

Abstract
Cell-based respirometers, such as the Seahorse Extracellular Flux Analyzer, are valuable tools to assess the functionality of mitochondria within adherent neurons, as well as other cell types. The Mito Stress Test is the most frequently employed protocol of drug additions to evaluate mitochondrial bioenergetic function. Sequential exposure of cells to an ATP synthase inhibitor such as oligomycin and an uncoupler such as FCCP cause changes in oxygen consumption rate that allow estimation of the cellular efficiency and capacity for mitochondrial ATP synthesis. While a useful first step in assessing whether an experimental treatment or genetic manipulation affects mitochondrial energetics, the Mito Stress Test does not identify specific sites of altered respiratory chain function. This article discusses limitations of the Mito Stress Test, proposes a refined protocol for comparing cell populations that requires independent drug titrations at multiple cell densities, and describes a stepwise series of respirometry-based assays that “map” locations of electron transport deficiency. These include strategies to test for cytochrome c release, to probe the functionality of specific electron transport chain complexes within intact or permeabilized cells, and to measure NADH oxidation by the linked activity of Complexes I, III, and IV. To illustrate utility, we show that although UK5099 and ABT-737 each decrease the spare respiratory capacity of cortical neurons, the stepwise assays reveal different underlying mechanisms consistent with their established drug targets: deficient Complex I substrate supply induced by the mitochondrial pyruvate carrier inhibitor UK5099 and cytochrome c release induced by the anti-apoptotic BCL-2 family protein inhibitor ABT-737.
Keywords: Seahorse, oxygen, respiration, bioenergetics, cytochrome c, pyruvate, BCL-2, BH3, ABT-737, UK5099
Graphical abstract
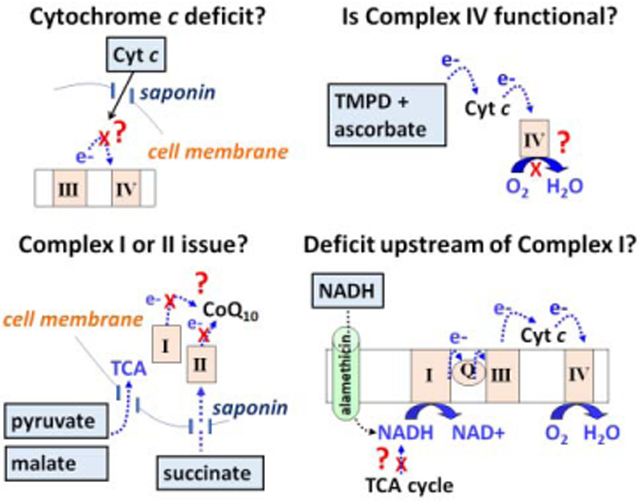
Introduction
The study of mitochondrial biology across a wide spectrum of research areas has accelerated in recent years. These areas include cancer, diabetes, cardiovascular disease, neurodegenerative disorders, and normal brain physiology. The advent of novel cell-based respirometers was crucial to the expansion of neural bioenergetic studies (Jekabsons & Nicholls 2004, Wu et al. 2007). Prior to cell respirometers, oxygen consumption by neurons or astrocytes was measured in suspension. However, cell detachment from culture surfaces caused undesirable functional alterations. Furthermore, the sensitivity of polarographic techniques precluded the study of less abundant brain cells like microglia. Now, multi-well respirometers such as the Seahorse Extracellular Flux (XF1) Analyzers (Agilent Technologies) that were specifically designed for adherent cells allow determination of multiple aspects of mitochondrial function in situ using carefully designed serial drug injection protocols.
One such protocol, the “Mito Stress Test” assay, describes a series of drug additions consisting of an ATP synthase inhibitor, an uncoupler, and one or more electron transport chain inhibitors. This experiment yields basal respiration rate and estimates of i) the fraction of mitochondrial oxygen consumption used for ATP synthesis, ii) the fraction driven by proton leak, iii) maximal respiratory capacity, iv) spare respiratory capacity, and v) non-mitochondrial respiration. Although this protocol is widely employed in the literature to identify differences in mitochondrial function due to disease conditions, genetic alterations, or toxin treatments, further exploration of the mechanistic bases for observed differences is often neglected. The purpose of this article is to provide recommendations for validating the results of the Mito Stress Test and to describe additional assays that probe the root cause(s) of electron transport chain deficiencies. Although the descriptions below are tailored to the multi-well Seahorse XF respirometers, the same tests are applicable to other cell-based respirometers or to oxygen-sensitive fluorophores used in standard microplate readers.
Materials and Methods
Materials
Reagents were from purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated. Cell culture supplies were obtained from Invitrogen (Grand Island, NY).
Cell culture
Rat cortical neurons and established MCF10A cell lines were cultured as described below. Because we could not achieve a high transfection efficiency using neurons, the established cell lines were employed to demonstrate the comparison of genetically manipulated cell populations.
Primary cultures of cortical neurons were prepared from a pool of embryonic day 18 rat cortices (~10 per preparation) that were obtained from both males and females (Stoica et at. 2005, Yakovlev et at. 2001). The University of Maryland Institutional Animal Care and Use Committee (IACUC) approved all procedures, which were in accordance with the NIH Guide for the Care and Use of Laboratory Animals. A cell density of 60,000-100,000 cells/well (0.32 cm2) was used in V7-PS or V28-PS microplates (Agilent Technologies, Santa Clara, CA). Cytosine arabinofuranoside (5 μM, Sigma-Aldrich) was added to the cultured cells after four days in vitro to inhibit glial proliferation. The neurons were maintained for 11-15 days in vitro as described (Clerc & Polster 2012), with this method yielding a >95% neuron-enriched culture at the time when experiments were performed (Laird et al. 2013).
MCF10A human mammary epithelial cells stably overexpressing BCL-2 or harboring an empty vector (pcDNA3) were described previously (Martin & Leder 2001). These cells were cultured in a 1:1 mixture of DMEM and F12 medium (DMEM-F12) containing 5% horse serum and the following supplements: hydrocortisone (0.5 μg/ml), insulin (10 μg/ml), epidermal growth factor (20 ng/ml), penicillin (100 IU/ml), and streptomycin (100 μg/ml). Cells were maintained at 37°C in a humidified atmosphere of 95% air/5% CO2.
Oxygen consumption rate (OCR) measurements by cell-based respirometry
Oxygen consumption by intact or permeabilized cells was measured at 37°C using a Seahorse XF24 Extracellular Flux Analyzer (Wu et al. 2007, Gerencser et al. 2009). Cells cultured in V7-PS or V28-PS plates were washed twice in 0.5 ml of artificial cerebrospinal fluid (aCSF) assay medium and then incubated in 0.675 ml aCSF at 37°C in a CO2-free incubator for one hour prior to measurements. The aCSF consisted of 120 mM NaCl, 3.5 mM KCl, 1.3 mM CaCl2, 0.4 mM KH2PO4, 1 mM MgCl2, 5 mM HEPES, 4 mg/ml fatty acid free bovine serum album (BSA, Sigma-Aldrich, catalogue No. A6003), and 15 mM glucose, pH 7.4. As described previously (Clerc & Polster 2012), experiments in which cells were permeabilized during the course of the assay were performed in aCSF containing an increased concentration of HEPES (20 mM instead of 5 mM) to limit the extent of acidification caused by the binding of calcium to the EGTA in the permeabilization mixture (see below).
Respirometry assays consisted of cycles of 2-3 min mix, 3 min wait, and 2 min measure for experiments with intact cells and of 2.5 min mix, 0.5 min wait, and 2 min measure for experiments in which cells were permeabilized. The shortened measurement cycle allowed for a greater number of measurements following permeabilization before the time-dependent decline of mitochondrial function. The compounds described in the figure legends were injected sequentially from stocks prepared in 75 μl assay medium at 10X, 11X, 12X, and 13X the final working concentration for injections 1-4, respectively. Drug concentrations described throughout the article are the final working concentration unless otherwise specified.
To measure oxygen consumption by permeabilized cells, saponin (25 μg/ml) was co-injected with 3.6 mM K2HPO4, 1 mM ADP, 5 mM EGTA, and either a combination of pyruvate and malate (5 mM each) or succinate (5 mM) and rotenone (0.5 μM). The saponin concentration was optimized both for cell type and the individual saponin lot, as the purity of the active saponins (amphipathic glycosides) depends on the source. Both saponin and pyruvate were prepared freshly from powder in aCSF for all experiments. EGTA, which releases a proton when it binds to calcium (Patton et al. 2004), was diluted from a 250 mM stock that was pH-adjusted to 7.4. The aCSF pH was reduced to 7.0 following injection of the permeabilization mixture, modeling a normal cytoplasmic pH.
For the Complex I-III-IV-linked NADH oxidation assay, the bacterial pore-forming peptide alamethicin (40 μg/ml) was added in conjunction with saponin to porate the mitochondrial inner membrane, and NADH (2 mM, prepared freshly in aCSF) replaced the Complex I or ll-linked substrates in the permeabilization mixture. Purified horse heart cytochrome c (100 μM) was included to replace endogenous cytochrome c loss. Neurons cultured in V28-PS microplates were used for this assay. V28-PS microplates have a measurement volume of 28 μl, in contrast to the 7 μl measurement volume of V7-PS plates. The greater volume prevents excessive oxygen depletion of the media during the measurement period (e.g. when rates exceed 1000 pmol O2/min), extending the upper limit of accurate OCR measurements. These plates are very useful for respirometry assays using mitochondria-rich cells like neurons and cardiomyocytes that have high respiratory capacity.
Data analysis
The percent change rather than the absolute OCR is of the most interest when comparing OCR after drug exposure to OCR pre-exposure within a single population of cells. Therefore, to reduce variability primarily due to well-to-well differences in cell plating/survival, results are generally depicted as normalized OCR (% baseline rate) as previously described (Wu et al. 2007, Schuh et al. 2011b). Both absolute and baseline-normalized OCR values are shown in the Fig. 1 optimization example. All data, with the exception of this example in Fig. 1, are representative of at least two independent experiments performed in at least duplicate.
Fig. 1. Comparison of cell respiratory capacities.

BCL-2 overexpressing MCF10A cells were compared with vector controls (CON) at the indicated plating cell densities (30,000 vs. 60,000 cells/well). Oxygen consumption rates (OCR) were measured following overnight cell proliferation. FCCP was added sequentially at 4 μM and 2 μM, respectively, followed by pyruvate (pyr, 10 mM) and antimycin A (Anti A, 1 μM). A & C) Absolute OCR values are shown (mean ± SD, n=3 wells). B & D) Baseline-normalized OCR values relative to the third rate, which was set at 100%, are displayed.
Results and Discussion
Comparing the respiratory capacity of two or more cell populations
A common application of the Mito Stress Test is to compare the bioenergetic properties of two or more cell populations that differ by at least one experimental condition, frequently a drug pre-treatment or a genetic manipulation (e.g. gene overexpression or deletion). In the test, the two cell populations are sequentially exposed to the ATP synthase inhibitor oligomycin, an uncoupler such as carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP) or 2,4-dinitrophenol (DNP), and one or more electron transport chain inhibitors, typically the Complex I inhibitor rotenone and the Complex III inhibitor antimycin A. With this test, it is easy to detect major (>50%) differences across multiple aspects of mitochondrial function. Thus, it is a good first choice for the test of mitochondria-centered hypotheses, particularly when the amount of biological material is limiting. However, alterations detected by the Mito Stress Test may result from both direct and indirect effects on the oxidative phosphorylation system or reflect non-optimal assay conditions, which must be further delineated by additional experiments.
Assay conditions that influence accurate determination of respiratory capacity, potentially leading to false negative or positive conclusions if the Mito Stress Test is the only protocol/test applied, include i) the dose of uncoupler, ii) the use of oligomycin, and iii) the choice of energy substrate. Specifically, some uncouplers like FCCP exhibit bell-shaped dose-response curves, necessitating optimization for independent cell populations. This is because FCCP stimulates respiration due to uncoupling up to a certain concentration, but inhibits respiration when the optimal uncoupling concentration relative to cell number is exceeded (Dranka et al. 2011). Second, oligomycin is added prior to uncoupler in the Mito Stress Test. However, oligomycin impairs the subsequent response to uncoupler in many cell types (Ruas et al. 2016, Compton et al. 2011, Kim et al. 2014), leading to an underestimation of the maximal respiration rate. Third, maximal respiration rate in many cell types is limited by the endogenous supply of NADH and FADH2 from the tricarboxylic acid (TCA) cycle to the electron transport chain, particularly when glucose is the only energy substrate provided (Clerc et al. 2012, Clerc et al. 2013, Laird et al. 2013).
We recommend the following to more rigorously compare the respiratory capacities of two or more cell populations: i) titrate the uncoupler (e.g. FCCP or DNP) in the absence of oligomycin at multiple cell densities and ii) supply exogenous pyruvate (10 mM) together with the uncoupler. An uncoupler causes many secondary changes within cells that, over time, may feedback to decrease respiration. Therefore, titrating the uncoupler using parallel wells rather than sequentially within individual wells is preferred. The uncoupler titration and pyruvate supplementation ensure both that the optimal dose of uncoupler is applied and that any limitation in pyruvate supply from glycolysis to the TCA cycle is overcome. Although pyruvate is often included in the Mito Stress Test assay medium at 1 mM, in neurons this concentration is insufficient to support maximal uncoupled respiration (Laird et al. 2013). Supplying pyruvate acutely, and only to uncoupled cells, avoids any effects of exposing cells to a non-physiological level of pyruvate for a prolonged period of time (e.g. on the NADH/NAD+ redox state). In some cases, one may want to estimate respiratory capacity both with and without limitation by endogenous substrate supply. This can be done by injecting pyruvate after uncoupling the cells, as we will see below.
Fig. 1 illustrates the utility of these recommendations using a genetically manipulated cell line. MCF10A BCL-2 overexpressing cells were compared with vector controls at multiple cell densities on the same plate. Large differences in basal and FCCP-stimulated respiration were observed between control cells and cells overexpressing the anti-apoptotic protein BCL-2 when cells were plated the night before the experiment at 30,000 cells per well (Fig. 1A). BCL-2 overexpressing cells proliferate at a faster rate than control cells and were present at a higher density at the time of assay (data not shown), likely leading to the difference in basal OCR. For reasons that will be made clear below, it is best practice to compare cell populations when they are at similar densities. This is easiest to achieve when cells are plated at multiple densities on the same plate.
Various methods of normalization can reduce differences in absolute OCR that are due to different cell numbers at the time of assay. Although normalization to total cellular protein is commonly used, this type of normalization is difficult if BSA is included in the assay medium because BSA protein complicates cellular protein measurements. We included BSA in our assays here due to the several advantages discussed previously (Jekabsons & Nicholls 2004, Yadava & Nicholls 2007, Schuh et al. 2011a, Bordt et al. 2017), which include modeling the extracellular protein concentration and promoting optimal drug responses. While directly imaging and counting cells is perhaps the best method of normalization, this method is very time consuming without expensive equipment to automate the process. An alternative method is to normalize respiration compared to the baseline rate (i.e. prior to any drug additions). Although this method does not allow one to compare basal respiration rates, it still enables comparisons of coupling and spare respiratory capacities.
Notably, BCL-2 overexpressing cells exhibited far greater uncoupled OCR relative to the vector control cells even after OCR was normalized to basal respiration rate (Fig. 1B), which is suggestive of higher spare respiratory capacity. This difference was not due to a disparity in the supply of mitochondrial substrate from glucose, as exogenous pyruvate (10 mM) did not abolish the difference despite stimulating uncoupled respiration in the BCL-2 overexpressing cells. Note that from the uncoupler titration in Fig. 1 it is not clear that maximal respiration was obtained since an OCR increase after the second addition of FCCP was observed. This was determined in subsequent experiments (data not shown).
Markedly different results were observed for control and BCL-2 overexpressing cells plated at 60,000 cells per well on the same plate at the same time. Basal respiration by MCF10A BCL-2 overexpressing cells was the same regardless of the plating density (60,000 vs. 30,000 cells per well, see Fig. 1C compared with Fig. 1A), as in both cases the cells were a confluent monolayer at the time of assay. However, the vector control cells plated at 60,000 cells per well exhibited the same basal respiration rate as the BCL-2 overexpressing cells. In addition, their response to the uncoupler FCCP was suggestive of a higher spare respiratory capacity relative to the cells plated in parallel at the lower density, and this response failed to differ from the response in the BCL-2 overexpressing cells (Fig. 1C and D). Visual inspection confirmed that the MCF10A control cell density at the time of assay was the same as that of the cells overexpressing BCL-2 (data not shown). Thus, the interpretation of the results from this experiment depends on plating density. If one had only plated the cells at 30,000 cells per well, one might have concluded that BCL-2 overexpression increased mitochondrial spare respiratory capacity whereas if one had only plated the cells at 60,000 cells per well, one might have concluded that BCL-2 overexpression had no effect on spare respiratory capacity.
Additional experiments are clearly needed before making a generalized conclusion as to whether BCL-2 affects mitochondrial respiration, including further cell density and drug titrations, multiple BCL-2 overexpressing cell lines, and complementary ways of evaluating mitochondrial respiratory chain function. BCL-2 overexpression was initially reported to increase mitochondrial membrane potential based on the extent of cationic fluorescent dye uptake (Shimizu et al. 1998). It was later demonstrated that increased mitochondrial volume and structural complexity in BCL-2 overexpressing cells was likely responsible for the increased uptake of membrane potential-sensitive fluorescent dyes (Kowaltowski et al. 2002). No difference in respiration between BCL-2 overexpressing and vector control-transfected PC12 cells was observed across a range of uncoupler concentrations in that study (Kowaltowski et al. 2002), consistent with what we found under our 60,000 cells per well plating condition. Therefore, it is possible that there is a purely technical explanation for the different results when cells were plated at 30,000 cells per well, e.g. the vector control cells at the lower density failed to show increased respiration in response to FCCP because the optimal moles of uncoupler per cell number was already exceeded by the first 4 μM FCCP addition. However, alternative factors may have contributed to the cell density-specific result, such as the relative concentrations of secreted signaling factors at the different cell densities. Complementary techniques, for example, measuring mitochondrial volume and structural complexity to determine whether other effects of BCL-2 on the mitochondria are cell density-dependent, would be helpful in distinguishing technical explanations from true biological effects. Comparison of bioenergetics at multiple cell densities may be particularly important for immune cells such as microglia, which may show distinct phenotypes at high and low densities due to the levels of signaling molecules such as nitric oxide and superoxide that are produced.
The Mito Stress Test as a first-pass drug screening tool
As discussed above, the Mito Stress Test provides information on the resting respiration of cells, the percentage of respiration linked to ATP production, oxygen consumption due to mitochondrial inner membrane proton leak, total and spare respiratory capacities, and oxygen consumption due to non-mitochondrial processes (such as by enzymes that produce reactive oxygen species). Therefore, it is often used in drug screening to identify novel compounds with therapeutic potential or to eliminate drugs with potential dose-limiting mitochondrial toxicity. In Fig. 2, we show an example of two drugs, ABT-737 and UK5099, that were each identified by the Mito Stress Test to specifically impair the spare respiratory capacity of primary rat cortical neurons within less than two hours of drug addition when added at the same concentration. To highlight the wider applicability of cell-based respirometry, in the remainder of this article we illustrate how it can be determined that although the two drugs behave the same way in the Mito Stress Test, they limit mitochondrial respiration by very different mechanisms of action. Although an order for performing these tests (which are summarized in Table 1) is suggested, the tests can be performed in any order. The order in which they are employed should be dictated by the hypothesis or goal of the study.
Fig. 2. Identification of drugs that alter neuronal bioenergetics by the Mito Stress Test.

A & B) OCR of primary cortical neurons following the injection of 10 μM drug or vehicle control (CON), with ABT-737 added in A and UK5099 in B. The Mito Stress Test was conducted subsequent to the injection of the test drug, which consisted of the sequential addition of oligomycin (Oligo, 0.3 μg/ml), followed by FCCP (3 μM) or 2,4-dinitrophenol (DNP, 200 μM), and then antimycin A (Anti A, 1 μM). OCR values are means ± SD from three wells (with the exception of CON in B, which is the mean of two wells). OCR are baseline-normalized as in Fig. 1.
Table 1.
Assays for mapping respiratory chain deficiencies by respirometry.
| Assay | Protocol | When to use | Information obtained |
|---|---|---|---|
| Mito Stress Test | Sequential addition of ATP synthase inhibitor (e,g, oligomycin), uncoupler (e.g. FCCP), and electron transport chain inhibitor (e.g. antimycin A) | Screening for phenotype or when biological material is limiting | May reveal differences in mitochondrial coupling, respiratory capacity, and non-mitochondrial respiration |
| Cell density and uncoupler titrations | Determination of optimal uncoupler concentration ± pyruvate in parallel wells (ideally done on cells plated at multiple densities) | Accurate comparisons of mitochondrial respiratory capacity between two (or more) cell populations are desired | Enables quantitative comparisons of respiratory capacities and may reveal differences that are cell density-dependent |
| Cytochrome c test | Addition of plasma membrane permeabilizing agent (e.g. saponin) plus ADP and mitochondrial substrate ± purified cytochrome c | A reduction in spare respiratory capacity is observed in the Mito Stress Test or uncoupler titration | Evaluates whether cytochrome c loss-of-function/release has occurred |
| Complex IV test | Addition of TMPD + ascorbate + antimycin A | A reduction in spare respiratory capacity is observed that is not rescued by exogenous cytochrome c | Measures Complex IV function |
| Complex I/II test | Comparison of Complex I-vs. II-linked substrates in permeabilized cells | A reduction in spare respiratory capacity is observed that is not rescued by exogenous cytochrome c or addition of TMPD+ascorbate | Evaluates the functionality of Complexes I and II |
| Complex I-IV linked activity assay | Addition of saponin plus the mitochondrial inner membrane pore-forming peptide alamethicin and the direct Complex I substrate NADH | A reduction in spare respiratory capacity is observed and other tests suggest that all components of the respiratory chain are functional | Evaluates whether the respiratory deficit lies at Complex I or somewhere upstream (e.g. in a substrate transporter or TCA cycle dehydrogenase) |
Test 1: Is the respiratory impairment due to a mitochondrial cytochrome c deficit?
Cytochrome c is a soluble mitochondrial intermembrane space protein that transfers electrons between Complex III and Complex IV (Brand & Nicholls 2011). A cytochrome c deficit could potentially occur due to reduced expression, increased degradation, post-translational modification, or release from the mitochondria. Apoptotic cell death stimuli trigger mitochondrial outer membrane permeabilization, releasing cytochrome c into the cytoplasm. The amount of cytochrome c accessible to the electron transport chain limits mitochondrial respiratory capacity, but, importantly, not always basal respiration, following its release (Clerc et al. 2012). As many drugs cause toxicity through mitochondrial membrane compromise, testing for cytochrome c release is a logical first step.
A simple way to determine whether mitochondrial cytochrome c content limits mitochondrial respiratory capacity is by testing whether addition of exogenous purified cytochrome c rescues the respiratory deficit (Krippner et al. 1996, Polster et al. 2001, Mootha et al. 2001, Clerc et al. 2012, Clerc et al. 2014, Kim et al. 2014). Because cytochrome c, a protein of ~13 kilodaltons, is not membrane-permeable, a method of delivering cytochrome c to the mitochondria is needed. Membrane cholesterol-depleting agents such as saponin (Fiskum et al. 1980, Safiulina et al. 2004, Clerc & Polster 2012) or perfringolysin O (Kim et al. 2014, Divakaruni et al. 2017) can serve this purpose by selectively permeabilizing the plasma membrane of cells without compromising the mitochondrial outer or inner membranes. The selectivity of these agents for the plasma membrane is based on the far higher cholesterol content of the plasma membrane compared with the mitochondrial membranes. However, saponin should still be titrated to determine the minimal amount necessary to achieve plasma membrane permeabilization, as the mitochondrial outer membrane will become compromised at a high enough concentration [(~10-fold excess, (Clerc & Polster 2012)], confounding the assay.
Once the optimal concentration of saponin (or similar permeabilizing agent) is determined for the cell type of interest, saponin is added with or without excess exogenous cytochrome c after the desired time of incubation with the test compounds, in this case ABT-737 or UK5099 treatment for one hour (Fig. 3). Also included together with saponin in the injection is the calcium chelator EGTA to maintain extramitochondrial calcium concentration near the cytoplasmic calcium level (100 nM) following permeabilization (Clerc & Polster 2012) and a combination of mitochondrial substrate, ADP, and excess phosphate to induce maximal phosphorylating respiration. Any respiratory substrate(s) can be used for this experiment, provided that electrons enter the respiratory chain prior to cytochrome c. In Fig. 3A we demonstrate use of the Complex II substrate succinate for this assay, added in conjunction with the Complex I inhibitor rotenone, and in Fig. 3B we demonstrate use of the Complex I-linked substrates pyruvate and malate. A benefit to using succinate in combination with rotenone is that, unlike the Complex l-linked substrate pyruvate, succinate is not cell permeable. Therefore, it is very easy to detect whether an insufficient concentration of saponin is used, as the Complex I inhibitor rotenone will inhibit mitochondrial respiration if the plasma membrane has not been permeabilized.
Fig. 3. Respirometry test for a drug-induced cytochrome c deficit.

Cytochrome c (Cyt c) supplementation prevents the effect of ABT-737 (10 μM) on neuronal OCR (A) but does not rescue the UK5099 (10 μM)-induced respiratory deficit (B). To test for cytochrome c-limited respiration, neurons were permeabilized using saponin (SAP, 25 μg/ml), enabling delivery of purified Cyt c (100 μM), together with respiratory chain substrates, ADP, phosphate, and EGTA (see the Materials and Methods section for details). The substrate combinations were succinate and ADP in the presence of rotenone (S/R/A) in A and pyruvate, malate, and ADP (P/M/A) in B. To confirm that the OCR rescue by Cyt c was due to restored electron transfer to Complex IV, the Complex IV inhibitor azide (5 mM) was added. In B, antimycin A (Anti A, 1 μM) was also added subsequently to azide. CON, vehicle control. OCR values are means ± SD from three wells.
The data in Fig. 3A show that exogenous cytochrome c addition prevents ABT-737-induced respiratory inhibition, strongly suggesting that the impairment was caused by mitochondrial cytochrome c release. The Complex IV inhibitor azide inhibits the cytochrome c-rescued OCR, confirming that electron transfer to Complex IV occurred. In contrast to the result obtained with ABT-737, exogenous cytochrome c had no effect on the attenuation of respiration caused by UK5099 (Fig. 3B). This finding indicates that although ABT-737 and UK5099 both impair spare respiratory capacity in the Mito Stress Test, they do so by different mechanisms, with further tests required to understand the action of UK5099.
If confirmation of cytochrome c release (vs. an alternative cytochrome c loss-of-function mechanism) is desired, a cytochrome c enzyme-linked immunosorbant assay (ELISA) can be multiplexed to cell-based respirometry (Clerc et al. 2012). This assay, which we described previously, allows correlation between the amount of cytochrome c release in individual respirometry wells to the extent of respiratory suppression observed.
Test 2: Is the respiratory impairment due to Complex IV inhibition?
Once cytochrome c is excluded as the respiration-limiting factor, other components of the electron transport chain should be systematically investigated. This can be done by establishing experimental conditions that limit electron entry to one part of the chain and determining whether the deficit can still be observed.
In Fig. 4, we demonstrate how Complex IV function can be isolated by providing cells with the cytochrome c electron donor, N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD) and the TMPD reducing agent, ascorbate. Cells were first treated with the drug UK5099, followed by the uncoupler DNP and pyruvate to stimulate maximal respiration. As we saw in the Mito Stress Test (see Fig. 2), maximal respiration is impaired by UK5099 and, following injection of the Complex III inhibitor antimycin A, mitochondrial respiration is abolished (Fig. 4). However, for both the vehicle and UK5099 treatment groups, respiration in the presence of the Complex III inhibitor is rescued by TMPD plus ascorbate addition. Azide inhibits this rescue, indicating that oxygen consumption is due to complex IV activity. It is important to note that TMPD and ascorbate cause chemical consumption of oxygen, introducing a background oxygen consumption rate. This background rate can be subtracted by injecting the TMPD/ascorbate combination in a neighboring “blank” well containing no cells. Although azide-resistant TMPD/ascorbate-driven OCR remains higher than the antimycin A-insensitive OCR of cells even after background subtraction, azide is a low-affinity inhibitor, making it difficult to completely inhibit Complex IV activity in cells. We demonstrate this in the experiment depicted in Fig. 3B by adding the higher-affinity Complex III inhibitor antimycin A (1 μM) after 5 mM azide, finding that additional suppression of oxygen consumption by the electron transport chain is observed.
Fig. 4. Respirometry test for Complex IV activity.

Complex IV functionality was assessed by monitoring azide-sensitive neuronal OCR supported by the exogenous electron donor TMPD in the presence of the TMPD-reducing agent ascorbate and the Complex III inhibitor antimycin A. UK5099 (10 μM) or vehicle (CON) was added first, followed by 200 μM DNP and 10 mM pyruvate (DNP+Pyr) to induce maximal respiration. Anti A (1 μM) was injected ± TMPD/ascorbate (TMPD/Asc, 0.4 mM/8 mM) as indicated. Finally, azide (5 mM) was added to confirm the specificity of the assay. OCR values are means ± SD from three wells.
Test 3: Is the respiratory impairment due to inhibition of Complex I or II?
After establishing that UK5099 leaves Complex IV function intact, the cell permeabilization technique described above can then be used to drive respiration specifically through Complex I or Complex II. This was done in the experiment depicted in Fig. 5 by adding saponin, EGTA, phosphate, and ADP with either the Complex I-linked substrates pyruvate and malate or the Complex II substrate succinate, together with rotenone to inhibit Complex I. Either the UK5099 drug or its vehicle was then injected. We observed that in the presence of UK5099, an immediate respiration decrease occurs in the cells provided with the Complex I-linked substrates (Fig. 5A) but not those provided with succinate (Fig. 5B). These findings suggest that UK5099 impairs respiration either by directly inhibiting Complex I enzyme activity or by interfering with the conversion of pyruvate and malate to the Complex I substrate NADH.
Fig. 5. Permeabilized cell assay to test for a Complex I- or II-linked respiration deficit.

Neurons were permeabilized by saponin (SAP) in the presence of either the Complex I-linked substrates pyruvate and malate, as well as ADP (P/M/A) (A), or the Complex II-linked substrate succinate, with the Complex I inhibitor rotenone and ADP included (S/R/A) (B). Subsequently UK5099 (10 μM) or vehicle (CON) was added to test for substrate-specific effects on respiration. See the Materials and Methods section for full details on the permeabilization mixture. OCR values are means from two wells.
Note that because the cytochrome c test described by Fig. 3 also relies on permeabilized cells, one can “multiplex” the test to probe for specific effects on Complex I or II-linked respiration by repeating the test with different substrate combinations. For example, based on the results in Fig. 5B, if succinate had been used for the experiment in Fig. 3B instead of pyruvate plus malate, we would have failed to observe UK5099-induced respiratory inhibition.
Test 4: Does the respiratory impairment lie upstream of the electron transport chain?
Although we found that UK5099 inhibits Complex I-dependent, but not Complex II-dependent respiration, we cannot conclude that UK5099 is a Complex I inhibitor. This is because the possibility that the drug acts upstream of Complex I, for example, by interfering with the transport of the Complex I-linked substrates into the mitochondrial matrix or by inhibiting the matrix TCA cycle dehydrogenases that supply Complex I with NADH substrate, was not excluded. To evaluate this possibility, we need to directly measure Complex I-dependent NADH oxidation, which can be done in a cell respirometer by a Complex I-III-IV linked activity assay (Bordt et al. 2017).
To use NADH as a substrate, permeabilization of not only the plasma membrane, but also of the inner mitochondrial membrane, is required. In this protocol, saponin is added to permeabilize the plasma membrane, as in the Complex I-dependent respiration assay described above, but now the bacterial pore-forming peptide alamethicin is also included to enable NADH mitochondrial matrix penetration (Gostimskaya et al. 2003, Ji et al. 2012, Divakaruni et al. 2017). Because mitochondrial membranes are permeabilized in this assay, it is necessary to add exogenous cytochrome c, as described above, to maintain electron transfer to Complex IV, the oxygen-consuming component of the electron transport chain. Using this test (Fig 6), we observed that although UK5099 inhibits Complex I-dependent respiration (see Fig. 5), it has absolutely no effect on NADH-fueled Complex I-III-IV dependent oxygen consumption, in contrast to the classical Complex I inhibitor rotenone, which was used as a positive control. Thus, through a series of tests it was determined that UK5099 is not an electron transport chain inhibitor, but instead acts on a target upstream of mitochondrial Complex I to inhibit respiration. While not completely defining the drug target, cell-based respirometry was able to eliminate several likely possibilities and suggest the next logical steps: i) measurement of substrate transport across the mitochondrial inner membrane and ii) measurement of pyruvate and malate dehydrogenase activities in the presence and absence of UK5099. These experiments would have revealed UK5099 as a mitochondrial pyruvate carrier inhibitor (Halestrap & Denton 1975, Herzig et al. 2012, Bricker et al. 2012).
Fig. 6. Complex I-III-IV linked respirometry assay to evaluate the ability of Complex I to oxidize NADH.

Complex I activity was measured directly in permeabilized neurons by monitoring OCR supported by the substrate NADH. The neurons were permeabilized by saponin (25 μg/ml) and the pore-forming peptide alamethicin (40 μg/ml), which was used to deliver NADH across the mitochondrial inner membrane. Exogenous cytochrome c (100 μM) was added to compensate for the cytochrome c loss caused by alamethicin, allowing Complex I-III-IV electron transfer to occur so that OCR could be measured as a readout of Complex I activity. UK5099 (10 μM), vehicle (CON), or the positive control rotenone (Rot, 0.5 μM) was added together with NADH (2 mM) and the other permeabilization components (see Material and Methods section for full details). Subsequently, azide (5 mM) was added to block oxygen consumption by Complex IV. OCR values are means from two wells.
Additional considerations on the use of cell-based respirometry
Cellular metabolism is plastic and can be highly sensitive to changes in cell culture conditions. In addition to primary neurons, cell lines, e.g. mouse hippocampal HT-22 cells (Grohm et al. 2012) and human SH-SY5Y neuroblastoma cells (Schneider et al. 2011), are sometimes used to investigate neural cell bioenergetics. When performing cell line comparisons, we recommend passaging cells at a consistent cell density and tracking passage number, while carefully monitoring for any changes in cell morphology, growth rate, or other properties that may impact results. Over-confluence can cause bioenergetic changes due to medium acidification and inhibition of proliferation while plating cells too sparsely can impact their health due to an insufficient concentration of secreted survival/growth factors, potentially selecting for individual cells with more robust survival properties. When performing respirometry, visually inspecting cells before and after the assays, as well as subsequently to any wash steps, is critical for troubleshooting. Whether using cell lines or primary cell cultures, it is important to think carefully about the assay conditions employed in culture and how they relate to the environment the cells would normally experience in vivo, as well as to how they might influence the experimental question one is trying to address. Finally, it is important to recognize that in a mixed population of cells, even in cells derived from single clones, mitochondrial defects may vary among individual cells. Cell-based respirometry, even with improved protocols, can only deliver the average response of the cell population. Individual cell differences may be particularly important when investigating changes due to a prolonged drug exposure or genetic deletion/overexpression. Only single cell imaging or specialized techniques for measuring oxygen consumption by single cells can reveal the heterogeneity of responses (Connolly et al. 2018, Gleichmann et al. 2009).
Conclusions
Cell-based respirometry can provide a wealth of mechanistic information when multiple drug addition protocols are employed, but experiments need to be carefully optimized to avoid erroneous conclusions. The Mito Stress Test, while providing valuable information on mitochondrial bioenergetics in intact cells, might be better considered a starting point rather than the endpoint of functional analysis. The bioenergetic “blueprints” described herein should be broadly useful for deconstructing mitochondrial defects in a variety of cell types and diseases.
Acknowledgments
Funding: This work was supported by the National Institutes of Health [grant numbers R01NS085165 and R21NS096538]. The funding source had no role in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Footnotes
Declarations of interest: N.Y. receives royalties from Agilent Technologies for U.S. Patent US9915647B2. Agilent Technologies had no role in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
The abbreviations used in the paper are: aCSF, artificial cerebrospinal fluid; anti A, antimycin A; BSA, bovine serum albumin; CON, control; cyt c, cytochrome c; DNP, 2,4-dinitrophenol; FCCP, carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone; OCR, oxygen consumption rate; oligo, oligomycin; P/M/A, pyruvate + malate + ADP; pyr, pyruvate; SAP, saponin; S/R/A, succinate + rotenone + ADP; TCA, tricarboxylic acid; TMPD, N,N,N′,N′-tetramethyl-p-phenylenediamine; XF, Extracellular Flux
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bordt EA, Clerc P, Roelofs BA et al. (2017) The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev Cell, 40, 583–594 e586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD and Nicholls DG (2011) Assessing mitochondrial dysfunction in cells. Biochem J, 435, 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricker DK, Taylor EB, Schell JC et al. (2012) A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science, 337, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerc P, Carey GB, Mehrabian Z, Wei M, Hwang H, Girnun GD, Chen H, Martin SS and Polster BM (2012) Rapid detection of an ABT-737-sensitive primed for death state in cells using microplate-based respirometry. PLoS One, 7, e42487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerc P, Ge SX, Hwang H, Waddell J, Roelofs BA, Karbowski M, Sesaki H and Polster BM (2014) Drp1 is dispensable for apoptotic cytochrome c release in primed MCF10A and fibroblast cells but affects Bcl-2 antagonist-induced respiratory changes. Br. J. Pharmacol, 171, 1988–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerc P and Polster BM (2012) Investigation of mitochondrial dysfunction by sequential microplate-based respiration measurements from intact and permeabilized neurons. PLoS One, 7, e34465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerc P, Young CA, Bordt EA, Grigore AM, Fiskum G and Polster BM (2013) Magnesium sulfate protects against the bioenergetic consequences of chronic glutamate receptor stimulation. PLoS One, 8, e79982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton S, Kim C, Griner NB, Potluri P, Scheffler IE, Sen S, Jerry DJ, Schneider S and Yadava N (2011) Mitochondrial dysfunction impairs tumor suppressor p53 expression/function. J Biol Chem, 286, 20297–20312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly NMC, Theurey P, Adam-Vizi V et al. (2018) Guidelines on experimental methods to assess mitochondrial dysfunction in cellular models of neurodegenerative diseases. Cell Death Differ, 25, 542–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divakaruni AS, Andreyev AY, Rogers GW and Murphy AN (2017) In situ measurements of mitochondrial matrix enzyme activities using plasma and mitochondrial membrane permeabilization agents. Anal. Biochem, 552, 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranka BP, Benavides GA, Diers AR et al. (2011) Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic. Biol. Med, 51, 1621–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiskum G, Craig SW, Decker GL and Lehninger AL (1980) The cytoskeleton of digitonin-treated rat hepatocytes. Proc Natl Acad Sci U S A, 77, 3430–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerencser AA, Neilson A, Choi SW, Edman U, Yadava N, Oh RJ, Ferrick DA, Nicholls DG and Brand MD (2009) Quantitative microplate-based respirometry with correction for oxygen diffusion. Anal Chem, 81, 6868–6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleichmann M, Collis LP, Smith PJ and Mattson MP (2009) Simultaneous single neuron recording of O2 consumption, [Ca2+]i and mitochondrial membrane potential in glutamate toxicity. J Neurochem, 109, 644–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gostimskaya IS, Grivennikova VG, Zharova TV, Bakeeva LE and Vinogradov AD (2003) In situ assay of the intramitochondrial enzymes: use of alamethicin for permeabilization of mitochondria. Anal Biochem, 313, 46–52. [DOI] [PubMed] [Google Scholar]
- Grohm J, Kim SW, Mamrak U, Tobaben S, Cassidy-Stone A, Nunnari J, Plesnila N and Culmsee C (2012) Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ, 19, 1446–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP and Denton RM (1975) The specificity and metabolic implications of the inhibition of pyruvate transport in isolated mitochondria and intact tissue preparations by alpha-Cyano-4-hydroxycinnamate and related compounds. Biochem J, 148, 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, Kunji ER and Martinou JC (2012) Identification and functional expression of the mitochondrial pyruvate carrier. Science, 337, 93–96. [DOI] [PubMed] [Google Scholar]
- Jekabsons MB and Nicholls DG (2004) In situ respiration and bioenergetic status of mitochondria in primary cerebellar granule neuronal cultures exposed continuously to glutamate. J Biol Chem, 279, 32989–33000. [DOI] [PubMed] [Google Scholar]
- Ji F, Sharpley MS, Derbeneva O et al. (2012) Mitochondrial DNA variant associated with Leber hereditary optic neuropathy and high-altitude Tibetans. Proc Natl Acad Sci U S A, 109, 7391–7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Patel P, Gouvin LM, Brown ML, Khalil A, Henchey EM, Heuck AP and Yadava N (2014) Comparative Analysis of the Mitochondrial Physiology of Pancreatic beta Cells. Bioenergetics, 3, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowaltowski AJ, Cosso RG, Campos CB and Fiskum G (2002) Effect of Bcl-2 overexpression on mitochondrial structure and function. J Biol Chem, 277, 42802–42807. [DOI] [PubMed] [Google Scholar]
- Krippner A, Matsuno-Yagi A, Gottlieb RA and Babior BM (1996) Loss of function of cytochrome c in Jurkat cells undergoing fas-mediated apoptosis. J Biol Chem, 271, 21629–21636. [DOI] [PubMed] [Google Scholar]
- Laird MD, Clerc P, Polster BM and Fiskum G (2013) Augmentation of normal and glutamate-impaired neuronal respiratory capacity by exogenous alternative biofuels. Transl. Stroke Res, 4, 643–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SS and Leder P (2001) Human MCF10A mammary epithelial cells undergo apoptosis following actin depolymerization that is independent of attachment and rescued by Bcl-2. Mol Cell Biol, 21, 6529–6536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Wei MC, Buttle KF, Scorrano L, Panoutsakopoulou V, Mannella CA and Korsmeyer SJ (2001) A reversible component of mitochondrial respiratory dysfunction in apoptosis can be rescued by exogenous cytochrome c. EMBO J, 20, 661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton C, Thompson S and Epel D (2004) Some precautions in using chelators to buffer metals in biological solutions. Cell Calcium, 35, 427–431. [DOI] [PubMed] [Google Scholar]
- Polster BM, Kinnally KW and Fiskum G (2001) BH3 death domain peptide induces cell type-selective mitochondrial outer membrane permeability. J. Biol. Chem, 276, 37887–37894. [DOI] [PubMed] [Google Scholar]
- Ruas JS, Siqueira-Santos ES, Amigo I, Rodrigues-Silva E, Kowaltowski AJ and Castilho RF (2016) Underestimation of the Maximal Capacity of the Mitochondrial Electron Transport System in Oligomycin-Treated Cells. PLoS. ONE, 11, e0150967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safiulina D, Kaasik A, Seppet E, Peet N, Zharkovsky A and Seppet E (2004) Method for in situ detection of the mitochondrial function in neurons. J Neurosci Methods, 137, 87–95. [DOI] [PubMed] [Google Scholar]
- Schneider L, Giordano S, Zelickson BR, M SJ, G AB, Ouyang X, Fineberg N, Darley-Usmar VM and Zhang J (2011) Differentiation of SH-SY5Y cells to a neuronal phenotype changes cellular bioenergetics and the response to oxidative stress. Free Radic Biol Med, 51, 2007–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuh RA, Clerc P, Hwang H, Mehrabian Z, Bittman K, Chen H and Polster BM (2011a) Adaptation of microplate-based respirometry for hippocampal slices and analysis of respiratory capacity. J Neurosci Res, 89, 1979–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuh RA, Clerc P, Hwang H, Mehrabian Z, Bittman K, Chen H and Polster BM (2011b) Adaptation of microplate-based respirometry for hippocampal slices and analysis of respiratory capacity. J Neurosci Res, 89, 1979–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S, Eguchi Y, Kamiike W, Funahashi Y, Mignon A, Lacronique V, Matsuda H and Tsujimoto Y (1998) Bcl-2 prevents apoptotic mitochondrial dysfunction by regulating proton flux. Proc Natl Acad Sci U S A, 95, 1455–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica BA, Movsesyan VA, Knoblach SM and Faden AI (2005) Ceramide induces neuronal apoptosis through mitogen-activated protein kinases and causes release of multiple mitochondrial proteins. Mol Cell Neurosci, 29, 355–371. [DOI] [PubMed] [Google Scholar]
- Wu M, Neilson A, Swift AL et al. (2007) Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol Cell Physiol, 292, C125–C136. [DOI] [PubMed] [Google Scholar]
- Yadava N and Nicholls DG (2007) Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. J Neurosci, 27, 7310–7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakovlev AG, Ota K, Wang G, Movsesyan V, Bao WL, Yoshihara K and Faden AI (2001) Differential expression of apoptotic protease-activating factor-1 and caspase-3 genes and susceptibility to apoptosis during brain development and after traumatic brain injury. J Neurosci, 21, 7439–7446. [DOI] [PMC free article] [PubMed] [Google Scholar]