

Abstract
Hepatitis B virus (HBV) chronically infects >250 million people and kills nearly a million annually, and current antivirals cannot clear the infection or adequately suppress disease. The virus replicates by reverse transcription, and the dominant antiviral drugs are nucleos(t)ide analogs that target the viral reverse transcriptase. We are developing antivirals targeting the other essential viral enzymatic activity, the ribonuclease H (RNaseH). HBV RNaseH inhibitors with efficacies in the low micromolar to nanomolar range against viral replication in culture have been identified in the α-hydroxytropolone and hydroxyimide chemotypes. Here, we review the promise of RNaseH inhibitors, their current structure–activity relationships, and challenges to optimizing the inhibitors into leads for clinical assessment.
■ HEPATITIS B VIRUS, DISEASE, AND TREATMENT
Hepatitis B virus (HBV) is a hepatotropic DNA virus that replicates by reverse transcription and chronically infects >250 million people worldwide.1 Viral replication induces hepatic inflammation that leads to a decades-long disease progression from asymptomatic infection to chronic hepatitis, hepatic fibrosis, and cirrhosis. Infection often terminates in death from liver failure or hepatocellular carcinoma, and HBV kills >880 000 people annually.2
Treatment for HBV infection is dominated by monotherapy with a nucleos(t)ide analogs (lamivudine, adefovir, telbivudine, entecavir, or tenofovir) that target the reverse transcriptase of the multifunctional HBV polymerase protein (Figure 1A). These drugs suppress HBV replication by 4–5 log10 in most patients, often to below the limit of detection. Therapy can also suppress the nuclear form of the HBV genome, the covalently closed circular DNA (cccDNA) that templates all HBV RNAs (Figure 1B), by ~1 log10 after 1–2 years.3 However, HBV is cleared in only 3–6% of patients even after years of treatment, and treatment reduces chances of liver failure or hepatocellular carcinoma by only 2- to 4-fold after 10 years.4 The costs of this partial suppression of disease progression are indefinite drug administration and potential side effects from decades of drug exposure.
Figure 1.
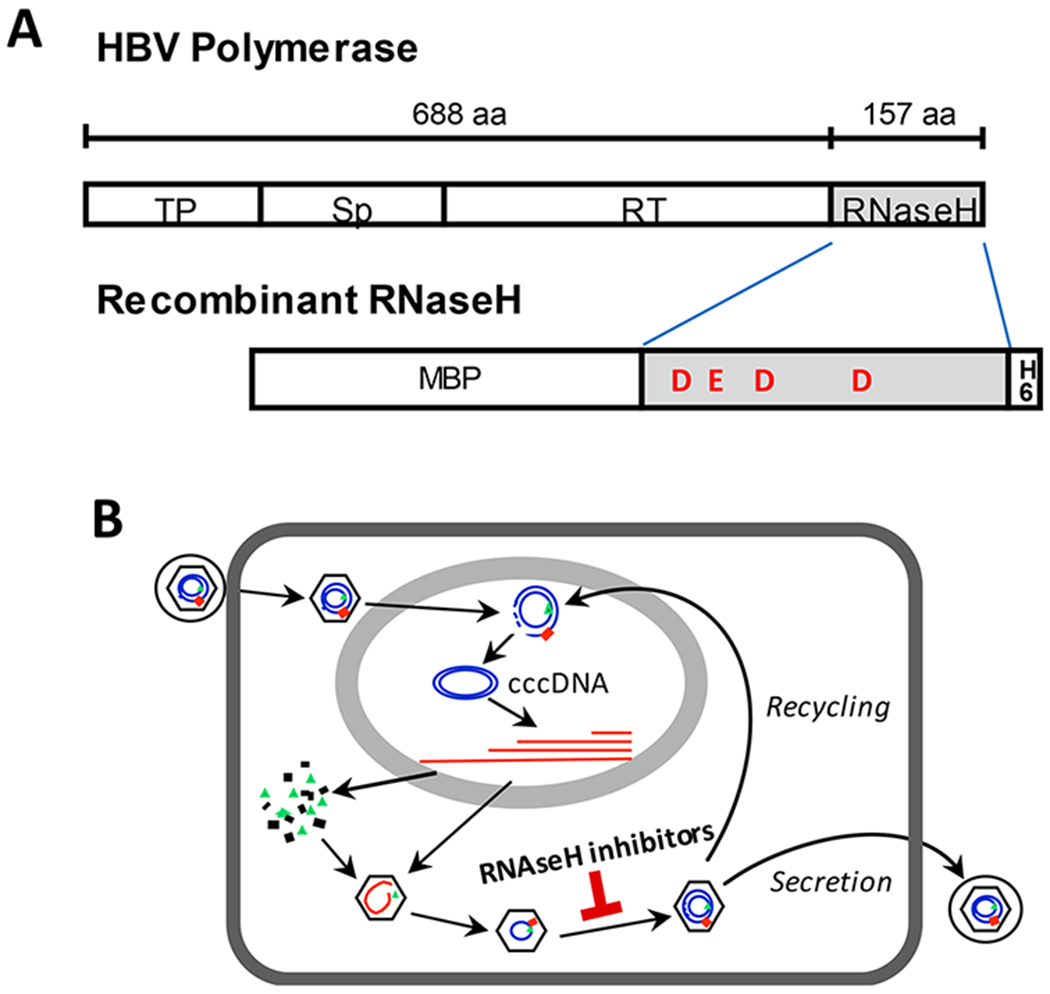
The HBV RNaseH. (A) The RNaseH is the C-terminal domain of the multifunctional HBV polymerase protein. The RNaseH can be expressed as a functional recombinant protein with N-terminal maltose-binding protein (MBP) and C-terminal hexahistidine (H6) tags. TP, terminal protein domain that primes DNA synthesis; Sp, spacer domain; RT, reverse transcriptase domain; RNaseH, RNase H domain. The relative locations of the carboxylic amino acids (D and E) that presumably coordinate the catalytic Mg2+ ions are shown for the recombinant RNaseH. (B) HBV replication cycle. Newly synthesized genomes can be secreted as mature virions or converted via “recycling” to the nuclear cccDNA. DNA is in blue and RNA is in red. The stage at which RNaseH inhibitors act is indicated. Panel B reprinted with permission from Tavis, J. E., Cheng, X., Hu, Y., Totten, M., Cao, F., Michailidis, E., Aurora, R., Meyers, M. J., Jacobsen, E. J., Parniak, M. A., and Sarafianos, S. G. (2013) The hepatitis B virus ribonuclease H is sensitive to inhibitors of the human immunodeficiency virus ribonuclease H and integrase enzymes. PLoS Pathog. 9 (1), e1003125 (ref 7). Copyright 2013 Tavis, Cheng, Hu, Totten, Cao, Michailidis, Aurora, Meyers, Jacobsen, Parniak, Sarafianos.
The failure of monotherapy with very potent nucleos(t)ide analogs to clear HBV implies that eliminating HBV will require combination therapy with compounds that act by different mechanisms, analogous to treatment for human immunodeficiency virus (HIV) infection. Many approaches to identifying drugs that act by novel mechanisms are being explored, including efforts targeting the HBV ribonuclease H (RNaseH).
■ HBV RIBONUCLEASE H
RNaseHs cleave RNA in a RNA/DNA heteroduplex, and the role of the HBV RNaseH is to destroy the HBV RNA after it has been copied into DNA by the reverse transcriptase1 (Figure 1B). RNaseHs belong to the nucleotidyl transferase superfamily that contains host and retroviral RNaseHs, including the HBV and HIV RNaseHs and human RNaseH 1 and 2. Hydrolysis of RNA by RNaseHs requires two Mg2+ ions in the enzyme active site that are bound to a “DEDD” motif.
Difficulties in expressing recombinant HBV RNaseH have severely restricted study of the enzyme and hampered anti-RNaseH drug screening. Therefore, work with the HBV RNaseH has been based on studies with the more tractable HIV RNaseH. Unfortunately, the HBV RNaseH and the HIV enzyme share only 23% amino acid identity, and the HBV RNaseH acts within a polymerase monomer compared to the HIV enzyme being part of a heterodimer. The HIV RNaseH structure is known, but no structural information exists for the HBV enzyme, and the HBV structure cannot be confidently modeled on other RNaseHs due to limited homologies. Therefore, the degree to which the HIV enzyme can serve as a model for the HBV RNaseH is limited.
■ HBV RNaseH AS A DRUG TARGET
HBV reverse transcription is catalyzed by coordinated function of the reverse transcriptase and RNaseH activities of the HBV polymerase protein. Inhibiting the RNaseH causes premature truncation of minus-polarity DNA strands, accumulation of RNA/DNA heteroduplexes within viral capsids, and failure to synthesize the viral plus-polarity DNA strand. This lethally damages the genome, rendering it unable to function in virions or be converted to cccDNA. Therefore, monotherapy with RNaseH inhibitors could be as effective as inhibiting the reverse transcriptase with nucleos(t)ide analogs. As novel inhibitors targeting a distinct essential enzymatic activity of the virus, RNaseH inhibitors would be good candidates for use in combination therapies with nucleos(t)ide analog and other HBV drugs against new targets that are under development.
■ IDENTIFICATION OF RNaseH INHIBITORS
We screened >500 compounds, primarily based on their similarity to inhibitors of the HIV RNaseH, in biochemical HBV RNaseH assays and/or cell-based HBV replication inhibition assays. Over 100 inhibitors that suppress HBV replication in culture with 50% effective concentration (EC50) values from ~100 nM to the low micromolar range were identified. 50% cytotoxic concentrations (CC50) range from 3 to >100 μM by MTS assays, leading to therapeutic indexes (TIs, CC50/EC50) of up to 700. These inhibitors are found mostly in two chemotypes, the α-hydroxytropolones (αHTs; Figure 2B) and N-hydroxyimides (Figure 2C).5–9 The N-hydroxyimides can be subdivided into three classes we have studied: N-hydroxyisoquinolinediones (HIDs), N-hydroxypyridinediones (HPyDs), and N-hydroxynapthyridinones (HNOs). In addition, Huber et al. recently screened 52 structurally related N-hydroxypyrimidinediones and found two hits, the most potent of which suppressed HBV replication in cells with an EC50 of 5.5 μM and a CC50 > 100 μM10 (17 in Huber et al., Figure 2C).
Figure 2.
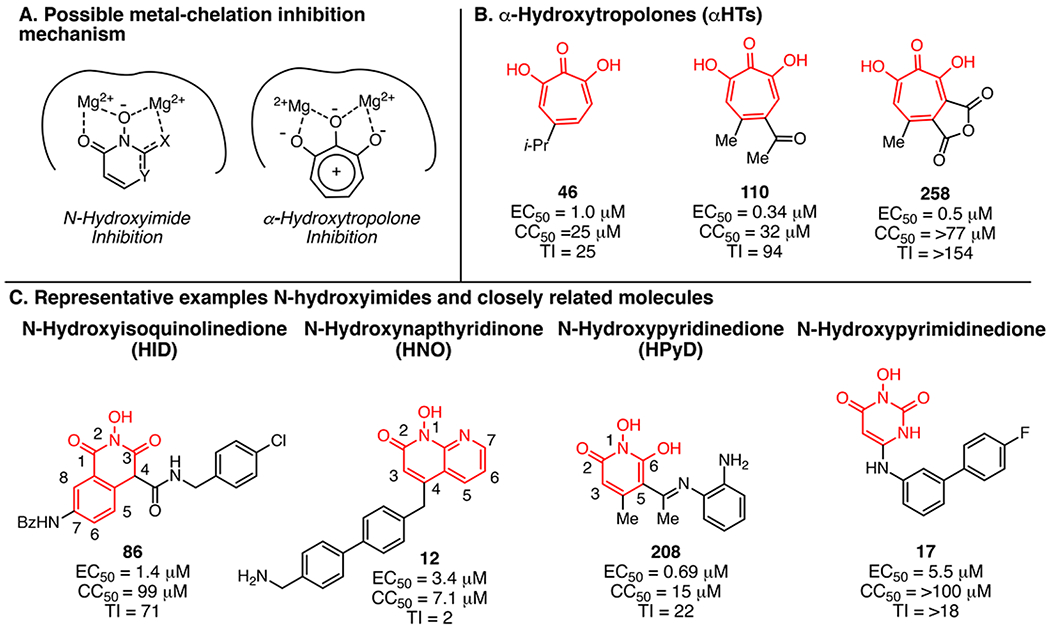
Presumed mechanism of RNaseH inhibition and exemplary compounds. (A) Coordination of two Mg2+ ions in the geometry in which they are found in RNaseH active sites by the N-hydroxyimide and α-hydroxytropolone pharmacophores. (B) Example α-hydroxytropolone RNaseH inhibitors. (C) Example N-hydroxyimide and closely related classes of inhibitors.
Most HIV RNaseH inhibitors coordinate the two catalytic Mg2+ ions in the RNaseH active site, usually through a trident array of Lewis basic atoms or electron donors. Likewise, all HBV RNaseH inhibitors identified to date contain a trident of oxygen or nitrogen atoms. Disrupting the trident ablates inhibition, implying a similar inhibition mechanism between HBV and HIV RNaseH inhibitors. Furthermore, hydroxytropolones and N-hydroxyimides are relatively acidic, which may allow them to chelate to the metals in an ionic or even dianioinic state (Figure 2A).
The compounds inhibit HBV RNaseHs from three genotypes, indicating that HBV’s high genetic diversity probably will not hinder drug development. The RNaseH inhibitors are synergistic with lamivudine and additive with the HBV capsid protein assembly modifier Hap12 without exacerbating cytotoxicity.11 Two RNaseH inhibitors, an HPyD (208) and an αHT (110), can significantly suppress HBV replication in chimeric mice carrying humanized livers.12 These data validate the RNaseH as a drug target.
■ α-HYDROXYTROPOLONE INHIBITORS
αHTs are highly oxygenated troponoids that can inhibit dinuclear metalloenzymes (Figure 2B),13 including viral nucleases such as the HIV, HBV, and xenotropic murine leukemia-related virus RNaseHs. Antiviral activity is believed to depend on their ionic character at physiological pH due to the acidity of both hydroxyls and the Lewis basic nature of the tropolone carbonyl that promotes coordination of the active site Mg2+ ions.
Most of what is known about the biological activity of αHTs has been driven by studies of natural product troponoids. The first αHT found to inhibit the HBV RNaseH was the natural product β-thujaplicinol (46; Figure 2B), which has an EC50 of 1.0 μM and a CC50 of 25 μM. There are only a few sources of natural αHTs, and until recently, there have been few strategies available to make synthetic αHTs. As a result, development of therapeutic αHTs has been stifled beyond the discovery phase.
The Murelli group pioneered new approaches for αHT synthesis, focusing on an oxidopyrylium cycloaddition/ring-opening strategy.14 165 structurally diverse αHTs and related troponoids have been assessed against HBV, and some αHTs have been synthesized on multigram scales. Eighty αHTs are active against HBV replication at ≤20 μM, with EC50’s as low as 0.11 μM. These studies have led to structurally novel synthetic αHTs such as 110 that have improved potency, TI values, and pharmacokinetic properties. While a clear structure—activity relationship (SAR) is still being established, it appears that small electron-withdrawing appendages are beneficial, and cytotoxicity increases with lipophilicity and the number of aromatic rings attached to the αHT pharmacophore.8
■ N-HYDROXYIMIDE AND CLOSELY RELATED INHIBITORS: N-HYDROXYISOQUINOLINEDIONES, N-HYDROXYPYRIDINEDIONES, AND N-HYDROXYNAPTHYRIDINONES
Three chemotypes containing a hydroxyimide or N-hydroxyimide-like moiety can inhibit the HBV RNaseH: the HIDs, HPyDs, and HNOs (Figure 2C).7,9 Like the αHTs, both the HPyD and HID scaffolds have an oxygen trident that is essential for activity. The HNO scaffold, exemplified by 12, lacks this trident because one of the oxygens is replaced with an aromatic nitrogen that also has a lone pair of electrons available to coordinate a Mg2+ cation. The major stabilizing interactions of the HBV RNaseH—inhibitor complexes are probably electrostatic, and they appear to be formed primarily between the chelating moiety of the ligands (N-hydroxyimide group) and the polar catalytic center of the enzyme. In each case, strong ionic interactions along with charge-assisted hydrogen bonds presumably anchor the chelating moiety to the Mg2+ ions.
The various N-hydroxyimide cores provide different directionality for branching, and empirically developed preliminary SARs for these appendages to some of the cores have been generated. For example, placing an alkyl groups at C4 on the HID scaffold is not tolerated, but including a carbonyl group as either an ester or amide at C4 (e.g., 86) is acceptable. This is believed to be due to the increased acidity of the α-hydrogen and therefore the ability to coordinate the Mg2+ ions in the ionic form. As is also illustrated in 86, extension of large groups in the 7-position is also tolerated. In contrast, adding a third ring that condenses positions 6 and 7 and a second nitrogen on the HID scaffold to create indole-flutimide analogues is not tolerated.9 Molecules generated from the HNO class focus mostly on the C4 position, which for this molecule is para or 180° to the N-hydroxyimide. The top molecule of this class to date, 12, has a methylenebiarylmethyl-amine appendage, although this activity is only slightly better than truncated versions possessing an aminobenzyl group. At present, the HPyDs appear to be the most promising N-hydroxyimides for further development.
■ SCREENING FOR NOVEL INHIBITORY CHEMOTYPES
The existing HBV RNaseH inhibitors were discovered by screening compounds similar to HIV RNaseH inhibitors. Expanding this repertoire will require exploring novel chemical space. Unfortunately, high-throughput biochemical screening is currently not feasible due to limitations of the recombinant HBV RNaseH. Although the enzyme can be used for mechanistic confirmation of RNaseH inhibition, it underestimates efficacy compared to replication inhibition assays by 50- to 100-fold and has an unacceptably high false-negative rate. Phenotypic screens measuring suppression of viral replication are feasible, but they must measure reduction of the viral plus-polarity DNA strand because the truncated minus-polarity DNA strands produced in the absence of RNaseH activity make standard PCR detection of RNaseH inhibitors very insensitive. Therefore, a strand-preferential screening protocol was developed8,9 that is amenable to a 96-well format, but its low throughput and high cost limit use to midthroughput screening at present. This continues to necessitate hypothesis-based screening rather than unbiased sampling of chemical diversity.
■ OUTLOOK FOR RNaseH INHIBITORS
Drugs to improve control and hopefully eliminate HBV will have to address the unique features of this infection. Patients typically begin treatment after decades of chronic infection, when serious liver damage has usually already occurred. In addition, treatment will take many months to years to clear or stably control HBV due to the unusual stability of the cccDNA that is the key molecule to be eradicated or inactivated to achieve a cure. As the cccDNA, like virions, is produced by reverse transcription1 (Figure 1B), control of HBV replication will need to be continuous and profound to prevent replenishment of the cccDNA. Therefore, new drugs against HBV will have to have very low toxicity, act by a variety of mechanisms, have outstanding pharmacokinetic properties to ensure long-lasting efficacy, be active against a wide range of HBV’s diverse genotypes, and be compatible with many other drugs. It is unlikely that any one drug will fully meet these demands, emphasizing the need for a broad approach for developing new anti-HBV drugs.
In this light, RNaseH inhibitors are promising candidates for further development given their relative insensitivity to HBV’s genetic diversity and ability to synergize with drugs from other classes.11 Key challenges to be addressed for RNaseH inhibitors to achieve their promise include developing high-throughput screening assays to access more chemical space, advancing synthetic methods to more rapidly derivatize the pharmacophores already identified, and exhaustively assessing the pharmacological and toxicological profiles of the inhibitors. If successful, RNaseH inhibitors may one day take their place in the cocktail(s) of drugs that will be deployed against HBV.
■ ACKNOWLEDGMENTS
We thank Elena Lomonosova for constructive discussions. Writing this Viewpoint was supported by NIH Grants R01 AI122669 and R21 AI124672.
■ ABBREVIATIONS
- αHT
α-hydroxytropolone
- CC50
50% cytotoxic concentration
- cccDNA
covalently closed circular DNA
- EC50
50% effective concentration
- HBV
hepatitis B virus
- HID
N-hydroxyisoquinolinedione
- HIV
human immunodeficiency virus
- HNO
N-hydroxynapthyridinone
- HPyD
N-hydroxypyridinedione
- RNaseH
ribonuclease H
- SAR
structure—activity relationship
- TI
therapeutic index
Footnotes
The authors declare the following competing financial interest(s): J.E.T., M.J.M., and R.P.M. are inventors on pending patent applications covering use of the RNaseH inhibitors as treatments for HBV infection. J.E.T. is a consultant for Seventh Wave Laboratories, Inc. regarding HBV biology.
■ REFERENCES
- (1).Seeger C, Zoulim F, and Mason WS (2013) Hepadnaviruses In Fields Virology (Knipe DM, and Howley PM, Eds.) 6 ed., pp 2185–2221, Lippincott Williams & Wilkins, Philadelphia PA. [Google Scholar]
- (2).WHO. (2017) Hepatitis B vaccines: WHO position paper - July 2017. Wkly. Epidemiol. Rec 92 (27), 369–392. [PubMed] [Google Scholar]
- (3).Cheng PN, Liu WC, Tsai HW, Wu IC, Chang TT, and Young KC (2011) Association of intrahepatic cccDNA reduction with the improvement of liver histology in chronic hepatitis B patients receiving oral antiviral agents. J. Med. Virol 83 (4), 602–607. [DOI] [PubMed] [Google Scholar]
- (4).Lok AS (2011) Does antiviral therapy for hepatitis B and C prevent hepatocellular carcinoma? J. Gastroenterol. Hepatol 26 (2), 221–227. [DOI] [PubMed] [Google Scholar]
- (5).Cai CW, Lomonosova E, Moran EA, Cheng X, Patel KB, Bailly F, Cotelle P, Meyers MJ, and Tavis JE (2014) Hepatitis B virus replication is blocked by a 2-hydroxyisoquinoline-1,3(2H,4H)-dione (HID) inhibitor of the viral ribonuclease H activity. Antiviral Res 108, 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lu G, Lomonosova E, Cheng X, Moran EA, Meyers MJ, Le Grice SF, Thomas CJ, Jiang JK, Meck C, Hirsch DR, D’Erasmo MP, Suyabatmaz DM, Murelli RP, and Tavis JE (2015) Hydroxylated Tropolones Inhibit Hepatitis B Virus Replication by Blocking the Viral Ribonuclease H Activity. Antimicrob. Agents Chemother 59 (2), 1070–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Tavis JE, Cheng X, Hu Y, Totten M, Cao F, Michailidis E, Aurora R, Meyers MJ, Jacobsen EJ, Parniak MA, and Sarafianos SG (2013) The hepatitis B virus ribonuclease h is sensitive to inhibitors of the human immunodeficiency virus ribonuclease h and integrase enzymes. PLoS Pathog 9 (1), e1003125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lomonosova E, Daw J, Garimallaprabhakaran AK, Agyemang NB, Ashani Y, Murelli RP, and Tavis JE (2017) Efficacy and cytotoxicity in cell culture of novel alpha-hydroxytropolone inhibitors of Hepatitis B virus ribonuclease H. Antiviral Res 144, 164–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Edwards TC, Lomonosova E, Patel JA, Li Q, Villa JA, Gupta AK, Morrison LA, Bailly F, Cotelle P, Giannakopoulou E, Zoidis G, and Tavis JE (2017) Inhibition of hepatitis B virus replication by N-hydroxyisoquinolinediones and related polyoxygenated heterocycles. Antiviral Res 143, 205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Huber AD, Michailidis E, Tang J, Puray-Chavez MN, Boftsi M, Wolf JJ, Boschert KN, Sheridan MA, Leslie MD, Kirby KA, Singh K, Mitsuya H, Parniak MA, Wang Z, and Sarafianos SG (2017) 3-Hydroxypyrimidine-2,4-Diones as Novel Hepatitis B Virus Antivirals Targeting the Viral Ribonuclease H. Antimicrob. Agents Chemother 61 (6), e00245–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lomonosova E, Zlotnick A, and Tavis JE (2017) Synergistic Interactions between Hepatitis B Virus RNase H Antagonists and Other Inhibitors. Antimicrob. Agents Chemother 61 (3), e02441–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Long KR, Lomonosova E, Li Q, Ponzar NL, Villa JA, Touchette E, Rapp S, Liley RM, Murelli RP, Grigoryan A, Buller RM, Wilson L, Bial J, Sagartz JE, and Tavis JE (2018) Efficacy of hepatitis B virus ribonuclease H inhibitors, a new class of replication antagonists, in FRG human liver chimeric mice. Antiviral Res 149, 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Meck C, D’Erasmo MP, Hirsch DR, and Murelli RP (2014) The biology and synthesis of alpha-hydroxytropolones. MedChemComm 5 (7), 842–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Meck C, Mohd N, and Murelli RP (2012) An oxidopyrylium cyclization/ring-opening route to polysubstituted alpha-hydroxytropolones. Org. Lett 14 (23), 5988–5991. [DOI] [PMC free article] [PubMed] [Google Scholar]