

Abstract
Background. Given the apparent high mortality associated with the novel swine-origin influenza A/H1N1 virus (S-OIV) in Mexico, we aimed to study the cytokine profiles induced by S-OIV and the effect of immunomodulators.
Methods. We assayed cytokines and their messenger RNA (mRNA) levels in culture supernatants of human macrophages infected with H5N1, S-OIV California/04/2009 (S-OIV-CA), S-OIV Hong Kong/415742 (S-OIV-HK), or seasonal H1N1 with or without celecoxib and mesalazine.
Results. Among the 12 cytokines showing detectable levels, levels of 8 proinflammatory cytokines (interleukin [IL] 2R, IL-6, interferon [IFN] α, macrophage inflammatory protein [MIP] α, MIP-1b, IFN-induced protein 10, regulated on activation, normal T cell expressed and secreted [RANTES], and monocyte chemotactic protein [MCP] 1) were higher in cells infected by H5N1 but similar among cells infected with H1N1, S-OIV-CA, or SOIV- HK. The levels of the other 4 cytokines were similar for H5N1, H1N1, S-OIV-CA and S-OIV-HK. Among the 8 cytokines induced by H5N1, 6 were suppressed by celecoxib and mesalazine. The mRNA levels of tumor necrosis factor a, IFN-γ, IL-6, and MCP-1 induced by H5N1 were higher than the levels of other cytokines at 12 and/or 24 h.
Conclusions. No major cytokine storm, as seen in H5N1 infection, is associated with S-OIV infection of cell lines. The mainstay of treatment for uncomplicated S-OIV infections should be antiviral agents without immunomodulators. For individual S-OIV-infected patients with severe primary viral pneumonia, severe sepsis, and multiorgan failure, immunomodulators may be considered as an adjunctive therapy in clinical trials.
While everybody has been worrying about when the influenza A/H5N1 virus (H5N1) will adapt well enough in human to cause a pandemic, the emergence of a novel swine-origin influenza A/H1N1 virus (S-OIV) has shocked the world [1–3]. On 11 June 2009, the WorldHealth Organization raised the pandemic alert level from phase 5 to phase 6. As of 3 July, there were 89,921 laboratory-confirmed cases of S-OIV infections and 382 confirmed deaths globally. The S-OIV genome is made up of a unique combination of gene segments that had not been observed in previously known influenza A viruses. Although the sequences of all the 8 gene segments are most closely related to those of swine isolates in the last 10–20 years, some are also homologous to those of previously found human or avian isolates, but with lower similarities. The first report on the clinical features of 642 confirmed cases of S-OIV infections in the United States showed that most patients presented with upper respiratory and systemic symptoms similar to those of seasonal influenza [1]. Most of the cases were self-limited, with 9% of patients hospitalized and only 2 deaths [1]. On the other hand, more recent reports on the epidemic in the Mexican population described apparently higher number of deaths, espe cially in persons !60 years old, although the total number of cases of S-OIV infection, the denominator for calculating the mortality rate, was not certain [4, 5].
Influenza viruses associated with seasonal influenza cause tissue damage mainly through virus-induced cell death. On the other hand, H5N1 infections in humans were associated with a massive cytokine storm, which had resulted in a high number of deaths [6–8]. Recently, we discovered that treatment with the triple combination of zanamivir, celecoxib, and mesalazine significantly decreased the mortality in mice infected by H5N1 when compared with zanamivir alone [9]. Moreover, the triple combination of zanamivir, celecoxib, and mesalazine was also associated with marked reduction in inflammatory markers [9]. Therefore, it is of both biological interest and practical importance to understand the cytokine profiles induced by S-OIV versus H5N1 and seasonal influenza virus, because this knowledge will aid in the design of treatment strategies for future cases of S-OIV infections. In this study, we compared the cytokine profiles of macrophages infected by a highly pathogenic strain of H5N1, 2 strains of S-OIV, or a strain of influenza A/ H1N1 virus (H1N1) and also evaluated the paracrine effects on A549 cells of macrophages infected by the different viruses. The implications of our results are discussed.
Methods
Viruses. The 4 viruses used in this study were A/Vietnam/ HN3028, S-OIV California/04/2009 (S-OIV-CA), S-OIV Hong Kong/415742 (S-OIV-HK) (S-OIV isolated from the first case of S-OIV infection in Hong Kong Special Administrative Region) [10], and A/Hong Kong/403946. Each virus was inoculated to Madin-Darby canine kidney (MDCK) cell monolayer in 1 mL of minimal essential medium (Invitrogen) containing tosylsulfonyl phenylalanyl chloromethyl ketone-treated trypsin (2 µg/mL) (Sigma), penicillin (0.6 µg/mL), and streptomycin (60 µg/mL) and incubated at 35°C for 1 h. Viruses were harvested when ∼75% of the cell monolayer manifested cytopathic effect. The median tissue culture infective dose (TCID50) was determined by using MDCK cells according to the Reed- Muench method [11]. All experimental protocols followed the standard operating procedures of the approved biosafety level 3 facilities.
Harvest of peripheral blood mononuclear cells and macrophage differentiation. Unpooled buffy coats of 3 healthy blood donors were obtained from the Hong Kong Red Cross Blood Transfusion Service, Hong Kong Special Administrative Region. Peripheral blood mononuclear cells (PBMCs) were separated from the buffy coats by centrifugation on a Ficoll-Paque density gradient (GE Healthcare) and purified by adherence [12]. The PBMCs were seeded onto 24-well tissue culture plates, at 1×107 cells per well, in macrophage serum-free medium (Invitrogen), penicillin (0.6 µg/mL), and streptomycin (60 µg/ mL) and incubated at 37°C and 5% carbon dioxide for 14 days for differentiation into macrophages.
Influenza virus infection of macrophages. One hour before virus infection, celecoxib (400 µg/mL), mesalazine (200 µg/ mL), celecoxib (400 µg/mL) plus mesalazine (200 µg/mL), or macrophage serum-free medium was added to the wells with differentiated macrophages. Each group of drug-treated differentiated macrophages was further divided into 5 subgroups and then infected with H5N1, S-OIV-CA, S-OIV-HK, or H1N1 (multiplicity of infection [MOI], 2) or mock-infected. After virus adsorption for 30 min, the virus inocula were removed, the macrophages were washed, and warm macrophage serumfree medium was added. Culture supernatants were collected at 12 and 24 h for cytokine assays, and macrophages were harvested for RNA extraction. All experiments were performed in triplicate.
Interaction between macrophages and epithelial cells. A549 (lung epithelial adenocarcinoma) cells were seeded onto 24- well tissue culture plates, at 7×104 cells per well, in minimal essential medium (Invitrogen) supplemented with 10% fetal calf serum and incubated at 37°C and 5% carbon dioxide for 24 h before the experiment. Drug treatment and virus infection of differentiated macrophages were performed as described above. At 12 h, culture supernatant from each well was collected, filtered with a 100-kDa-pore filter (Millipore), and added to the uninfected fresh A549 cells. After further incubation for 12 and 24 h, culture supernatants were collected for cytokine assays. All experiments were performed in triplicate.
Cytokine assays. Culture supernatants of macrophages and A549 cells, collected at 12 and 24 h, were assayed for 25 cytokines by using the Human Cytokine Twenty-Five-Plex Panel according to the manufacturer's instructions (Invitrogen). The assay plate was analyzed with the Bio-Plex Luminex 200 XYP instrument (Bio-Rad Laboratories). Cytokine concentrations were calculated using Bio-Plex Manager software (version 5.0; Bio-Rad Laboratories).
RNA extraction. Total RNA was extracted from the macrophages using the RNeasy Mini Spin Column (Qiagen). The RNA was eluted in 50 µL of RNase-free water and was used as the template for real-time reverse transcription-polymerase chain reaction (RT-PCR).
Real-time RT-PCR. Reverse transcription was performed using the SuperScript III kit (Invitrogen). Real-time RT-PCR assays for tumor necrosis factor (TNF)-α, interferon (IFN)-γ, interleukin (IL)-6, IL-8, monocyte chemotactic protein (MCP)-1, and cyclooxygenase (COX)-2 were performed as described elsewhere, with the primers listed in Table 1 and with glyceraldehyde 3-phosphate dehydrogenase for normalization [10, 13]. Complementary DNA (cDNA) was amplified in a LightCycler 2.0 instrument (Roche) with 20-µL reaction mixtures containing FastStart DNA Master SYBR Green I Mix reagent kit (Roche), 2 µL of cDNA, 2- or 4-mmol/L magnesium chloride, and 0.5 mmol/L primers at 95°C for 10 min, followed by 50 cycles of denaturation, annealing, and extension (Table 1).
Table 1.
Primers and Conditions for Real-Time Reverse Transcription-Polymerase Chain Reaction (PCR)

Results
Virus titers. The titers of viruses released from MDCK cells infected with H5N1, S-OIV-CA, S-OIV-HK, or H1N1 infected cells were measured when ∼75% of the cell monolayer manifested cytopathic effects. The titers of S-OIV-CA, S-OIV-HK, and H1N1 were 108 TCID50/mL, and that of H5N1 was 107 TCID50/mL.
Cytokine levels in macrophages infected with H5N1, S-OIVCA, S-OIV-HK, or H1N1 and lung epithelial cells inoculated with supernatants of virus-infected macrophages. Among the 25 cytokines assayed using the Human Cytokine Twenty- Five-Plex Panel, 12 showed detectable levels at 12 and/or 24 h in macrophages and/or A549 cells (Figures 1 and 2). Among these 12 cytokines, the levels of 8 (IL-2 receptor [IL-2R], IL- 6, IFN-α, macrophage inflammatory protein [MIP]-1a, MIP- 1β, IFN-γ-induced protein 10, regulated on activation, normal T cell expressed and secreted [RANTES], and MCP-1) were markedly higher for H5N1 than for H1N1, S-OIV-CA, or SOIV- HK (Figures 1A-1H and 2A-2F). The levels of each of these 8 cytokines were similar among cells infected with H1N1, S-OIV-CA, or S-OIV-HK. For the other 4 cytokines (IL-1 receptor antagonist, IL-12p40/p70, IL-8, and monokine induced by IFN-γ ), their levels were similar among cells infected with H5N1, H1N1, S-OIV-CA, or S-OIV-HK (Figures 1I-1L and 2G-2J). Six of the 8 cytokines induced by H5N1 (IL-6, IFNa, MIP-1b, IFN-γ-induced protein 10, RANTES, and MCP- 1) were suppressed by celecoxib (400 µg/mL) plus mesalazine (200 µg/mL).
Messenger RNA levels in macrophages infected with H5N1, S-OIV-CA, S-OIV-HK, or H1N1. Among the 6 cytokines assayed, the messenger RNA (mRNA) levels of 3 (TNF-α, IFN-γ and IL-6) were markedly higher for H5N1 than for H1N1, S-OIV-CA, or S-OIV-HK at 12 h, and the levels of 4 (TNF-α, IFN-γ, IL-6, and MCP-1) were markedly higher for H5N1 at 24 h (Figure 3). The mRNA levels of all 6 cytokines induced by S-OIV-CA were higher than those induced by S-OIV-HK at 24 h (Figure 3).
Figure 3.

Induction of cytokine genes expression in macrophages infected with H1N1, swine-origin influenza A/H1N1 virus California/04/2009 (SOIV- CA), S-OIV Hong Kong/415742 (S-OIV-HK), or H5N1 (multiplicity of infection, 2). Expression of tumor necrosis factor-a (A), interferon-γ (B), interleukin (IL)-6 (C), IL-8 (D), monocyte chemotactic protein 1 (E), and cyclooxygenase 2 (F) were assayed by real-time reverse-transcription polymerase chain reaction. Data shown are n-fold changes of gene expression relative to mock-infected macrophages, after normalization to glyceraldehyde 3- phosphate dehydrogenase in each sample. Means of triplicate assays are shown.
Discussion
We documented that no major cytokine storm, as in H5N1 infection, is associated with S-OIV infection. Since its first report in 1998, H5N1 has been associated with very high case fatality rates of 160% [6, 8]; the cytokine storm produced by the NS gene segment of H5N1 leads to severe systemic inflammatory response and multiorgan failure [7]. Although the data available so far suggested that the present S-OIV is not associated with particular high mortality rates, the rate associated with the ancestor of S-OIV, the classic swine influenza A (H1N1) virus, was higher than that associated with seasonal influenza [14]. In a review of 50 cases of apparent zoonotic swine influenza virus infection, a case fatality rate of 14% was observed [14]. Because no animal model was available for SOIV infection during the time of our study, we used human macrophages harvested from healthy blood donors to determine whether S-OIV infection is associated with more marked cytokine induction than seasonal influenza, although not as severe as in H5N1 infection.
With this model, both mRNA and cytokine assays showed that H5N1 infection was associated with major cytokine storms at 12 and 24 h in macrophages and lung epithelial cells. Among the 5 cytokines for which both mRNA and cytokine levels were assayed, only 3 (IL-6, IL-8, and MCP-1) had detectable levels with the Luminex assay, all with good correlation between the 2 assays. For IL-6 and MCP-1, levels were highest in H5N1- infected macrophages (Figures 1B, 1H, 3C, and 3E), but theIL-8 level was highest in S-OIV-CA-infected macrophages (Figures 1K and 3D), which may imply strong neutrophil recruitment in S-OIV-CA infection. Among the 12 detectable cytokines, all 8 markedly induced by H5N1 are proinflammatory cytokines, whereas 1 of the other 4 that showed similar levels among the 4 viruses, IL-1 receptor antagonist, is a major antiinflammatory cytokine. On the other hand, neither S-OIV strain was associated with such a cytokine storm, with overall mRNA and cytokine levels similar to those seen in seasonal H1N1 (Figures 1–3). This finding is in line with the apparent relatively low mortality observed currently in most countries outside Mexico in the present influenza pandemic.
Interestingly, a study using a recently developed animal model, showed significant induction of IFN-γ, IL-4, IL-5, and IL-10 in the lungs of mice infected with S-OIV-CA compared with animals infected with seasonal H1N1 strain KUTK-4 [15]. In addition, markedly higher levels of MCP-1, MIP-1a, IL-6, and IL-18 were observed in the lungs of 2 of 3 macaques infected with S-OIV-CA [15]. The apparent difference between these in vitro data and ours could reflect different viral loads used in the 2 studies. We used an MOI of 2, but high doses of viruses were used in the animal studies. It is possible that high cytokine levels, reactive hemophagocytosis, and multiorgan failure could be present in patients with severe S-OIV infections with high viral loads. Additional experiments using higher MOIs would be necessary to determine whether higher cytokine responses could be associated with higher viral load in the present cell line model. Additional experiments using PBMCs from more healthy blood donors will also reveal whether there is much interindividual variability in immune and cytokine responses of human macrophages in response to influenza virus infections.
Our results imply that the mainstay of treatment for uncomplicated S-OIV infections should be antiviral agents instead of antiviral plus immunomodulators. Recently, we documented that zanamivir plus celecoxib (COX-2 inhibitor) and mesalazine (inhibitor of inflammation widely used for treatment of inflammatory bowel disease) significantly decreased the mortality associated with H5N1 infection, compared with zanamivir alone in a mouse model [9]. This was due to a modulation of the cytokine storm associated with H5N1 infection, indicated by a suppression of the inflammatory markers [9]. This finding is in line with those in our study, which showed that celecoxib plus mesalazine suppressed 6 of the 8 proinflammatory cytokines markedly induced by H5N1 (Figures 1 and 2). Others have also shown that selective COX-2 inhibitors suppressed the hyperinduction of cytokines in the proinflammatory cascade in H5N1-infected macrophages [16]. Because our study showed that S-OIV was not associated with a major cytokine storm and S-OIV infections were not clinically associated with major mortality, most patients should be treated with oseltamivir or zanamivir. However, for individual patients with S-OIV infection who have severe primary viral pneumonia with severe sepsis and multiorgan failure, immunomodulators may be considered as an adjunctive therapy in clinical trials. Because we previously showed that the addition of immunomodulators 48 h after H5N1 challenge were very effective in decreasing both the mortality rate and cytokine storm in a mouse model [9], such adjunctive treatment may theoretically be useful even after the start of a severe systemic inflammatory response.
There may have been attenuation of S-OIV during the early phase of the pandemic when the virus was adapting to the host. S-OIV-CA was isolated from a patient in California on 1 April 2009, whereas S-OIV-HK was isolated from a patient in Hong Kong on 30 April [10]. For the 6 cytokines with mRNA expression levels assayed, the levels of all 6 were higher for SOIV- CA-infected macrophages than for S-OIV-HK infected macrophages (Figure 3). Although this phenomenon was shown by the cytokine assay only for the IL-8 level, this may be because relatively small differences can be picked up only by the more sensitive RT-PCR assay. This explanation is in line with the fact that TNF-α and IFN-γ expressions were detected only by RT-PCR and not by the cytokine assays for all 4 viruses. We speculate that this attenuation of the virus may explain the relatively higher number of deaths in the Mexican population during the early phase of the pandemic. Sequencing the S-OIVHK genome and comparative genomic analysis would reveal whether the virus was rapidly evolving in April 2009. Close monitoring of S-OIV strains as the pandemic evolves is important. It has been observed that the 1918 Spanish flu pandemic was also mild initially, but the mortality was markedly increased in the second wave during the influenza season [17]. As a temporal series of the virus is not available, we do not know if this was due to an increase in virulence of the virus. However, the reverse genetically reconstructed 1918 H1N1 virus was shown to induce lethal pneumonia with proinflammatory damage and cytokine storm similar to that of the H5N1 virus in mouse, ferrets and monkey models [18, 19]. The present S-OIV was a result of multiple reassortment. Therefore, it would not be surprising if there is further reassortment and/or mutation, giving rise to more virulent phenotypes as the pandemic evolves in the coming months.
One major concern at the moment is the acquisition of antiviral resistance, and in recent weeks oseltamivir resistance has emerged in S-OIV [20]. This is similar to the situation in the 2008–2009 influenza season, when most H1N1 viruses that circulated were resistant to oseltamivir though still susceptible to zanamivir [21, 22]. Another scenario that would be at least as disastrous would be acquisition of virulence genes, such as NS or PB1-F2, from viruses such as H5N1, giving rise to a highly infectious and fatal virus. Therefore, as the pandemic evolves, strains of S-OIV should be monitored not only for antiviral resistance but also for their virulence potential, using the present cell line model as an alternative to the less available animal models [23, 24]. Because ferret and mice models of SOIV infection showed marked viral replication in lungs and seasonal influenza replicated mainly in the nasopharynx, there is reason to believe that more cytokine induction may occur in S-OIV, because the lung is a large organ with many reticuloendothelial cells. Because mRNA levels can be detected more sensitively than the corresponding cytokine levels, detection of mRNA levels with RT-PCR would be more sensitive for monitoring the evolving the virulence potential of the virus. Monitoring its genome sequence in the coming second wave in winter is also important to determine whether there is continued attenuation or increased virulence in correlation with the clinical scenario.
Figure 1.
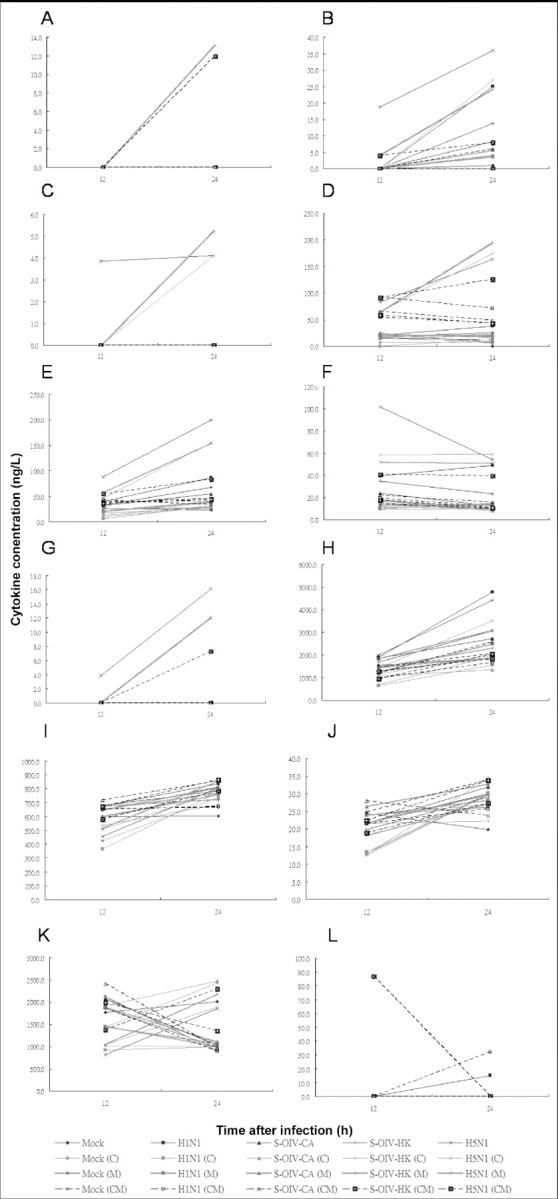
Induction of cytokines in mock-infected macrophages and macrophages infected with H1N1, swine-origin influenza A/H1N1 virus California/ 04/2009 (S-OIV-CA), S-OIV Hong Kong/415742 (S-OIV-HK), or H5N1 (multiplicity of infection, 2). Levels of interleukin (IL)-2R (A), IL-6 (B), interferon (IFN)-a (C), macrophage inflammatory protein (MIP)-1a (D), MIP-1b (E), IFN-γ-induced protein 10 (F), regulated on activation, normal T cell expressed and secreted (RANTES), (G), monocyte chemotactic protein 1 (H), IL-1 receptor antagonist (I), IL-12p40/p70 (J), IL-8 (K), and monokine induced by IFN-γ (L) were assayed by Luminex assay. Means of triplicate assays are shown. C, celecoxib; CM, celecoxib plus mesalazine; M, mesalazine.
Figure 2.
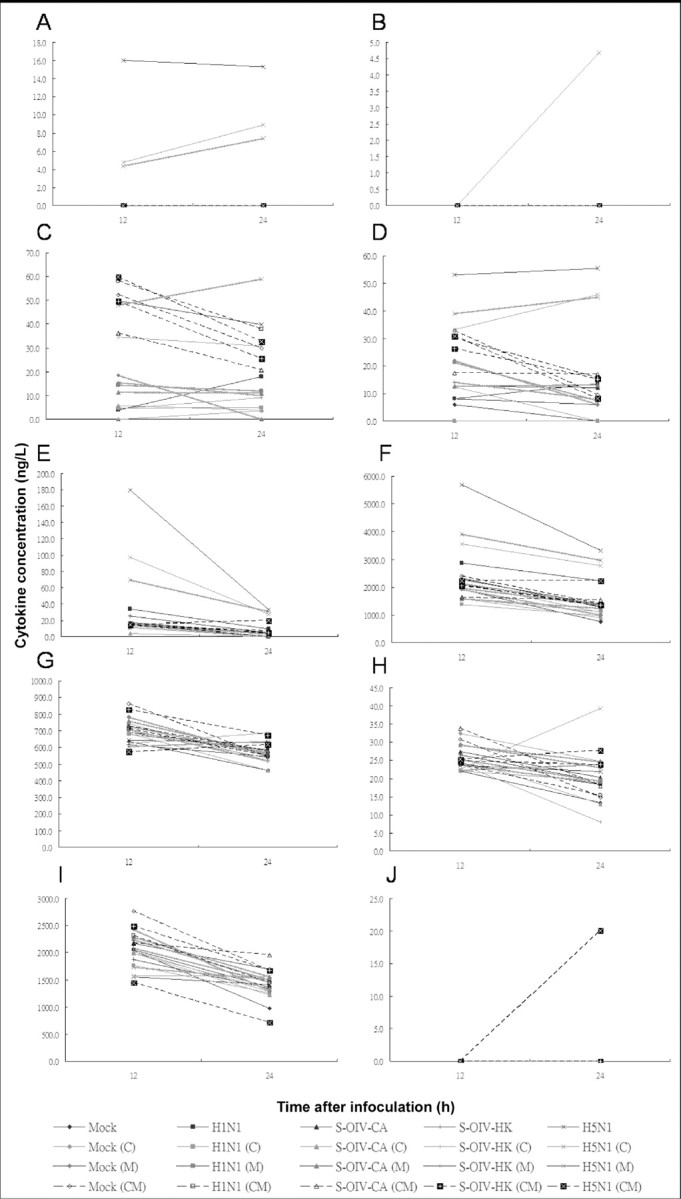
Induction of cytokines in lung epithelial cells by soluble mediators secreted by mock-infected macrophages and macrophages infected with H1N1, swine-origin influenza A/H1N1 virus California/04/2009 (S-OIV-CA), S-OIV Hong Kong/415742 (S-OIV-HK), or H5N1 (multiplicity of infection, 2). Expression of interleukin (IL)-6 (A), interferon (IFN)-α (B), macrophage inflammatory protein (MIP)-1α (C), MIP-1b (D), IFN-γ-induced protein 10 (E), monocyte chemotactic protein 1 (F), IL-1 receptor antagonist (G), IL-12p40/p70 (H), IL-8 (I), and monokine induced by IFN-γ (J) were assayed by Luminex assay. Levels of IL-2R and regulated on activation, normal T cell expressed and secreted (RANTES) were not detectable and are not shown. Means of triplicate assays are shown. C, celecoxib; CM, celecoxib plus mesalazine; M, mesalazine.
Acknowledgments
We are grateful to the Genome Research Centre, University of Hong Kong, for the Bio-Plex Luminex 100 XYP platform.
Footnotes
Potential conflicts of interest: none reported.
Financial support: Research Fund for the Control of Infectious Diseases, Health, Welfare and Food Bureau, Hong Kong Special Administrative Region; Research Grants Council; University Development Fund and Outstanding Young Researcher Award, University of Hong Kong. This study was partially supported by the Providence Foundation Limited in memory of the late Dr Lui Hac Minh, an HKV award for CAE membership, Dr Hector T. G. Ma, and the Food and Health Bureau of the Hong Kong SAR Government.
References
- 1.Dawood FS, Jain S, Finelli L, et al. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. Novel Swine-Origin Influenza A (H1N1) Virus Investigation Team. N Engl J Med. 2009;360:2605–2615. doi: 10.1056/NEJMoa0903810. [DOI] [PubMed] [Google Scholar]
- 2.Fraser C, Donnelly CA, Cauchemez S, et al. Pandemic potential of a strain of influenza A (H1N1): early findings. Science. 2009;324:1557–1561. doi: 10.1126/science.1176062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garten RJ, Davis CT, Russell CA, et al. Antigenic and genetic characteristics of swine-origin 2009 A (H1N1) influenza viruses circulating in humans. Science. 2009;325:197–201. doi: 10.1126/science.1176225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chowell G, Bertozzi SM, Colchero MA, et al. Severe respiratory disease concurrent with the circulation of H1N1 influenza. N Engl J Med. 2009;361:674–679. doi: 10.1056/NEJMoa0904023. [DOI] [PubMed] [Google Scholar]
- 5.Perez-Padilla R, de la Rosa-Zamboni D, Ponce de Leon S, et al. Pneumonia and respiratory failure from swine-origin influenza A (H1N1) in Mexico. INER Working Group on Influenza. N Engl J Med. 2009;361:680–689. doi: 10.1056/NEJMoa0904252. [DOI] [PubMed] [Google Scholar]
- 6.Yuen KY, Chan PK, Peiris M, et al. Clinical features and rapid viral diagnosis of human disease associated with avian influenza A H5N1 virus. Lancet. 1998;351:467–471. doi: 10.1016/s0140-6736(98)01182-9. [DOI] [PubMed] [Google Scholar]
- 7.Cheung CY, Poon LL, Lau AS, et al. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet. 2002;360:1831–1837. doi: 10.1016/s0140-6736(02)11772-7. [DOI] [PubMed] [Google Scholar]
- 8.Beigel JH, Farrar J, Han AM, et al. Avian influenza A (H5N1) infection in humans. N Engl J Med. 2005;353:1374–1385. doi: 10.1056/NEJMra052211. [DOI] [PubMed] [Google Scholar]
- 9.Zheng BJ, Chan KW, Lin YP, et al. Delayed antiviral plus immunomodulator treatment still reduces mortality in mice infected by high inoculum of influenza A/H5N1 virus. Proc Natl Acad Sci USA. 2008;105:8091–8096. doi: 10.1073/pnas.0711942105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lau SK, Chan KH, Yip CC, et al. Confirmation of the first Hong Kong case of human infection by novel swine-origin influenza A (H1N1) virus using ultra-rapid & real-time RT-PCR. J Clin Microbiol. 2009;47:2344–2346. doi: 10.1128/JCM.00924-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reed LJ, Muench H. A simple method of estimating fifty per cent endpoints. Am J Hyg. 1938;7:493–497. [Google Scholar]
- 12.Montaner LJ, Collin M, Herbein G. Human monocytes: isolation cultivation and its applications. In: Herzenberg LA, editor. Weir's handbook of experimental immunology. 5th ed. Vol IV. Cambridge, MA: Blackwell Science; 1996. pp. 1–11. [Google Scholar]
- 13.Woo PC, Lau SK, Chu CM, et al. Characterization and complete genome sequence of a novel coronavirus coronavirus HKU1 from patients with pneumonia. J Virol. 2005;79:884–895. doi: 10.1128/JVI.79.2.884-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Myers KP, Olsen CW, Gray GC. Cases of swine influenza in humans: a review of the literature. Clin Infect Dis. 2007;44:1084–1088. doi: 10.1086/512813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Itoh Y, Shinya K, Kiso M, et al. In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature. 2009;460:1021–1025. doi: 10.1038/nature08260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee SM, Cheung CY, Nicholls JM, et al. Hyperinduction of cyclooxygenase-2-mediated proinflammatory cascade: a mechanism for the pathogenesis of avian influenza H5N1 infection. J Infect Dis. 2008;198:525–535. doi: 10.1086/590499. [DOI] [PubMed] [Google Scholar]
- 17.Taubenberger JK, Morens DM. 1918 Influenza: the mother of all pandemics. Emerg Infect Dis. 2006;12:15–22. doi: 10.3201/eid1201.050979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kobasa D, Jones SM, Shinya K, et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature. 2007;445:319–323. doi: 10.1038/nature05495. [DOI] [PubMed] [Google Scholar]
- 19.Tumpey TM, Basler CF, Aguilar PV, et al. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science. 2005;310:77–80. doi: 10.1126/science.1119392. [DOI] [PubMed] [Google Scholar]
- 20.Chen H, Cheung CL, Tai H, et al. Oseltamivir resistant influenza A pandemic (H1N1) 2009, Hong Kong, China. Emerg Infect Dis. 2009;15:1970–1972. doi: 10.3201/eid1512.091057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Centers for Disease Control and Prevention. FluView: a weekly influenza surveillance report prepared by the Influenza Division. Available at http://www.cdc.gov/flu/weekly/
- 22.World Health Organization. 2009. Influenza A(H1N1) virus resistance to oseltamivir-2008/2009 influenza season, northern hemisphere. 18 March 2009. Available at http://www.who.int/csr/disease/influenza/H1N1webupdate20090318%20ed_ns.pdf. [Google Scholar]
- 23.Munster VJ, de Wit E, van den Brand JM, et al. Pathogenesis and transmission of swine-origin 2009 A (H1N1) influenza virus in ferrets. Science. 2009;325:481–483. doi: 10.1126/science.1177127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maines TR, Jayaraman A, Belser JA, et al. Transmission and pathogenesis of swine-origin 2009 A (H1N1) influenza viruses in ferrets and mice. Science. 2009;325:484–487. doi: 10.1126/science.1177238. [DOI] [PMC free article] [PubMed] [Google Scholar]