study were not within this age range and had normal to high BMD. It is well‐known that the bone of db/db mice has increased susceptibility to fractures given these factors.44
On the other hand, the TALLYHO/JngJ mouse is a polygenic murine model of T2DM that is characterized by increased body weight, moderate hyperglycemia, and hyperinsulinemia45; obesity and hyperglycemia were recapitulated in this study. Like all polygenic strains, TALLYHO/JngJ has no exact genetic control strains45, 46; TALLYHO/JngJ seems to be Swiss‐derived and shares 86.8% genotype homology with SWR/J, which is most often utilized as a control.45 Additionally, Devlin and colleagues showed that TALLYHO/JngJ mice have increased Ct.Th and decreased BV/TV, which this study verified; however, Devlin and colleagues showed more severe deficits in BV/TV than the present study and showed decreased Tb.N and Tb.Th in comparison to this study. BMD was also determined to be decreased in TALLYHO/JngJ by Devlin and colleagues, which was reiterated in the present study.47 The differences in the skeletal fragility phenotypes of TALLYHO/JngJ between the present study and the established literature are likely because of known discrepancies in the phenotypes of the strain based on acquisition location and environmental factors.45
With respect to humans, it is well‐known that although BMD is normal or increased in T2DM, patients are, paradoxically, at risk of increased bone fractures.7, 32, 48, 49, 50 However, the remainder of the literature regarding skeletal fragility in human T2DM shows vastly varied findings with respect to cortical and trabecular bone compartments.51, 52, 53, 54 Given these facts, we suggest TALLYHO/JngJ is the best model for the study of skeletal fragility in T2DM and fracture healing because of its polygenic background much like that of the human disease and increases in cortical bone compartment. Furthermore, db/db mice have deficiencies in the cortical and trabecular compartments that are different from and do not explicitly fit the human phenotype. Moreover, leptin is responsible for reducing bone fragility by inducing osteoblast proliferation and inhibiting osteoclastogenesis.55 Therefore, the skeletal fragility found in db/db mice is likely primarily caused by the loss of leptin's effects, which does not mechanistically mimic the proinflammatory state of diabetes that induces osteoclastogenesis and the subsequent skeletal fragility found in diabetic humans.56
hPTH (1–34) rescues bone fracture healing in murine models of T2DM bone fragility
Multiple studies in humans57, 58, 59 and animal models60, 61, 62, 63 have determined the benefits of systemic recombinant human PTH in fracture healing and age‐related osteoporosis. In Sprague–Dawley rats, Holzer and colleagues showed that hPTH (1–34), when administered intermittently, enhanced fracture‐healing62; utilizing the same rat strain, this fact was recapitulated by Alkhiary and colleagues(60) and Nakajima and colleagues.63 However, literature concerning the effects of hPTH (1–34) in the T2DM state is relatively limited. Indeed, Liu and colleagues is the only study, to our knowledge, presently available that studies the effects of fracture healing in diabetic rodents and how to rescue the phenotype: They were able to show that exogenous PTH‐related peptide was able to accelerate fracture healing in db/db mice.64 Conversely, Ohuchi and colleagues showed that hPTH (1–34) improved BMD and bone strength in Akita diabetic mice, but did not test fracture healing.65 Suzuki and colleagues also tested the effects of hPTH (1–34) on streptozotocin‐induced diabetic Wistar rats and found that the treatment increased BMD, but failed to test fracture healing as well.66 Therefore, the present study represents one of the few studies concerning systemic hPTH (1–34) administration in diabetic fractures and explores untested rodent strains. Based on histological, μCT, and mechanical stress testing data, intermittent systemic hPTH (1–34) improves the delayed fracture‐healing phenotype of T2DM.
Indirect fracture healing recapitulates both the endochondral and intramembranous ossification that are paramount in embryological skeletal formation.67 Indirect fracture healing progresses through four distinct stages: hematoma formation, generation of a cartilaginous callus (soft callus), resorption of cartilage with mineralization of the callus (hard callus), and bone remodeling.67 Ultimately, the endochondral ossification required in indirect fracture healing is dependent on the mineralization of a soft callus; therefore, the greater the deposition of cartilage in the soft callus, the larger the callus and more accelerated and robust the ultimate fracture healing. Previous literature, however, has elucidated the fact that fracture calluses in diabetics are significantly smaller in size as compared with those generated in a normal metabolic state.68, 69, 70, 71 The present study shows that intermittent systemic hPTH (1–34) increases cartilage formation in and size of the soft callus at 7 days postfracture, the peak of soft callus formation in animal models.72 Moreover, Ko and colleagues showed that the excessive production of inflammatory cytokines that is pathognomonic of the diabetic state, specifically increased expression of TNFα, reduces MSC numbers in fracture callus areas by decreasing proliferation and increasing the apoptotic rate of MSCs.73 The present study shows that intermittent systemic hPTH (1–34) administration ameliorates the decreased number of CD51+ MSCs in the chosen diabetic murine models. Furthermore, multiplex immunohistochemistry shows that CD51+ MSC coexpress membrane PTHr1 and the nuclear marker of cell proliferation, Ki67, suggesting that hPTH (1–34) is directly responsible for restoring MSC proliferative capacity in T2DM mouse models.
Previous studies have shown that the fracture callus mineralization—development of the hard fracture callus—is reduced and delayed in diabetes.68, 69, 74 Likewise, patients with T2DM have been shown to have decreased mechanical strength of bone following fracture healing.70, 71, 75 Therefore, the diabetic state not only attenuates early fracture healing by decreasing cartilaginous callus generation, it also decreases the mineralization and formation of new bone in late fracture healing. Data from the present study shows that treatment with hPTH (1–34) increases the BV/TV in fracture calluses at day 28, the peak of mineralization and beginning of woven bone deposition in rodent models72; these data suggest reversal of the delayed late fracture healing phenotype found in T2DM. Furthermore, data collected on mechanical stress testing conducted during this study illustrate that an increased amount of force is required to induce a repeat fracture after completion of bone fracture healing in the chosen murine models. Increased Fmax to induce a fracture is demonstrative of further increased callus mineralization and bone formation. Therefore, it is evident that hPTH (1–34) treatment increases both early and late fracture healing in a mouse model with T2DM. hPTH (1–34) shows promise in reversing the negative sequelae of T2DM on bone fracture healing; however, clinical trials are needed moving forward to assess the replicability of our findings and efficacy of treatment in a human model.
Disclosure
MDR is a consultant for Paragon 28 Inc. No other author has a conflict of interest.
Acknowledgments
Funding provided by R01AR073607 (FYL), R01AR056246 (FYL), and Musculoskeletal Transplantation Foundation (FYL).
Authors' roles: Study design: KDA, JB, H‐KK, and FYL; Study conduct: KDA, AHAW, Y‐HC, and IL; Data collection: KDA, AHAW, Y‐HC, IL, H‐KK, SC, ZH, LL, and SL; Data analysis: JB; Data interpretation: KDA, JB, and MDR; Drafting manuscript: KDA and AHAW; Revising manuscript content: KDA, AHAW, JB, H‐KK, SC, MDR, and FYL. All authors approved the final manuscript. JB takes responsibility for the integrity of the data analysis.
References
- 1. Ren Y, Liu B, Feng Y, et al. Endogenous PTH deficiency impairs fracture healing and impedes the fracture‐healing efficacy of exogenous PTH (1‐34). PLoS One. 2011;6(7):e23060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Oxlund H, Ejersted C, Andreassen T, Tørring O, Nilsson M. Parathyroid hormone (1‐34) and (1‐84) stimulate cortical bone formation both from periosteum and endosteum. Calcif Tissue Int. 1993;53(6):394–9. [PubMed] [Google Scholar]
- 3. Jilka RL. Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone. 2007;40(6):1434–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lin EA, Liu C‐J, Monroy A, Khurana S, Egol KA. Prevention of atrophic nonunion by the systemic administration of parathyroid hormone (PTH 1–34) in an experimental animal model. J Orthop Trauma. 2012;26(12):719–23. [DOI] [PubMed] [Google Scholar]
- 5. Andreassen TT, Ejersted C, Oxlund H. Intermittent parathyroid hormone (1–34) treatment increases callus formation and mechanical strength of healing rat fractures. J Bone Miner Res. 1999;14(6):960–8. [DOI] [PubMed] [Google Scholar]
- 6. Hodsman AB, Bauer DC, Dempster DW, et al. Parathyroid hormone and teriparatide for the treatment of osteoporosis: a review of the evidence and suggested guidelines for its use. Endocr Rev. 2005;26(5):688–703. [DOI] [PubMed] [Google Scholar]
- 7. Vestergaard P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes—A meta‐analysis. Osteoporos Int. 2007;18(4):427–44. [DOI] [PubMed] [Google Scholar]
- 8. Janghorbani M, Van Dam RM, Willett WC, Hu FB. Systematic review of type 1 and type 2 diabetes mellitus and risk of fracture. Am J Epidemiol. 2007;166(5):495–505. [DOI] [PubMed] [Google Scholar]
- 9. Yamamoto M, Yamaguchi T, Yamauchi M, Kaji H, Sugimoto T. Diabetic patients have an increased risk of vertebral fractures independent of BMD or diabetic complications. J Bone Miner Res. 2009;24(4):702–9. [DOI] [PubMed] [Google Scholar]
- 10. Becker DJ, Kilgore ML, Morrisey MA. The societal burden of osteoporosis. Curr Rheumatol Rep. 2010;12(3):186–91. [DOI] [PubMed] [Google Scholar]
- 11. Dempster DW. Osteoporosis and the burden of osteoporosis‐related fractures. Am J Manag Care. 2011;17(6):S164. [PubMed] [Google Scholar]
- 12. Orsini LS, Rousculp MD, Long SR, Wang S. Health care utilization and expenditures in the United States: a study of osteoporosis‐related fractures. Osteoporos Int. 2005;16(4):359–71. [DOI] [PubMed] [Google Scholar]
- 13. Burge R, Dawson‐Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis‐related fractures in the United States, 2005–2025. J Bone Miner Res. 2007;22(3):465–75. [DOI] [PubMed] [Google Scholar]
- 14. Ray NF, Chan JK, Thamer M, Melton LJ 3rd. Medical expenditures for the treatment of osteoporotic fractures in the United States in 1995: report from the National Osteoporosis Foundation. J Bone Miner Res. 1997;12(1):24–35. [DOI] [PubMed] [Google Scholar]
- 15. Centers for Disease Control and Prevention . National diabetes statistics report, 2017. Atlanta, GA: Centers for Disease Control and Prevention, US Department of Health and Human Services; 2017. [Google Scholar]
- 16. Rőszer T. Inflammation as death or life signal in diabetic fracture healing. Inflamm Res. 2011;60(1):3–10. [DOI] [PubMed] [Google Scholar]
- 17. Lim JC, Ko KI, Mattos M, et al. TNFα contributes to diabetes impaired angiogenesis in fracture healing. Bone. 2017;99:26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tryggestad JB, Willi SM. Complications and comorbidities of T2DM in adolescents: findings from the TODAY clinical trial. J Diabetes Complications. 2015;29(2):307–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McNair P, Christensen M, Madsbad S, Christiansen C, Transbøl I. Hypoparathyroidism in diabetes mellitus. Eur J Endocrinol. 1981;96(1):81–6. [DOI] [PubMed] [Google Scholar]
- 20. Inaba M, Nagasue K, Okuno S, et al. Impaired secretion of parathyroid hormone, but not refractoriness of osteoblast, is a major mechanism of low bone turnover in hemodialyzed patients with diabetes mellitus. Am J Kidney Dis. 2002;39(6):1261–9. [DOI] [PubMed] [Google Scholar]
- 21. Berg C, Neumeyer K, Kirkpatrick P. Teriparatide. Nature Reviews Drug Discovery. 2003;2(4):257–8. [DOI] [PubMed] [Google Scholar]
- 22. Quattrocchi E, Kourlas H. Teriparatide: a review. Clin Ther. 2004;26(6):841–54. [DOI] [PubMed] [Google Scholar]
- 23. Saag KG, Shane E, Boonen S, et al. Teriparatide or alendronate in glucocorticoid‐induced osteoporosis. N Engl J Med. 2007;357(20):2028–39. [DOI] [PubMed] [Google Scholar]
- 24. Saag KG, Zanchetta JR, Devogelaer JP, et al. Effects of teriparatide versus alendronate for treating glucocorticoid‐induced osteoporosis: thirty‐six–month results of a randomized, double‐blind, controlled trial. Arthritis Rheum. 2009;60(11):3346–55. [DOI] [PubMed] [Google Scholar]
- 25. Migliaccio S, Brama M, Malavolta N. Management of glucocorticoids‐induced osteoporosis: role of teriparatide. Ther Clin Risk Manag. 2009;5:305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cheung A, Seeman E. Teriparatide therapy for alendronate‐associated osteonecrosis of the jaw. N Engl J Med. 2010;363(25):2473–4. [DOI] [PubMed] [Google Scholar]
- 27. Lau AN, Adachi JD. Resolution of osteonecrosis of the jaw after teriparatide [recombinant human PTH‐(1‐34)] therapy. J Rheumatol. 2009;36(8):1835–7. [DOI] [PubMed] [Google Scholar]
- 28. Lee JJ, Cheng SJ, Jeng JH, Chiang CP, Lau HP, Kok SH. Successful treatment of advanced bisphosphonate‐related osteonecrosis of the mandible with adjunctive teriparatide therapy. Head Neck. 2011;33(9):1366–71. [DOI] [PubMed] [Google Scholar]
- 29. Narongroeknawin P, Danila MI, Humphreys LG Jr, Barasch A, Curtis JR. Bisphosphonate‐associated osteonecrosis of the jaw, with healing after teriparatide: a review of the literature and a case report. Spec Care Dentist. 2010;30(2):77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ivers RQ, Cumming RG, Mitchell P, Peduto AJ. Diabetes and risk of fracture: the Blue Mountains eye study. Diabetes Care. 2001;24(7):1198–203. [DOI] [PubMed] [Google Scholar]
- 31. Schwartz AV, Sellmeyer DE, Ensrud KE, et al. Older women with diabetes have an increased risk of fracture: a prospective study. J Clin Endocrinol Metabol. 2001;86(1):32–8. [DOI] [PubMed] [Google Scholar]
- 32. De Liefde I, Van der Klift M, De Laet C, Van Daele P, Hofman A, Pols H. Bone mineral density and fracture risk in type‐2 diabetes mellitus: the Rotterdam study. Osteoporos Int. 2005;16(12):1713–20. [DOI] [PubMed] [Google Scholar]
- 33. Forsen L, Meyer H, Midthjell K, Edna T‐H. Diabetes mellitus and the incidence of hip fracture: results from the Nord‐Trøndelag health survey. Diabetologia. 1999;42(8):920–5. [DOI] [PubMed] [Google Scholar]
- 34. Nelson DA, Jacober SJ. Why do older women with diabetes have an increased fracture risk? J Clin Endocrinol Metabol. 2001;86(1):29–31. [DOI] [PubMed] [Google Scholar]
- 35. Yamamoto M, Yamaguchi T, Nawata K, Yamauchi M, Sugimoto T. Decreased PTH levels accompanied by low bone formation are associated with vertebral fractures in postmenopausal women with type 2 diabetes. J Clin Endocrinol Metabol. 2012;97(4):1277–84. [DOI] [PubMed] [Google Scholar]
- 36. Pedrazzoni M, Ciotti G, Pioli G, et al. Osteocalcin levels in diabetic subjects. Calcif Tissue Int. 1989;45(6):331–6. [DOI] [PubMed] [Google Scholar]
- 37. Coleman DL. Obese and diabetes: two mutant genes causing diabetes‐obesity syndromes in mice. Diabetologia. 1978;14(3):141–8. [DOI] [PubMed] [Google Scholar]
- 38. Iwaniec U, Dube M, Boghossian S, et al. Body mass influences cortical bone mass independent of leptin signaling. Bone. 2009;44(3):404–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cao JJ. Effects of obesity on bone metabolism. J Orthop Surg Res. 2011;6(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Reid IR, Ames R, Evans MC, et al. Determinants of total body and regional bone mineral density in normal postmenopausal women‐a key role for fat mass. J Clin Endocrinol Metabol. 1992;75(1):45–51. [DOI] [PubMed] [Google Scholar]
- 41. Williams GA, Callon KE, Watson M, et al. Skeletal phenotype of the leptin receptor–deficient db/db mouse. J Bone Miner Res. 2011;26(8):1698–709. [DOI] [PubMed] [Google Scholar]
- 42. Lorentzon R, Alehagen U, Boquist L. Osteopenia in mice with genetic diabetes. Diabetes Res Clin Pract. 1986;2(3):157–63. [DOI] [PubMed] [Google Scholar]
- 43. Takeshita N, Mutoh S, Yamaguchi I. Osteopenia in genetically diabetic db/db mice and effects of 1α‐hydroxyvitamin D3 on the osteopenia. Life Sci. 1995;56(13):1095–101. [DOI] [PubMed] [Google Scholar]
- 44. Ealey KN, Fonseca D, Archer MC, Ward WE. Bone abnormalities in adolescent leptin‐deficient mice. Regul Pept. 2006;136(1–3):9–13. [DOI] [PubMed] [Google Scholar]
- 45. Kim JH, Saxton AM. The TALLYHO mouse as a model of human type 2 diabetes. Methods Mol Biol. 2012;933:75–87. [DOI] [PubMed] [Google Scholar]
- 46. Leiter EH. Selecting the “right” mouse model for metabolic syndrome and type 2 diabetes research. Methods Mol Biol. 2009;560:1–17. [DOI] [PubMed] [Google Scholar]
- 47. Devlin M, Van Vliet M, Motyl K, et al. Early‐onset type 2 diabetes impairs skeletal acquisition in the male TALLYHO/JngJ mouse. Endocrinology. 2014;155(10):3806–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dhaliwal R, Cibula D, Ghosh C, Weinstock R, Moses A. Bone quality assessment in type 2 diabetes mellitus. Osteoporos Int. 2014;25(7):1969–73. [DOI] [PubMed] [Google Scholar]
- 49. Kao WL, Kammerer CM, Schneider JL, Bauer RL, Mitchell BD. Type 2 diabetes is associated with increased bone mineral density in Mexican‐American women. Arch Med Res. 2003;34(5):399–406. [DOI] [PubMed] [Google Scholar]
- 50. Moseley KF. Type 2 diabetes and bone fractures. Curr Opin Endocrinol Diabetes Obes. 2012;19(2):128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Paccou J, Ward KA, Jameson KA, Dennison EM, Cooper C, Edwards MH. Bone microarchitecture in men and women with diabetes: the importance of cortical porosity. Calcif Tissue Int. 2016;98(5):465–73. [DOI] [PubMed] [Google Scholar]
- 52. Shanbhogue VV, Hansen S, Frost M, et al. Compromised cortical bone compartment in type 2 diabetes mellitus patients with microvascular disease. Eur J Endocrinol. 2016;174(2):115–24. [DOI] [PubMed] [Google Scholar]
- 53. Osima M, Kral R, Borgen TT, et al. Women with type 2 diabetes mellitus have lower cortical porosity of the proximal femoral shaft using low‐resolution CT than nondiabetic women, and increasing glucose is associated with reduced cortical porosity. Bone. 2017;97:252–60. [DOI] [PubMed] [Google Scholar]
- 54. Burghardt AJ, Issever AS, Schwartz AV, et al. High‐resolution peripheral quantitative computed tomographic imaging of cortical and trabecular bone microarchitecture in patients with type 2 diabetes mellitus. J Clin Endocrinol Metabol. 2010;95(11):5045–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cornish J, Callon K, Bava U, et al. Leptin directly regulates bone cell function in vitro and reduces bone fragility in vivo. J Endocrinol. 2002;175(2):405–16. [DOI] [PubMed] [Google Scholar]
- 56. Jiao H, Xiao E, Graves DT. Diabetes and its effect on bone and fracture healing. Curr Osteoporos Rep. 2015;13(5):327–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Finkelstein JS, Hayes A, Hunzelman JL, Wyland JJ, Lee H, Neer RM. The effects of parathyroid hormone, alendronate, or both in men with osteoporosis. N Engl J Med. 2003;349(13):1216–26. [DOI] [PubMed] [Google Scholar]
- 58. Neer RM, Arnaud CD, Zanchetta JR, et al. Effect of parathyroid hormone (1‐34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344(19):1434–41. [DOI] [PubMed] [Google Scholar]
- 59. Dempster DW, Cosman F, Kurland ES, et al. Effects of daily treatment with parathyroid hormone on bone microarchitecture and turnover in patients with osteoporosis: a paired biopsy study. J Bone Miner Res. 2001;16(10):1846–53. [DOI] [PubMed] [Google Scholar]
- 60. Alkhiary YM, Gerstenfeld LC, Krall E, et al. Enhancement of experimental fracture‐healing by systemic administration of recombinant human parathyroid hormone (PTH 1‐34). J Bone Joint Surg Am. 2005;87(4):731–41. [DOI] [PubMed] [Google Scholar]
- 61. Barnes GL, Kakar S, Vora S, Morgan EF, Gerstenfeld LC, Einhorn TA. Stimulation of fracture‐healing with systemic intermittent parathyroid hormone treatment. J Bone Joint Surg Am. 2008;90(Suppl_1):120–7. [DOI] [PubMed] [Google Scholar]
- 62. Holzer G, Majeska RJ, Lundy MW, Hartke JR, Einhorn TA. Parathyroid hormone enhances fracture healing: a preliminary report. Clin Orthop Relat Res. 1999;366:258–63. [DOI] [PubMed] [Google Scholar]
- 63. Nakajima A, Shimoji N, Shiomi K, et al. Mechanisms for the enhancement of fracture healing in rats treated with intermittent low‐dose human parathyroid hormone (1–34). J Bone Miner Res. 2002;17(11):2038–47. [DOI] [PubMed] [Google Scholar]
- 64. Liu A, Li Y, Wang Y, Liu L, Shi H, Qiu Y. Exogenous parathyroid hormone‐related peptide promotes fracture healing in Lepr (−/−) mice. Calcif Tissue Int. 2015;97(6):581–91. [DOI] [PubMed] [Google Scholar]
- 65. Ohuchi K, Miyakoshi N, Kasukawa Y, et al. Effects of teriparatide on bone in autochthonous transgenic model mice for diabetes mellitus (Akita mice). Osteoporos Sarcopenia. 2019;5(4):109–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Suzuki K, Miyakoshi N, Tsuchida T, Kasukawa Y, Sato K, Itoi E. Effects of combined treatment of insulin and human parathyroid hormone (1–34) on cancellous bone mass and structure in streptozotocin‐induced diabetic rats. Bone. 2003;33(1):108–14. [DOI] [PubMed] [Google Scholar]
- 67. Marsell R, Einhorn TA. The biology of fracture healing. Injury. 2011;42(6):551–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gandhi A, Beam HA, O'Connor JP, Parsons JR, Lin SS. The effects of local insulin delivery on diabetic fracture healing. Bone. 2005;37(4):482–90. [DOI] [PubMed] [Google Scholar]
- 69. Beam HA, Russell Parsons J, Lin SS. The effects of blood glucose control upon fracture healing in the BB Wistar rat with diabetes mellitus. J Orthop Res. 2002;20(6):1210–6. [DOI] [PubMed] [Google Scholar]
- 70. Gooch HL, Hale JE, Fujioka H, Balian G, Hurwitz SR. Alterations of cartilage and collagen expression during fracture healing in experimental diabetes. Connect Tissue Res. 2000;41(2):81–91. [DOI] [PubMed] [Google Scholar]
- 71. Herbsman H, Powers JC, Hirschman A, Shaftan GW. Retardation of fracture healing in experimental diabetes. J Surg Res. 1968;8(9):424–31. [DOI] [PubMed] [Google Scholar]
- 72. Einhorn TA. The cell and molecular biology of fracture healing. Clin Orthop Relat Res. 1998;355:S7–S21. [DOI] [PubMed] [Google Scholar]
- 73. Ko KI, Coimbra LS, Tian C, et al. Diabetes reduces mesenchymal stem cells in fracture healing through a TNFα‐mediated mechanism. Diabetologia. 2015;58(3):633–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Follak N, Klöting I, Merk H. Influence of diabetic metabolic state on fracture healing in spontaneously diabetic rats. Diabetes Metab Res Rev. 2005;21(3):288–96. [DOI] [PubMed] [Google Scholar]
- 75. Macey LR, Kana S, Jingushi S, Terek R, Borretos J, Bolander M. Defects of early fracture‐healing in experimental diabetes. J Bone Joint Surg Am. 1989;71(5):722–33. [PubMed] [Google Scholar]