

1 Introduction
When sudden death (SD) occurs in adults and elderly persons, coronary atherosclerosis is the usual cause [1,2]. On the contrary, a large spectrum of cardiovascular diseases, both congenital and acquired, may account for SD in the young [3–10]. These diseases are frequently concealed and discovered with surprise only at postmortem by means of a thorough macroscopic and microscopic investigation. This review will address the spectrum of structural substrates of cardiac SD with particular emphasis given to the possible role of molecular biology techniques in identifying subtle or even merely functional disorders accounting for electrical instability.
2 Epidemiology, pathophysiology and substrates of cardiac SD
SD is defined as a natural, unexpected fatal event occurring within 1 h of the beginning of symptoms, in an apparently healthy subject or one whose disease was not so severe enough as to predict such an abrupt outcome [11]. In the USA, the annual incidence of SD in people aged 35–74 years is 191/100 000 in men and 57/100 000 in women; almost half of all SDs occur in people with known coronary artery disease [12]. In the Veneto region, Northeast of Italy, we recently calculated an overall prevalence of SD of 0.8/100 000/year in the young, based only upon autopsy reports [13]. When focusing the attention only on young athletes the prevalence was twice that in young non athletes, i.e. 1.6/100 000/year; these figures are explained by the existence of cardiovascular diseases which cause a risk of SD during effort, such as hypertrophic cardiomyopathy (HCM), arrhythmogenic right ventricular cardiomyopathy (ARVC) and congenital coronary anomalies.
As far as pathophysiology is concerned, cardiac arrest may be either mechanical, when the heart and circulatory functions are suddenly impeded by mechanical factors (i.e. cardiac tamponade, pulmonary thromboembolism, etc.) or arrhythmic (mostly ventricular fibrillation) [9].
Based upon the Veneto Region study project on juvenile SD, cardiovascular diseases accounted for more than 80% of cases and about one third of events were due to a congenital heart defect present since birth [8,9,13]. Table 1 reports the main causes of cardiovascular SD in the young in major series which have been published in English. In our experience, the most common causes include premature coronary atherosclerosis (21%), ARVC (14%), mitral valve prolapse (12%), non-atherosclerotic coronary artery disease (11%), myocarditis (10%), conduction system disease (9%) and HCM (7%). It is noteworthy that 6% of all SDs remained unexplained even after thorough macroscopic and histologic examination. Whether these are truly idiopathic or unexplained due to an inability to identify subtle pathologic substrates remains to be elucidated. It may be that structural abnormality resides at molecular level, thus enhancing the need for molecular biology investigation [14].
Table 1.
Major causes of cardiac SD in young people
| Authors, time interval | Population | Age (years) | Total no. | Causesa (%) |
|---|---|---|---|---|
| Drory et al. 1976–85 | Israel | 9–39 | 118 | Atherosclerotic CAD (58) |
| Myocarditis (25) | ||||
| HCM (13) | ||||
| Conduction system (4) | ||||
| Neuspiel and Kuller 1972–80 | Allegheny County, US | 1–21 | 51 | Myocarditis (27) |
| DCM (24) | ||||
| Conduction system (12) | ||||
| Aortic dissection (6) | ||||
| CAA (6) | ||||
| Atherosclerotic CAD (4) | ||||
| Topaz and Edwards 1960–83 | St. Paul, Minnesota, US | 7–35 | 50 | MVP (24) |
| Myocarditis (25) | ||||
| HCM (12) | ||||
| CAA (4) | ||||
| Aortic stenosis (4) | ||||
| Phillips et al. 1965–85 | American forces, US | 17–28 | 20 | Myocarditis (42) |
| CAA (15) | ||||
| HCM (10) | ||||
| MVP (5) | ||||
| Atherosclerotic CAD (5) | ||||
| Aortic stenosis (5) | ||||
| Kramer et al. 1974–86 | Soldiers, Israel | 17–30 | 24 | Myocarditis (29) |
| HCM (25) | ||||
| MVP (13) | ||||
| Atherosclerotic CAD (13) | ||||
| Aortic dissection (8) | ||||
| CAA (4) | ||||
| DCM (4) | ||||
| Conduction system (4) | ||||
| Basso (present study) 1979–1999 | Veneto region Italy | 1–35 | 273 | Atherosclerotic CAD (21) |
| ARVC (14) | ||||
| Valve disease (12) | ||||
| CAA (11) | ||||
| Myocarditis (10) | ||||
| Conduction system disease (9) | ||||
| HCM (7) |
Abbreviations: ARVC, arrhythmogenic right ventricular cardiomyopathy; CAA, coronary artery anomaly; CAD, coronary artery disease; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; MVP, mitral valve prolapse.
3 New molecular assays: perspectives of application in the study of SD
The principle of all molecular hybridization assays is the complementary base pairing between two nucleic acid strands. In situ hybridization provides the direct detection of nucleic acid in cellular material in which simultaneous morphological analysis can be performed. Polymerase chain reaction (PCR) allows millions of copies of any specific DNA sequence to be generated in a few hours. The reaction consists of an in vitro enzymatic amplification of defined DNA sequence by repeated rounds of heat denaturation, primer annealing and DNA polymerase-mediated primer extension. The amplified DNA can then be seen as a distinct band after standard agarose gel electrophoresis, and the specificity of detection can be increased by subsequent hybridization or DNA sequencing. According to the nested PCR technique, a second pair of primers ‘internal’ to the original primer pair used in a subsequent series of amplification cycles. Using this strategy, the sensitivity is enhanced from 100 to 1000 times, to such an extent that even a single copy target can be detected in a complex background of 300 000 cells or more.
One of the most important and frequent applications of these novel techniques is for the identification of microbial pathogens. Given the extreme sensitivity of these techniques, particularly of PCR, a single copy of a gene can be readily detected even from an extremely small amount of tissue. The decision to develop and apply PCR for routine diagnosis of myocarditis must be considered in relation to the low cost, speed, sensitivity and reliability of more conventional culture and/or serological methods [15]. Different molecular strategies have now been developed to recognize and distinguish an infective from a latent state of virus. In addition to qualitative analysis and genome sequencing different methods have been developed for the quantification of PCR product. Quantification of different viral genome has now successfully introduced into routine diagnostic virology as a prognostic marker of severe infective disease. Genome quantification may also allow to accurately assess the risk of infection and to monitor the therapeutic response [16].
Moreover, PCR may detect virtually all the common genetically inherited diseases in which the defective gene has been identified such as Duchenne muscular dystrophy, Marfan syndrome and HCM.
PCR can also trace the inheritance of diseases in which the defective locus has only been defined in terms of linkage to other cellular genes. Using PCR, allelic forms of many cellular genes can be identified by sizing selected introns between the coding exons. This approach should provide much more detailed linkage studies than are presently possible, using restriction fragment length polymorphism analysis.
A variety of types of samples may be used for PCR analysis. Extracted nucleic acid may be easily amplified, even in a partially purified sample. Extracted DNA or RNA from formalin-fixed, paraffin-embedded samples obtained either at autopsy or at surgery, are successfully used as templates for the PCR. This could permit an ever more frequent application not only for perspective but also for retrospective studies of either genetically determined or infective cardiovascular diseases. The recent description of a successful identification 22 years later of inherited long QT syndrome in a 12-year-old girl who had died suddenly, through molecular testing of archived paraffin-embedded myocardial samples, is emblematic [17]. First of all, this result has potentially great importance for forensic science by providing a plausible mechanism for cardiac arrest in the setting of an otherwise structurally normal heart. Moreover, thanks to this molecular biology finding, the presence of the disease-causing mutation was subsequently demonstrated in one of the decedent's parents who also had a diagnostic QTc with inherent treatment and prevention strategies.
However, it is important to stress that random search of any gene in formalin-fixed material is not affordable and the results are often disappointing. Thus, such investigation should be performed only in selected cases in which clinical or morphologic phenotype should suggest a peculiar genotype.
In the following sections we will analyze those cardiovascular diseases either acquired from an infectious pathogenesis or genetically determined, where availability of molecular biology techniques will open new avenues of investigation. In some instances, such as in the setting of dilated and ARVC, molecular pathology techniques will be of help for both genetic screening and the search for viral genomes.
4 Cardiovascular diseases at risk of SD: potential targets of molecular pathology investigation
4.1 Infective cardiac disease
4.1.1 Myocarditis
Myocarditis accounted for up to 44% of fatal events in major series on cardiac SD in the young [18]. The strongest evidence that subclinical myocarditis can be a cause of ventricular fibrillation comes from an autopsy series on USA army recruits in which 42% of those who died suddenly had histological evidence of myocarditis [19]. Whether the high incidence of myocarditis in this series is due to a selection bias towards more tropical diseases as well as to an overestimation of inflammatory infiltrates due to the absence of standardized diagnostic criteria remains intriguing. Although myocarditis usually presents with signs of pump failure and ventricular dilatation, ventricular arrhythmias have been described in patients with myocarditis and apparently normal hearts. A recent flu-like illness is common, although the symptoms may be mild and clinical signs of heart failure subtle or absent. Cardiac involvement is unpredictable and may affect the conduction system, causing heart block, or the ordinary myocardium, causing ventricular arrhythmias. SD may occur both in the active or healed phases as a consequences of life-threatening ventricular arrhythmias that develop mostly in the setting of an unstable myocardial substrate, namely inflammatory infiltrate, interstitial edema, myocardial necrosis and fibrosis. The gross appearance of the heart is not distinctive and its weight may be within normal values. A major limitation in the autopsy diagnosis of myocarditis in several series has been the lack of standardized histological criteria. A ‘starry-like sky’ feature (>14 leucocytes/mm2) or a patchy inflammatory infiltrate, either polymorphous or lymphocytic, sometimes no more than 3 foci at a magnification of 6×, and not necessarily associated with myocyte necrosis, are typical features. This subtle substrate, together with the possible inflammatory involvement of the conduction system, seems highly arrhythmogenic and may account for unexpected arrhythmic cardiac arrest. To increase diagnostic sensitivity of histology, use of immunohistochemistry by means of a large panel of monoclonal and polyclonal antibodies is mandatory to identify and characterize the inflammatory infiltrate.
Evidence of myocardial infection, whether bacterial or viral, has been rarely found. Chlamydia pneumoniae myocarditis was implicated in the SDs of several young Swedish elite orienteerers following RNA detection of this organism in the heart of one of the victims [20]. A subsequent paper implicated this agent in one third of cardiac SDs among 15 Swedish orienteerers who died unexpectedly between 1979 and 1992 [21].
Nonetheless, viral infections are the most plausible cause. Although enterovirus is the most important causative agent in the pathogenesis of myocarditis, several studies have shown that various other viruses, such as adenovirus, herpesvirus (cytomegalovirus, herpes simplex virus, Epstein Barr virus) parvovirus, influenza virus A and B, and hepatitis C virus can be involved in myocardial infective disease, particularly in the pediatric population [15]. Viruses rarely cause evident cytopathic damage, so that classical morphology often fails to identify the etiological agents. The diagnosis of viral myocarditis has, for a long time, been based on viral culture and serology. However, these investigations are time consuming and generally fall short in specificity and sensitivity. More recently, molecular biology techniques as PCR and nested-PCR have been shown to rapidly detect the presence of infective agents, in specific and very sensitive way. Their application is now successfully carried out also on tissues in which nucleic acid could be partially degraded such as autoptic or paraffin embedded samples (Fig. 1) [22,23].
Fig. 1.
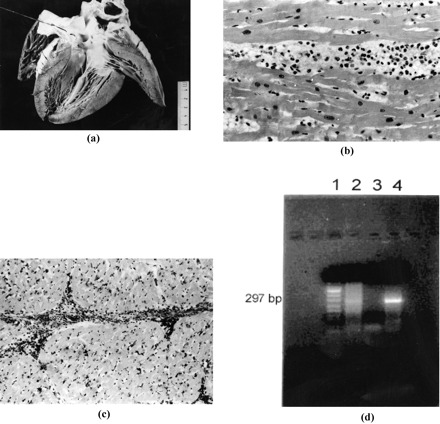
A 17-year-old, previously asymptomatic, boy who died suddenly at rest. View of the heart specimen removed at autopsy which is grossly normal. At histology, the left ventricular myocardium shows a polymorphous inflammatory infiltrate associated with myocyte necrosis (hematoxylin—eosin ×240). Immunohistochemistry reveals a massive T lymphocytes infiltrate (CD43 ×240). Agarose gel electrophoresis showing enteroviral RT-PCR results: Lanes: 1, DNA molecular-weight-marker (pUCBM21 DNA cleaved with Hpa II and Dra I plus Hind III) 2, RT-PCR amplified products of patient's specimen (coxsackievirus B3 KB infected cells) 3, negative control (uninfected cells) 4, RT-PCR amplified products of positive control (coxsackievirus B3 KB infected cells).
4.1.2 Unstable coronary plaque
The coronary artery pathology in SD adult victims consists of single, double or triple vessel atherosclerotic disease and usually includes a thrombotic occlusion of a coronary segment, which accounts for sharp interruption of the regional myocardial blood flow [1,2]. On the contrary, coronary SD in the young is usually due to a single subobstructive plaque, located at the first segment of the anterior descending coronary artery, mostly fibrocellular, devoid of atheroma, fissuring or thrombosis [7] (Fig. 2). In the setting of acute thrombosis, superficial erosion seems to be a peculiar mechanism precipitating plaque instability, unlike in adults where it is mainly due to rupture of the thin fibrous cap of an atheromatous plaque [24]. Endothelial erosion may be the consequence of either plaque inflammation or intimal smooth muscle cell proliferation [25]. The inflammatory nature of atherosclerotic plaque components prompted the postulation that either infection and/or autoimmune phenomena are involved in the onset and progression of the disease [26]. Several reports, using molecular hybridization assays, have shown a correlation between the occurrence of atherosclerosis and the presence of infective microorganisms, like herpesviruses and Chlamydia pneumoniae[27]. Although a cause—effect relationship is far from being demonstrated and these microorganisms could act simply as ‘innocent bystanders’, some microbial infections have been associated with acute coronary syndromes; in particular, an increase in enterovirus-specific antibodies at the time of myocardial infarction diagnosis suggests a role for this virus in triggering acute coronary syndromes [28,29], thus opening new avenues of research in unstable coronary plaques.
Fig. 2.
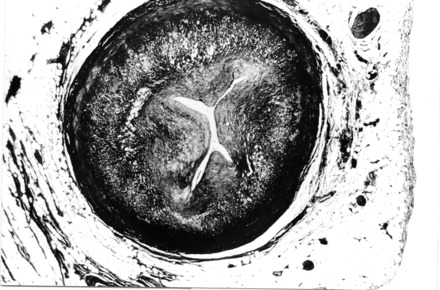
A 19-year-old athlete who died suddenly at rest. A transverse section at the level of first tract of left anterior descending coronary artery reveals a concentric, fibrocellular atherosclerotic plaque which is completely devoid of lipids (Heidenhain trichrome ×12).
4.2 Genetically determined cardiac disease
4.2.1 Cardiomyopathy
HCM is heart muscle disease phenotypically characterized by either symmetric or asymmetric left ventricular hypertrophy with a chaotic spatial arrangement of the myocytes (‘disarray’), often marked by SD along the natural history. Heart dysfunction appears more in the form of electrical instability than of impaired contractility, which, on the contrary, may be enhanced. Risk factors are considered to be youth, previous syncopal episodes, a malignant family history, myocardial ischemia, sustained ventricular tachycardia on electrophysiological test and ventricular tachycardia on holter monitoring, attenuated blood pressure changes on exercise and enhanced QRS fractionation of the extrasystole [30]. Recently, molecular genetic studies demonstrated that HCM is a heterogeneous disease, with several missense mutations in genes encoding for proteins of the cardiac sarcomere, among which β-myosin heavy chain, cardiac troponin T, α-tropomyosin, myosin binding C and actinin [31]. Despite data on genotype—phenotype correlations are still preliminary, it seems that the phenotype varies not only with the type of mutation but also within individuals carrying the same mutation. For instance the arginine-to-glutamine mutation in 403 codon of β-myosin is associated with a poor prognosis, whereas the arginine-to-tryptophan mutation appears more benign [32]. Moreover, the knowledge that myosin-binding protein C mutations appear to be associated with age-related penetrance in adulthood and that troponin T mutation is associated with up to 50% of non penetrance, no or mild hypertrophy (HCM ‘without hypertrophy’) and a high risk of SD, even in the absence of severe left ventricular hypertrophy, would have consequences for genetic counselling. The recent pointing out of a high frequency of hepatitis C virus in the hearts of patients with the apical form of HCM is intriguing and opens new avenues of investigation [33].
At postmortem, the heart shows asymmetric left ventricular hypertrophy, usually in the basal portion of the ventricular septum but also in the anterior free wall and apex [34] (Fig. 3). The septal bulging together with a septal endocardial plaque and anterior mitral valve leaflet thickening, may account for the left ventricular outflow gradient. The combination of myocardial disarray and interstitial fibrosis represent an ideal substrate of inhomogeneous intraventricular conduction with potential reentry phenomena. More recently, a detailed pathology study on young subjects dying suddenly demonstrated that the superimposition of ischemic damage, in the form of myocyte necrosis and large fibrous scars mimicking healed infarction [34]. The ischemic damage occurs in the absence of significant epicardial coronary artery disease, although small vessel disease as well as intramural course of the left anterior descending coronary artery have been noted. Elevated intramyocardial diastolic pressure may restrict intramural arteries during diastole thus impairing coronary filling and myocardial perfusion. The combination of myocardial disarray and replacement fibrosis has to be considered the malignant arrhythmogenic substrate in HCM.
Fig. 3.
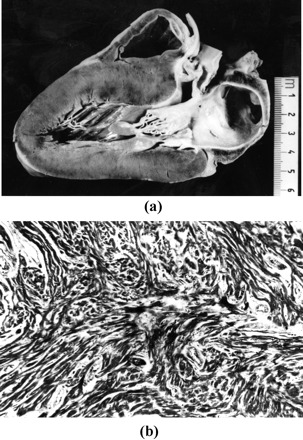
A 16-year-old boy with previous history of syncope who died suddenly on effort. Long axis cut of the heart specimen: note the asymmetric septal hypertrophy with subaortic bulging and septal endocardial fibrous plaque; histology of the interventricular septum reveals typical myocardial disarray with interstitial fibrosis (Heidenhain trichrome ×47).
ARVC, which is also known as right ventricular dysplasia, is one of the leading causes of SD in the young in our geographic area [6,35,36]. In these subjects the presence of ECG abnormalities, like an inverted T wave in the right precordial leads (V1–V3), increased QRS duration of >110 ms, late potentials detected by high resolution ECG and ventricular arrhythmias, even in the shape of single premature ventricular beats with left bundle branch block morphology, should raise suspicion of the disease and lead to further non-invasive and invasive investigations. Patients with a history of syncope, familial SD and precordial T wave inversion beyond V3 seem to have a worse prognosis.
The disease is characterized pathologically by a peculiar myocardial atrophy with fibro-fatty substitution of the RV free wall in an otherwise apparently normal heart (Fig. 4). Histology discloses the disappearance of the RV myocardium with the fibro-fatty or fatty replacement, extending from the epicardium towards the endocardium. The intraventricular conduction delay, consequent to fibro-fatty replacement, is a source of electrical instability, due to reentrant phenomena, in the shape of ventricular arrhythmias with left bundle branch block morphology, indicating a right ventricular origin. Familiarity with an autosomal dominant inheritance has been demonstrated in nearly 50% of cases [37]. Six loci have been identified by linkage analysis so far, two mapping to chromosome 14 and the remaining to chromosomes 1, 2, 3 and 10, one each. Recently, we identified a mutation of the cardiac ryanodine receptor gene in families who map to chromosome 1q42–q43 [38]. A recessive form associated with palmoplantar keratosis has also been also reported on chromosome 17 and a defective gene encoding for a cell-to-cell adhesion molecule, i.e. plakoglobin, has been found [39]. Apoptosis has been demonstrated to account for myocyte death both in autopsy and biopsy material [40]. RV aneurysms, left ventricular involvement, focal myocarditis as well as bouts of apoptosis, most probably worsen ventricular electrical vulnerability and lower the ventricular fibrillation threshold. Evidence of inflammatory infiltrates associate with myocyte death is a frequent finding in several pathological series [41]: whether inflammation is primary or secondary to cell death remains to be established. To test the infective etiopathogenetic theory, we recently did a nested PCR investigation which failed to detect any enteroviral genome in biopsies of ARVC patients with both recent and chronic clinical onset of the disease [42]. However, it may be that other types of virus are involved and need to be searched for.
Fig. 4.
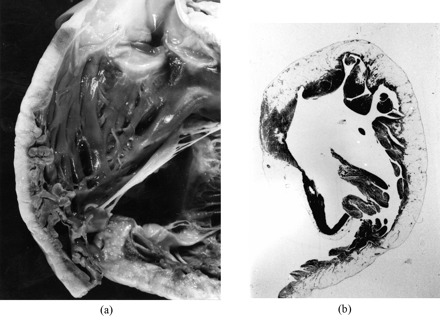
A 35-year-old man with a previous history of palpitations and negative T waves in V1–V2 who died suddenly at rest. Gross view of the RV outflow tract showing massive fatty replacement of the free wall myocardium; At histology, the RV infundibulum shows transmural fatty replacement of the atrophic myocardium (Heidenhain trichrome ×3).
Dilated cardiomyopathy is a genetically and clinically heterogeneous disease. The natural history demonstrates that death occurs not only due to progressive congestive heart failure or as a complication of thromboembolism, but also abruptly due to arrhythmic cardiac arrest. Although death is obviously expected along the natural history, in a few cases arrhythmic SD may be the first manifestation of the disease and the diagnosis is achieved only at postmortem by observing a heavy heart with dilated ventricles and no inflammatory myocardial or coronary artery disease. As far as molecular basis of dilated cardiomyopathy, at least 30% of cases are inherited, with a significant percentage of the remaining cases being acquired (i.e. myocarditis, autoimmune, etc.). Inherited forms may have autosomal dominant, autosomal recessive, X-linked, or mitochondrial transmission, with evident genetic heterogeneity [43]. The persistence of a viral infection is thought to have a pathogenetic role in different chronic myocardial diseases with unknown etiology [44,45]. Different molecular techniques have produced controversial results with respect to the rate of enteroviral positivity in myocardial samples of patients with dilated cardiomyopathy. Several studies showed no, or a very low percentage of, enteroviral PCR positivity in patients suffering from the end stage of dilated cardiomyopathy [22]. Viral clearance or involvement of other cardiotropic viruses, such as adenovirus, human cytomegalovirus, coronavirus and hepatitis C virus, could explain the apparently negative findings.
4.2.2 Marfan syndrome
The disease is familial in the majority of patients, whereas 30% of cases are sporadic. It maps to chromosome 15q15–q21.3 [46,47] and the defective gene encodes fibrillin-1, which is the major constituent of microfibrils of the extracellular matrix. Marfan patients usually die suddenly because of aortic dissection with cardiac tamponade (type I–II with rupture within the pericardial cavity) and exhibit typical cardiovascular features consisting of mitral valve prolapse, annuloaortic ectasia, with or without fusiform aneurysm of the ascending aorta, and aortic incompetence. Aortic dissection in Marfan syndrome may be also observed without dilatation of the aorta, so that its occurrence may be unpredictable on the clinical grounds. The basic defect of spontaneous laceration of the ascending aorta consists in elastic disruption and cystic medial necrosis in the tunica media leading to aortic wall fragility. A severe pattern of cystic medial necrosis, quite similar to that observed in the aortic tunica media of patients with Marfan's syndrome, has been reported in patients who died suddenly with bicuspid aortic valve, either isolated or associated with isthmal coarctation, suggesting a possible genetic background with a developmental defect involving both the aortic valve and the ascending aorta (Fig. 5). Although familial forms have been described the genetic defect has not been found so far.
Fig. 5.
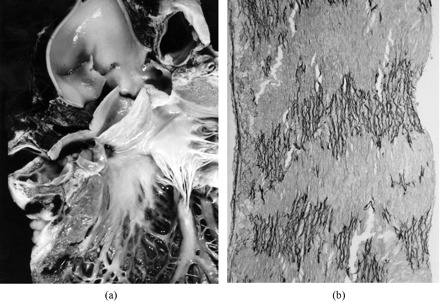
A 24-year-old boy who died suddenly at rest due to massive cardiac tamponade. Gross view of the ascending aorta: note the intimal tear just above a functionally normal bicuspid aortic valve; Histology of the ascending aorta shows severe atrophy and fragmentation of elastic lamellae (Weigert van Gieson ×60).
4.2.3 Supravalvular aortic stenosis
This is a rare genetic disease with autosomal dominant inheritance which has been linked to chromosome 7 and results from a defect in the elastin gene which accounts for an hour-glass obstruction of the ascending aorta and left ventricular hypertrophy [48,49]. The phenotype is marked by elastosis of the aortic tunica media, intimal thickening and dysplastic cusps [50]. Left ventricular hypertrophy, isolation of the coronary ostia because of fusion of semilunar cusps with the aortic wall, as well as stenotic intraaortic course of the proximal coronary arteries, are all factors that account for the coronary ischemia in these patients [51]. Williams syndrome is an autosomal dominant disorder that is characterized by supravalvular aortic stenosis, peripheral pulmonary stenosis, obstructive coronary lesions, abnormal facies and mental retardation. This syndrome was found associated with a deletion of a region of the same chromosome 7 (7q11.23) that includes the elastin gene and is thought to be a contiguous gene disorder caused by a deletion of multiple adjacent genes.
4.2.4 Ion channel disease
Arrhythmic SD may occur in the absence of gross structural cardiac pathology (idiopathic ventricular fibrillation or ‘mors sine materia’) due to ion channel disorders. In this setting, besides the availability of ECG tracings and full family and personal history, molecular investigation will allow the definite cause of SD to be found.
The long QT syndrome is a familial disease with high cardiac electrical instability, presenting with syncope due to ventricular tachyarrhythmias or with cardiac arrest on exercise or emotional stress, often under the age of 15 [52]. The cause of death at necropsy cannot be ascertained unless there are prior ECG data. Genetic analysis reveals multiple abnormalities in genes related to both cardiac potassium and cardiac sodium channels. At present, important genotype—phenotype correlations and the identification of gene-specific arrhythmogenic triggers are slowly emerging. Alterations of ion pumps and currents account for the lengthening of the action potential and prolonged QT interval on ECG, and the propensity to ventricular fibrillation. The mortality in untreated symptomatic cases exceeds 60% within 15 years. Clearly ECG screening of surviving relatives is the only clinical way to establish the diagnosis in asymptomatic carriers. On the basis of pattern of transmission, two major clinical syndromes have been described: the more common autosomal dominant form with a pure cardiac phenotype (Romano—Ward) [53] and the rarer autosomal recessive form characterized by the association with congenital deafness (Jervell and Lang-Nilesen) [54].
The Romano—Ward LQTS results from a mutation in genes encoding cardiac ion channels or auxiliary ion-channel subunits: KvLQT1 (at the LQT1 locus), HERG (at LQT2), SCN5A (at LQT3), HKCNE1 (encoding minimal potassium-channel β subunit [MinK] at LQT5), and HKCNE1 (encoding MinK-related peptide 1 [MiRP1] at LQT6) [55]. Moreover, families linked to none of these genes have been described thus suggesting the existence of other disease genes. The autosomal recessive variant of LQTS arises in patients who inherit abnormal KvLQT1 or minK alleles from both parents and expresses itself with remarkably long QT intervals.
The Brugada syndrome is a clinical and ECG syndrome, characterized by apparent right bundle branch block with right precordial ST segment elevation and an apparently normal heart, which has been described in cases of SD by Brugada and Brugada, unfortunately without postmortem reports [56]. These ECG characteristics may depend on exaggerated transmural differences in action potential configuration, especially in the right ventricular outflow tract. By studying a family with a SD case, confirmation of an organic substrate was given by Corrado et al. [57], who reported not only fibro-fatty dystrophy in the RV free wall but also involvement of the conduction system with sclerotic interruption of the right bundle branch. The coexistence of both ‘septal’ and ‘parietal’ right conduction defects might account for the ECG pattern of right bundle branch block and persistent ST segment elevation with ventricular electrical instability.
Conversely, in the absence of structural heart disease, the ECG abnormalities could arise from ion currents dysfunction, such as Ito L-type Ca2+ channel [ICa (L)] and INa. At least one variant of the Brugada syndrome is caused by mutations in cardiac sodium channels gene SCN5A, the same gene which is implicated in the LQT3 syndrome [58]. The mutation of SCN5A causes loss of function in the Brugada syndrome and gain of function in the LQT3. In particular, it results in the Brugada syndrome in reduced sodium current density, thus accounting for the loss of the epicardial action potential dome with transmural dispersion of repolarization, which in turn may predispose to the development of ventricular fibrillation. However, other families with Brugada syndrome have been tested showing no defects on the cardiac sodium channel, thus suggesting genetic heterogeneity in analogy with other inherited cardiac diseases.
5 Final consideration on the pathologist's role in the diagnosis of causes and prevention of SD
An accurate diagnosis of the underlying morbid entity at risk of SD either at postmortem or in living patients with aborted SD and life-threatening arrhythmias, is the prerequisite to adopt therapeutic and preventive strategies, by establishing whether the disease is acquired due to infectious agents or hereditary.
In the latter situation, the final diagnosis may be the starting point for a widespread investigation of the family, to detect asymptomatic carriers and to reassure non-carriers. ECG and echocardiographic studies are essential preliminary investigations carried out in all first degree family members. Unfortunately, they can detect only a small proportion of gene carriers, particularly in children, where phenotypic expression may still be absent. Genetic screening is theoretically the most effective tool in the early recognition of asymptomatic genetic carriers. The potential benefit for patients and family members is self-evident, since in the majority of cases SD may be the first and sole manifestation of a familial disease. Moreover, the risk of cardiac arrest is demonstrably higher with some specific mutation. However, there are some concerns about genetic screening. First, at present it is an expensive, laborious procedure, and can be done only at a few tertiary referral centers, with a specific research interest in molecular genetics. There are pressures to study families because of competition in science. Some people question whether genetic knowledge is beneficial, neutral or harmful, and whether parents have the right to make decisions for their children, especially when a disease is not manifest until middle age and little can be offered in terms of treatment.
As a consequence, pathologists who see most of the index cases have a great responsibility. If nothing is done and another SD occurs, the family rightly feels aggrieved. If the pathological diagnosis is wrong, many expensive and time-consuming investigations are carried out without benefit. This implies that all young SDs should undergo autopsy by expert pathologists, whether or not working for a coroner or medical examiner. Moreover, by applying molecular biology techniques to the study of blood and fresh tissue obtained through endomyocardial biopsy, the pathologist will also have an essential role in providing a correct in vivo diagnosis of genetically determined or infective heart diseases potentially at risk of SD, thus allowing the more rational management of patients.
Having achieved a precise diagnosis and recognized a potentially hereditary disease as the cause of life-threatening arrhythmias and/or SD, it is then advisable to inform through the general practitioner and to start a sequence of investigations from a detailed family history to referral of parents, siblings and offspring to a cardiologist for screening. A full genetic study should be carried out only when the family asks for it. HCM, long QT syndrome and other ion channels diseases, Marfan's syndrome and ARVC are, at present, cardiac familial disease at risk of SD; all are theoretically amenable to genetic screening and all have the potential to devastate families by expected SD and the families have the right to the best available advice. A precise pathological diagnosis of the underlying heart disease, which also takes advantage of advanced molecular biology techniques, will be the source of vital information for the community, relatives and future generations.
Acknowledgements
Supported by MURST, Rome; Veneto Region, Venice; and Fondazione Cassa di Risparmio, Padova, Italy.
Time for primary review 29 days.
References
- [1].Baroldi G, Falzi G, Mariani F. Sudden coronary death: a postmortem study in 208 selected cases compared to 97 ‘control’ subjects. Am Heart J. 1979;98:20–31. doi: 10.1016/0002-8703(79)90316-8. [DOI] [PubMed] [Google Scholar]
- [2].Davies M.J, Thomas A. Thrombosis and acute coronary artery lesions in sudden cardiac ischemic death. New Engl J Med. 1984;310:1137–1140. doi: 10.1056/NEJM198405033101801. [DOI] [PubMed] [Google Scholar]
- [3].Drory Y, Turetz Y, Hiss Y, et al. Sudden unexpected death in persons less than 40 years of age. Am J Cardiol. 1991;68:1388–1392. doi: 10.1016/0002-9149(91)90251-f. [DOI] [PubMed] [Google Scholar]
- [4].Neuspiel D.R, Kuller L.H. Sudden and unexpected natural death in childhood and adolescence. J. Am. Med. Assoc. 1985;254:1321–1325. [PubMed] [Google Scholar]
- [5].Topaz O, Edwards J.E. Pathologic features of sudden death in children, adolescents, and young adults. Chest. 1985;87:476–482. doi: 10.1378/chest.87.4.476. [DOI] [PubMed] [Google Scholar]
- [6].Thiene G, Nava A, Corrado D, et al. Right ventricular cardiomyopathy and sudden death in young people. New Engl J Med. 1988;318:129–133. doi: 10.1056/NEJM198801213180301. [DOI] [PubMed] [Google Scholar]
- [7].Corrado D, Basso C, Poletti A, et al. Sudden death in the young. Is coronary thrombosis the major precipitating factor? Circulation. 1994;90:2315–2323. doi: 10.1161/01.cir.90.5.2315. [DOI] [PubMed] [Google Scholar]
- [8].Basso C, Frescura C, Corrado D, et al. Congenital heart disease and sudden death in the young. Human Pathol. 1995;26:1065–1072. doi: 10.1016/0046-8177(95)90267-8. [DOI] [PubMed] [Google Scholar]
- [9].Basso C, Corrado D, Thiene G. Cardiovascular causes of sudden death in young individuals including athletes. Cardiol. Rev. 1999;7:127–135. doi: 10.1097/00045415-199905000-00009. [DOI] [PubMed] [Google Scholar]
- [10].Kramer M.R, Drory Y, Lev B. Sudden death in young Israeli soldiers: analysis of 83 cases. Isr J Med Sci. 1989;25:620–624. [PubMed] [Google Scholar]
- [11].Goldstein S. The necessity of a uniform definition of sudden coronary death: witnessed death within 1 h of the onset of acute symptoms. Am Heart J. 1982;103:156–159. doi: 10.1016/0002-8703(82)90552-x. [DOI] [PubMed] [Google Scholar]
- [12].Gillum R.F. Sudden coronary death in the United States: 1980–1985. Circulation. 1989;79:756–765. doi: 10.1161/01.cir.79.4.756. [DOI] [PubMed] [Google Scholar]
- [13].Corrado D, Basso C, Schiavon M, et al. Screening for hypertrophic cardiomyopathy in young athletes. New Engl J Med. 1998;339:364–369. doi: 10.1056/NEJM199808063390602. [DOI] [PubMed] [Google Scholar]
- [14].Calabrese F, Basso C, Thiene G, et al. Molecular pathology of cardiac diseases liable to cause sudden death. Cardiac arrhythmias. In: Raviele A, editor. Springer Verlag; Milan: 1999. pp. 6–33. [Google Scholar]
- [15].Martin A.B, Webber S, Fircker F.J, et al. Acute myocarditis. Rapid diagnostic by PCR in children. Circulation. 1994;90:330–339. doi: 10.1161/01.cir.90.1.330. [DOI] [PubMed] [Google Scholar]
- [16].Preiser W, Elzinger B, Brink N.S. Quantitative molecular virology in patients management. J Clin Pathol. 2000;53:76–83. doi: 10.1136/jcp.53.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ackerman M.J, Tester D.J, Porter C.J. Swimming, a gene-specific arrhythmogenic trigger for inherited long-QT syndrome. Mayo Clin Proc. 1999;74:1094–1098. doi: 10.4065/74.11.1088. [DOI] [PubMed] [Google Scholar]
- [18].Basso C, Calabrese F, Corrado D, et al. Myocarditis: an underestimated cause of sudden cardiac death. Fighting sudden cardiac death: a worldwide challenge. In: Aliot E, Clementy J, Prystowsky E.N, editors. Futura; Armonk, NY: 2000. pp. 447–458. [Google Scholar]
- [19].Philips M, Robinowitz M, Higgins J.R, et al. Sudden cardiac death in Air Force recruits: a 20-year review. J. Am. Med. Assoc. 1986;256:2696–2699. [PubMed] [Google Scholar]
- [20].Wesslen L, Pahlson C, Friman G. Myocarditis caused by Chlamydia pneumoniae (TWAR) and sudden unexpected death in a Swedish elite orienteerer. Lancet. 1992;240:427–428. doi: 10.1016/0140-6736(92)91509-7. [DOI] [PubMed] [Google Scholar]
- [21].Wesslen L, Pahlson C, Lindquist O, et al. An increase in sudden unexpected cardiac deaths among young Swedish orienteerers during 1979–1992. Eur Heart J. 1996;17:902–910. doi: 10.1093/oxfordjournals.eurheartj.a014972. [DOI] [PubMed] [Google Scholar]
- [22].Calabrese F, Valente M, Thiene G, et al. Presence of enteroviral genome in native hearts may influence outcome of transplanted patients. Diagn Mol Pathol. 1999;8:39–46. doi: 10.1097/00019606-199903000-00007. [DOI] [PubMed] [Google Scholar]
- [23].Ou J, Sole M.J, Butany J.W, et al. Detection of enterovisrus RNA in biopsies from patients with myocarditis and cardiomyopathy using gene amplification by polymerase chain reaction. Circulation. 1990;82:8–16. doi: 10.1161/01.cir.82.1.8. [DOI] [PubMed] [Google Scholar]
- [24].Davies M.J. Stability and Instability: two faces of coronary atherosclerosis. The Paul Dudley With Lecture 1995. Circulation. 1996;94:2013. doi: 10.1161/01.cir.94.8.2013. [DOI] [PubMed] [Google Scholar]
- [25].Basso C, Thiene G. Sudden death in the young due to coronary artery thrombosis: the role of endothelial erosion as a mechanism of plaque instability. Eur Heart J. 2000;21:39. (Abstr Suppl) [Google Scholar]
- [26].Ross R. Atherosclerosis. An inflammatory disease. New Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- [27].Rosenfeld M.E, Blessing E, Lin T.M, et al. Chlamydia, inflammation and atherogenesis. J Infect Dis. 2000;181:S492–497. doi: 10.1086/315618. [DOI] [PubMed] [Google Scholar]
- [28].Nicholls A.C, Thomas M. Coxsackie virus infection in acute myocardial infarction. Lancet. 1977;1:883–884. doi: 10.1016/s0140-6736(77)91203-x. [DOI] [PubMed] [Google Scholar]
- [29].Roivanen M, Alfthan G, Jousilahti P, et al. Enterovirus infections as a possible risk factor for myocardial infarction. Circulation. 1998;98:2534–2537. doi: 10.1161/01.cir.98.23.2534. [DOI] [PubMed] [Google Scholar]
- [30].Spirito P, Seidman C.E, McKenna W.J, et al. The management of hypertrophic cardiomyopathy. New Engl J Med. 1997;336:775–785. doi: 10.1056/NEJM199703133361107. [DOI] [PubMed] [Google Scholar]
- [31].Bonne G, Carrier L, Richard P, et al. Familial hypertrophic cardiomyopathy. From mutations to functional defects. Circ Res. 1998;83:580–593. doi: 10.1161/01.res.83.6.580. [DOI] [PubMed] [Google Scholar]
- [32].Epstein N.D, Cohn G.M, Cyran F, et al. Differences in clinical expression of hypertrophic cardiomyopathy associated with two distinct mutations in the β-myosin heavy chain gene. A 908Leu—Val mutation and a 403Arg—GLN mutation. Circulation. 1992;86:345–352. doi: 10.1161/01.cir.86.2.345. [DOI] [PubMed] [Google Scholar]
- [33].Matsumori A, Yutani C, Ikeda Y, et al. Hepatitis C virus from the hearts of patients with myocarditis and cardiomyopathy. Lab Invest. 2000;80:1137–1142. doi: 10.1038/labinvest.3780120. [DOI] [PubMed] [Google Scholar]
- [34].Basso C, Thiene G, Corrado D, et al. Hypertrophic cardiomyopathy and sudden death in the young. Pathologic evidence of myocardial ischemia. Hum Pathol. 2000;31:988–998. doi: 10.1053/hupa.2000.16659. [DOI] [PubMed] [Google Scholar]
- [35].Nava A, Rossi L, Thiene G, editors. Elsevier; Amsterdam: 1997. Arrhythmogenic right ventricular cardiomyopathy/dysplasia. [Google Scholar]
- [36].Basso C, Thiene G, Corrado D, et al. Arrhythmogenic right ventricular cardiomyopathy: dysplasia, dystrophy, or myocarditis? Circulation. 1996;94:983–991. doi: 10.1161/01.cir.94.5.983. [DOI] [PubMed] [Google Scholar]
- [37].Nava A, Bauce B, Basso C, et al. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2000;36:2226–2231. doi: 10.1016/s0735-1097(00)00997-9. [DOI] [PubMed] [Google Scholar]
- [38]. [Tiso N, Stephan DA, Nava A, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Gen, 2001, in press.]
- [39].McKoy G, Protonotarios N, Crosby A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease) Lancet. 2000;355:2119–2124. doi: 10.1016/S0140-6736(00)02379-5. [DOI] [PubMed] [Google Scholar]
- [40].Valente M, Calabrese F, Thiene G, et al. In vivo evidence of apoptosis in arrhythmogenic right ventricular cardiomyopathy. Am J Pathol. 1998;152:479–484. [PMC free article] [PubMed] [Google Scholar]
- [41].Thiene G, Corrado D, Nava A, et al. Right ventricular cardiomyopathy: is there evidence of an inflammatory aetiology? Eur Heart J. 1991;12:22–25. doi: 10.1093/eurheartj/12.suppl_d.22. [DOI] [PubMed] [Google Scholar]
- [42].Calabrese F, Angelini A, Thiene G, et al. No detection of enteroviral genome in the myocardium of patients with arrhythmogenic right ventricular cardiomyopathy. J Clin Pathol. 2000;53:382–387. doi: 10.1136/jcp.53.5.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Schultz K.R, Garjarski R.J, Pignatelli R, et al. Genetic heterogeneity in familial dilated cardiomyopathy. Biochem Mol Med. 1995;56:87–93. doi: 10.1006/bmme.1995.1061. [DOI] [PubMed] [Google Scholar]
- [44].Giacca M, Severini G.M, Mestroni L, et al. Low frequency of detection by nested polymerase chain reaction of Enterovirus ribonucleic acid in endomyocardial tissue of patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 1994;24:1033–1040. doi: 10.1016/0735-1097(94)90866-4. [DOI] [PubMed] [Google Scholar]
- [45].Andreoletti L, Hober D, Decoene C, et al. Detection of enteroviral RNA by polymerase chain reaction in endomyocardial tissue of patients with chronic cardiac diseases. J Med Virol. 1996;48:53–59. doi: 10.1002/(SICI)1096-9071(199601)48:1<53::AID-JMV9>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- [46].Kainulainen K, Pulkkinen K, Savolainen A, et al. Location on chromosome 15 of the gene defect causing Marfan syndrome. New Engl J Med. 1990;323:935–939. doi: 10.1056/NEJM199010043231402. [DOI] [PubMed] [Google Scholar]
- [47].Dietz H.C, Outting G.R, Pyeritz R.E, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- [48].Ewart A, Morris C, Ensing G, et al. A human vascular disease, supravalvular aortic stenosis maps to chromosome 7. Proc Natl Acad Sci USA. 1993;90:3226–3230. doi: 10.1073/pnas.90.8.3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Curran M.E, Atkinson D, Ewart A, et al. The elastin gene is disrupted by a translocation associated with supravalvular aortic stenosis. Cell. 1993;73:159–168. doi: 10.1016/0092-8674(93)90168-p. [DOI] [PubMed] [Google Scholar]
- [50].Williams J.C.P, Barratt Boyes B.G, Lowe J.B. Supravalvular aortic stenosis. Circulation. 1961;24:1311–1318. doi: 10.1161/01.cir.24.6.1311. [DOI] [PubMed] [Google Scholar]
- [51].Thiene G, Ho S.Y. Aortic root pathology and sudden death in youth: review of anatomical varieties. Appl Pathol. 1986;4:237–245. [PubMed] [Google Scholar]
- [52].Schwartz P.J, Locati E.H, Napolitano C, et al. The long QT syndrome. Cardiac electrophysiology: from cell to bedside. In: Zipes D.P, Jalife J, editors. 2nd ed. W.B. Saunders; Philadelphia, PA: 1995. p. 788. [Google Scholar]
- [53].Romano C, Gemme G, Pongiglione R. Aritmie cardiache rare dell'età pediatrica. Clin Pediatr. 1963;45:658–683. [PubMed] [Google Scholar]
- [54].Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the QTc interval and sudden death. Am Heart J. 1957;54:59–68. doi: 10.1016/0002-8703(57)90079-0. [DOI] [PubMed] [Google Scholar]
- [55].Priori S, Barhanin J, Hauer R.N.W, et al. Genetic and molecular basis of cardiac arrhythmias. Impact on clinical management. Eur Heart J. 1999;20:174–195. doi: 10.1053/euhj.1998.1220. [DOI] [PubMed] [Google Scholar]
- [56].Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. J Am Coll Cardiol. 1992;20:1391–1396. doi: 10.1016/0735-1097(92)90253-j. [DOI] [PubMed] [Google Scholar]
- [57].Corrado D, Nava A, Buja G, et al. Familial cardiomyopathy underlies syndrome of right bundle branch block ST segment elevation and sudden death. J Am Coll Cardiol. 1996;27:443–448. doi: 10.1016/0735-1097(95)00485-8. [DOI] [PubMed] [Google Scholar]
- [58].Chen Q, Kirsch G.E, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–296. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]