

Abstract
In OA chondrocytes, there is diminished mitochondrial production of ATP and diminished extracellular adenosine resulting in diminished adenosine A2A receptor (A2AR) stimulation and altered chondrocyte homeostasis which contributes to the pathogenesis of OA. We tested the hypothesis that A2AR stimulation maintains or enhances mitochondrial function in chondrocytes. The effect of A2AR signaling on mitochondrial health and function was determined in primary murine chondrocytes, a human chondrocytic cell line (T/C-28a2), primary human chondrocytes, and a murine model of OA by transmission electron microscopy analysis, mitochondrial stress testing, confocal live imaging for mitochondrial inner membrane polarity, and immunohistochemistry. In primary murine chondrocytes from A2AR−/− null mice, which develop spontaneous OA by 16 weeks, there is mitochondrial swelling, dysfunction, and reduced mitochondrial content with increased reactive oxygen species (ROS) burden and diminished mitophagy, as compared to chondrocytes from WT animals. IL-1-stimulated T/C-28a2 cells treated with an A2AR agonist had reduced ROS burden with increased mitochondrial dynamic stability and function, findings which were recapitulated in primary human chondrocytes. In an obesity-induced OA mouse model, there was a marked increase in mitochondrial oxidized material which was markedly improved after intraarticular injections of liposomal A2AR agonist. These results are consistent with the hypothesis that A2AR ligation is mitoprotective in OA.
Keywords: adenosine receptor, ATP, chondrocytes, mitochondria, osteoarthritis
1 ∣. INTRODUCTION
With aging many people suffer chronic, sterile, low-grade inflammation, termed inflammaging, which is thought to contribute to the pathogenesis of age-related diseases. Although reaction to misfolded and damaged proteins from damaged cells contributes to the low-grade inflammatory response at the cellular level other cellular and molecular derangements have been described that contribute to inflammaging including diminished autophagy and mitophagy, accumulation of oxidized mitochondrial proteins with mitochondrial dysfunction, dysregulation of protein expression due to epigenetic changes in the aging cell and cellular senescence.1-3 Cellular responses to endogenous and exogenous stress, such as reactive oxygen species (ROS) and overnutrition, result in a compensatory activation of the cellular quality control that requires coordination among the nucleus, cytoplasm, endoplasmic reticulum, and mitochondria.1,4,5 Inflammaging and mitochondrial dysfunction have been studied in several animal models to better understand pathogenesis of degenerative diseases and to find targets for antiaging therapies.6,7 Inflammaging and mitochondrial dysfunction have also been implicated in several musculoskeletal degenerative diseases such as osteoarthritis, osteopenia, and poor bone healing.8-10
Osteoarthritis (OA) etiology is multi-factorial and includes aging, obesity, trauma to the joint, and in more rare cases, heredity, significantly, that of certain mitochondrial haplogroups.11 One mechanism by which chondrocytes maintain cartilage and cellular homeostasis is through release of ATP which is converted extracellularly to adenosine; adenosine then binds to its A2A receptor (A2AR) to regulate chondrocyte function.12 A2AR null mice develop spontaneous OA by 16 weeks.12 With aging or in the setting of OA, intracellular ATP levels drop in chondrocytes13 leading to a reduction in ATP release and extracellular adenosine resulting in diminished endogenous A2AR stimulation and dysregulation of chondrocyte function.12 Application of liposomal preparations of either adenosine or the selective A2AR agonist CGS21680, abrogates development of OA in models of post-traumatic OA (PTOA) in rats12 and obesity-induced OA in mice.14,15
Oxidant-mediated injury of mitochondria with diminished mitochondrial repair and replacement likely play a role in mitochondrial dysfunction in aging chondrocytes.13 Indeed, over a dozen mitochondrial enzymes are capable of ROS production and subsequently capable of signaling and inducing pro-inflammatory responses on their own, which include the activation of transcription factors such as hypoxia-inducible factor-1 (HIF-1), metabolic adaptation, differentiation, proliferation, and cell death.16 Furthermore, there are reduced levels of oxidative phosphorylation (OXPHOS) complexes in OA chondrocytes, which might lower the coupling capacity in these cells, thus, reducing ATP production17 and adenosine availability for endogenous A2AR ligation. We hypothesized the lack of A2AR ligation and resulting inflammation derives from the cell’s inability to tolerate ROS burden and produce sufficient ATP via mitochondrial respiration to be exported onto the extracellular matrix (ECM) and converted into adenosine for endogenous A2AR ligation.
We here report that A2AR stimulation appears to diminish inflammaging, the pathologic process strongly associated with aging and inflammation, which leads to cellular dysfunction in many organs and tissues. At the center of this process, there appears to be both a reduction in mitochondrial function, increased mitochondrial ROS burden and diminished ATP content and release into the extracellular space—all which appear to be mitigated in turn by ligation of the A2AR. A2AR null (A2AR−/−) mice have increased mitochondrial dysfunction, which is responsible for increased ROS burden and reduced ATP production via OXPHOS. Our model of spontaneous OA shows mitochondrial changes in receptor null chondrocytes, including loss of mitochondrial function, depolarization, mitochondrial swelling, fragmentation, and ultimately, mitophagy. Liposomal adenosine or CGS21680 reduce ROS burden in an obesity-induced OA murine model in vivo.14,15 Similar findings were also made in the human chondrocyte cell line (T/C28a-2) and in primary human chondrocytes, where we further studied the bioenergetic effects of A2AR ligation. A2AR ligation improved several traditional markers of inflammaging, including ROS burden and mitochondrial swelling in IL-1β-stimulated T/C28a-2 cells. A2AR ligation of IL-1β-stimulated fully differentiated primary human chondrocytes expanded mitochondrial networks indicating translational value in humans. These effects on mitochondria correlated with increased ATP release and adenosine availability for further A2AR ligation in primary human chondrocytes. To the best of our knowledge, these results are the first to suggest that A2AR ligation can diminish inflammaging in chondrocytes by altering mitochondrial dynamics and function, likely through the previously described capacity of A2AR ligation to increase cAMP/protein kinase A (PKA) signaling18 and inhibit NF-κB nuclear translocation.19
2 ∣. METHODS
2.1 ∣. Materials, reagents, and software used in this study
Antibodies: Rb anti-Adenosine A2 Receptor (Abcam Cat# ab3461, RRID:AB_303823), Ms anti-ATP5A (Abcam Cat# ab14748, RRID:AB_301447), Go anti-8 hydroxyguanosine (Abcam Cat# ab10802, RRID:AB_297482), Ms anti-alpha smooth muscle actin (Abcam Cat# ab7817, RRID:AB_262054), MS anti-VDAC (Abcam Cat# ab14734, RRID:AB_443084), Total OxPhos Rodent WB Antibody Cocktail (Abcam Cat# ab110413, RRID:AB_2629281), Anti-Rabbit IgG-FITC Ab produced in goat (Sigma-Aldrich Cat# F9887, RRID:AB_259816), Anti-Mouse IgG-TRITC Ab produced in goat (Sigma-Aldrich Cat# T7782, RRID:AB_261768), IRDye 800CW Goat Anti-Rabbit IgG (LI-COR Biosciences Cat# 926-32211, RRID:AB_621843), IRDye 680CW Goat Anti-Mouse IgG (LI-COR Biosciences Cat# 926-68070, RRID:AB_10956588). Reagents: CGS21680 (A2AR agonist; Tocris (MI, USA), 1063), ZM241385 (A2AR antagonist; Tocirs, 1063), rhIL-1β (R&D systems (MN, USA), 201-LB), rmIL-1β (R&D systems, 401-ML); PARAFORMALDEHYDE 16% SOLUTION, EM GRADE (Electron Microscopy Sciences, 15710-S), Reagents from Sigma-Aldrich (MO, USA): RIPA Buffer (R0278), Glycerol (G6279), Triton (G6279), EDTA (E9884), bovine serum albumin (BSA) (A3294-100g), collagenase D (11088866001), Poly_1-Lysine (P4707), 2-deoxy-D-glucose (D6134), D-glucose (G7021), sodium pyruvate (P5280), hydrogen peroxide (216763), Fluoroshield with DAPI (4′,6-diamidino-2-phenylindole; F60557-20ML). Reagents from Life Technologies (NY, USA): DMEM (11695-092), fetal bovine serum (FBS) (10082147), HBSS (14175-079); from Thermo Fisher (MA, USA): PrestoBlue Cell Viability Reagent (A13261), MitoTracker Green FM (M7514), tetramethylrhodamine (TMRM, T668); from other sources: 1XPBS and TE (10 mM Tris, 1 mM EDTA) buffer (NYU Langone Health DART Reagent Prep Service), Tween 20 (Bio-Rad, 170-6431), Xylene (Acros, 42268-0040), EtOH (Fisher Scientific, 04 355 226), Proteinase K (Promega, v302b), clear bottom culture slides (greiner bio-one, 543079), prediluted Protein Assay Standards: BSA set (Thermo Scientific, 23208), Mini-PROTEAN TGXTM Gels 8%-16% (Bio-Rad, 456-1104), NuPAGE 3%-8% of Tris-Acetate (Invitrogen, EA0375BOX), Tris-Acetate SDS Running Buffer (Invitrogen, LA0041), 10X SDS-PAGE (NYU Langone Health DART Reagent Prep Service), Immun-Blot PVDF Membrane for Protein Blotting (Bio-Rad, 162-0177), CAPS, 3-(Cyclohexylamino)-1-propanesulfonic acid (Sigma-Aldrich (MO, USA), C2632-25G), MuLV Reverse Transcriptase PCR Kit (Applied Biosystems, 10289614), Brilliant II SYBR Green qPCR Master Mix (Agilent, 600828), Seahorse XFe24 FluxPak (sensor cartridge, culture plates, calibrant solution; Agilent Technologies (Tx, USA), 102340-100), Seahorse XF Cell Mito Stress Test Kit (Agilent Technologies (Tx, USA), 103015-100) ATP determination kit (Life Technologies, A22066), Plasma Membrane Protein Extraction Kit (Abcam, ab65400), Detergent Compatible (DC) Protein Assay (Bio-Rad, 5000111), QIAshredder columns (Qiagen, Invitrogen 74104), T/C-28a2 human chondrocyte cell line (Dr. Miguel Otero, HSS Research Institute, Hospital for Special Surgery, New York, NY, 10021, USA OteroM@hss.edu), RRID:CVCL_6850), WT C57BL/6J mice (Jackson labs, Cat# JAX:000664 RRID:IMSR_JAX:000664), Adenosine A2 Receptor KO, C57BL/6J background mice (Dr. Jiang Fan Chen (Boston University School of Medicine, Boston, MA)) PRIMERS:A2AR forward (human) 5′GGGCGCAGTATGAGAGGGC3′ Sigma-Aldrich (MO, USA); A2AR reverse (human) 5′GCTCTGCGCATTGTTGTCAC3′ Sigma-Aldrich (MO, USA); GAPDH forward (human) 5′GGATTTGGTCGTATTGGG3′ Sigma-Aldrich (MO, USA); GAPDH reverse (human) 5′GGAAGATGGTGATGGGATT3′ Sigma-Aldrich (MO, USA). Software: Seahorse Wave (Agilent Technologies, Seahorse Wave, RRID:SCR_014526), FIJI (Fiji, RRID:SCR_002285), PRISM (PRISM, RRID:SCR_005375), Image Studio Software (LI-COR Image Studio Software, RRID:SCR_015795). Instruments: Zeiss AxioObserver.Z1 and Zeiss 880 confocal microscope (NYU Langone Health Microscopy Core) FlexStation 3 Multi-Mode Microplate Reader (NYU Langone Health Small Instrument Fleet).
2.2 ∣. Animals and primary chondrocyte harvest
Mice employed in this study were kept under regular lighting conditions (12 hours light/dark cycles) and given food and water ad libitum. Adenosine A2A receptor knockout (A2AR−/−) mice, bred on a C57BL/6 background (referred to here as wild type, WT), were kindly provided by Dr. Jiang Fan Chen (Boston University School of Medicine, Boston, MA). Mice were bred and raised in the same facility (Science Building Animal Facility, NYU Langone Medical Center), receiving the same food and care. Phenotype had been confirmed by genotyping as previously reported.20 Neonatal pups were sacrificed by decapitation and used for chondrocyte extraction from knee and hip joint. A2ARKO and WT pups from 1-5 days of age were sacrificed and their knees and femoral heads were collected and digested in two subsequent incubations of 3 mg/mL of collagenase D in serum-free DMEM for 45 minutes each at 37°C. The cartilage chunks were agitated then incubated overnight with 0.5 mg/mL of collagenase D in serum-free DMEM at 37°C. The following day, the digested cartilage was run through a 48 μm nylon mesh and centrifuged to pull down chondrocytes. Harvested chondrocytes were then cultured with full media (DMEM + FBS). The New York University School of Medicine Institutional Animal Care and Use Committee approved all protocols for experimental procedures involving the use of animals in accordance to U.S. law.
2.3 ∣. Histology, immunohistochemistry, and immunofluorescence
Mouse and rat knees were decalcified after sacrifice, removal of soft tissue, and fixation (4% ofPFA for 48 hours) in 10% of EDTA (7.4 pH) for 4 weeks. Parafilm-embedded histological sections (5 μm) were cut, mounted, and prepared for immunohistochemistry for mitochondrial markers (anti-ATPase (1/1000 dilution), anti-8-OHg (1/200 dilution)) as previously described [14]. Briefly, slides are incubated at 60°C for 30 minutes and deparaffinized by soaking in pure xylene for 10min three times. Slides are hydrated by soaking in decreasing concentrations of ethanol (100%-70%) for 2 minutes each. and distilled water. Antigen retrieval is achieved by soaking in 1× Proteinase K solution (in TE buffer) for 20 minutes at 37°C. Slides were blocked in a phosphate buffered saline (PBS)-BSA3%, Triton1%, FBS 5% solution for 1 hours at room temperature, washed in PBS-BSA3%, and incubated overnight at 4°C with respective primary antibodies dissolved in PBS%-BSA3%. The next morning, slides are washed with PBS and incubated for 1 hours at room temperature with according fluorescent secondary antibody. For experiments with T/C-28a2 human chondrocyte cell line and primary murine WT and A2AR−/− chondrocytes, cells were plated in tissue culture slides treated accordingly and fixed with 4% of PFA for 15 minutes at room temperature. Cells were washed with PBS (1×) and permeabilized with 0.25% of Triton in PBS for 10 minutes. After washing with PBS. Cells are blocked using 1% of BSA in PBST for 30min and incubated overnight with primary antibodies of interest in 1% of BSA in PBST overnight at 4°C. The following day, cells were washed with PBS and incubated with according fluorescent secondary antibody in 1% of BSA in PBS-Tween for 1 hours in room temperature. Cells were washed and mounted using Fluoroshield with DAPI. Slides and cells were visualized with a Zeiss AxioObserver.Z1.
2.4 ∣. Seahorse mitochondrial stress assay
Chondrocytes (primary from WT and A2AR−/− mice, T/C-28a2 chondrocytic cells) were plated on seahorse assay plates at equal cell densities (30K cells/well). After cells reached ~80% confluency, they were treated with vehicle (DMEM, 10% of FBS), IL-1β (5 ng/mL for 4 hours) with and without the A2AR agonist, CGS21680 (1 μM for 1 hours, CGS) or with CGS alone. Cells were then placed for 1 hours in a non-CO2 incubator with seahorse stress media. Cells were subsequently placed onto a SeahorseXFe24 analyzer for a mitochondrial stress test which measured extracellular (media) acidification rate (ECAR) oxygen consumption rates (OCR) at baseline and after subsequent port injections of 1uM oligomycin (inhibition of ATP synthase), 2 μM FCCP (p-triflouromethoxyphenylhydrazone, mitochondrial uncoupling), and 0.5 μM rotenone/antimycin a (electron transport chain inhibitors by blocking electron transfer from complex I to CoQ). FCCP titrations were performed to determine optimal concentration of 2 μM in both human and murine chondrocytes. Glycolytic measurements during mitochondrial stress were normalized by ECAR measurements after an additional port injection of 2-deoxy-D-glucose. Cell count normalization was performed by either total protein amount or by determining cell viability using PrestoBlue Cell Viability Reagent. Fisher. Agilent MitoStressTest Report Generators and PRISM were subsequently used to analyze the data.
2.5 ∣. ATP content and release from cell culture
TC28a2 cells were plated in 24 well plates at a 25 000 cell/well density, grown overnight, and treated with vehicle (DMEM, 10% of FBS), IL-1β (5 ng/mL for 4 hours) with and without the A2AR agonist, CGS21680 (1 μM for 1 hours, CGS) or with CGS alone (n = 3). Cells were then incubated with either warm PBS or warm 100 mM hydrogen peroxide (Sigma-Aldrich, 216763) for 1 hours at 37°C to extract released and total ATP, respectively. Supernatants were then transferred to 1.5 mL tubes and centrifuged at 600g for 5 minutes at 4°C. 500 μL of supernatant was then transferred to a fresh tube and kept on ice. ATP determination from each sample was then assessed using an ATP determination kit according to manufacturer’s instruction, reading luminescence at 560 nm (integration time 10 000 msec) using a FlexStation three Multi-Mode Microplate Reader.
2.6 ∣. Live cell imaging
Chondrocytes were cultured in clear bottom culture slides. Primary murine and T/C-28a2 cells were incubated with medium or IL-1β (5 ng/mL) for 4 hours and CGS21680 (1 mM) was added to relevant wells for the last hour prior to analysis. Cells were then washed and stained using different probes for mitochondrial content (MitoTracker Green FM Thermo Fisher, MA, USA, at 37°C for 30 minutes, then washed) and membrane potential (equilibrated with TMRM at 37°C for 20 minutes at 60 nM and imaged at 15 nM) using manufacturer’s instructions. Cells were imaged at 37°C in HBSS at NYU Langone Health Microscopy Core using a Zeiss 880 confocal microscope. Images were quantitated by ImageJ software. The results shown represent the mean (+/−SD) of at least four separate determinations. TMRM total intensity/cell was determined using ImageJ software using manufacturer’s instructions considering 10 cells/image of eight images in three separate experiments.
2.7 ∣. Transmission electron microscopy
8-well chamber cultured mouse or human chondrocyte cells were fixed in 2.5% of glutaraldehyde and 2% of paraformaldehyde in 0.1M sodium cacodylate buffer (pH 7.2) for 2 hours at 4°C and postfixed with 1% of osmium tetroxide with 1% of potassium ferrocyanide in sodium cacodylate buffer for 1 hour at 4°C. The cells were block stained with 0.25% of uranyl acetate, processed in a standard manner, and embedded in Araldite 502 epoxy resin (Electron Microscopy Sciences, Hatfield, PA). Ultrathin sections (60 nm) were cut, mounted on copper grids, and stained with uranyl acetate and lead citrate by standard methods. Stained grids were examined under Talos120C electron microscope and photographed with a Gatan OneView camera. 20 random cells in each sample were imaged for morphological analysis.
2.8 ∣. Protein extraction and Western blotting assay
After cell harvest and/or treatments, the total protein extracts were collected and stored at −80°C. Total protein fractions were quantified using the Protein Assay Kit (Bio-Rad). For determination of cytosolic and plasma membrane (PM) expression of the A2AR, PM protein was isolated using Plasma Membrane Protein Extraction Kit according to manufacturer’s instructions. Western blotting was performed by electrophoresing 20 μg/mL protein through 3%-8% of tris-acetate (OPA1) or 8%-16% of polyacrylamide gels followed by transfer of proteins to polyvinylidene difluoride (PDVF) membranes according to suggestions of antibody manufacturer (ie, high pH CAPS (N-cyclohexyl-3-aminopropanesulfonic acid) buffer for OxPhos complexes). PDVF membranes were incubated overnight at 4°C with the specific primary antibody (1:1000 or as recommended by manufacturer), and after washing, incubated with goat anti-rabbit IRDye 800 CW and goat anti-mouse IRDye 680 RD (1:5000). Membranes were scanned with Li-cor Odyssey equipment and the intensities of the protein bands were quantified by densitometric analysis using Image Studio 2.0.38 software.
2.9 ∣. Reverse transcription and real time PCR
RNA extraction was performed from mouse primary chondrocytes using RNeasy Mini Kit (Qiagen, Invitrogen) and QIAshredder columns (Qiagen, Invitrogen), following the manufacturer’s protocol. RNA reverse transcription was performed using the MuLV Reverse Transcriptase PCR Kit (Applied Biosystems). After RNA reverse transcription to cDNA, real time PCR reactions were performed for a relative quantification of human A2AR (normalized to human GAPDH) StepOne Real-Time PCR Sysyem (Thermo Fisher Scientific) with Brilliant SYBR Green Kit QPCR Master Mix, according to the manufacturer’s protocol.
2.10 ∣. Primary human mesenchymal stem cell culture and chondrogenic differentiation
Tissue samples were collected from children with non-syndromic sagittal craniosynostosis or other craniofacial diseases undergoing surgical reconstruction whereby excess tissue is routinely discarded (IRB-approved, s17-01599—“Effect of A2AR Agonists on Osteogenic Potential of Stem Cells”). These samples were retrieved immediately after harvesting from the operating theater in sterile 50 mL falcon tubes containing around 15 mL of Liebovitz 15 medium and transferred to the laboratory on ice. Samples were either processed, immediately or occasionally stored in the cold room (4 degrees Celsius) overnight prior to processing, in a sterile container. After washing in sterile PBS ×1 in a laminar flow hood, samples were reserved for either stem cell isolation (human chondroprogenitor, hCP). Cells were cultured in humidified incubators at 37°C with 5% of CO2 in “expansion medium” which consisted of DMEM containing GlutaMAX and supplemented with 10% of embryonic stem cell-qualified serum (ES-FBS) and 1% of penicillin/streptomycin. Cells were plated as appropriate for each experiment with “chondrogenic differentiation medium” (expansion medium with 0.1 μM dexamethasone, 10 ng/mL transforming growth factor β1, insulin transferrin selenium, and 50 μg/mL ascorbate-2-phosphate) for 1 week (early differentiation) or 3 weeks (mature differentiation) before the experiments were performed.
2.11 ∣. Statistics
Mitochondrial fragmentation was assessed by determining lengths of mitochondria from confocal images on ImageJ software using manufacturer’s instructions. Briefly, continuous mitochondria with homogenous TMRM staining were traced using freehand-line and measuring line lengths. Data from at least 5 mitochondria of at least 20 cells per condition were measured in 3 separate experiments. TEM cristae analysis was performed manually using ImageJ; at least 30 mitochondria per condition were analyzed. Other data were analyzed by 1-Way ANOVA or t test (as appropriate) using PRISM, and we here show representative of at least three experiments.
3 ∣. RESULTS
3.1 ∣. Diminished A2AR stimulation leads to increased mitochondrial ROS burden in vivo.
Prior reports indicate that chondrocytes from patients with OA have a loss of mitochondrial content and increased oxidative damage and although chondrocytes sit in an avascular and aneural matrix, up to 25% of their ATP content is the product of OXPHOS with metabolites that diffuse through the cartilage matrix to the chondrocyte.13 Because A2AR stimulation restores chondrocyte homeostasis, we decided to evaluate whether lack of A2AR stimulation affects mitochondrial dynamics or function in null chondrocytes. We hypothesized that the absence of A2AR in chondrocytes of A2AR−/− mice, which develop spontaneous OA,12 resulted in ROS accumulation and mitochondrial dysfunction.. Sections from previously reported12 12 week old A2AR−/− (OARSI 1.4 ± 1.2) and wild-type (WT, OARSI 0.2 ± 0.2739) mouse knees were prepared for histologic examination and stained for oxidized mitochondrial DNA (8-hydroxyguanosine (8-OHg)) and ATPase (Complex V) which showed increased ROS burden and reduced mitochondrial content in A2AR−/− knees in vivo (Figure 1A). Primary WT and A2AR−/− chondrocytes were isolated and subjected to live imaging with Mitotracker Green for assessment of mitochondrial content after treatment with vehicle or IL-1β treatment (5 ng/mL for 4 hours) with and without the A2AR agonist, CGS21680 (1 μM for 1 hours, CGS) or CGS alone. Primary chondrocytes from A2AR−/− and WT pups had no significant difference in mitochondrial content at baseline (control, CTRL), but mitochondrial content is increased in WT chondrocytes that received CGS21680 after IL-1β exposure, while the effect is lost in A2AR−/− chondrocytes (Figure 1B,C).
FIGURE 1.
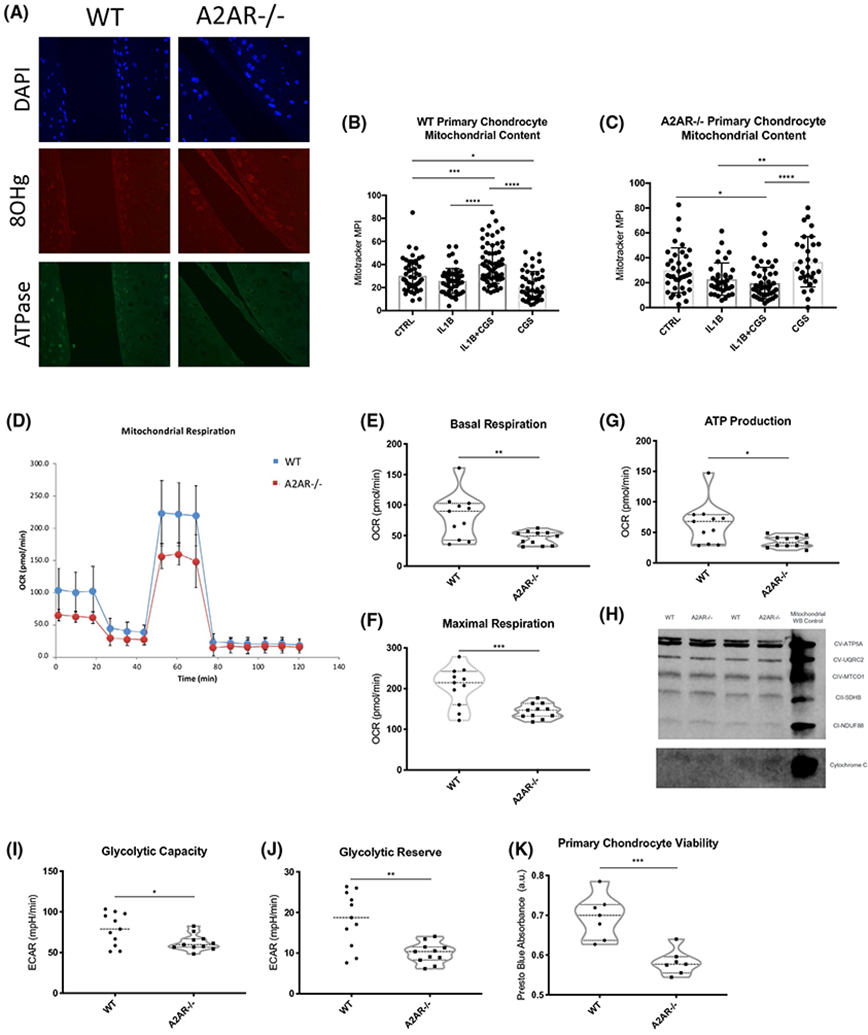
Lack of A2AR signaling results in ROS burden and reduction of mitochondrial content in vivo and in vitro which reduces mitochondrial function and viability in primary chondrocytes. A, Histologic sections of aged-matched A2AR−/− (OARSI 1.426 ± 1.199) and WT (OARSI 0.2 ± 0.2739) mice show lack of A2AR signaling lead to an increase of ROS burden and reduction in mitochondrial content. B, Primary WT and A2AR−/− chondrocytes were isolated and subjected to live imaging with Mitotracker Green for assessment of mitochondrial content after treatment with vehicle or IL-1β treatment (5 ng/mL for 4 hours) with and without the A2AR agonist, CGS21680 (1 μM for 1 hours, CGS) or CGS alone. (A) IL-1β-treated WT primary chondrocytes increase their mitochondrial content after A2AR ligation (*P = .0157, ***P = .0007, ****P < .0001 ONE-Way ANOVA followed by Bonferroni post hoc testing). C, IL-1β treatment of null primary chondrocytes decreased mitochondrial content, which cannot be abrogated by A2AR ligation (*P = .0268, **P = .0037, ****P < .0001 ONE-Way ANOVA followed by Bonferroni post hoc testing). D, Isolated primary WT and A2AR−/− chondrocytes were treated as described above and subjected to Seahorse mitochondrial stress test to evaluate mitochondrial and metabolic function. A2AR−/− chondrocytes had significantly reduced E, basal respiration OCR (P = .0055), F, maximal respiration OCR (P = .0009), G, ATP production extrapolated from oxygen consumption rate (OCR; P = .0100). H, These changes are not due to differential expression of any OXIPHOS Complex. I, Glycolytic capacity (P = .0223), and J, glycolytic reserve (P = .0011) were also reduced in null chondrocytes. K, Reduction in metabolic rates resulted in loss of viability (P = .0004). Statistics: unpaired, two-tailed t tests
ROS accumulation and loss of mitochondrial content limits mitochondrial function and overall oxygen consumption for ATP production, limiting ATP/adenosine availability for anti-inflammatory A2AR ligation and directly promoting matrix loss and secondary cartilage mineralization in human chondrocytes.21 Moreover, we have previously reported that primary WT chondrocytes stimulated with IL-1β and chondrocytes harvested from rats with PTOA have decreased ATP and adenosine content and release.12 We, therefore, determined whether diminished A2AR signaling in primary chondrocytes lacking A2AR resulted in impaired mitochondrial functionality as a result of the ROS burden or the reduction of mitochondrial content. Chondrocytes isolated from WT mice had significantly greater oxygen consumption rates than A2AR−/− chondrocytes which was associated with greater ATP production (Figure 1D-G). These findings were not due to a decrease in total amounts of OXPHOS complexes from total cell lysate western blot (Figure 1H) or RNA sequencing analysis (data not shown). Interestingly, lack of A2AR signaling also reduced extracellular acidification rates (ECAR) indicating significant differences in glycolic capacity and glycolic reserve (Figure 1I,J). Mitochondrial and glycolytic loss of function resulted in significant reduction in viability of primary A2AR−/− chondrocytes, as measured by Presto Blue absorbance (Figure 1K). Thus, diminished A2AR stimulation results in increased accumulation of mitochondrial oxidation products and loss of mitochondrial function exacerbating already described pro-inflammaging signals that ultimately compromise cell viability. These findings are consistent with the hypothesis that diminished endogenous A2AR stimulation alters mitochondrial oxidation and function, as seen in human OA.13,17
3.2 ∣. Primary A2AR−/− chondrocytes have reduced ability to meet metabolic demands because mitochondria are dysfunctional and subsequently marked for mitophagy
Since accumulation of ROS leads to the gradual dysfunction and loss of mitochondria,8,13 we next asked what mitochondrial structural changes could be responsible for ROS burden and loss of functionality in A2AR−/− chondrocytes. The live cell fluorescent probe, TMRM, stains mitochondria when they possess physiological membrane potential. Mean pixel intensity (MPI) for TMRM live imaging staining was significantly reduced in unstimulated A2AR−/− chondrocytes, consistent with the in vivo findings of mitochondrial dysfunction and indicating a propensity for depolarization in absence of A2AR signaling (Figure 2A,B). WT primary chondrocytes elongate their mitochondria after stimulation with CGS21680 with or without IL-1β (Figure 2C). There were no significant differences in mitochondrial lengths between unstimulated WT or null chondrocytes (Figure 2D), but lack of A2AR signaling prevented mitochondrial elongation after stress, and treatment of A2AR−/− chondrocytes with CGS alone further reduced mitochondrial lengths (Figure 2E). Markers of oxidative damage and mitophagy were subsequently found to be affected by spontaneous depolarization of mitochondria (data not shown).
FIGURE 2.

In A2AR−/− primary chondrocytes mitochondria depolarize spontaneously, fusion upon stimuli is impaired and subsequently mitochondria are marked for mitophagy. A, Primary WT and A2AR−/− chondrocytes were isolated from neonatal mice, treated with medium (CTRL) or IL-1β (5 ng/mL for 4 hours) with and without the A2AR agonist, CGS21680 (1 μM for 1 hours, CGS) or CGS alone, and then, stained with TMRM to evaluate mitochondrial membrane polarity using live microscopy under constant conditions. B, Untreated A2AR−/− chondrocytes had significantly reduced MPI for TMRM, indicating susceptibility for spontaneous membrane potential collapse (P = .0004, unpaired, two-tailed t test). Mitochondrial size or lengths were calculated using the tracing and measuring tool of ImageJ of over 30 mitochondria per condition, C, WT chondrocytes treated with CGS with or without IL-1β had increased mitochondrial lengths than cells that received medium or IL-1β alone (P < .0001, ONE-Way ANOVA followed by Bonferroni post hoc testing). D, There were no significant differences in mitochondrial lengths of untreated WT or null chondrocytes (unpaired, two-tailored t test), but (E) lack of A2AR signaling prevented mitochondrial length expansion upon stimuli (P = .0377, ONE-Way ANOVA followed by Bonferroni post hoc testing; MPI: mean pixel intensity)
Primary chondrocytes isolated from WT and A2AR−/− mice were prepared for transmission electron microscopy (TEM) and at least 30 mitochondria per condition were analyzed as previously described22 to assess mitochondrial ultrastructure (Figure 3A). A2AR−/− primary chondrocytes had significant mitochondrial swelling (P < .0001, t test) as measured by increased cristae widths (Figure 3B). There were no significant changes to the number of cristae per mitochondria or the ratio of number of cristae junctions to number of cristae, suggesting that the observed mitochondrial dysfunction results from increased cristae width and subsequent uncoupling23 and not due to reduced area for OXPHOS or a decreased number of contact sites for metabolite exchange with the endoplasmic reticulum (ER), respectively (Figure 3C-E). Cristae ultrastructure is controlled by careful regulation of various proteins and protein complexes,19,22 some of which have been reported to be regulated by the NF-κB pathway19 and modulated by PKA,24 therefore, likely downstream of A2AR signaling.
FIGURE 3.

A2AR−/− primary chondrocytes have altered mitochondrial ultrastructure under TEM. Primary WT and A2AR−/− chondrocytes were isolated from neonatal mice and (A) submitted to TEM to evaluate mitochondrial structure, at least 30 mitochondrion per condition were analyzed (scale = 0.5 μm). (B) Cristae widths were significantly greater in A2AR−/− chondrocytes’ mitochondria while there were no significant changes in (C) number of cristae per mitochondria, (D) cristae junction (CJ) per mitochondria or (E) in the ratio of the number of CJ to the number of cristae (P < .0001, unpaired, two-tailed t test)
3.3 ∣. ROS burden is mitigated in vivo by intra-articular injections of liposomal adenosine or CGS21680 in a mouse model of obesity-induced OA
C57BL/6 WT mice were fed a high fat diet (HFD, 60% of calories from fat) which has been established to induce OA and metabolic inflammation.25 After being on this diet for 12 weeks, mice developed OA (HFD-OA OARSI 5.2 ± 1.8), and subsequently received six intraarticular injections (every 10 days) of empty liposomes (OARSI 4.2 ± 1.6) or preparations of either liposomal adenosine or CGS21680. Both liposomal adenosine and CGS21680 halted and reversed proteoglycan catabolism and chondrocyte viability compromise with OARSI scores of 1.3 ± 0.8 and 1.8 ± 1.0, respectively (P < .0001 versus HFD-OA; Manuscript in Revision;15). Knees were excised after sacrifice and prepared for histology. Immunofluorescence of histologic sections was performed to determine mitochondrial content (anti-ATPase) and ROS burden (anti-8-OHg). ROS burden was significantly reduced in knees that received liposomal adenosine or CGS21680 with modest increases in mitochondrial content in these groups (Figure 4). As indicated by arrows, obese mice with established OA have increased ATPase and 8-OHg staining, but mice that received liposomal CGS21680 had increased ATPase staining without increased ROS burden (8-OHg stain).
FIGURE 4.

Mitochondrial markers in HFD-OA mice are improved by exogenous A2AR agonist. WT mice were fed a high fat diet (HFD, 60% of calories from fat) for 12 weeks to promote obesity and to induce OA (HFD-OA; OARSI 5.167 ± 1.835). After OA induction, mice were subjected to six intraarticular injections (every 10 days) of empty liposomes (LIPO; OARSI 4.167 ± 1.602) or liposomes containing either adenosine (LIPO + Ade; OARSI 1.333 ± 0.8165 ) or CGS21680 (LIPO + CGS; OARSI 1.833 ± 0.9832). Mice were then sacrificed, and knees were prepared for histology. LIPO + Ade and LIPO+CGS OARSI scores have P < .0001 versus HFD-OA. Age-matched mice that were fed regular chow were used as healthy controls (healthy CTRL; OARSI 0.2 ± 0.4472). Histologic sections were stained to determine mitochondrial content (anti-ATPase) and ROS burden (oxidized guanosine residues, 8-OHg). Liposomal adenosine and CGS21680 enhance mitochondrial content while diminishing ROS burden as marked by arrows
3.4 ∣. A2AR stimulation with CGS21680 reverses ROS burden and increases mitochondrial functionality in IL-1β-treated human chondrocytic, T/C-28a2, cells
After initial evaluation of the mitochondrial phenotype of A2AR−/− chondrocytes, we decided study the potential effect of A2AR ligation in human chondrocytes on mitochondrial function and ultrastructure. Because A2AR stimulation restores chondrocyte homeostasis,12 we decided to assess whether A2AR stimulation also affects mitochondrial dynamics or function in vitro. A human chondrocyte cell line, T/C-28a2, was tested for ROS burden by immunofluorescent staining of oxidized mitochondrial DNA (8-hydroxydeoxyguanosine (8-OHg)) and mitochondrial content (ATP synthase) after vehicle or IL-1β treatment (5 ng/mL for 4 hours) with and without the A2AR agonist, CGS21680 (1 μM for 1 hours, CGS) or CGS alone. IL-1β treatment significantly increased intracellular oxidized mitochondrial DNA, reflecting the ROS burden induced by cytokine stress (Figure 5A,B). IL-1β treatment followed by A2AR ligation (CGS) reverses accumulation of ROS, significantly reducing ROS burden to a comparable level to that of the untreated cells. Notably, mitochondrial content is not significantly modulated by any of the treatments within this time frame, except for CGS versus IL-1β alone, suggesting longer time frames might yield more significant changes in mitochondrial content (Figure 5C).
FIGURE 5.
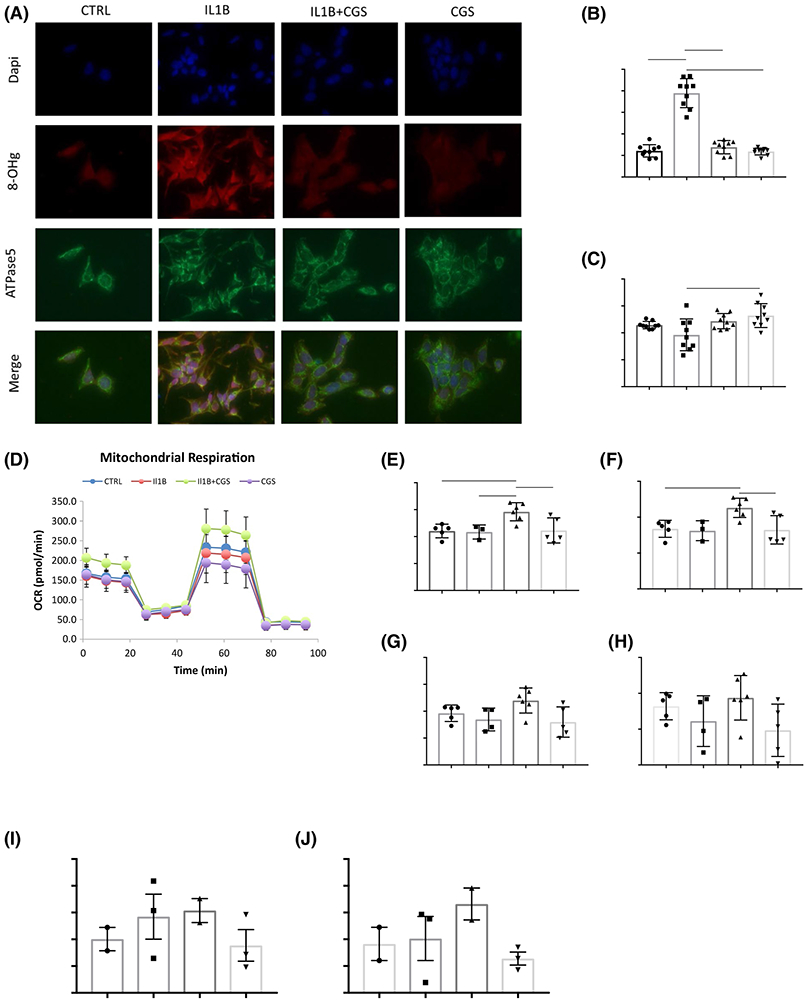
CGS2180 improves ROS burden and mitochondrial functionality in human chondrocytes in vitro. T/C28a2 cells were treated with vehicle or IL-1β treatment (5 ng/mL for 4 hours) with and without the A2AR agonist, CGS21680 (1 μM for 1 hours, CGS) or CGS alone. (A) Cells were then fixed and stained for 8-OHg and ATPase as markers of (B) ROS burden and (C) mitochondrial content, respectively. (D) Cells were treated and subsequently submitted to Seahorse Mitochondrial Stress Test which demonstrated cells that received A2AR ligation after IL-1β treatment had increased (E) basal respiration, (F) ATP production, (G) maximal respiration, and (H) spare respiratory capacity. (I-J) Total ATP content and ATP release by cells that received A2AR ligation after IL-1β treatment were quantitatively increased as determined by bioluminescence assay. Mean Pixel Intensity (MPI) and oxygen consumption rates (OCR) statistics: PRISM Graph Pad, One way-ANOVA, Bonferroni, *P < .05, **P = .0040, ****P < .0001
To evaluate the effects of A2AR ligation in human chondrocyte mitochondrial function, we submitted T/C-28a2 cells to SeahorseXFe24 mitochondrial stress testing to assess mitochondrial function via oxygen consumption rates (OCR). Neither IL-1β treatment nor A2AR stimulation alone affected mitochondrial respiration in T/C-28a2 cells, but A2AR stimulation of IL-1β-treated cells increased basal rates of mitochondrial respiration and ATP production, as well as maximal respiration after uncoupling and spare respiratory capacity (Figure 5D-H). We used a bioluminescence assay for quantitative determination of ATP to confirm that A2AR ligation after IL-1β treatment increased ATP release and total ATP content in T/C-28a2 chondrocytes (Figure 5I,J).
Mitochondrial dynamics are modulated by PKA downstream of A2AR agonism.18 T/C-28a2 cells were incubated for 4 hours with myristoylated PKA inhibitor and subsequently exposed to medium or IL-1β (5 ng/mL for 4 hours) with and without the A2AR agonist, CGS (1 μM for 1 hours) or CGS alone. Cells were then stained for ROS burden (anti-8-OHg) and mitochondrial content (anti-ATPase). PKA inhibition increases ROS burden and reduces mitochondrial content in all conditions and A2AR ligation of IL-1β-treated chondrocytes no longer results in significant reduction of ROS burden (Supplemental Figure 1).
3.5 ∣. A2AR stimulation stabilizes mitochondrial membrane potential and reduces mitochondrial swelling after IL-1β exposure in human chondrocytic, T/C-28a2, cells
We next determined whether changes to mitochondrial membrane polarity (as determined by TMRM staining mean pixel intensity (MPI)) could explain the effects of A2AR stimulation in human chondrocytic cells. As others have previously shown, IL-1β reduces TMRM MPI suggesting a disruption to mitochondrial membrane potential.26 A2AR stimulation of IL-1β-treated cells increases TMRM MPI as compared to that of cells that were treated with vehicle or CGS alone (Figure 6A,B). Thus, A2AR stimulation stabilizes mitochondrial membrane potential after IL-1β treatment.
FIGURE 6.
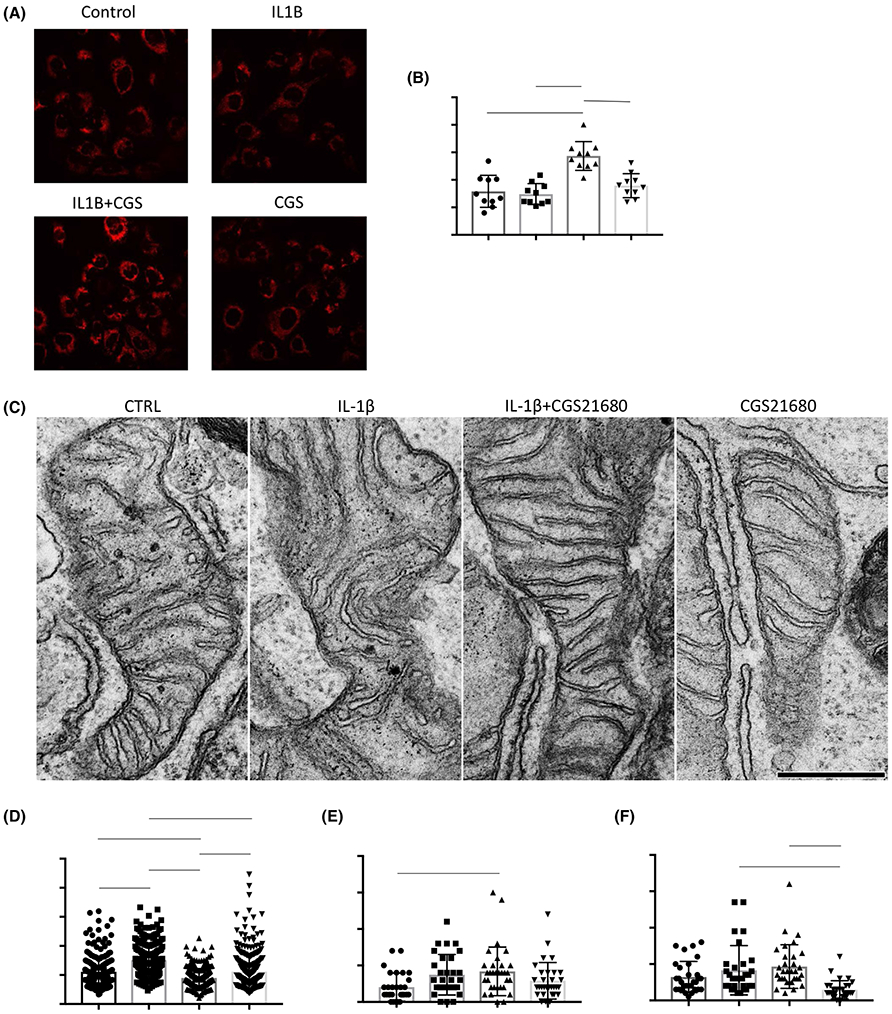
A2AR stimulation of IL-1β-treated human chondrocytic cells stabilizes mitochondrial membrane potential and reduces mitochondrial swelling. T/C-28a2 cells were incubated with medium or IL-1β (5 ng/mL for 4 hours) with and without the A2AR agonist, CGS21680 (1 μM for 1 hours, CGS) or CGS alone and then (A) stained with tetramethylrhodamine (TMRM) which revealed (B) significantly increased MPI in cells that received both IL-1β and CGS compared to cells that received either treatment alone or untreated cells (***P = .0001, ****P < .001, ONE-Way ANOVA followed by Bonferroni post hoc testing). (C) Cells were treated as above and submitted to TEM to evaluate mitochondrial structure (scale = 0.5 μm). Cells that received CGS21680 after IL-1β exposure had significantly (D) reduced cristae widths and (E) increased number of cristae junctions per mitochondria. (F) A2AR ligation alone, reduced the number of cristae per mitochondria than that seen in cells that received IL-1β or IL-1β+CGS (*P = .0126, ***P = .0005, ****P < .0001, ONE-Way ANOVA followed by Bonferroni post hoc testing). MPI: mean pixel intensity
Moreover, when we analyzed mitochondria by TEM, we found that T/C-28a2 cells that received IL-1β and subsequent CGS21680 treatment (as described above) had significantly decreased mitochondrial swelling as evidenced by reduced cristae widths and had increased number of cristae junctions per mitochondria than untreated controls (Figure 6C-E). Reduced cristae widths and increased number of cristae junctions have, respectively, been shown to correlate with increased coupling and sites for metabolite exchange with the ER, crucial for the export of newly synthesized ATP out of mitochondria.23,27 Respiratory capacity and overall cell viability are dependent on changes in cristae number and shape.28 Notably, cells that were treated with the A2AR agonist, CGS21680, alone had a reduced number of cristae per mitochondria compared to untreated controls with no significant change in cristae widths or number of cristae junctions (Figure 6F). This is perhaps because ligation of the receptor in lieu of IL-1β is perceived as excess availability of ATP precursors and lack of metabolic demand results in a reduction of cristae number, the site of OXPHOS. These data support our finding that A2AR ligation without previous exposure to IL-1β resulted in reduced ATP release in human T/C-28a2 chondrocytes (Figure 5J).
3.6 ∣. A2AR ligation enhances mitochondrial membrane potential and expands mitochondrial networks in IL-1β-stimulated primary human chondrocytes
Primary human pre-chondroblasts were harvested from discarded surgical specimens following reconstructive surgery and differentiated in vitro as previously established.29 Fully differentiated chondrocytes were incubated with IL-1β (5 ng/mL, 4 hours) and subsequently exposed to CGS21680 (1 μM, 1 hours) with or without the A2AR antagonist ZM241385 (1 μM, 1 hours, ZM) (Figure 7A). Nuclei are shown for clarity since fully differentially chondrocytes undergo condensation and cells overlap with one another. As in human chondrocytic cells, the effects of A2AR ligation were potentiated if cells were preexposed to IL-1β, suggesting A2AR upregulation or increased sensitization. CGS21680 treatment of IL-1β-exposed, fully differentiated, primary human chondrocytes resulted in a significant expansion of mitochondrial networks that appear to run the lengths of actin filaments in chondrocytes away from the nucleus (reduced perinuclear localization) and increased TMRM MPI (Figure 7B) suggesting there is a greater surface area of inner mitochondrial membrane with physiological potential gradient.
FIGURE 7.

A2AR ligation after IL-1β stimulation expands mitochondrial networks in primary human chondrocytes. A, Fully differentiated human chondrocytes were treated with vehicle (CTRL) or IL-1β (5 ng/mL, 4 hours) with and without CGS21680 (1 μM, 1 hours, CGS) or CGS+ZM241385 (1 μM, 1 hours). CGS and ZM241385 (ZM) alone also tested (both at 1 μM for 1 hours). Cells were then stained with TMRM and live imaged in a confocal microscope to asses mitochondrial inner membrane potential. (B) A2AR ligation after stimulation with IL-1β increased tetramethylrhodamine (TMRM) mean pixel intensity (MPI) and promoted the greatest expansion of mitochondrial network from control. Cells treated with IL-1β, CGS, and ZM (IL1B+CGS+ZM) or ZM alone had increased mitochondrial membrane potential collapse (reduced TMRM MPI) further demonstrating that the mitoprotective effects of adenosine act at the A2AR. Mean Pixel Intensity of images was calculated using Fiji/ImageJ; one-way ANOVA: CTRL versus IL1B, P = .0009, CTRL versus IL1B+CGS P < .0001, CTRL versus IL1B+CGS+ZM P = .0125, CTRL versus CGS P = .0075, CTRL versus ZM P = .0042, IL1B versus IL1B+CGS P < .0001, L1B versus CGS P < .0001, IL1B+CGS versus IL1B+CGS+ZM P < .0001, IL1B+CGS versus ZM P < .0001, IL1B+CGS+ZM versus CGS P < .0001, CGS versus ZM P < .0001. (C) Mitochondrial volumes were evaluated using Amira software analysis of representative Z-stack images from fully differentiated chondrocytes from three3 different donors. Imaging conditions were kept constant for all confocal imaging and thresholds were kept constant throughout analyses for all conditions. Mitochondrial volume is significantly increased in chondrocytes that received IL-1β+CGS compared to all other treatments (CTRL P < .0001; IL-1β P = .0009; IL-1β+CGS+ZM P = .0002; CGS P = .0001; ZM P = .0018; one-way ANOVA). (D) Fully differentiated human chondrocytes were treated with vehicle (CTRL) or IL-1β (5 ng/mL, 4 hours) with and without CGS21680 (1 μM, 1 hours, CGS) or CGS+ZM241385 (1 μM, 1 hours). ATP release was determined with bioluminescent assay and extracellular adenosine and inosine levels were determined by HPLC. Although not statistically significant (ONE-Way ANOVA), A2AR ligation with or without IL-1β exposure, yielded increased levels of ATP, adenosine and inosine in the extracellular space. Each reading is normalized to cell viability (Presto Blue Absorbance) and results are shown normalized to vehicle treated cells (control, CTRL)
Contrary to cells that received both IL-1β+CGS, human chondrocytes that received IL-1β, IL-1β+CGS+ZM or ZM alone exhibited mitochondrial network condensation, reduced TMRM MPI and seemingly increased perinuclear localization, suggesting both IL-1β and A2AR antagonism promote mitochondrial and cellular oxidative stress and that the improvements on mitochondrial health are, indeed, due to A2AR signaling. Mitochondrial volumes were determined using Amira software analysis of stacked confocal images. A threshold was set to identify mitochondria in representative stacks of cells differentiated from three different donors and volumes were normalized to total volume within stack. Imaging conditions were kept constant for all confocal imaging and thresholds were kept constant throughout conditions. There was a significant enhancement of the volume of mitochondria in chondrocytes that received IL-1β+CGS from control (Figure 7C). Moreover, cells that received IL-1β+CGS or CGS alone had the highest levels of ATP release and of adenosine and inosine at the extracellular space (Figure 7D). These data further demonstrate the mitoprotective role of A2AR and its translational value in humans.
3.7 ∣. IL-1β stimulates increased A2AR expression, allowing for greater response after CGS21680 treatment
We and otherS have previously reported cytokine (TNF-α, IL-1β) stimulation of macrophages and endothelial cells increases expression and function of A2AR by preventing receptor desensitization.30,31 Similarly, our group has observed increased A2AR expression on chondrocytes in osteoarthritic cartilage in humans and rats.12 Thus, it was not surprising that the effect of A2AR stimulation on mitochondrial dynamics and function was greater in chondrocytes that had been treated with IL-1β. To better understand the basis for the enhanced effect of A2AR stimulation on mitochondrial function in IL-1β-stimulated cells, we treated with IL-1β overnight and evaluated mRNA and protein expression of the A2AR. Indeed, T/C-28a2 cells that are exposed to IL-1β had increased mRNA and protein expression of the A2AR (Figure 8A).
FIGURE 8.

IL-1β increases mRNA levels and total protein expression of the A2AR in both cytoplasm and plasma membrane which potentiates the effects of receptor ligation in the setting of inflammation. T/C-28a2 cells were incubated with medium (IL1B−) or IL-1β (5ng/mL overnight, IL1B+). Whole cell lysates were prepared to isolate protein from the cytosol and plasma membrane compartments, where IL-1β exposure increased protein levels. mRNA levels for A2AR were also upregulated (normalized to GAPDH)
4 ∣. DISCUSSION
The results presented here demonstrate a role for adenosine, acting at the A2AR, in preventing mitochondrial injury in chondrocytes as a mechanism for the preservation of chondrocyte homeostasis. Moreover, these results support the hypothesis that endogenously generated adenosine plays a similar role in preventing other manifestations of inflammaging. Adenosine, both endogenously generated and exogenously applied, inhibits production of inflammatory reactants and cytokines by stimulated neutrophils, T cells, B cells, and macrophages as well as inhibiting responses to inflammatory cytokines by inhibiting NFκB activation (Reviewed in32). Moreover, adenosine A2AR and A2B receptor (A2BR) promote bone growth and inhibit bone loss in various models.33,34 Thus, we speculate that adenosine, produced by the extracellular dephosphorylation of ATP, maintains cellular and tissue homeostasis, and when adenosine production is reduced as a result of aging or injury, inflammation, and responses to inflammatory cytokines increase, leading to inflammaging.
Mitochondrial dysfunction has been implicated in a number of metabolic and rheumatologic diseases such as OA and is thought to be the basis for inflammaging, a pathologic aging response dependent on unchecked nitric oxide (NO) and ROS burden.17,35,36 The aim of this study was to understand the relationships between adenosine signaling via the A2AR and mitochondrial function, and dynamics in chondrocytes. Mitoprotective therapies such as antioxidants, caspase inhibitors, and Szeto-Schiller mitoprotective peptides have had promising results in experimental OA models,37-39 but little is understood about their potential roles on the reversal of chronic inflammaging in vivo. Before this publication, no other group has been able to demonstrate the mitoprotective effects on chondrocytes of a compound that has already been shown to modulate OA progression by increasing cartilage volumes in animal models.14,15
Our previously reported animal model of spontaneous OA, the A2AR null mouse, has a mitochondrial pathology similar to that reported in human OA tissues,8,13,35,36 including increased pro-inflammatory and catabolic mediators,12 significant ROS burden, reduction of mitochondrial content and function, and disrupted mitochondrial dynamics. Ongoing work in our laboratory suggests that A2AR disruption also compromises autophagy and mitophagy (Unpublished data, B Friedman, and BN Cronstein). These findings suggest that autocrine activation of A2AR modulates mitochondrial dynamics and function in chondrocytes in vivo particularly, because we found that A2AR ligation in HFD-OA mice improved mitochondrial markers in vivo. Moreover, A2AR−/− mice had increased ROS and reduced mitochondrial content in vivo at 12 weeks, which precedes the development of overt OA in vivo.12
Primary neonatal chondrocytes from A2AR−/− mice have significantly reduced mitochondrial content and function, namely reduced OXPHOS and glycolysis rates, resulting in loss of viability. Interestingly, these observations occur in chondrocytes harvested from intact knees in neonatal null mice, suggesting that mitochondrial dysfunction and oxidative stress begin early life in A2AR−/− mice and that these changes eventually compromise tissue homeostasis. Along these findings, loss of cellular protein quality control has been shown to occur in settings of chronic ROS burden and pro-inflammatory stimuli, thus accelerating aging and reducing longevity.1 Moreover, these findings provide further evidence for the role of mitochondrial dysfunction leading to articular cartilage compromise.35,40,41 The hallmarks of inflammaging have been recapitulated in the A2AR null mouse cartilage and it would be interesting to investigate whether there is accelerated aging in other organ systems and tissues in these mice, particularly because these changes are present at birth.
Of note, A2AR−/− cells did not seem to have impaired oxygen consumption rates due to a decrease of OXPHOS complex content, so it was pertinent to investigate mitochondrial ultrastructure and dynamics to better understand how lack of adenosine signaling via the A2AR affects mitochondria. Lack of A2AR signaling resulted in increased mitochondrial swelling and mitochondrial membrane potential collapse. It remains unclear what are the exact mechanisms by which mitochondrial swelling develops in A2AR null chondrocytes, but increased cleavage of OPA1 likely is a hallmark of this process (data not shown). OPA1 cleavage is driven by metalloproteases after mitochondrial membrane potential collapse.42 Mitochondrial dysfunction is associated with increased OPA1 cleavage which also leads to inactivation of mitochondrial fusion (elongation)—which we have also observed in null chondrocytes.43 Mitochondrial fusion leads to the elongation of mitochondria which has been described as a protective measure to maximize the surface area devoted to OXPHOS and to prevent loss of mitochondria by autophagy under stress.44 That is, precisely what is observed in murine WT and human chondrocytes that received CGS21680 after IL-1β stimulation. All treatments of A2AR−/− chondrocytes resulted in a more fragmented mitochondrial network which could be accredited to failure to undergo fusion. Reduced cAMP/PKA signaling in A2AR−/− chondrocytes appears to play a role in the fragmentation of the mitochondrial network, likely through reduced PKA phosphorylation of dynamin-related protein 1 (DRP1),24 but more work needs to be done to confirm this hypothesis. Further work on cristae structure complex formation in null chondrocytes may provide evidence for the mechanism(s) by which reduced A2AR signaling results in disrupted ultrastructure, but it is likely that these effects are due to reduced activation of NF-κB.19
Optimal mitochondrial membrane potential is an established marker for mitochondrial health. Moreover, mitochondrial swelling allows for sustained proton leak, further depolarization of mitochondria and subsequent labeling of dysfunctional mitochondria for mitophagy.45 This sequence of events is recapitulated in the A2AR−/− chondrocyte. Null cells have increased PINK1 expression without changes to Parkin protein levels (data not shown). NO has been shown to disrupt the recruitment of Parkin by PINK146 and loss of Parkin-mediated clearance of dysfunctional mitochondria has been shown to further upregulate ROS burden and reduce cell viability in human chondrocytes.47 Consistent with our observations, previous reports indicate that primary human OA chondrocytes have disrupted autophagic response to oxidative damage.45
After initial evaluation of mitochondria in the animal models of OA, we wanted to determine if A2AR agonism could enhance mitochondrial function and dynamics in human T/C28a2 chondrocytic cells and in primary human chondrocytes. A2AR ligation of IL-1β-treated T/C28a2 chondrocytes reduced ROS burden and increased OCR and ATP production by improving mitochondrial polarization and reversing mitochondrial swelling. Indeed, increased stability of cristae structure allows spaciotemporal organization of OXPHOS complexes48 that result in maximal respiration in inflamed chondrocytes after A2AR ligation as seen by increased OCR and ATP content and release in vitro. In fully differentiated primary human chondrocytes, A2AR stimulation provoked the greatest expansion of the mitochondrial network and enhanced mitochondrial membrane potential only after IL-1β stimulation, an effect abrogated by co-administration of a selective A2AR antagonist. Both IL-1β and A2AR blockade with ZM241385 increased perinuclear localization of mitochondria which has been linked to increased oxidative signaling in the nucleus49 and explains how ROS burden and mitochondrial dysfunction interconnect in the overall inflammaging phenotype seen in OA.50
Surprisingly, we found that only following IL-1β stimulation, A2AR ligation with CGS21680 improved mitochondrial markers. Th1 inflammatory cytokines potentiate the effeci of A2AR by limiting receptor desensitization and decreasing G-protein-coupled receptor kinase 2 association with the plasma membrane.51,52 We hypothesized that IL-1β potentiated these effects by increasing the number of A2AR available for stimulation, as we have previously described.52,53 Indeed, we found IL-1β increased mRNA and protein levels in the cytosol and at the plasma membrane for the A2AR. Consistent with these findings, we have previously reported that rats with post-traumatic OA (PTOA) and human OA tissues have increased chondrocyte A2AR expression in vivo.12
Adequate integration of stress responses promote cell survival, tissue homeostasis and longevity, but eventually fail in the setting of aging or chronic inflammation as damage overwhelms each cellular compartment’s ability to cope with stress (Reviewed in1). Pharmacological interventions to promote protein quality control, autophagy, and mitochondrial fitness have therefore become of particular interest in the treatment of age-related degenerative diseases, including OA.54 OA is a common and chronic rheumatologic, metabolic, and degenerative disease for which medical therapies are limited, expensive, and ineffective.55 Liposomal preparations of adenosine or the A2AR-specific, CGS21680 have been shown to modulate OA progression by restoring cartilage volumes in established OA models of PTOA and obesity-induced OA.12,14 The results here presented suggest that A2AR agonism also improves mitochondrial dynamics and function in inflamed human chondrocytic cells which promotes chondrocyte homeostasis, healing, and subsequent preservation of cartilage. Preliminary data from our group suggests A2AR ligation also modulates mitophagy and autophagy. We believe that A2AR ligation may serve as a novel disease-modifying OA drug as it modulates multiple aspects of inflammaging, namely pro-inflammatory cytokine expression, oxidative damage, mitochondrial dysfunction, and autophagy.
5 ∣. MATERIALS
Raw data will become available after acceptance at https://figshare.com/account/home#/projects/63962.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Miguel Otero, PhD for providing T/C28-a2 chondrocytes. Primary human pre-chondroblasts were made available by our collaborators at NYU School of Medicine (NYUSoM) and NYU School of Dentistry: Drs. Roberto Flores, Paulo Coelho, Amel Ibrahim, and MD/MS candidate Maxime Wang with their IRB-approved human study (s17-01599 - Effect of A2AR Agonists on Osteogenic Potential of Stem Cells). Thank you to the patients and their families who participated in Dr. Flores’ human study. We would like to thank the DART Microscopy Laboratory (Chris Petzold, Kristen Dancel-Manning, and Michael Cammer), the Histology Core and the High Throughput Biology Laboratory at NYU Langone Health for their assistance. This work was supported by grants from the National Institute of Arthritis, Musculoskeletal and Skin Diseases (RO1-AR054897 and RO1-AR056672), the NYU-HHC Clinical and Translational Science Institute (UL1-TR000038 from the National Center for Advancing Translational Sciences), and the Arthritis Foundation. CMC would like to thank the Medical Scientist Training Program at NYU School of Medicine (T32GM007308) and the National Institutes of Health (3RO1AR056672-07S1) for her training and funding.
Funding information
HHS | NIH | National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), Grant/Award Number: RO1-AR054897 and RO1-AR056672; Arthritis Foundation (AF); HHS | NIH | National Institute of General Medical Sciences (NIGMS), Grant/Award Number: T32GM007308; NYU-HHC Clinical and Translational Science Institute, Grant/Award Number: UL1-TR000038
Abbreviations:
- A2AR
Adenosine A2A Receptor
- ECM
extracellular matrix
- FCCP
p-triflouromethoxyphenylhydrazone
- hCP
human chondrogenic precursors
- LIPO
liposome
- OA
osteoarthritis
- OXPHOS
oxidative phosphorylation
- PKA
protein kinase A
- PINK1
PTEN-induced kinase 1
- ROS
reactive oxygen species
- TEM
transmission electron microscopy
- TMRM
tetramethylrhodamine
Footnotes
COMPETING INTERESTS
Drs. Corciulo and Cronstein have received a patent for the use of intra-articular injection of liposomal preparations of adenosine and A2AR agonists for the treatment of osteoarthritis. In addition, Drs. Corciulo and Cronstein are among the founders of Regenosine, a company incorporated to develop adenosine receptor-related therapies for osteoarthritis.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
REFERENCES
- 1.Higuchi-Sanabria R, Frankino PA, Paul JW 3rd, Tronnes SU, Dillin A. A futile battle? protein quality control and the stress of aging. Dev Cell. 2018;44:139–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmad A, Banerjee S, Wang Z, Kong D, Majumdar AP, Sarkar FH. Aging and inflammation: etiological culprits of cancer. Curr Aging Sci. 2009;2:174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anson RM, Bohr VA. Mitochondria, oxidative DNA damage, and aging. J Am Aging Assoc. 2000;23:199–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chandel NS. Mitochondria as signaling organelles. BMC Biol. 2014;12:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mendez AS, Alfaro J, Morales-Soto MA, et al. Endoplasmic reticulum stress-independent activation of unfolded protein response kinases by a small molecule ATP-mimic. Elife. 2015;4:e05434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inoue K, Nakada K, Hayashi J, Isobe K. Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Tanpakushitsu Kakusan Koso. 2001;46:829–837. [PubMed] [Google Scholar]
- 7.Wallace DC. Animal models for mitochondrial disease. Methods Mol Biol. 2002;197:3–54. [DOI] [PubMed] [Google Scholar]
- 8.Loeser RF. The role of aging in the development of osteoarthritis. Trans Am Clin Climatol Assoc. 2017;128:44–54. [PMC free article] [PubMed] [Google Scholar]
- 9.Demontiero O, Vidal C, Duque G. Aging and bone loss: new insights for the clinician. Ther Adv Musculoskelet Dis. 2012;4:61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibon E, Lu L, Goodman SB. Aging, inflammation, stem cells, and bone healing. Stem Cell Res Ther. 2016;7:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soto-Hermida A, Fernandez-Moreno M, Oreiro N, et al. Mitochondrial DNA (mtDNA) haplogroups influence the progression of knee osteoarthritis. Data from the osteoarthritis Initiative (OAI). PloS One. 2014;9:e112735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corciulo C, Lendhey M, Wilder T, et al. Endogenous adenosine maintains cartilage homeostasis and exogenous adenosine inhibits osteoarthritis progression. Nat Commun. 2017;8:15019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Terkeltaub R, Johnson K, Murphy A, Ghosh S. Invited review: the mitochondrion in osteoarthritis. Mitochondrion. 2002;1:301–319. [DOI] [PubMed] [Google Scholar]
- 14.Corciulo C, Castro C, Coughlin T, et al. A2A adenosine receptor stimulation regenerates cartilage in osteoarthritis animal model. Osteoarthritis Cartilage. 2019;27:S174. [Google Scholar]
- 15.Corciulo C, Castro C, Jacob S, Fenyo D, Kennedy O, Cronstein BN. Regenerating cartilage and reversing osteoarthritis (OA) stimulation of adenosine A2A receptors (A2AR) increases cartilage volume and matrix in vitro and in vivo In: American College of Rheumatology Annual Meeting. Vol. 69 San Diego, CA, USA; 2017. [Google Scholar]
- 16.McElroy GS, Chandel NS. Mitochondria control acute and chronic responses to hypoxia. Exp Cell Res. 2017;356:217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blanco FJ, Lopez-Armada MJ, Maneiro E. Mitochondrial dysfunction in osteoarthritis. Mitochondrion. 2004;4:715–728. [DOI] [PubMed] [Google Scholar]
- 18.Dagda RK, Gusdon AM, Pien I, et al. Mitochondrially localized PKA reverses mitochondrial pathology and dysfunction in a cellular model of Parkinson’s disease. Cell Death Differ. 2011;18:1914–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laforge M, Rodrigues V, Silvestre R, et al. NF-kappaB pathway controls mitochondrial dynamics. Cell Death Differ. 2016;23:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen JF, Moratalla R, Impagnatiello F, et al. The role of the D2 dopamine receptor (D2R) in A2A adenosine receptor (A2AR)-mediated behavioral and cellular responses as revealed by A2A and D2 receptor knockout mice. Proc Natl Acad Sci. 2001;98:1970–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson K, Jung A, Murphy A, Andreyev A, Dykens J, Terkeltaub R. Mitochondrial oxidative phosphorylation is a downstream regulator of nitric oxide effects on chondrocyte matrix synthesis and mineralization. Arthritis Rheum. 2000;43:1560–1570. [DOI] [PubMed] [Google Scholar]
- 22.Quintana-Cabrera R, Quirin C, Glytsou C, et al. The cristae modulator optic atrophy 1 requires mitochondrial ATP synthase oligomers to safeguard mitochondrial function. Nat Commun. 2018;9:3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cogliati S, Frezza C, Soriano ME, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8:939–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Griffin TM, Huebner JL, Kraus VB, Yan Z, Guilak F. Induction of osteoarthritis and metabolic inflammation by a very high-fat diet in mice: effects of short-term exercise. Arthritis Rheum. 2012;64:443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lopez-Armada MJ, Carames B, Martin MA, et al. Mitochondrial activity is modulated by TNFalpha and IL-1beta in normal human chondrocyte cells. Osteoarthritis Cartilage/OARS, Osteoarthritis Res Soc. 2006;14:1011–1022. [DOI] [PubMed] [Google Scholar]
- 27.Rampelt H, Zerbes RM, van der Laan M, Pfanner N. Role of the mitochondrial contact site and cristae organizing system in membrane architecture and dynamics. Biochim Biophys Acta Mol Cell Res. 2017;1864:737–746. [DOI] [PubMed] [Google Scholar]
- 28.Quintana-Cabrera R, Mehrotra A, Rigoni G, Soriano ME. Who and how in the regulation of mitochondrial cristae shape and function. Biochem Biophys Res Comm. 2018;500:94–101. [DOI] [PubMed] [Google Scholar]
- 29.Guasti L, Prasongchean W, Kleftouris G, et al. High plasticity of pediatric adipose tissue-derived stem cells: too much for selective skeletogenic differentiation? Stem Cells Transl Med. 2012;1:384–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khoa ND, Montesinos CM, Williams AJ, Kelly M, Cronstein BN. Th1 cytokines regulate adenosine receptors and their downstream signalling elements in human microvascular endothelial cells. J Immunol. 2003;171:3991–3998. [DOI] [PubMed] [Google Scholar]
- 31.Khoa ND, Postow M, Danielsson J, Cronstein BN. Tumor necrosis factor-{alpha} prevents desensitization of G{alpha}s-coupled receptors by regulating GRK2 association with the plasma membrane. Mol Pharmacol. 2006;69:1311–1319. [DOI] [PubMed] [Google Scholar]
- 32.Cronstein BN, Sitkovsky M. Adenosine and adenosine receptors in the pathogenesis and treatment of rheumatic diseases. Nat Rev Rheumatol. 2017;13:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corciulo C, Wilder T, Cronstein BN. Adenosine A2B receptors play an important role in bone homeostasis. Purinergic Signal. 2016;12:537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mediero A, Kara FM, Wilder T, Cronstein BN. Adenosine A(2A) receptor ligation inhibits osteoclast formation. Am J Pathol. 2012;180:775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blanco FJ, Rego I, Ruiz-Romero C. The role of mitochondria in osteoarthritis. Nat Rev Rheumatol. 2011;7:161–169. [DOI] [PubMed] [Google Scholar]
- 36.Maneiro E, Martin MA, de Andres MC, et al. Mitochondrial respiratory activity is altered in osteoarthritic human articular chondrocytes. Arthritis Rheum. 2003;48:700–708. [DOI] [PubMed] [Google Scholar]
- 37.Delco ML, Bonnevie ED, Szeto HS, Bonassar LJ, Fortier LA. Mitoprotective therapy preserves chondrocyte viability and prevents cartilage degeneration in an ex vivo model of posttraumatic osteoarthritis. J Orthopaedic Res. 2018;36:739–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.D'Lima D, Hermida J, Hashimoto S, Colwell C, Lotz M. Caspase inhibitors reduce severity of cartilage lesions in experimental osteoarthritis. Arthritis Rheum. 2006;54:1814–1821. [DOI] [PubMed] [Google Scholar]
- 39.Kurz B, Lemke A, Kehn M, et al. Influence of tissue maturation and antioxidants on the apoptotic response of articular cartilage after injurious compression. Arthritis Rheum. 2004;50:123–130. [DOI] [PubMed] [Google Scholar]
- 40.Cillero-Pastor B, Martin MA, Arenas J, Lopez-Armada MJ, Blanco FJ. Effect of nitric oxide on mitochondrial activity of human synovial cells. BMC Musculoskelet Disord. 2011;12:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cillero-Pastor B, Rego-Perez I, Oreiro N, Fernandez-Lopez C, Blanco FJ. Mitochondrial respiratory chain dysfunction modulates metalloproteases −1, −3 and −13 in human normal chondrocytes in culture. BMC Musculoskelet Disord. 2013;14:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guillery O, Malka F, Landes T, et al. Metalloprotease-mediated OPA1 processing is modulated by the mitochondrial membrane potential. Biol Cell. 2008;100:315–325. [DOI] [PubMed] [Google Scholar]
- 43.Griparic L, Kanazawa T, van der Bliek AM. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol. 2007;178:757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goutas A, Syrrou C, Papathanasiou I, Tsezou A, Trachana V. The autophagic response to oxidative stress in osteoarthritic chondrocytes is deregulated. Free Radic Biol Med. 2018;126:122–132. [DOI] [PubMed] [Google Scholar]
- 46.Oh CK, Sultan A, Platzer J, et al. S-Nitrosylation of PINK1 attenuates PINK1/Parkin-dependent mitophagy in hiP-SC-based Parkinson’s disease models. Cell Rep. 2017;21:2171–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ansari MY, Khan NM, Ahmad I, Haqqi TM. Parkin clearance of dysfunctional mitochondria regulates ROS levels and increases survival of human chondrocytes. Osteoarthritis Cartilage/OARS, Osteoarthritis Res Soc. 2018;26:1087–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lizana L, Bauer B, Orwar O. Controlling the rates of biochemical reactions and signaling networks by shape and volume changes. Proc Natl Acad Sci USA. 2008;105:4099–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Al-Mehdi AB, Pastukh VM, Swiger BM, et al. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci Signal. 2012;5:ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thomas WD, Zhang XD, Franco AV, Nguyen T, Hersey P. TNF-related apoptosis-inducing ligand-induced apoptosis of melanoma is associated with changes in mitochondrial membrane potential and perinuclear clustering of mitochondria. J Immunol. 2000;165:5612–5620. [DOI] [PubMed] [Google Scholar]
- 51.Khoa ND, Montesinos MC, Reiss AB, Delano D, Awadallah N, Cronstein BN. Inflammatory cytokines regulate function and expression of adenosine A(2A) receptors in human monocytic THP-1 cells. J Immunol. 2001;167:4026–4032. [DOI] [PubMed] [Google Scholar]
- 52.Khoa ND, Postow M, Danielsson J, Cronstein BN. Tumor necrosis factor-alpha prevents desensitization of Galphas-coupled receptors by regulating GRK2 association with the plasma membrane. Mol Pharmacol. 2006;69:1311–1319. [DOI] [PubMed] [Google Scholar]
- 53.Nguyen DK, Montesinos MC, Williams AJ, Kelly M, Cronstein BN. Th1 cytokines regulate adenosine receptors and their downstream signaling elements in human microvascular endothelial cells. J Immunol. 2003;171:3991–3998. [DOI] [PubMed] [Google Scholar]
- 54.Ren ZL, Wang CD, Wang T, et al. Ganoderma lucidum extract ameliorates MPTP-induced parkinsonism and protects dopaminergic neurons from oxidative stress via regulating mitochondrial function, autophagy, and apoptosis. Acta Pharmacol Sin. 2019;40:441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bitton R The economic burden of osteoarthritis. Am J Manag Care. 2009;15:S230–S235. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.