

Abstract
The protease ADAM17 (a disintegrin and metalloproteinase 17) catalyzes the shedding of various transmembrane proteins from the surface of cells, including tumor necrosis factor (TNF) and its receptors. Liberation of TNF receptors (TNFRs) from cell surfaces can dampen the cellular response to TNF, a cytokine that is critical in the innate immune response and promotes programmed cell death but can also promote sepsis. Catalytically inactive members of the rhomboid family of proteases, iRhom1 and iRhom2, mediate the intracellular transport and maturation of ADAM17. Using a genetic screen, we found that the presence of either iRhom1 or iRhom2 lacking part of their extended amino-terminal cytoplasmic domain (herein referred to as ΔN) increases ADAM17 activity, TNFR shedding, and resistance to TNF-induced cell death in fibrosarcoma cells. Inhibitors of ADAM17, but not of other ADAM family members, prevented the effects of iRhom-ΔN expression. iRhom1 and iRhom2 were functionally redundant, suggesting a conserved role for the iRhom amino termini. Cells from patients with a dominantly inherited cancer susceptibility syndrome called tylosis with esophageal cancer (TOC) have amino-terminal mutations in iRhom2. Keratinocytes from TOC patients exhibited increased TNFR1 shedding compared with cells from healthy donors. Our results explain how loss of the amino terminus in iRhom1 and iRhom2 impairs TNF signaling, despite enhancing ADAM17 activity, and may explain how mutations in the amino-terminal region contribute to the cancer predisposition syndrome TOC.
INTRODUCTION
A disintegrin and metalloproteinase 17 (ADAM17) [also known as TNFα converting enzyme (TACE)] is a membrane-anchored metalloproteinase, capable of processing a wide array of cell surface/membrane proteins, and is a central regulator of epidermal growth factor receptor (EGFR) and tumor necrosis factor receptor (TNFR) signaling pathways, which control cell proliferation, survival, oncogenesis, and immunity (1). TNF is liberated from its membrane anchor by ADAM17 to produce a soluble proinflammatory cytokine (2–4). However, ADAM17 can also modulate responses to this cytokine by catalyzing shedding of TNF-binding receptors p55 (TNFR1) and p75 (TNFR2) (5, 6). TNFR1 signaling is a key component of innate immunity, host defense, and septic shock (7, 8), yet TNFR1 engagement can also induce cell death through signaling leading to activation of caspase-8 (9).
ADAM17 is controlled by catalytically inactive members of the rhomboid protease family: iRhom1 and iRhom2. These integral membrane proteins promote the maturation and transport of ADAM17 to the cell surface (10–13). Absence of iRhom2 abolishes ADAM17 activity in immune cells thereby blocking TNF secretion, resulting in susceptibility toward bacterial infections but resistance to septic shock and rheumatoid arthritis (11–14). In nonhematopoietic cells, ADAM17 appears to be controlled by a combination of iRhom2 and iRhom1 (10). The essential role of iRhoms in regulating the function of ADAM17 is highlighted by recent iRhom1 and iRhom2 double knockout studies demonstrating fully impaired ADAM17 maturation across all tissues examined (15) and striking similarity between iRhom1−/−iRhom2−/− mice and ADAM17-deficient mice (15). However, much remains to be learned about how iRhoms accomplish their regulation of ADAM17 and what features of iRhoms are important for their function.
Reports have identified familial dominantly acting mutations in the N-terminal cytoplasmic tail of iRhom2 (also known as RHBDF2 in humans) as causative for a rare syndrome named tylosis with esophageal cancer (TOC), which is characterized by palmoplantar keratoderma and up to 95% lifetime risk of malignancy of the esophagus (16, 17). These TOC-associated mutations in iRhom2 are associated with an increase in the maturation and activity of ADAM17 in patient-derived epidermal keratinocytes, resulting in significantly up-regulated shedding of ADAM17 substrates, including EGF family growth factors and proinflammatory cytokines (18, 19). Furthermore, studies of two separate spontaneous mouse mutants (Cub and Uncv) reveal hair and skin abnormalities also associated with deletions in the N terminus of iRhom2 (11–14, 20). Although the immediate consequences of these murine mutations are not entirely clear, especially in the case of cub mice, all studies in both humans and mice suggest phenotypes involving misregulation of ADAM17 substrates and offer a clue that the N-terminal domain of iRhom2 may be important for controlling its activity (21).
Our initial identification of a connection between iRhom2 and ADAM17 involved a cyclic packaging rescue (CPR) screen for TNF resistance, which identified a version of iRhom2 with a truncated N terminus (12). Here, we report a separate TNF resistance CPR screen, which identified two versions of iRhom1, both also missing parts of their N termini. To gain more insight into how iRhoms operate, we examined the ability of truncated and full-length iRhoms to regulate ADAM17 activity in a well-defined cellular context. We observed a high degree of functional overlap for iRhom1 and iRhom2 and demonstrate that deletion of parts of the cytoplasmic N terminus of iRhom2 or iRhom1 results in specific enhancement of ADAM17 activity, TNFR shedding, and resistance to TNF-induced cell death. Our results support the link of N-terminal iRhom mutants with constitutive activity of ADAM17.
RESULTS
Truncation of iRhom2 or iRhom1 cytoplasmic domains triggers resistance against TNF-induced cell death
L-929 murine fibrosarcoma cells are highly sensitive to TNF-induced cell death through engagement of their cognate cell surface receptors (22–24). Complementary DNAs (cDNAs) capable of conferring resistance to L-929 cell killing by TNF were identified from a mouse 3T3 cell–derived cDNA library through enrichment in a CPR screen (25). Three different cDNAs were isolated after six successive rounds of infection, cell killing, and rescue of viral particles from surviving cells (Fig. 1A). Sequencing revealed the identity of these hits as c-FLIP, an established negative regulator of TNF-induced cell death (26), along with two cDNAs corresponding to nucleotides 249 to 2571 and 618 to 2571 of native iRhom1 (the latter referred to henceforth as iRhom1-ΔN) (Fig. 1B and fig. S1A). The similarity of this result to the identification of an N-terminally truncated version of iRhom2 we previously reported using a separate CPR screen (12) and recent literature concerning mutations in the N terminus of iRhom2 (20, 21, 27) led us to investigate whether there was a selective advantage for removal of part of the extended cytoplasmic N terminus, a hallmark feature of iRhoms (28).
Fig. 1. N truncated iRhom1-ΔN confers TNF resistance as identified by CPR screening.
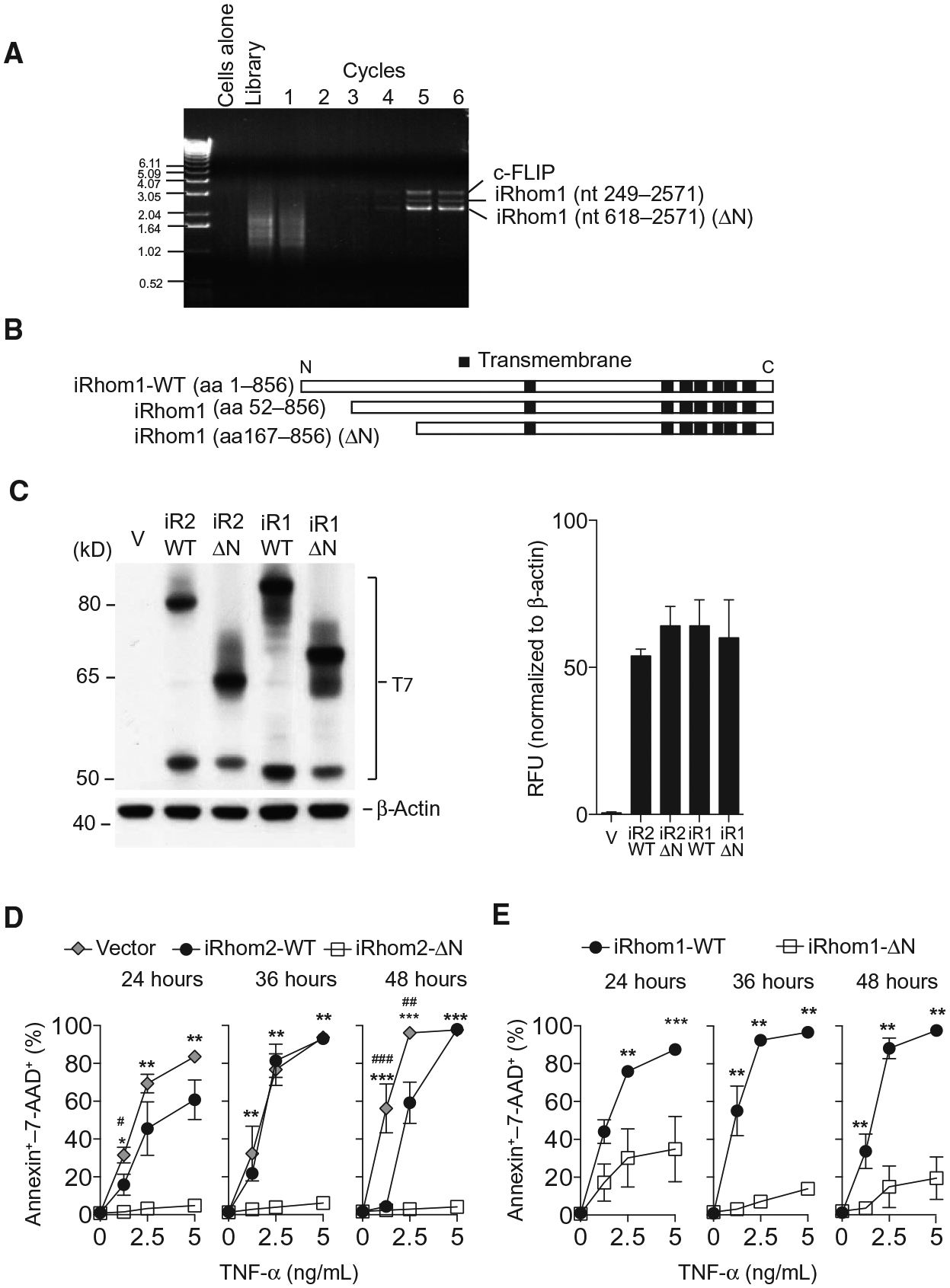
(A) Polymerase chain reaction (PCR) results from each round in the CPR screen (see Materials and Methods) showing enrichment for c-FLIP and two short versions of iRhom1 in TNF-resistant cells. (B) Systematic representation of wild-type (WT) and short versions of iRhom1 identified by CPR relative to their predicted transmembrane domain structures. (C) Immunoblotting and densitometry for T7 in lysates from L-929 cells expressing a control vector, T7-tagged WT or ΔN-iRhom1 (iR1) or iRhom2 (iR2) (n = 3). (D and E) Cell death, assessed by annexin V binding and 7-AAD staining using flow cytometry, in 1 × 105 L-929 cells transfected as indicated and treated with recombinant TNF for up to 48 hours (n ≥ 5). Data are means ± SEM from the number of experiments (n) indicated; *P < 0.05, **P < 0.01, ***P < 0.001 against ΔN; #P < 0.05, ##P < 0.01 against vector. nt, nucleotide; aa, amino acid.
Full-length and ΔN truncated iRhom proteins were detected at predicted molecular weights, and in similar abundance, in cells stably overexpressing tagged iRhom constructs (Fig. 1C). We first compared the ability of cells overexpressing either full-length wild-type iRhom2, iRhom2-ΔN, or vector control to withstand exposure to recombinant TNF (Fig. 1D). We observed only a slight reduction in cell death assessed by annexin Vand 7-AAD staining for cells expressing wild-type iRhom2, whereas iRhom2-ΔN cells had markedly lower levels of annexin V, 7-AAD–positive cells (Fig. 1D). We found similar effects when comparing cells expressing wild-type iRhom1 versus iRhom1-ΔN, which were also strongly resistant to TNF cytotoxicity (Fig. 1E). Next, we confirmed these observations using a separate assay to measure cells remaining adherent after 48 hours of TNF treatment. Consistently, we observed a slight protection against TNF-mediated cell death in wild-type iRhom2–expressing cells, but major protection was triggered by iRhom2-ΔN (fig. S1B). Moreover, iRhom1-ΔN–expressing cells were also protected against TNF-induced cell death compared to wild-type iRhom1 (fig. S1C). Cells expressing full-length and ΔN-iRhoms retained a similar capacity to undergo cell death resulting from other stimuli, such as staurosporine (fig. S1D). These data indicate that deletion of part of the cytoplasmic domain of either iRhom1 or iRhom2 confers a selective advantage over their full-length counterparts in TNF resistance.
Truncated iRhoms curb TNFR signaling through release of TNFRs
We next investigated the mechanism by which iRhom2-ΔN confers resistance toward TNF-induced cytotoxicity by examining the status of signaling downstream of the TNFRs. TNFR engagement in L-929 cells results in activation of both cell survival and cell death signaling pathways, causing activation of cell survival–associated nuclear factor κB (NF-κB) and cleavage of cell death–associated poly(adenosine diphosphate–ribose) polymerase (PARP) (29–32). When we examined PARP levels by immunoblot, we detected cleaved PARP in TNF-treated control cells (Fig. 2A). However, PARP cleavage was reduced in cells expressing wild-type iRhom2 and was not detected in cells expressing iRhom2-ΔN (Fig. 2A). Next, we looked for hallmarks of TNF-mediated NF-κB activation (33) and detected inhibitor of κB-α (IκB-α) phosphorylation and degradation as well as Ser536 phosphorylation of p65 in vector and wild-type iRhom2–expressing cells but not in iRhom2-ΔN–expressing cells (Fig. 2B). These findings indicate that TNFR signaling was blocked by iRhom2-ΔN upstream of both survival and death signaling branches (9).
Fig. 2. iRhom2-ΔN induces TNFR shedding.
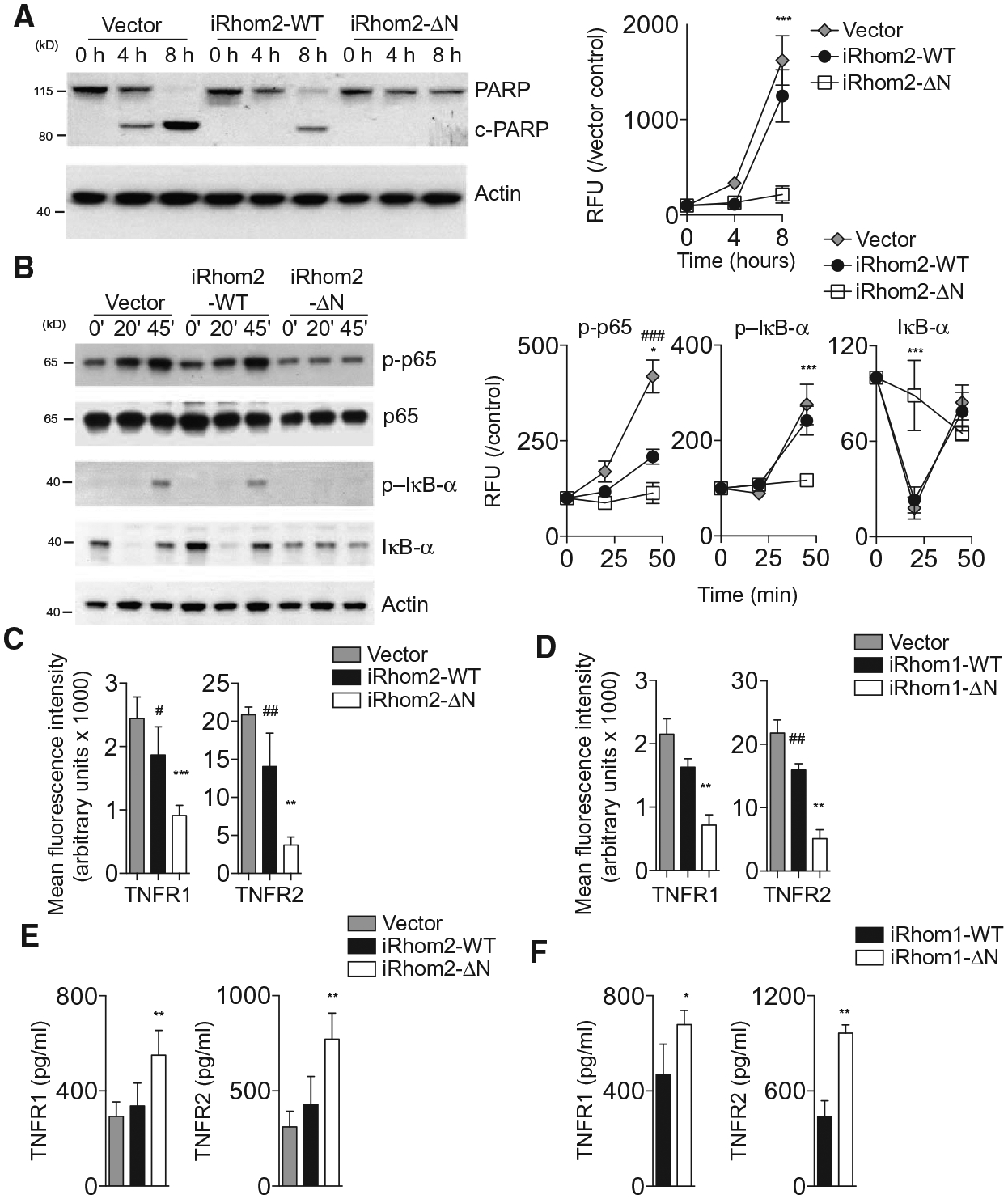
(A and B) Immunoblotting and densitometry for PARP (A) or total and phosphorylated NF-κB pathway proteins (B) in lysates from L-929 cells overexpressing a vector control, iRhom2-WT, or iRhom2-ΔN and exposed to recombinant TNF (2.5 ng/ml) for up to 8 hours (A) or 45 min (B) (n = 3, normalized to control and actin). (C and D) Flow cytometry analysis of TNFR1 and TNFR2 surface abundance on L-929 cells overexpressing WT or truncated iRhom2 (C) or iRhom1 (D) (n = 5 or 6, respectively). (E and F) Amount of TNFR1 and TNFR2 in supernatants from 1 × 105 L-929 cells expressing WT or truncated iRhom2 (E) or iRhom1 (F) (n = 5). Data are means ± SEM from the number of experiments (n) indicated; *P < 0.05, **P < 0.01, ***P < 0.001 against ΔN; #P < 0.05, ##P < 0.01, ###P < 0.001 against vector. RFU, relative fluorescence units.
To test whether surface TNFR abundance itself was affected by different iRhom2 isoforms, we used flow cytometry to measure TNFR abundance on the surface of L-929 cells transduced with vector, wild-type iRhom2, or iRhom2-ΔN. TNFR1 and TNFR2 abundance was highly reduced on cells expressing iRhom2-ΔN or iRhom1-ΔN compared to cells expressing vector or either wild-type iRhom (Fig. 2, C and D). When we measured the concentration of soluble TNFRs in conditioned media from these cultures, we observed greater concentrations of soluble TNFR1 and TNFR2 in conditioned media from cells expressing iRhom2-ΔN or iRhom1-ΔN than in those from control cells or cells expressing either wild-type iRhom (Fig. 2, E and F). These effects were not associated with either decreased TNFR transcript expression (fig. S2, A and B) or large differences in total cellular abundance of TNFRs (fig. S2, C to E).
Together, the data indicate that the blockade in TNFR signaling after ΔN-iRhom expression was caused by reduced surface abundance and enhanced shedding of TNFRs. Previous work suggested that shedding of TNFRs was responsible for iRhom2-mediated resistance toward TNF by blocking TNFR signaling (11, 12). We now show that this shedding mechanism is true not only for truncated iRhom2 but also for truncated iRhom1. Furthermore, we confirm that TNFR signaling is blocked by ΔN-iRhom expression by showing that both downstream cell survival and cell death signaling responses are impaired in these cells.
iRhom-regulated TNFR shedding depends on ADAM17
TNFRs are cleaved from the cell surface by membrane proteases such as ADAM17 (fig. S3A) (5, 6). To investigate whether increased TNFR shedding into the supernatant of iRhom-ΔN–expressing cells was dependent on metalloproteinases in L-929 cells, we investigated shedding in the presence of several ADAM family inhibitors. Increased release of TNFRs into the supernatant of iRhom2-ΔN–expressing cells relative to control or wild-type iRhom2 was blocked by the broad-spectrum metalloproteinase inhibitor marimastat (Fig. 3A). Furthermore, shedding induced by iRhom2-ΔN or iRhom1-ΔN was unaffected by an ADAM10-selective inhibitor, GI254023X (GI) (34), but was abolished by an inhibitor of both ADAM10 and ADAM17, GW280264X (GW) (Fig. 3, B and C). Consistently, culturing iRhom2-ΔN–expressing cells with marimastat restored the ability of recombinant TNF to trigger cell death to a similar extent as that observed in cells expressing wild-type iRhom2 (Fig. 3D). TNF itself had little effect on TNFR shedding (fig. S3, B and C).
Fig. 3. Effects of ΔN-iRhoms can be blocked by ADAM17 inhibitors.
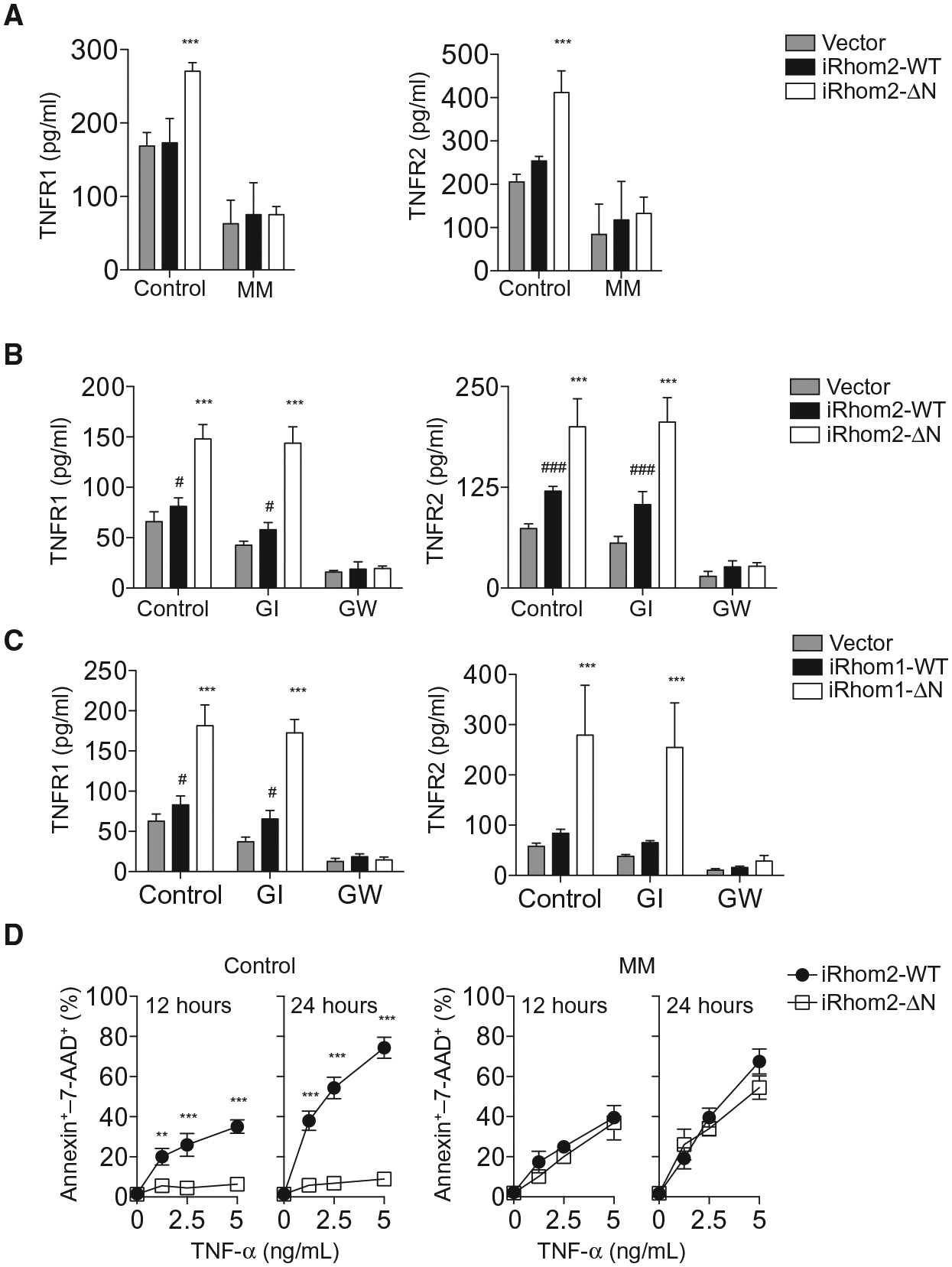
(A and B) Abundance of TNFR1 and TNFR2 in the culture supernatants from 1 × 105 L-929 cells expressing WT or truncated iRhom2 cultured in presence or absence of marimastat (MM; 20 μM) (A) or GI or GW (each 3 μM) (B) for 6 hours (n = 5 or 6, respectively). (C) As in (B) in 1 × 105 L-929 cells expressing WT or truncated iRhom1. (D) Cell death as a percentage of annexin–7-AAD+ cells in cultures (n = 3) of 1 × 105 L-929 cells expressing WT or truncated iRhom2 treated with recombinant TNF in the presence or absence of marimastat (20 μM). Data are means ± SEM from the number of experiments (n) indicated; **P < 0.01, ***P < 0.001 against WT; #P < 0.05, ###P < 0.001 against vector.
To firmly establish that ΔN-iRhoms exert their effects through ADAM17, we stably expressed either a scrambled control or one of two ADAM17-specific short hairpin RNA (shRNAs) (Fig. 4A) in L-929 cells transduced with vector, wild-type iRhom2, and iRhom2-ΔN. Surface TNFR1 and TNFR2 abundance was reduced in cells expressing iRhom2-ΔN or iRhom1-ΔN relative to wild-type or vector-transduced controls; however, these effects were rescued by ADAM17 silencing (Fig. 4B and fig. S4, A and B), suggesting that truncated iRhoms enhance ADAM17-dependent shedding. Next, we examined whether ADAM17 silencing would disrupt the survival advantage of ΔN-iRhoms against TNF. As expected, knocking down ADAM17 abolished the survival advantage caused by expression of ΔN-iRhoms conferred in response to TNF (Fig. 4, C and D). Together, the data indicate that TNFR shedding and TNF resistance phenotypes associated with expression of N-terminally truncated iRhoms can be blocked by knockdown or inhibition of ADAM17, establishing ADAM17 as a mechanism through which iRhoms mediate their effects on TNF signaling, at least in the cell context studied.
Fig. 4. Stable knockdown of ADAM17 prevents increased TNFR shedding and resistance to TNF.
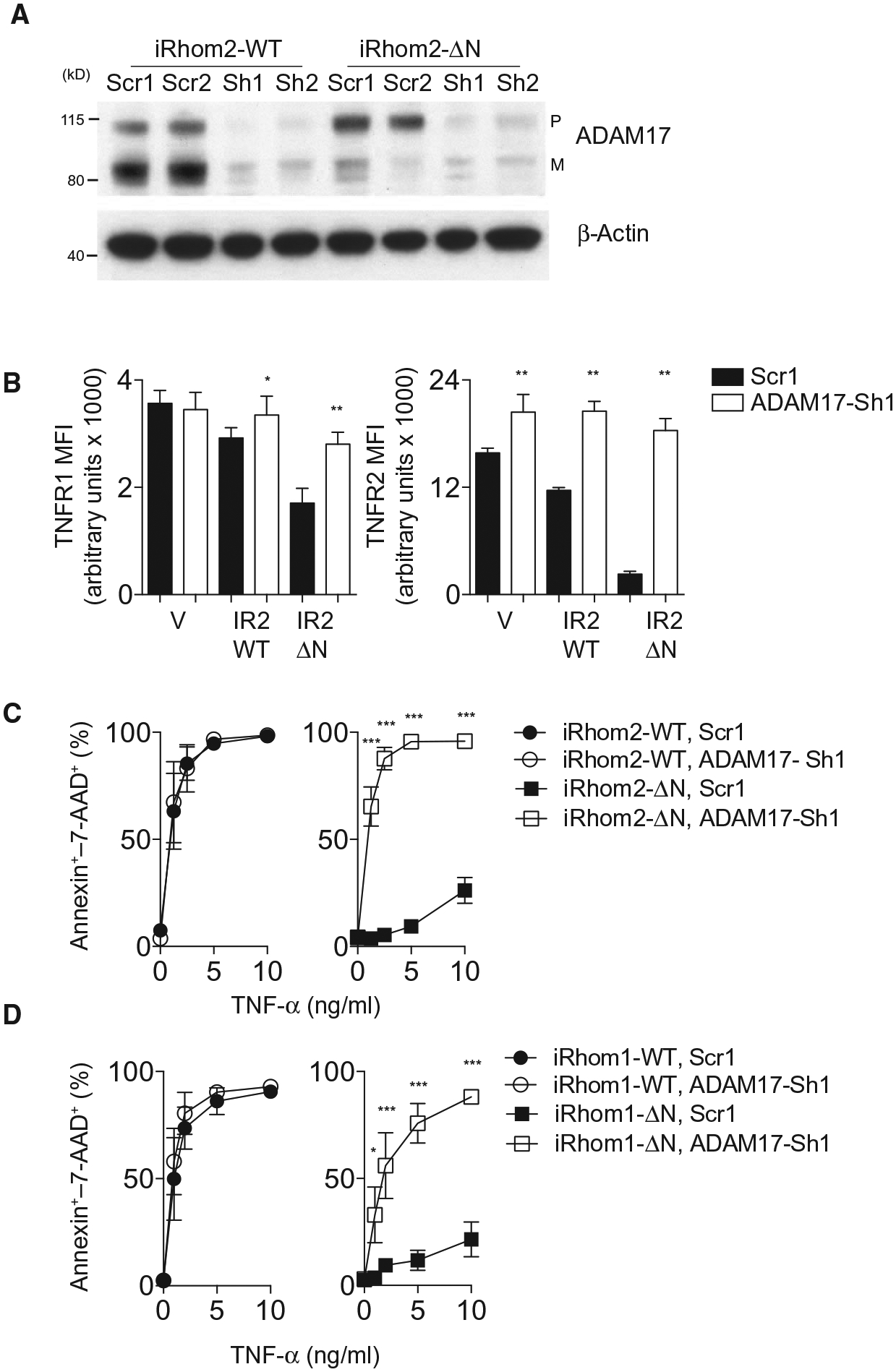
(A) Immunoblot for pro (P) and mature (M) ADAM17 in lysates from L-929 cells expressing full-length and ΔN-iRhom2 transfected with either a scrambled control (Scr1) or an ADAM17-targeted shRNA (ADAM17-Sh1). Blot is representative of three experiments. (B) Surface abundance of TNFR1 and TNFR2 as determined by flow cytometry on L-929 cells expressing a vector control, full-length (WT) iRhom2 or iRhom2-ΔN in the presence of either control or ADAM17 shRNA (n = 6). (C and D) Cell death as a proportion of annexin–7-AAD+ cells in cultures of 1 × 105 L-929 cells stably expressing full-length or ΔN-iRhom2 (C) or iRhom1 (D) and either scrambled or ADAM17 shRNA treated with recombinant TNF for 48 hours (n = 4 or 6, respectively). Data are means ± SEM from the number of experiments (n) indicated; *P < 0.05, ***P < 0.001. MFI, mean fluorescence intensity.
Truncation of the cytoplasmatic tail results in enhanced presence of iRhom2 at the cell surface
We next asked how wild-type iRhoms and their ΔN counterparts might differentially affect ADAM17 activity. Binding of iRhom2 to ADAM17 is thought to be important for ADAM17 maturation and activation (11–13). When we pulled down either wild-type or truncated iRhom2, we detected ADAM17 in both immunoprecipitated lysates (Fig. 5A). Reciprocally, when ADAM17 was pulled down, both wild-type iRhom2 and iRhom2-ΔN were detected (Fig. 5A). Similar data were obtained in wild-type iRhom1– and iRhom1-ΔN–expressing cells (fig. S5A). These data indicated that both isoforms of iRhom1 and iRhom2 are capable of binding ADAM17, consistent with previous reports (12, 35).
Fig.5. Truncation of the cytoplasmatic tail results in increased surface expression of iRhom2.
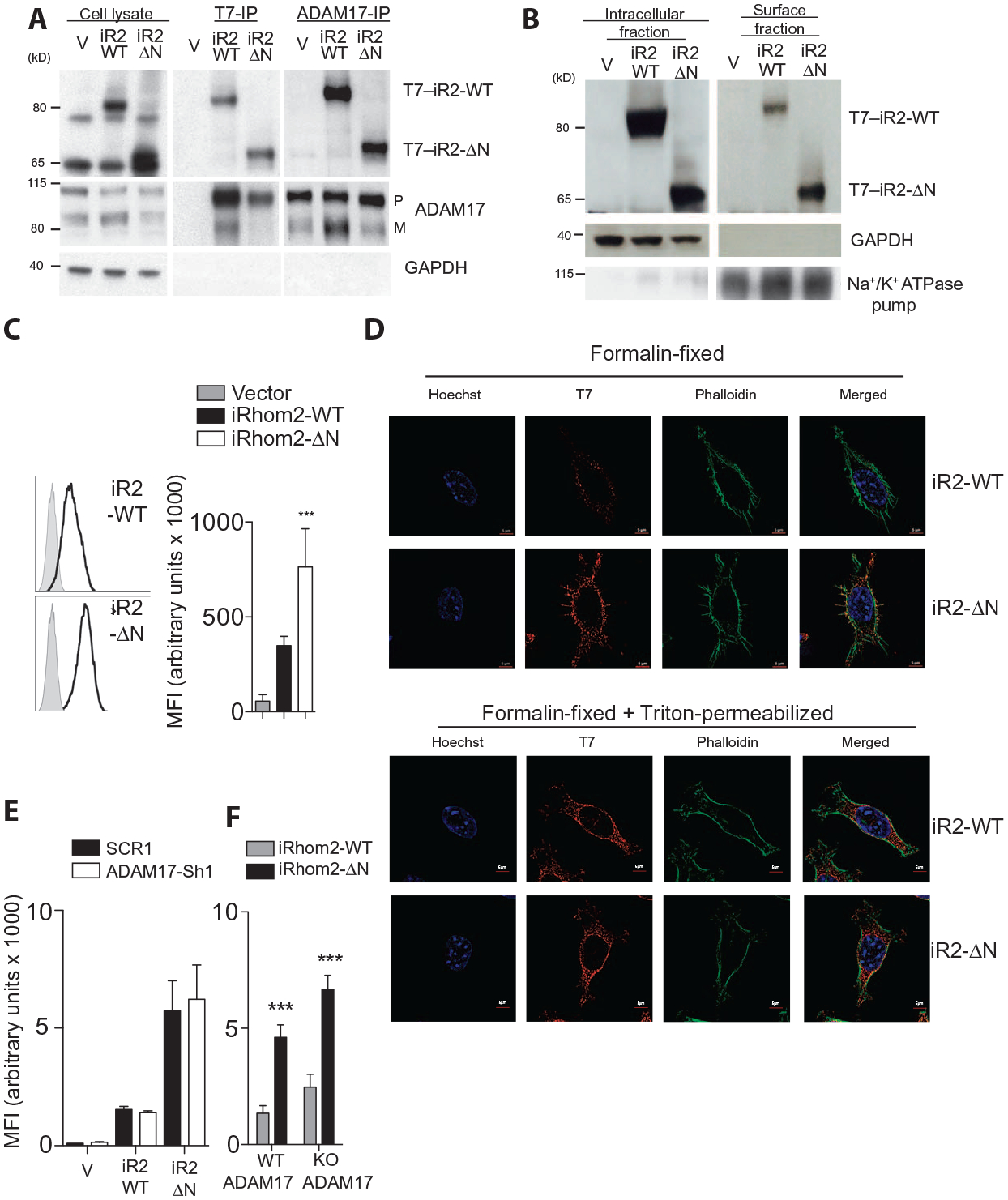
(A) Immunoprecipitation (IP) for T7 or ADAM17 followed by immunoblotting for the same in lysates from L-929 cells stably expressing T7-tagged WT or truncated iRhom2. Blot is representative of three experiments. (B) Immunoblotting for iRhom2 using a T7 antibody in intracellular and cell surface fractions from L-929 cells stably expressing WT or truncated iRhom2. Blot is representative of three experiments. (C) Surface abundance of iRhom2, determined using an antibody against T7, on stably transfected L-929 cells (n = 8). (D) Immunocytochemistry for iRhom2 using T7 antibodies (Cy3), phalloidin–fluorescein isothiocyanate (FITC), and Hoechst staining in stably transfected L-929 cells, fixed, and/or permeabilized as indicated (n ≥ 3 experiments). (E and F) Flow cytometry analysis of MFI of the surface abundance of iRhom2 on unpermeabilized stably transfected L-929 cells expressing either scrambled or ADAM17 shRNA (n ≥ 4 experiments) (E) or immortalized WT or Adam17 knockout (KO) mouse embryonic fibroblasts (MEFs) (n = 12 experiments) (F). Data are means ± SEM from the number of experiments (n) indicated; ***P < 0.001. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. Na+/K+ ATPase, Na+- and K+-dependent adenosine triphosphatase.
When we enriched for cell surface proteins, we noticed that a larger proportion of iRhom2-ΔN was present in surface fractions versus intracellular fractions and relative to wild-type iRhom2 (Fig. 5B). These findings were further confirmed by T7 surface antibody staining and flow cytometry, where the MFI for iRhom2-ΔN was significantly higher than that for wild-type iRhom2 (Fig. 5C). Consistently, immunofluorescence of tagged iRhom2 revealed greater staining intensity for iRhom2-ΔN versus wild-type iRhom2 on formalin-fixed cells, differences which were not apparent after 1% Triton permeabilization (Fig. 5D). Furthermore, in permeabilized cells, subcellular localization of both wild-type iRhoms and ΔN-iRhoms in the proximity of Golgi marker GM130 (fig. S5B) was consistent with previous reports (12, 36). Increased surface localization of iRhom2-ΔN compared with wild-type iRhom2 persisted after ADAM17 knockdown and in ADAM17 knockout fibroblasts (Fig. 5, E and F) (4), indicating that ADAM17 is not strictly required for iRhom2-ΔN trafficking.
Truncated and wild-type iRhom2 did not exhibit significantly different rates of protein degradation after inhibition of protein synthesis with cycloheximide, as measured by immunoblotting (fig. S5C). This argues against a role for differential protein stability affecting iRhom2 surface abundance. Increased surface abundance of iRhom2-ΔN is also unlikely to be the result of greater RNA expression because transcript abundance was comparable between iRhom2-ΔN and wild-type iRhom2 (fig. S5D). We detected mature ADAM17 on the cell surface of cells expressing vector, wild-type iRhom2, or iRhom2-ΔN (fig. S5E). However, a puzzling overall reduction in the total quantity of mature ADAM17 was observed in immunoblots of cells expressing iRhom-ΔN (Fig. 5A and fig. S5, A and E), an effect that was prevented by marimastat treatment (fig. S5F). These data indicate that ΔN-iRhoms are capable of binding to ADAM17 in a similar manner to that of wild-type iRhoms and that the specific effects of iRhom2-ΔN may be related to its enhanced abundance at the cell surface.
N-terminal iRhom mutations increase constitutive activity of ADAM17
ADAM17 can be rapidly activated in response to stimuli including phorbol 12-myristate 13-acetate (PMA) (37, 38). We therefore wondered whether the effects of ΔN-iRhoms might influence the rapid activation of ADAM17. When we stimulated L-929 cells with PMA, we observed an expected increase in TNFR1 and TNFR2 shedding from cells expressing either vector or wild-type iRhom2 (fig. S6A). In contrast, we did not detect PMA-induced TNFR shedding from cells expressing iRhom2-ΔN (fig. S6A), and the amount of TNFRs in the stimulated cells expressing iRhom2-ΔN was not increased when compared to cells expressing wild-type iRhom2 (fig. S6A). In all cases, PMA-stimulated TNFR shedding was blocked by marimastat (fig. S6A) or GW, which both inhibit ADAM17, but not by the ADAM10-selective inhibitor GI (Fig. 6A). Furthermore, similar data using cells expressing iRhom1-ΔN or wild-type iRhom1 indicated that both truncated iRhoms may induce a constitutively active TNFR shedding state, resembling PMA-stimulated shedding state in control cells (Fig. 6B).
Fig. 6. N-terminal mutations in iRhom2 trigger constitutive activity of ADAM17.
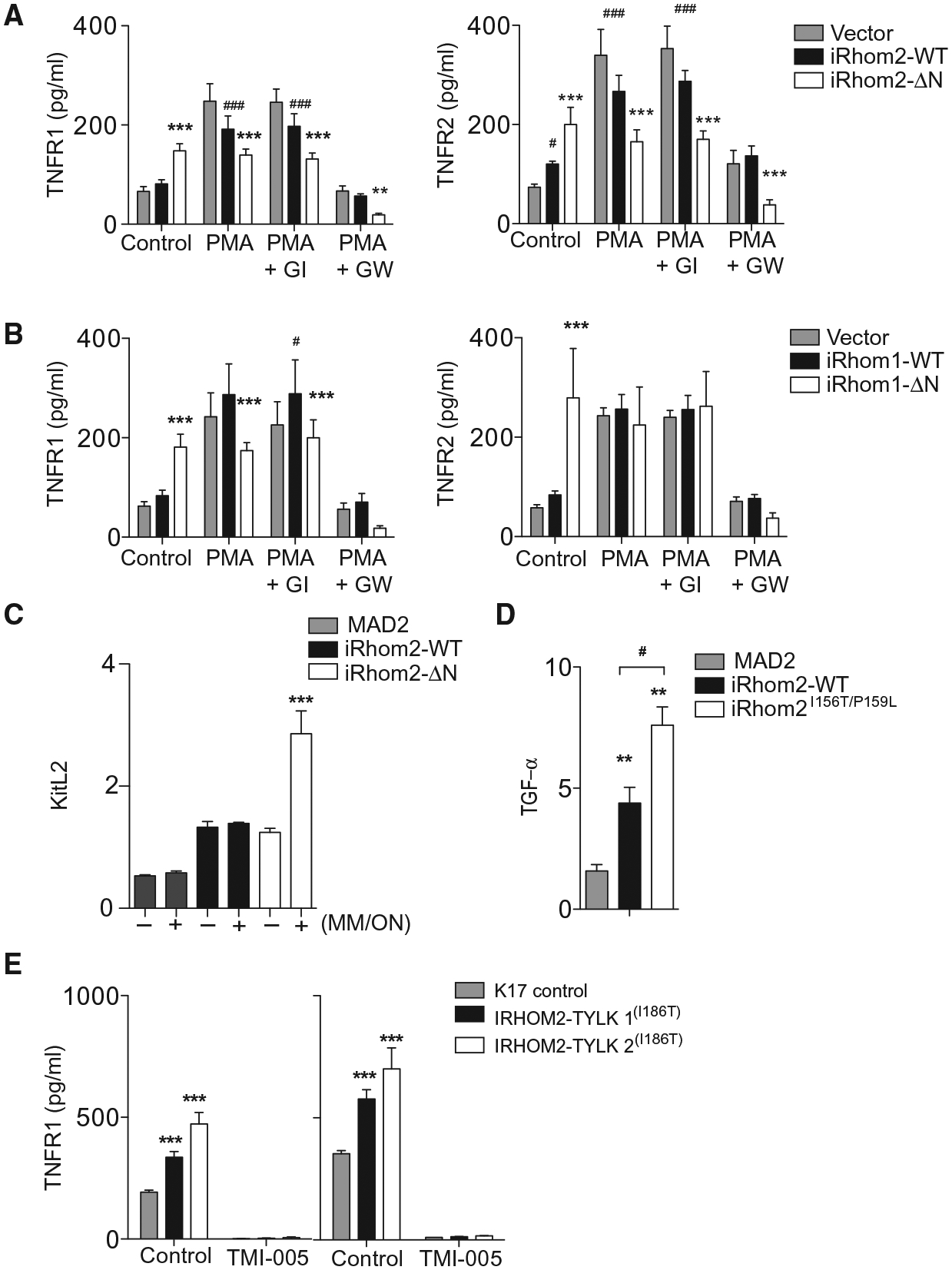
(A and B) TNFR1 and TNFR2 in conditioned media of 1 × 105 L-929 cells expressing WT or ΔN-iRhom2 (A) or iRhom1 (B) cultured with PMA (100 ng/ml) in the presence (where indicated) of GI or GW (3 μM each) (n = 6). (C) KitL2 shedding in immortalized MEFs (iMEFs) genetically lacking iRhom2 transfected with alkaline phosphatase (AP)–tagged KitL2 along with MAD2 (control), iRhom2-WT, or iRhom2-ΔN, and (where indicated, +) preincubated with marimastat overnight (MM/ON), followed by washout of the inhibitor (n = 3). (D) Transforming growth factor–α (TGF-α) shedding from iMEFs genetically lacking iRhom1 and iRhom2 and transfected with AP-tagged TGF-α along with MAD2 (control), murine WT iRhom2, or iRhom2I156T/P159L (n ≥ 5). (E) Concentration of soluble TNFR1 in supernatants from 2 × 105 keratinocytes from either healthy donors (K17 cells) or tylosis patients who have mutations in IRHOM2(l186T/WT) [TYLK1 and TYLK2 treated with vehicle (dimethyl sulfoxide; DMSO) or TMI-005 ADAM17 inhibitor for 24 (left) or 48 (right) hours]. Data are means ± SEM from the number of experiments (n) indicated; *P < 0.05, **P < 0.01, ***P < 0.001 against WT (A and B) or MAD2 (D and E); #P < 0.05 against vector (A and B) or mutant construct (D and E).
To investigate these effects in a different cellular context, we coexpressed wild-type or truncated iRhom2 or as control an unrelated cytoplasmic protein MAD2 (mitotic arrest deficient 2) along with the iRhom2-selective AP-tagged ADAM17 substrate KitL2 in iRhom2−/− iMEFs (10, 12). To improve the detection of potential differences in shedding, we took advantage of the reversible nature of ADAM17 inhibition with marimastat to block shedding overnight, enabling uncleaved substrate to accumulate, and then observed constitutive shedding immediately after washout of the inhibitor. Compared to control cells, only cells expressing iRhom2-ΔN exposed overnight to marimastat exhibited greatly enhanced shedding of KitL2 (Fig. 6C). In a separate experiment, we used an irreversible inhibitor of ADAM17 [diphenylamine carboxylate (DPC)], which selectively binds to active ADAM17. Thus, if ADAM17 is not activated while cells are exposed to DPC, ADAM17 can be fully activated after the inhibitor has been removed; however, if ADAM17 is activated in the presence of DPC, ADAM17 will not be able to recover activity after the inhibitor is removed (37). Incubation of cells expressing iRhom2-ΔN with DPC for as little as 30 min prevented the increased KitL2 shedding even after the inhibitor was removed, whereas cells expressing wild-type iRhom2 required overnight exposure to DPC to exhibit ADAM17 inactivation (fig. S6B). These data suggest that ADAM17 is constitutively in a more active state in the iRhom2-ΔN setting.
Dominantly acting familial mutations in the N-terminal cytoplasmatic tail of iRhom2 cause a cancer susceptibility disorder called TOC (16, 17). These mutations increase constitutive ADAM17 activity in keratinocytes (18). Compared to overexpression of wild-type murine iRhom2 in iRhom1 and iRhom2 double knockout iMEFs, expression of a construct bearing mouse homologs of two causative iRhom2I156T/P159L TOC mutations caused increased constitutive TGF-α shedding (Fig. 6D). Finally, we examined human immortalized keratinocyte cell lines TYLK1 and TYLK2 derived from TOC patients bearing heterozygous mutation in iRHOM2(l186T/WT) (16, 17) and control keratinocyte cell lines (K17). Significantly higher amounts of constitutive TNFR1 shedding into culture supernatants were detected from cells generated from TOC patients compared to cells derived from healthy controls (Fig. 6E), shedding that could be blocked by the ADAM17 inhibitor TMI-005. These data are consistent with enhanced ADAM17-dependent shedding of amphiregulin, TGF-α, and heparin-binding EGF-like growth factor that is observed in TOC patient–derived keratinocytes (18). These results indicate that ADAM17 activity is enhanced by N-terminal iRhom mutations and may provide an explanation for why cells that have ΔN-iRhom proteins have a selective advantage over their wild-type counterparts in promoting resistance to TNF-induced cell death.
DISCUSSION
Here, we have identified a high degree of functional overlap for iRhom1 and iRhom2 in controlling the activity of ADAM17. Starting from unbiased genetic screens, we have established that deletion of part of the cytoplasmic N terminus of either iRhom1 or iRhom2 results in a gain of function by promoting constitutive ADAM17-dependent shedding of TNFRs from L-929 cells, thereby blocking downstream signaling and protecting cells from TNF-induced cytotoxicity. We demonstrate that iRhom1- and iRhom2-dependent effects are conserved for at least two endogenous ADAM17 substrates (TNFR1 and TNFR2), and using gene silencing shows these effects to be specific to ADAM17.
Little is known about how the iRhoms are functionally controlled. Our results suggest that the extended cytoplasmic N-terminal domain is of conserved importance for the actions of both iRhom1 and iRhom2. Dominant inherited human N-terminal mutations in iRhom2 result in overgrowth of epithelial tissues and predisposition to esophageal cancer (39). Our results involving dominant N-terminal mutated mouse iRhoms and enhanced TNFR shedding from TOC patient–derived keratinocytes suggest that these human mutations could similarly result in greater or constitutive ADAM17 activity. This could result in direct consequences for neoplasia, such as overproduction of EGFR ligands, strongly implicated in cancer (40) and previously observed in TOC keratinocytes (18), or shedding of TNFRs as an evasion mechanism for tumor cells to escape cell death induced by TNF. Increased abundance of ADAM17 is also known to be associated with breast, ovarian, kidney, colon, and prostate cancers (41).
Recent studies involving two separate spontaneous mouse mutants with deletions in the N terminus of iRhom2 describe hair and skin abnormalities in these animals (20, 21, 27, 42). Discerning the consequences of these mutations has not been entirely straightforward; Hosur et al. report enhanced stability of mutant iRhom2 in cub mice and elevated secretion of ADAM17 substrate amphiregulin, observations not corroborated by Siggs et al. (20) in the same mice. Mice bearing a separate Uncv allele display aberrancies in hair follicles, which may be connected to misregulation of ADAM17 substrates. In the case of both cub and Uncv, expression of mutant alleles results in an apparent decrease in mature ADAM17 by Western blotting (20, 27). Paradoxically, we also see an apparent reduction in mature ADAM17 by immunoblot after expression of ΔN-iRhoms, despite strong evidence of enhanced ADAM17 activity. Because this effect could be reversed by inhibiting ADAM17 with marimastat, we hypothesize that the apparent reduction of mature ADAM17 may be an artifact of activation-associated autocatalytic degradation of ADAM17 known to occur when it is strongly activated, such as by high concentration of PMA (43). Rapid inhibition of ADAM17 activity with DPC in iRhom2-ΔN–expressing cells provides further evidence for enhanced constitutive ADAM17 activity in this context.
Aside from the mouse and human mutations listed above, the in vivo relevance of alternative iRhom1 or iRhom2 isoforms has not yet been described. However, expression of C-terminally tagged iRhom2 and iRhom1 frequently results in lower–molecular weight bands, which could correspond to posttranslationally processed iRhoms lacking N-terminal regions (11, 44). Whether these versions exist for endogenous proteins, and whether they represent activated iRhom states, warrants investigation in future studies, including the generation of high-quality antibodies. Similar to our results, a previous study involving expression of human iRhom1 (44) (then referred to as p100hRho) in the fly wing identified a phenotype for an N-terminally truncated but not full-length version of the protein. Because iRhom1 has been previously suggested to play a role in human cancer (45, 46), and considering functional overlap, we have identified between N-terminal mutations in iRhom1 and iRhom2 the potential existence of human mutations in iRhom1, and their contributions to cancer warrants further study.
MATERIALS AND METHODS
CPR screening
L-929 selector cell line was established by transfection of expression plasmids coding for mCAT1 (murine cationic amino acid transporter, mediating ecotropic retrovirus infection) and hCAR (human coxsackie-virus and adenovirus receptor), and selected for high infectivity to green fluorescent protein–expressing ecotropic retrovirus and human adenovirus and high susceptibility to TNF. CPR was carried out as described previously (25). The Phoenix ecotropic packaging cell line was transfected with retroviral mouse 3T3 cell cDNA library by using calcium phosphate coprecipitation. Retroviral supernatants were obtained 2 days after transduction and used to transduce the selector cells. Two days after retrovirus transduction, cells were cultured with TNF (2 ng/ml) for 24 hours. Surviving cells were allowed to recover from TNF stress and expanded in culture for 3 days, and then infected with adenoviruses expressing gag-pol and env (amounts of adenoviruses were determined empirically). Rescued retroviruses were harvested 2 days after adenovirus infection and used to transduce new batches of selector cells. Genomic DNA was extracted from a fraction of the surviving cells at each round of screening. A portion of cDNA inserts were amplified by PCR using primers 5′-AGCCCTCACTCCTTCTCTAG-3′ and 5′-ACCTACAGGTGGGGTCTTTCATTCCC-3′ and sequenced.
Generation and maintenance of cells expressing iRhom isoforms
Constructs of wild-type iRhom1 and iRhom1-ΔN, and wild-type iRhom2 and iRhom2-ΔN were constructed using a three-step ligation. The 5′ and 3′ portions were amplified, and restricted middle portion was ligated into modified version of pMSCVpuro (Clontech) containing a C-terminal T7 tag vector. Retrovirus encoding wild-type iRhom1 and iRhom1-ΔN, wild-type iRhom2 and iRhom2-ΔN, and empty pMSCVpuro alone (BD Biosciences) was produced in Phoenix ecotropic packaging cells and used to infect L-929 cells. Stable L-929 cells were generated through selection using puromycin (10 μg/ml). Unless otherwise indicated, cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 2 μM of l-glutamine, 0.1 U of penicillin, streptomycin (0.1 μg/ml), antibiotics, and 10% fetal calf serum (FCS).
MEFs were isolated from E13.5 wild type, ADAM17−/−(4), iR2−/− iR1−/−, and iR1/2−/− (10, 12, 15) embryos to generate primary MEFs for immortalization. Briefly, head and viscera were removed, and the remaining tissues were subjected to trypsin treatment for 15 min at 37°C. Cells were collected and immortalized by transducing the plasmid expressing SV40 large Tantigen maintained in DMEM supplemented with anti-biotics and 10% FCS.
Immunofluorescence
Intracellular immunofluorescence staining was performed in L-929 cells stably expressing T7-iRhom2, T7–iRhom2-ΔN, T7-iRhom1, T7–iRhom1-ΔN, and control vector, grown on glass coverslips, fixed with 4% formalin, and were permeabilized with 1% Triton X-100. All iRhom versions were detected using a T7 tag rabbit primary monoclonal antibody (mAb) at 1:300 (Abcam) and donkey anti-rabbit Cy3-labeled secondary Ab (Jackson ImmunoResearch). The Golgi marker GM130 (BD eBioscience) was recognized with a donkey anti-mouse Cy2-labeled secondary Ab (Jackson ImmunoResearch). Surface immunofluorescence staining of T7-iRhom2 and T7–iRhom2-ΔN cells was performed after fixation with 4% formalin. iRhom versions were detected using a T7 tag rabbit mAb at 1:300 (Abcam) and donkey anti-rabbit Cy3-labeled secondary Ab (Jackson ImmunoResearch). Slides were stained with phalloidin-FITC (Sigma) and Hoechst 34580 (Invitrogen). Images were captured using confocal microscopy (Zeiss ELYRA).
Stable knockdown of ADAM17
Lentiviral particles were generated by calcium phosphate transfection of subconfluent (50 to 60%) 293TV cells with 10 μg of shRNA (OriGene) and 5 μg each of pMDG1.vsvg, pRSV-Rev, and pMDLg/pRRE constructs. Lentiviral particles were collected 24 and 48 hours later, filtered through a 0.45-μm filter, and stored at −80°C. Parental L-929 cells were infected with lentiviral particles containing the indicated shRNAs, and cells were selected with puromycin selection (48 hours; 5 μg/ml).
ELISA detection of TNF and TNFR shedding
To analyze the TNFR shedding in L-929 cells overexpressing T7-iRhom2, T7–iRhom2-ΔN, T7-iRhom1, T7–iRhom1-ΔN, and control vector, cells were seeded (1 × 105) in 24-well plates. After 24 hours, supernatant was harvested and subjected to enzyme-linked immunosorbent assay (ELISA; DuoSet ELISA, R&D Systems), which was performed according to the manufacturer’s instructions. Cells were co-incubated with marimastat (20 μM; BB 2516, Tocris Bioscience) for 6 hours. For the treatment with GI or GW (Sigma), culture medium was removed, and the cells were washed with phosphate-buffered saline (PBS). Fresh FCS-free DMEM containing 3 μM of the inhibitors was added to the cells, and the TNFR shedding was analyzed after 6 hours.
Similarly, for TNFR shedding from TOC patient–derived keratinocytes, 2 × 105 cells per cell line were seeded into 24-well tissue culture plates. After 24 and 48 hours, supernatant was harvested and used for ELISA, performed according to the manufacturer’s instructions (as above). To enable blockade of ADAM17, cells were co-incubated with ADAM17 inhibitor TMI-005 (500 nM) or vehicle (DMSO). This medium was then collected after either 24 or 48 hours, and the concentration of TNFR therein was measured by ELISA. All experiments were carried out in triplicate.
Ectodomain shedding assay for AP-tagged KitL2
iRhom2-deficient fibroblasts were transfected with iRhom and AP-tagged KitL2 constructs using lipofectamine 2000 as previously described (47). Twenty-four hours after transfection, cells were incubated with marimastat (a gift from O. Ouerfelli, Sloan-Kettering Institute, New York, NY) or DPC-333/BMS-561392 (a gift from R. Waltermire, Bristol-Myers Squibb, New Brunswick, NJ) overnight or as indicated, followed by three brief washes. Constitutive shedding was measured after 2 hours of subsequent incubation. Quantification of AP activity culture supernatant and cell lysate was performed by colorimetric assays as previously described (47).
Viability assay
L-929 cells (1 × 105) overexpressing T7-iRhom2, T7–iRhom2-ΔN, T7-iRhom1, T7–iRhom1-ΔN, and control vector were seeded in 24-well plates. Same conditions were used for ADAM17 shRNA–treated cells. Cells were incubated with recombinant mouse TNF (R&D Systems), with or without 20 μM BB 2516 (Tocris Bioscience). Viability was determined at indicated time points using annexin V–7-AAD exclusion (eBioscience) by flow cytometry (48).
Clonogenic assay
L-929 cells (1 × 105) overexpressing T7-iRhom2, T7–iRhom2-ΔN, T7-iRhom1, T7–iRhom1-ΔN, and control vector were seeded in 24-well plates. Cells were treated with recombinant mouse TNF (R&D Systems) at the indicated concentrations. After 48 hours, cells were washed and stained with crystal violet at room temperature for 1 hour. The remaining crystal violet was dissolved with methanol after analysis.
Reverse transcription PCR
RNA was extracted using RNeasy Kit (Qiagen). mRNA expression of wild-type iRhom1 and iRhom1-ΔN, and wild-type iRhom2 and iRhom2-ΔN was performed by reverse transcription PCR (Bio-Rad). For the analysis, the expression of the entire target mRNA was normalized to β-actin or GAPDH expression. Gene expression values were then calculated on the ΔΔCt method. Relative quantities (RQ) were determined with the equation RQ = 2−ΔΔCt.
Cell surface protein isolation
Surface proteins from L-929 cells over expressing T7-iRhom2, T7–iRhom2-ΔN, or control vector were isolated using the sulfo-NHS-SS-biotin–based Cell Surface Protein Isolation Kit (Pierce) according to the manufacturer’s instructions, after addition of 20 μM BB 2516 (Tocris Bioscience) and 1,10-phenanthroline (Sigma) to the lysis and wash buffers. Cell surface fractions were compared to column flow-through (intracellular fractions) by immunoblotting.
Flow cytometry
Single-cell suspensions from cultured cells were stained for 20 min at 4°C with T7 antibody in PBS containing 1% FCS and 5 mM EDTA. Staining for annexin Vand 7-AAD (eBioscience) was performed in solution containing 5mMCa2+. Staining with TNFR1- and TNFR2-biotin antibodies (eBioscience) was performed in PBS containing 1% FCS and 5 mM EDTA for 1 hour at room temperature, followed by wash and incubation with phycoerythrin-Cy7–coupled streptavidin-labeled antibody (eBioscience) for 30 min at 4°C.
Immunoprecipitation and immunoblotting
Briefly, cells were lysed in PBS containing 1% Triton X-100 (PBS-T; Sigma), EDTA-free protease inhibitor cocktail (Roche), PhosSTOP (1 tablet/10 ml), and the inhibitors BB 2516 (20 μM; Tocris Bioscience) and 1,10-phenanthroline (10 mM; Sigma). The pulldown was performed using mouse mAbs recognizing the T7 tag or ADAM17 antibody (Abcam). To analyze the PARP cleavage, the cells were exposed to 2.5 ng of recombinant mouse TNF (R&D Systems) for indicated time points. Phospho-p65 or phospho–IκB-α activation was determined by harvesting cells from 60-mm dishes after treatment with 2.5 ng of recombinant mouse TNF for indicated time points. After lysis, the samples were used for immunoblotting using phospho-p65 and phospho–IκB-α antibodies (Cell Signaling).
Stability assay
L-929 cells (1 × 105) overexpressing T7-iRhom2, T7–iRhom2-ΔN, or control vector were seeded in 24-well plates. Cells were incubated with cycloheximide (100 μg/ml; Sigma) for indicated time points. After fixing cells using 4% formalin (Sigma) in PBS at room temperature for 40 min, cells were washed two times with and resuspended in PBS-T for 15 min at room temperature in the dark. After additional wash and suspension in PBS containing 1% FCS and 5 mM EDTA, cells were stained with a T7 antibody (eBioscience) on ice for 30 min, followed by measurement using flow cytometry.
Statistical analysis
Data are expressed as means ± SEM or SD. Statistical significance between two groups was analyzed using the Mann-Whitney U test. For experiments involving analysis of multiple time points, two-way analysis of variance (ANOVA) with an additional Bonferroni posttest was used. P values <0.05 were considered statistically significant. In all the experiments, n indicates the number of independent experiments performed.
Supplementary Material
www.sciencesignaling.org/cgi/content/full/8/401/ra109/DC1
Fig. S1. Cloning of CPR screen–identified iRhom versions.
Fig. S2. TNFR1 and TNFR2 abundance in iRhom-expressing cells.
Fig. S3. ADAM17 mediates cleavage of TNFRs.
Fig. S4. Stable knockdown of ADAM17 prevents ΔN-iRhom–dependent TNFR shedding.
Fig. S5. Localization patterns of truncated iRhoms and association with ADAM17.
Fig. S6. Increased ADAM17 activity associated with N-terminal truncated iRhom2.
Acknowledgments:
We are grateful for the technical assistance of E. Bäcker. We thank O. Ouerfelli (Sloan-Kettering Institute, New York, NY) and R. Waltermire (Bristol-Myers Squibb, New Brunswick, NJ) for the gifts of marimastat and DPC-333/BMS-561392, respectively.
Funding: This study was supported by the Alexander von Humboldt Foundation (SKA2010), the Medical Research Council (MR/L010402/1) and Cancer Research UK (C7570/A19107 to D.P.K.), the German Research Council (SFB974, LA2558/3-1, LA2558/5-1, TRR60, LA1419/5-1, and EV 206/1-1), the Forschungskommission of the Heinrich Heine University, the Canadian Institutes of Health Research Fellowship (201210MFE-289576-150035 to D.R.M.), and the NIH (GM64750 to C.P.B.). J.S. is supported by the German Research Council (SCHE907/3-1). G.P.N. is supported by grants from the NIH (U19 AI057229, U54CA149145, N01-HV-00242, 1U19AI100627, 5R01AI07372405, R01CA184968, 1 R33 CA183654, R33 CA183692, HHSF223201210194C, 41000411217, and 7500108142) and the U.S. Department of Defense (OC110674 and 11491122).
Footnotes
Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Blobel CP, ADAMs: Key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol 6, 32–43 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Moss ML, Jin S-L, Milla ME, Bickett DM, Burkhart W, Carter HL, Chen W-J,Clay WC, Didsbury JR, Hassler D, Hoffman CR, Kost TA, Lambert MH,Leesnitzer MA, McCauley P, McGeehan G, Mitchell J, Moyer M, Pahel G, Rocque W,Overton LK, Schoenen F, Seaton T, Su J-L, Warner J, Willard D, Becherer JD, Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-α. Nature 385, 733–736 (1997). [DOI] [PubMed] [Google Scholar]
- 3.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF,Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA,Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP, A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 385, 729–733 (1997). [DOI] [PubMed] [Google Scholar]
- 4.Horiuchi K, Kimura T, Miyamoto T, Takaishi H, Okada Y, Toyama Y, Blobel CP, Cutting edge: TNF-a-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J. Immunol 179, 2686–2689 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC,Russell WE, Castner BJ, Johnson RS, Fitzner JN, Boyce RW, Nelson N,Kozlosky CJ, Wolfson MF, Rauch CT, Cerretti DP, Paxton RJ, March CJ,Black RA, An essential role for ectodomain shedding in mammalian development. Science 282, 1281–1284 (1998). [DOI] [PubMed] [Google Scholar]
- 6.Reddy P, Slack JL, Davis R, Cerretti DP, Kozlosky CJ, Blanton RA, Shows D,Peschon JJ, Black RA, Functional analysis of the domain structure of tumor necrosis factor-α converting enzyme. J. Biol. Chem 275, 14608–14614 (2000). [DOI] [PubMed] [Google Scholar]
- 7.Pfeffer K, Matsuyama T, Kündig TM, Wakeham A, Kishihara K, Shahinian A,Wiegmann K, Ohashi PS, Krönke M, Mak TW, Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 73, 457–467 (1993). [DOI] [PubMed] [Google Scholar]
- 8.Rothe J, Lesslauer W, Lötscher H, Lang Y, Koebel P, Köntgen F, Althage A,Zinkernagel R, Steinmetz M, Bluethmann H, Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature 364, 798–802 (1993). [DOI] [PubMed] [Google Scholar]
- 9.Micheau O, Tschopp J, Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Maretzky T, McIlwain DR, Issuree PDA, Li X, Malapeira J, Amin S, Lang PA,Mak TW, Blobel CP, iRhom2 controls the substrate selectivity of stimulated ADAM17-dependent ectodomain shedding. Proc. Natl. Acad. Sci. U.S.A 110, 11433–11438 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adrain C, Zettl M, Christova Y, Taylor N, Freeman M, Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science 335, 225–228 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McIlwain DR, Lang PA, Maretzky T, Hamada K, Ohishi K, Maney SK, Berger T,Murthy A, Duncan G, Xu HC, Lang KS, Häussinger D, Wakeham A, Itie-Youten A,Khokha R, Ohashi PS, Blobel CP, Mak TW, iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science 335, 229–232 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siggs OM, Xiao N, Wang Y, Shi H, Tomisato W, Li X, Xia Y, Beutler B, iRhom2 is required for the secretion of mouse TNFα. Blood 119, 5769–5771 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Issuree PDA, Maretzky T, McIlwain DR, Monette S, Qing X, Lang PA,Swendeman SL, Park-Min K-H, Binder N, Kalliolias GD, Yarilina A, Horiuchi K,Ivashkiv LB, Mak TW, Salmon JE, Blobel CP, iRHOM2 is a critical pathogenic mediator of inflammatory arthritis. J. Clin. Invest 123, 928–932 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xue L, Maretzky T, Weskamp G, Monette S, Qing X, Issureea PDA, Crawford HC,McIlwain DR, Mak TW, Salmon JE, Blobel CP, iRhoms 1 and 2 are essential upstream regulators of ADAM17-dependent EGFR signaling. Proc. Natl. Acad. Sci. U.S.A 112, 6080–6085 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blaydon DC, Etheridge SL, Risk JM, Hennies H-C, Gay LJ, Carroll R, Plagnol V,McRonald FE, Stevens HP, Spurr NK, Bishop DT, Ellis A, Jankowski J, Field JK,Leigh IM, South AP, Kelsell DP, RHBDF2 mutations are associated with tylosis, a familial esophageal cancer syndrome. Am. J. Hum. Genet 90, 340–346 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saarinen S, Vahteristo P, Lehtonen R, Aittomäki K, Launonen V, Kiviluoto T,Aaltonen LA, Analysis of a Finnish family confirms RHBDF2 mutations as the underlying factor in tylosis with esophageal cancer. Fam. Cancer 11, 525–528 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Brooke MA, Etheridge SL, Kaplan N, Simpson C, O’Toole EA, Ishida-Yamamoto A,Marches O, Getsios S, Kelsell DP, iRHOM2-dependent regulation of ADAM17 in cutaneous disease and epidermal barrier function. Hum. Mol. Genet 23, 4064–4076 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Etheridge SL, Brooke MA, Kelsell DP, Blaydon DC, Rhomboid proteins: A role in keratinocyte proliferation and cancer. Cell Tissue Res. 351, 301–307 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Siggs OM, Grieve A, Xu H, Bambrough P, Christova Y, Freeman M, Genetic interaction implicates iRhom2 in the regulation of EGF receptor signalling in mice. Biol. Open 3, 1151–1157 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hosur V, Johnson KR, Burzenski LM, Stearns TM, Maser RS, Shultz LD, Rhbdf2 mutations increase its protein stability and drive EGFR hyperactivation through enhanced secretion of amphiregulin. Proc. Natl. Acad. Sci. U.S.A 111, E2200–E2209 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W,Grooten J, Fiers W, Vandenabeele P, Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med 187, 1477–1485 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsujimoto M, Yip YK, Vilcek J, Tumor necrosis factor: Specific binding and internalization in sensitive and resistant cells. Proc. Natl. Acad. Sci. U.S.A 82, 7626–7630 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vanlangenakker N, Bertrand MJM, Bogaert P, Vandenabeele P, Vanden Berghe T, TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis. 2, e230 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhattacharya D, Logue EC, Bakkour S, DeGregori J, Sha WC, Identification of gene function by cyclical packaging rescue of retroviral cDNA libraries. Proc. Natl. Acad. Sci. U.S.A 99, 8838–8843 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer J-L,Schröter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J, Inhibition of death receptor signals by cellular FLIP. Nature 388, 190–195 (1997). [DOI] [PubMed] [Google Scholar]
- 27.Liu B, Xu Y, Li WL, Zeng L, Proteomic analysis of differentially expressed skin proteins in iRhom2Uncv mice. BMB Rep. 48, 19–24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adrain C, Freeman M, New lives for old: Evolution of pseudoenzyme function illustrated by iRhoms. Nat. Rev. Mol. Cell Biol 13, 489–498 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Azijli K, Weyhenmeyer B, Peters GJ, de Jong S, Kruyt FAE, Non-canonical kinase signaling by the death ligand TRAIL in cancer cells: Discord in the death receptor family. Cell Death Differ. 20, 858–868 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kearney CJ, Cullen SP, Tynan GA, Henry CM, Clancy D, Lavelle EC,Martin SJ, Necroptosis suppresses inflammation via termination of TNF- or LPS-induced cytokine and chemokine production. Cell Death Differ 22, 1313–1327 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McIlwain DR, Berger T, Mak TW, Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol 5, a008656 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strelow A, Bernardo K, Adam-Klages S, Linke T, Sandhoff K, Krönke M, Adam D, Overexpression of acid ceramidase protects from tumor necrosis factor–induced cell death. J. Exp. Med 192, 601–612 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Legler DF, Micheau O, Doucey M-A, Tschopp J, Bron C, Recruitment of TNF receptor 1 to lipid rafts is essential for TNFα-mediated NF-κB activation. Immunity 18, 655–664 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Hartmann D,Fahrenholz F, Postina R, Matthews V, Kallen K-J, Rose-John S, Ludwig A, The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood 102, 1186–1195 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Christova Y, Adrain C, Bambrough P, Ibrahim A, Freeman M, Mammalian iRhoms have distinct physiological functions including an essential role in TACE regulation. EMBO Rep. 14, 884–890 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zettl M, Adrain C, Strisovsky K, Lastun V, Freeman M, Rhomboid family pseudo-proteases use the ER quality control machinery to regulate intercellular signaling. Cell 145, 79–91 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le Gall SM, Maretzky T, Issuree PDA, Niu X-D, Reiss K, Saftig P, Khokha R,Lundell D, Blobel CP, ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. J. Cell Sci 123, 3913–3922 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maretzky T, Evers A, Zhou W, Swendeman SL, Wong P-M, Rafii S, Reiss K,Blobel CP, Migration of growth factor-stimulated epithelial and endothelial cells depends on EGFR transactivation by ADAM17. Nat. Commun 2, 229 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nitoiu D, Etheridge SL, Kelsell DP, Insights into desmosome biology from inherited human skin disease and cardiocutaneous syndromes. Cell Commun. Adhes 21, 129–140 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Hirsch FR, Varella-Garcia M, Cappuzzo F, Predictive value of EGFR and HER2 overexpression in advanced non-small-cell lung cancer. Oncogene 28, S32–S37 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Mochizuki S, Okada Y, ADAMs in cancer cell proliferation and progression. Cancer Sci. 98, 621–628 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leilei Y, Bing L, Yang L, Shaoxia W, Yuan X, Dongping W, Huahu Y, Shichen S,Guangzhou Z, Ruiyun P, Lin Z, Wenlong L, iRhom2 mutation leads to aberrant hair follicle differentiation in mice. PLOS One 9, e115114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doedens JR, Black RA, Stimulation-induced down-regulation of tumor necrosis factor-α converting enzyme. J. Biol. Chem 275, 14598–14607 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Nakagawa T, Guichard A, Castro CP, Xiao Y, Rizen M, Zhang H-Z, Hu D, Bang A,Helms J, Bier E, Derynck R, Characterization of a human rhomboid homolog, p100hRho/RHBDF1, which interacts with TGF-α family ligands. Dev. Dyn 233, 1315–1331 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Zou H, Thomas SM, Yan Z-W, Grandis JR, Vogt A, Li L-Y, Human rhomboid family-1 gene RHBDF1 participates in GPCR-mediated transactivation of EGFR growth signals in head and neck squamous cancer cells. FASEB J. 23, 425–432 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yan Z, Zou H, Tian F, Grandis JR, Mixson AJ, Lu PY, Li L-Y, Human rhomboid family-1 gene silencing causes apoptosis or autophagy to epithelial cancer cells and inhibits xenograft tumor growth. Mol. Cancer Ther 7, 1355–1364 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maretzky T, Zhou W, Huang X-Y, Blobel CP, A transforming Src mutant increases the bioavailability of EGFR ligands via stimulation of the cell-surface metalloproteinase ADAM17. Oncogene 30, 611–618 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grusdat M, McIlwain DR, Xu HC, Pozdeev VI, Knievel J, Crome SQ,Robert-Tissot C, Dress RJ, Pandyra AA, Speiser DE, Lang E, Maney SK,Elford AR, Hamilton SR, Scheu S, Pfeffer K, Bode J, Mittrücker H-W, Lohoff M,Huber M, Häussinger D, Ohashi PS, Mak TW, Lang KS, Lang PA, IRF4 and BATF are critical for CD8+ T-cell function following infection with LCMV. Cell Death Differ. 21, 1050–1060 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
www.sciencesignaling.org/cgi/content/full/8/401/ra109/DC1
Fig. S1. Cloning of CPR screen–identified iRhom versions.
Fig. S2. TNFR1 and TNFR2 abundance in iRhom-expressing cells.
Fig. S3. ADAM17 mediates cleavage of TNFRs.
Fig. S4. Stable knockdown of ADAM17 prevents ΔN-iRhom–dependent TNFR shedding.
Fig. S5. Localization patterns of truncated iRhoms and association with ADAM17.
Fig. S6. Increased ADAM17 activity associated with N-terminal truncated iRhom2.