

Abstract
Despite mounting evidence implicating inflammation in cardiovascular diseases, attempts at clinical translation have shown mixed results. Recent preclinical studies have reenergized this field and provided new insights into how to favorably modulate cardiac macrophage function in the context of acute myocardial injury and chronic disease. In this review, we discuss the origins and roles of cardiac macrophage populations in the steady-state and diseased heart, focusing on the human heart and mouse models of ischemia, hypertensive heart disease, and aortic stenosis. Specific attention is given to delineating the roles of tissue-resident and recruited monocyte-derived macrophage subsets. We also highlight emerging concepts of monocyte plasticity and heterogeneity among monocyte-derived macrophages, describe possible mechanisms by which infiltrating monocytes acquire unique macrophage fates, and discuss the putative impact of these populations on cardiac remodeling. Finally, we discuss strategies to target inflammatory macrophage populations.
Keywords: monocyte, macrophage, dendritic cells, T cells, myocardial infarction, heart failure
INTRODUCTION
Despite recent advances in the treatment of cardiovascular disorders, heart failure remains a prevalent disease with significant morbidity, mortality, and burden on healthcare costs worldwide. This situation is likely to worsen as the global prevalence of atherosclerosis increases and the population ages, both of which are significant risk factors for the development of heart failure. Although current approaches to heart failure treatment including neurohormonal blockade, implantable defibrillators, and advanced heart failure therapies have improved survival (1), patient outcomes remain suboptimal. These observations highlight the opportunity and clinical importance of developing new modalities to prevent or treat heart failure.
An area of growing interest has been the role of inflammation in both the onset and progression of heart failure. Considerable similarities appear to exist between the roles of innate immune cells in heart failure and atherosclerosis (an area of heavy investigation). Atherosclerosis has been characterized as a chronic low-grade state of inflammation driven by excess cytokine [interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)] production and corresponding reduction in proresolving mediators (2). Patients with chronic heart failure tend to have higher levels of serum biomarkers associated with inflammation, including high-sensitivity C-reactive protein (hsCRP), IL-1β, IL-6, IL-8, and TNF, each of which has been found to correlate with clinical events (3–6). For example, the Controlled Rosuvastatin Multinational Trial in Heart Failure (CORONA) found that rosuvastatin lowered CRP levels, which correlated with fewer hospitalizations for cardiovascular causes in the 4,202 patients studied. Subgroup analysis of this study also found that IL-8 levels were correlated with all end points in 1,464 of the patients studied independent of N-terminal probrain natriuretic peptide (NT-proBNP), the most commonly used serum prognostic biomarker for heart failure severity (6).
Unfortunately, it has been challenging to translate these prognostic findings into clinically meaningful therapies (7). A prime example surrounds the use of TNF antagonists in two complementary clinical trials. The Randomized Etanercept Worldwide Evaluation (RENEWAL) program investigated a chimeric soluble TNF receptor (sTNFR2) in a population of 1,500 patients (8), and the Anti-TNFα Therapy Against Congestive Heart Failure (ATTACH) trial studied an anti-TNFα chimeric monoclonal antibody (infliximab) in 150 patients (9). Both trials enrolled patients with symptomatic heart failure with reduced ejection fraction (HFrEF) and elevated CRP levels. Neither trial observed a benefit, despite lowered CRP. In fact, the ATTACH trial was ended early due to higher rates of mortality and hospitalization in the high-dose group (8, 9). As a result, TNF blockade is considered contraindicated in HFrEF. These results were met with obvious disappointment, and many questioned the relevance of targeting inflammation in heart failure.
Recently, a sense of new hope has reinvigorated this field. Based on growing preclinical evidence and observational biomarker analyses, the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS) was designed as a pivotal phase III study to test the efficacy of IL-1β blockade in a cohort of high-risk coronary artery disease patients (10). This study demonstrated significant reductions in subsequent ischemic events and cardiovascular mortality. In addition to providing definitive support for the role of inflammation in atherothrombosis, subsequent analysis found that among the 10,061 patients enrolled, IL-1β blockade dose dependently reduced the rate of hospitalization due to heart failure (11). While the exact source of IL-1β and mechanism of action remain incompletely defined, these data provide compelling conceptual evidence that targeting inflammation is a beneficial approach for patients with atherosclerosis and resultant heart failure. Additional evidence for immunomodulation in ischemic heart disease stems from investigation of a monoclonal antibody against P-selectin (a cell-adhesion molecule important to immune cell trafficking), which was reported to reduce myocardial damage after percutaneous coronary intervention (PCI) in patients with non-ST-segment elevation myocardial infarction (SELECT-ACS Trial) (12, 13).
In this review, we discuss advances in our understanding of the initiation, propagation, and impact of inflammation on heart failure progression. In recent years, remarkable progress has been made in delineating the diversity and exploring the function of immune cell populations resident within and recruited to the heart. Although this review largely focuses on monocytes and macrophages as therapeutic targets, numerous other immune populations exist within the healthy and diseased myocardium and are likely to contribute to the heart failure pathogenesis.
CARDIAC MACROPHAGES RESIDENT IN THE STEADY-STATE HEART
The heart is an immunologically rich tissue, harboring more than 12 times as many immune cells as skeletal muscle (14). The majority of CD45+ (common leukocyte antigen) cells are macrophages as defined by F4/80, CD64, and myeloid-epithelial-reproductive protein tyrosine kinase (MerTK) expression (15). Until recently, cardiac macrophages were thought to be a relatively uniform population of cells enriched for markers belonging to a classification of macrophages referred to as M2 (16). This classification system was established based on an in vitro stimulation system that defined macrophages as either inflammatory (M1) or reparative (M2). However, this nomenclature is largely considered incomplete and difficult to apply to in vivo populations where cells not only adopt a multitude of fates between M1 and M2 but also may dynamically interconvert between phenotypes depending on environmental inputs (17–20). Indeed, tissue-specific cues can instruct organ-specific transcription profiles in macrophages when transplanted from one organ to another (21).
Macrophage Ontogeny
An alternative approach to categorize macrophages is to focus on their developmental origin, as cells that are derived from differing origins are likely to be functionally distinct. As tools emerged to perform genetic lineage tracing in the hematopoietic system of mice, it was observed that not all macrophages originated from FLT3+-definitive hematopoietic stem cells (HSCs). In fact, many organs contain HSC-independent macrophages that are derived from embryonic (primitive and erythromyeloid) progenitors located within the yolk sac and fetal liver. HSC-independent macrophages seed tissues during embryonic development and exist throughout adult life independent of monocyte input, defying years of dogma that described macrophages as derived from circulating monocytes (22–25). This approach was essential to delineating the heterogeneity of cardiac macrophages (26).
Recent studies utilized a combination of lineage tracing, parabiosis models, conventional flow cytometry, and single-cell transcriptomic techniques to rigorously define cardiac macrophage subpopulations in the steady-state mouse heart. These studies identified three subsets of cardiac macrophages that could be distinguished based on the cell surface expression of C-C chemokine receptor 2 (CCR2) and major histocompatibility complex class II (MHC-II) expression. Single-cell RNA sequencing of cardiac macrophages confirmed the presence of these three subsets using an unbiased methodology (27) (Figure 1). Analysis of parabiosis models and CCR2-deficient mice revealed that CCR2+MHC-IIhigh macrophages were derived from circulating monocytes, whereas CCR2−MHC-IIhigh and CCR2−MHC-IIlow macrophages existed independent of monocyte input. Selective labeling of resident cardiac macrophages (CX3CR1-CreERT2) confirmed that monocytes preferentially contribute to CCR2+MHC-IIhigh macrophages. Conversely, genetic fate mapping of embryonic-derived macrophages (FLT3-Cre and CSF1R-MerCreMer) demonstrated that CCR2−MHC-IIhigh and CCR2−MHC-IIlow macrophages likely arose from HSC-independent embryonic hematopoietic progenitors (15, 28–30).
Figure 1.

Resident macrophage subsets in the steady-state heart. In the mouse, three macrophage subsets are differentiated by CCR2 and MHC-II expression: CCR2−MHC-IIlow, CCR2−MHC-IIhigh, and CCR2+MHC-IIhigh. Humans have similar subsets but lack the CCR2−MHC-IIlow subset. HLA-DR is the human homologue of MHC-II. Predominant functional distinctions parallel macrophage ontogeny via expression of CCR2. Abbreviations: CCR, C-C chemokine receptor; MHC, major histocompatibility complex.
Temporally, CCR2−MHC-IIlow macrophages first appear in the developing heart at embryonic day 10.5–11.5. These cells are located adjacent to epicardial structures, including the coronary vasculature, and require signals from the epicardium to infiltrate the developing heart (31). CCR2−MHC-IIlow macrophages likely give rise to CCR2−MHC-IIhigh macrophages during postnatal development. CCR2+MHC-IIhigh macrophages appear to infiltrate the heart after the second week of life in a C-C chemokine ligand 2/7 (CCL2/CCL7)- and CCR2-dependent manner (32–34).
Resident Macrophage Functions
Under steady-state conditions, the predominant functional distinction is between CCR2+ macrophages and CCR2− macrophages. CCR2+MHC-IIhigh macrophages display markedly increased mRNA expression of inflammatory chemokines [CCL2, CCL7, CCL17, and C-X-C chemokine ligand 2 (CXCL2)] and cytokines (IL-1β, IL-6, TNF) and secrete higher levels of IL-1β protein. In contrast, CCR2−MHC-IIhigh and CCR2−MHC-IIlow macrophages express reduced levels of inflammatory mediators and, instead, express numerous growth factors and extracellular matrix components. CCR2− macrophages regulate coronary development and are required for neonatal cardiac regeneration likely through stimulation of coronary angiogenesis and cardiomyocyte proliferation (33, 34). As discussed in detail below, selective depletion of CCR2− macrophages or CCR2+ macrophages leads to divergent effects on inflammatory responses to sterile myocardial injury. The primary distinction between CCR2−MHC-IIhigh and CCR2−MHC-IIlow macrophages appears to be the ability to present antigens to primed T cells. CCR2+MHC-IIhigh, CCR2−MHC-IIhigh, and CCR2−MHC-IIlow macrophages display similar capacity for phagocytosis in vitro (15).
Cardiac macrophages interact closely with cardiomyocytes and may form functional connections between cell types. Within the atrioventricular node, cardiac macrophages express connexin 43 and form gap junctions with cardiomyocytes. Either macrophage depletion or selective deletion of connexin 43 in macrophages resulted in delayed atrioventricular node conduction and heart block (27). These findings illustrate that, in principle, cardiac macrophages can directly interact with cardiomyocytes, influence electrical conduction, and potentially participate in the pathogenesis of cardiac arrhythmias. Consistent with this notion, macrophage-derived IL-1β was sufficient to mediate electrical remodeling of atrial myocytes, leading to atrial fibrillation-like activity in vitro (35). Furthermore, these intriguing observations raise the possibility that electrical coupling with cardiomyocytes could influence the behavior or function of cardiac macrophages. Future studies will undoubtedly explore this interesting possibility.
Human Cardiac Macrophages
Importantly, concepts of cardiac macrophage diversity are similarly relevant in the human heart. Flow cytometric and immunohistochemical examination of left ventricular (LV) myocardial tissue obtained from discarded donors and heart failure patients at the time of LV assist device implantation or heart transplantation revealed that the human heart contains similar populations of CCR2+HLA-DRhigh and CCR2−HLA-DRhigh macrophages. In contrast to the mouse, the human heart lacked CCR2−HLA-DRlow macrophages. Combined immunohistochemical and in situ hybridization analysis of endomyocardial biopsies obtained from a cohort of sex-mismatched heart transplant recipients (female donors transplanted into male recipients) provided important insights into the contribution of monocytes to the maintenance of human cardiac CCR2− and CCR2+ macrophages. Using the presence of a Y chromosome as a marker for recruited monocyte-derived macrophages, it was evident that monocytes exclusively contributed to CCR2+ macrophages. Transcriptomic profiling and ex vivo functional assays demonstrated that human CCR2− and CCR2+ macrophages were functionally analogous to their mouse counterparts. Clinically, the abundance of CCR2+ cardiac macrophages was associated with and predictive of LV remodeling in a select group of heart failure patients that underwent implantation of an LV assist device (36).
These findings highlight the presence, developmental progression, and functional and clinical relevance of macrophage heterogeneity in the heart and identify CCR2+ macrophages as a particularly inflammatory subset. In the following sections, we discuss how CCR2+ macrophages are activated following myocardial injury, interact with other immune cell populations including infiltrating monocytes, and contribute to the pathogenesis of heart failure. We further discuss the plasticity of infiltrating monocytes and monocyte-derived macrophages and their respective roles in cardiac repair and LV remodeling.
CARDIAC MACROPHAGES IN THE DISEASED HEART
Sterile Inflammation
Although a variety of pathogens may infect and impose significant damage to the heart (infective myocarditis), this review focuses on sterile inflammation that typically results from ischemia, ischemia reperfusion injury, and hemodynamic stress.
Ischemia and reperfusion result in tissue necrosis, apoptosis, and necrotic forms of programmed cell death (necroptosis, ferroptosis). Among these cell death pathways, tissue necrosis, necroptosis, and ferroptosis appear to stimulate robust inflammatory responses through the release of alarmins and danger-associated molecular patterns (DAMPs) (37). It is clear that innate immune responses represent an important pathway to preserve tissue integrity through removal of necrotic debris and collagen deposition, as suppression of the innate immune response results in myocardial thinning and cardiac rupture after coronary ligature in mice. However, it is critical to note that exaggeration or failure to resolve inflammatory responses potentiates heart failure progression through collateral myocardial injury, adverse LV remodeling, and inhibition of tissue repair (38).
In the setting of hemodynamic stress, tissue necrosis is usually not evident. Instead, it is hypothesized that persistent mechanical stress and chronic neurohormonal activation compromises cellular autophagic functions. This leads to the accumulation of damaged organelle, protein, and DNA, triggering the release of DAMPs such as mitochondrial DNA (mtDNA), which continuously activate inflammatory pathways (39–42). These findings correlate clinically as well. In a small study of acute heart failure patients, circulating mtDNA levels at the time of admission to the ICU were predictive of mortality independent of NT-proBNP, age, gender, and APACHE II score (43). The potential roles for mtDNA and nuclear DNA (nDNA) as mediators of inflammatory responses and prognostically relevant biomarkers continue to be an active area of investigation (44).
The following discussion focuses on models of ischemic injury (coronary ligation and ischemia reperfusion injury), pressure overload [transaortic constriction (TAC)], and chronic neurohormonal activation [angiotensin II (Ang II)], primarily in mice.
Local Immune Cell Activation
As previously discussed, cardiac injury results in the release of alarmins (IL-1α, IL-1–8) and DAMPs [mtDNA, nDNA, high-mobility group protein B1 (HMGB1), S100A8/9, ATP, formyl peptides, and lipids] via multiple mechanisms (39–42). Resident innate immune cells are the first to sense and respond to the injury via nuclear-encoded receptors that specifically recognize alarmins and DAMPs, resulting in immune cell activation, proliferation, and trafficking as well as chemokine and cytokine production and generation of oxidative intermediates (Figure 2) (28).
Figure 2.
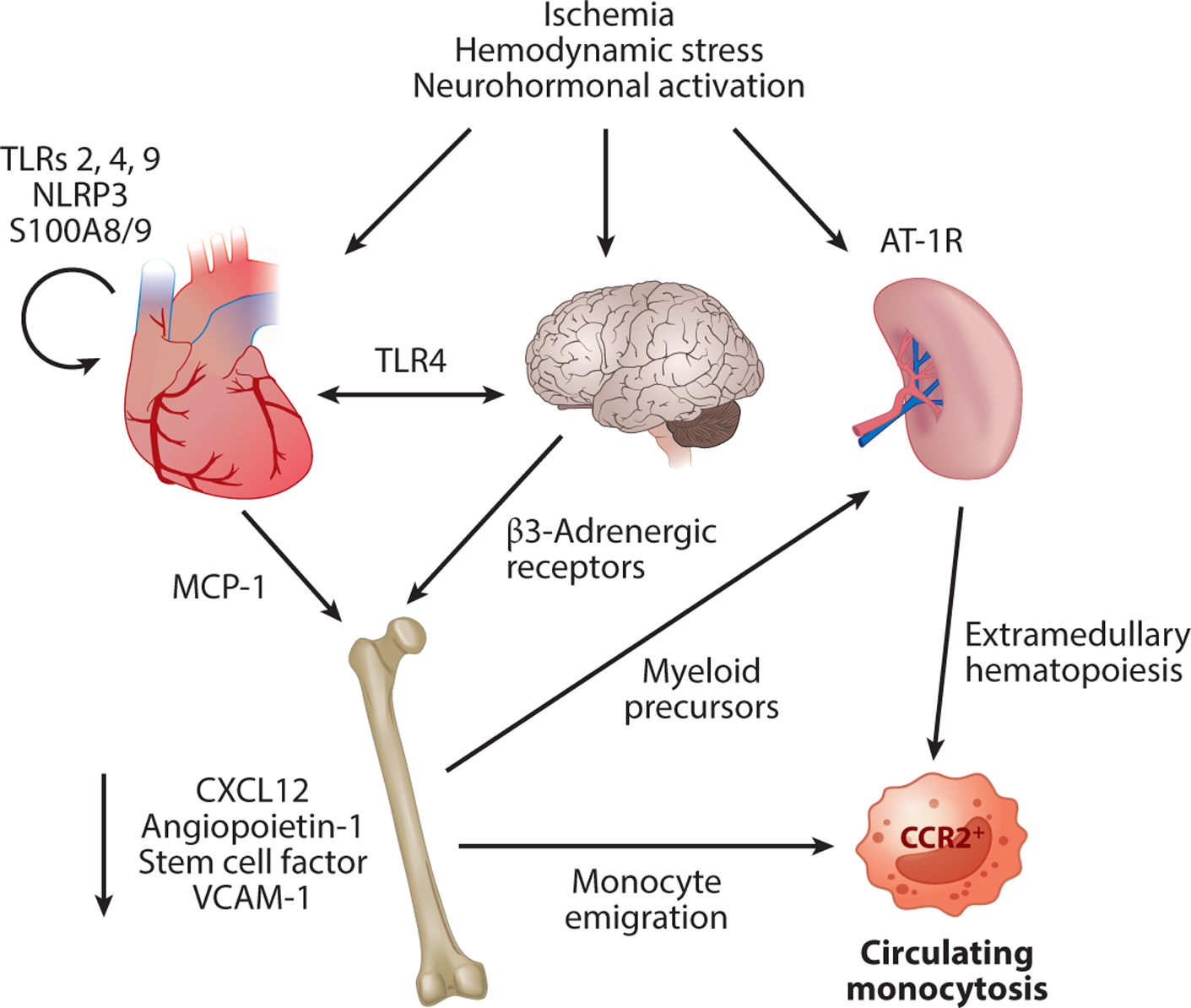
Immune cell activation and mobilization. Cardiac injury as the result of ischemia, hemodynamic stresses, and neurohormonal activation releases proteins such as DAMPs and alarmins that are recognized at the cellular level by receptors such as TLR2, TLR4, and TLR9. This results in cytokine and chemokine production favoring monocyte and myeloid precursor emigration from the bone marrow, extramedullary hematopoiesis in the spleen, and a circulating monocytosis. Neurohormonal activation amplifies these processes at each level, further exacerbating the monocytosis. Abbreviations: AT-1R, angiotensin II type 1 receptor; CCR, C-C chemokine receptor; DAMP, danger-associated molecular pattern; MCP-1, monocyte chemotactic protein 1; TLR, Toll-like receptor.
DAMP signaling.
Endosomal and cytosolic DNA (mtDNA, nDNA) are recognized by Toll-like receptor 9 (TLR9) and cGAS/STING (cyclic GMP-AMP synthase/stimulator of interferon genes), each of which is highly expressed in resident cardiac macrophages (45, 46). TLR9 and STING activation have been implicated in the pathogenesis of chronic heart failure and ischemic cardiomyopathy, respectively (40, 47). Interestingly, TLR9 activation may also have context-dependent protective roles. In a mouse model of isoproterenol-induced hypertrophy, TLR9 signaling led to activation of phosphoinositide 3-kinase (PI3K) and protein kinase B (PKB, Akt) in vivo and prevented isoproterenol-induced hypertrophy, indicating a possible protective role of TLR9 activation in cardiac inflammation (48). In line with these observations, pretreatment of mice with 1668-thioate (a CpG-containing TLR9 activator) 16 h before TAC attenuated subsequent hypertrophy and fibrosis and preserved cardiac function as measured by echocardiographic parameters (49).
HMGB1 and ATP are typically released following necrosis or nonapoptotic forms of programmed cell death and are detected by an array of receptors, including TLR2 and TLR4, purine receptors, and nod-like receptor family pyrin domain-containing 3 found on myeloid cells (50). Although necrosis is typically associated with ischemic forms of injury, activation of both TLR2 and TLR4 has been found to be critical for LV remodeling in nonischemic models of heart failure, indicating that cell death may also trigger immune cell activation in a variety of disease settings (15, 51–55). Treatment with the TLR4 antagonist eritoran attenuated hypertrophic changes and inflammatory gene expression in response to pressure overload (52). Similarly, following Ang II infusion, TLR4 deficiency suppressed LV remodeling, which was attributed to reduced monocyte chemotactic protein 1 (MCP-1) expression and less monocyte infiltration (54). Blockade of the TLR4 coreceptor, myeloid differentiation 2, protected against Ang II–induced cardiac remodeling independent of systolic blood pressure (56). TLR2 may also regulate LV remodeling. After one week of Ang II infusion, mice developed hypertrophy with fibrotic changes that were absent in TLR2 knockout or anti-TLR2 antibody-treated mice. These changes occurred in parallel with a significant decrease in infiltrating macrophages, attributed to a decrease in MCP-1, as well as significant decreases in cytokine production, including IL-1β, IL-6, TNF-α, and transforming growth factor-beta (TGF-β) (55). Interestingly, these changes were lost by adoptive transfer of wild-type bone marrow into TLR2-deficient mice, indicating an inflammatory and profibrotic role for TLR2-activated infiltrating immune cells during Ang II infusion.
TLR signaling may also have context-dependent protective roles. TLR2 deficiency accelerated the development of cardiac dysfunction in a pressure overload model, possibly by attenuating a compensatory cardiac hypertrophic response that was driven by stretch-dependent activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling, IL-1β, insulin-like growth factor 1 (IGF-1), and vascular endothelial growth factor (VEGF) production (53, 57–59). VEGF and IGF-1 are critical for physiologic cardiac hypertrophy (60–62). Using bone marrow transplant models, TLR2 deficiency only resulted in a cardioprotective phenotype when knocked out in the recipient mouse, indicating that TLR2 is required in resident cardiac cells (53).
Another pathway of interest involves myeloid proteins 8 and 14 (S100A8 and S100A9), which are released by necrosis and hypoxia and bind to the receptor for advanced glycation end products (RAGE) as well as TLR4. In mice postmyocardial infarction, it was found that RAGE was crucial for S100A8/9 signaling, resulting in NF-κB activation and increased production of inflammatory mediators including S100A8/9 themselves and representing a positive feedback loop that amplifies the immune response during hypoxic injury (63). RAGE signaling has been widely studied as a mechanism by which hyperglycemia causes systemic inflammation and may represent the link between diabetes and cardiovascular disease (64). This pathway was recently implicated in the ability of resident macrophages to “cloak” injured or dying cells and suppress neutrophil recruitment (65). Whether this mechanism is relevant to cardiac injury remains to be explored.
Resident cardiac macrophages.
The concept that resident cardiac macrophages may have distinct functions compared to macrophages derived from infiltrating monocytes was motivated by unique functions of cardiac macrophages in the context of neonatal myocardial injury (66). In response to myocardial infarction, apical resection, or diphtheria toxin–induced cardiomyocyte ablation, macrophages within the neonatal heart regulate tissue repair and regeneration. Macrophage depletion resulted in the loss of regenerative potential (cardiomyocyte proliferation and angiogenesis) and favored fibrotic scar formation and adverse LV remodeling. At this developmental stage, macrophage expansion occurred through local proliferation of tissue-resident macrophages rather than Ly6Chigh monocyte recruitment. Intriguingly, neonatal cardiac macrophages demonstrated minimal inflammatory chemokine or cytokine expression and, instead, expressed an array of growth factors previously impacted in cardiomyocyte proliferation and angiogenesis (33). The signals responsible for neonatal cardiac macrophage activation remained poorly defined.
Recent work has provided initial insights into the roles of resident cardiac macrophages in the injured adult heart. Numerous studies have documented that resident cardiac macrophages expand by local proliferation following myocardial infarction (30), TAC (67), and Ang II infusion (15). For example, resident cardiac macrophages proliferate in response to TAC through a KLF4-dependent pathway. Depletion of all resident cardiac macrophages using a temporal and cell type– specific diphtheria toxin receptor-mediated cell ablation system (CX3CR1-ertCre; Rosa26-DTR mice) demonstrated that, as a group, resident cardiac macrophages are protective because they are required to preserve LV systolic function and reduce LV remodeling following myocardial infarction (30). Selective depletion of resident CCR2− macrophages prior to myocardial infarction similarly demonstrated a protective role for this population and additionally revealed a role for CCR2− macrophages in suppressing monocyte and neutrophil infiltration (32).
Intriguingly, cardiac-resident CCR2+ macrophages appear to have functions that are quite distinct from resident CCR2− macrophages. Selective depletion of this small population of CCR2+ cells (10–15% of total resident cardiac macrophages) prior to myocardial ischemia reperfusion injury resulted in marked reductions in infarct size, improvements in LV systolic function, and reduced LV remodeling (32). Mechanistically, CCR2+ macrophages orchestrated the infiltration of neutrophils and monocytes into the myocardium through the expression of the chemoattractant chemokines CXCL2, CXCL5, CCL2, and CCL7. Resident CCR2+ macrophages also expressed numerous cytokines (IL-1β, IL-6, and TNF), generated reactive oxygen species (ROS), and potentiated the differentiation of monocytes into inflammatory macrophage subsets. Many of these appeared to be mediated through signaling of MYD88, an adapter molecule that is necessary for TLR and IL-1R signaling (32, 33, 46, 68). To date, the exact ligands responsible for resident CCR2+ macrophage activation remain poorly defined. However, recent studies have implicated ferroptotic cell death of cardiomyocytes as an early source of DAMPs and alarmins (69).
Although some effector functions of resident CCR2+ macrophages directly impart collateral damage to the myocardium (i.e., generation of cytokines and ROS), it is likely that their overriding effects are largely mediated through the recruitment of neutrophils and monocytes. Indeed, neutrophil depletion is sufficient to reduce infarct size and improve LV systolic function after myocardial infarction (69). Similarly, reductions in monocyte recruitment are also cardioprotective. In the following sections, we review the importance of infiltrating monocytes and neutrophils in more detail, highlight identified effector mechanisms, and discuss emerging concepts related to neutrophil and monocyte diversity and fate specification.
Peripheral Immune Cell Activation
In addition to the activation of resident cardiac macrophages, immune cell populations in other lymphoid tissues such as the spleen are also activated. As discussed previously, this can enhance the circulating pool of immune cells available to respond to the tissue injury, which can contribute to the progression from myocardial injury to heart failure. The following sections explore this in detail.
Myelopoiesis and immune cell mobilization.
Following myocardial injury, hematopoietic tissues (bone marrow and spleen) are activated to selectively produce and mobilize peripheral myeloid cells (29). In the context of either cardiac ischemic injury or hemodynamic stress, activation of hematopoietic tissues is mediated by the sympathetic nervous system triggering the release of adrenaline and noradrenaline. Engagement of β3-adrenergic receptors expressed on stromal bone marrow niche cells alters the expression of factors associated with HSC quiescence (CXCL12, angiopoietin-1) and bone marrow retention (stem cell factor, VCAM-1). β3-Adrenergic receptor signaling results in selective maturation and emigration of myeloid cells as well as myeloid precursors that take up residence in the spleen (29). Hematopoietic activation also favors the production of CCR2+ precursors, as their proliferative abilities postmyocardial infarction are vastly superior to their CCR2− counterparts. This effect was recapitulated with intravenous delivery of HMGB1, suggesting the role of DAMP signaling (70).
Mobilization of myeloid precursors to the spleen initiates extramedullary hematopoiesis that occurs following Ang II infusion and myocardial infarction (71, 72). Consistent with the spleen serving as an important source of monocytes, removal of the spleen prior to Ang II infusion significantly reduces cardiac macrophage infiltration (71). A similar scenario is evident after ischemic cardiac injury, including splenic immune cell expansion (72). In addition, splenectomy is sufficient to partially reverse heart failure phenotypes in a model of established ischemic cardiomyopathy. Moreover, adoptive transfer of mononuclear cells isolated from postmyocardial infarction murine spleens traffics to the heart and induces a heart failure phenotype (73). 18FDG-PET imaging of patients who suffered an acute coronary syndrome revealed significant tracer uptake in the spleen, providing indirect evidence of hematopoietic tissue activation in humans. Interestingly, tracer uptake correlated with increased inflammatory gene expression (74).
On the basis of these observations, it has been hypothesized that a cardio-splenic axis mediates myelopoiesis, immune cell mobilization, cardiac recruitment, and subsequent deterioration of LV function following cardiac injury or hemodynamic stress (71, 72). This hypothesis is consistent with the findings that central sympathetic nervous system activation is associated with LV hypertrophy in hypertensive patients (75) and that β-adrenergic receptor knockout mice display attenuated cardiac remodeling and systemic inflammation post-TAC (76). Furthermore, observational human studies suggest a relationship between sympathetic nervous activation and heart failure with preserved ejection fraction (HFpEF) (77), indicating that this may represent a central mechanism contributing to the progression of both HFrEF and HFpEF.
Neutrophils.
Numerous myeloid cell populations infiltrate the injured myocardium, including neutrophils, monocytes, and dendritic cells. Among these cell types, neutrophils appear to enter the heart at the earliest time points, beginning within 2 h following ischemia reperfusion injury. Intravital imaging of beating mouse hearts has revealed that neutrophils infiltrate the myocardium through a two-step process: endothelial cell adhesion and transendothelial migration. Endothelial cell adhesion is mediated through an endothelial TLR4-TRIF-type I interferon (IFN)-dependent pathway that is triggered by cardiomyocyte ferroptosis (69). Transendothelial migration is governed by a pathway that involves MYD88-dependent secretion of neutrophil chemokines (CXCL2 and CXCL5) from resident CCR2+ macrophages (46). Subsequent neutrophil recruitment appears to be regulated by infiltrating monocytes that differentiate into a cell type that resembles resident CCR2+ macrophages (32, 33).
In general, neutrophils are thought to impair myocardial repair and contribute to heart failure progression through generation of damaging cytokines and ROS and release of enzymes stored in secretory granules. However, neutrophils may also have cardioprotective properties (78). Mechanistically, neutrophil-derived ROS may stimulate the differentiation of reparative macrophages, as described in the liver following acetaminophen-induced liver injury (79). Additionally, neutrophil gelatinase-associated lipocalin was shown to upregulate MerTK production in macrophages, a receptor essential for efferocytosis (78).
Intriguingly, recent observations have highlighted that distinct populations of neutrophils may exist: CD206− and CD206+ neutrophils. Following myocardial infarction, infiltrating neutrophils are predominately CD206−. Over subsequent days CD206+ neutrophils appear to accumulate; however, the functional relevance of this finding is uncertain (80). In an atherosclerosis model, infusion of subclinical doses of lipopolysaccharide (LPS) increase the number of CD206− neutrophils correlating with larger atherosclerotic plaque size and reduced plaque stability. 4-Phenylbutyrate treatment, which targets peroxisome homeostasis, leads to increased numbers of CD206+ neutrophils, decreased plaque size, and increased plaque collagen (81). The full extent of neutrophil heterogeneity, signals that govern neutrophil fate specification, and the functional relevance of these cell populations remain poorly defined. Future studies are likely to focus on these questions as well as interactions between neutrophils and other immune cell populations.
Monocytes and monocyte-derived macrophages.
Monocytes are robustly recruited to the heart following ischemic injury or hemodynamic stress. By day 7 following cardiac injury, monocyte-derived macrophages vastly outnumber resident macrophage subsets (33, 82, 83). When viewed at the population level, monocyte-derived macrophages are generally inflammatory and produce high levels of inflammatory chemokines and cytokines, including CCL2, CCL7, IL-1β, and TNF (33). These cells are thought to recruit additional monocytes and neutrophils to the heart and contribute to ongoing collateral myocardial injury (50, 68). Blockade of monocyte recruitment through inhibition of CCR2 signaling improves outcomes in mouse models of myocardial infarction (84–86), demonstrating the overall impact of monocytes on disease outcomes.
Monocyte-derived macrophages also play a key role in response to hemodynamic stress (pressure overload, Ang II infusion), representing >50% of all cardiac immune cells (71, 87). Within the first week following TAC, there is a significant increase in Ly6Chigh blood monocytes and CD45+ cardiac leukocytes (88, 89). This time frame corresponds to an acute reduction in LV systolic function. Over the subsequent 1–2 weeks LV systolic function normalizes as a consequence of compensatory LV hypertrophy (88). During this time period cardiac macrophage abundance normalizes to near baseline. Macrophages then accumulate over 4–8 weeks following TAC in mice that transition to heart failure (67). While debate exists regarding whether early macrophage expansion is dependent on monocytes, it is clear that the second wave of macrophage expansion is dependent on infiltrating monocytes (67, 87–89). Inhibition of CCR2 signaling through administration of RS-504393 or using CCR2-knockout mice resulted in reduced macrophage abundance, preservation of LV systolic function, and increased coronary angiogenesis.
Monocytes display remarkable plasticity and have the capacity to differentiate into a variety of macrophage and dendritic cell subsets. Using bulk RNA sequencing, macrophage phenotypes following myocardial infarction vary over time. Day 1 macrophages expressed inflammatory chemokines/cytokines and matrix-degrading enzymes (matrix metalloproteinases, IL-1β, TNF), day 3 macrophages expressed inflammatory chemokines/cytokines and transcripts associated with phagocytosis and cell proliferation, and day 7 macrophages expressed genes associated with extracellular matrix remodeling (82). Importantly, none of these macrophage signatures fit into the historically defined M1 and M2 nomenclature.
Single-cell RNA sequencing has provided additional insights into the diverse phenotypes monocytes may acquire upon entering the myocardium (30, 32). These studies identified at least 7 distinct macrophage subsets and 3 distinct dendritic cell subsets that were present in the myocardium following myocardial infarction. Each macrophage subpopulation displayed unique morphology and tissue localization. The precise functions of these newly discovered immune cell subsets and the instructive cues that orchestrate these cell fate decisions are not fully understood. One exception is a subpopulation of macrophages that display a type I IFN signature. IFN-biased macrophages are localized in the peri-infarct region and appear to be a key mediator of inflammation following myocardial infarction (32, 47). Genetic disruption of interferon regulatory factor 3 (IRF3) or interferon alpha receptor (IFNAR) or administration of a neutralizing IFNAR antibody 48 h following myocardial infarction led to reduced mortality and LV remodeling. Cytosolic sensing of either mitochondrial or nuclear DNA through cGAS and STING signaling appears to be required to induce this phenotype. Although the exact sources of DNA are not well understood, dying cardiomyocytes are a likely possibility (47). Consistent with these findings, conditional deletion of cGAS in macrophages results in altered macrophage phenotypes, including increased numbers of reparative CD206+ macrophages (45).
A key observation from single-cell RNA sequencing studies in mice is that monocytes may be able to acquire reparative fates, including those that resemble resident macrophages found in the healthy heart. While the clinical implications of these findings are obvious, the exact mechanisms that might govern these events are unclear. Interactions between monocytes and their local environment are a likely contributor. Upon entering the injured heart, monocytes and monocyte-derived macrophages encounter and phagocytose cellular debris. As a result, phagocytosis and efferocytosis may represent one such mechanism. Suppression of efferocytosis through MerTK and milk fat globule epidermal growth factor 8 (MFGE8) knockout led to marked reductions in production of proangiogenic growth factor (VEGF-A) expression following myocardial infarction in mice. This resulted in accelerated adverse LV remodeling and recapitulated myeloid-specific VEGF-A deficiency (90). In humans, low MFGE8 levels correlate with poor cardiac function in dilated cardiomyopathy (91). Other possibilities include exposure to specific cytokines and growth factors, interaction with resident macrophages, mechanical strain, or even electrical coupling with cardiomyocytes. Granulocyte macrophage colony-stimulating factor (GM-CSF) signaling has recently been implicated in this context (92).
Dendritic cells.
Dendritic cells appear to adversely contribute to LV remodeling following myocardial infarction. Selective depletion of classical dendritic cells using ZBTB46-DTR bone marrow chimeric mice was cardioprotective, whereas selective depletion of plasmacytoid dendritic cells using BDCA2-DTR bone chimeric mice appeared to have no effect (93). Clinically, dendritic cell abundance negatively correlated with cardiac rupture following ST-segment elevation myocardial infarction (94). It is important to note that dendritic cells represent a heterogeneous immune cell population, and it is likely that distinct dendritic cell subsets may have differing effects on LV remodeling, heart failure progression, and cardiac tissue repair. Consistent with this concept, single-cell RNA sequencing has identified multiple classic dendritic cell subsets in the heart following ischemic myocardial injury (30). Furthermore, dendritic cell depletion reduced the abundance of CD206+ macrophages, suggesting that among their functions dendritic cells may also influence monocyte differentiation (95).
Interactions Between Cardiac Macrophages and T Lymphocytes
Functionally, interactions between monocytes, macrophages, and other immune populations likely represent important mechanisms by which immune cells adopt particular cell fates and impact tissue repair and remodeling within the heart. Among such immune cell populations, T cells play important roles in the response to cardiac injury (96–98). In both ischemic and nonischemic murine models of heart failure, both regulatory and effector CD4 T cells have been implicated, and their activation was at least partially dependent on antigen recognition (99–101). Antigen presentation and chemokine/cytokine signaling may mediate potential interactions between macrophages and T cells. Both CCR2−MHC-IIhigh and CCR2+MHC-IIhigh macrophages have the capacity to present antigen to T cells (15). Additionally, CCR2+MHC-IIhigh macrophages secrete chemokines and cytokines that impact T cell function (32, 102).
The role of T cells in ischemic cardiac injury was first explored in adoptive transfer experiments performed in RAG1 knockout mice (which lack both T and B cells) (103). In this context, only adoptive transfer of IFN-γ-competent effector CD4+ T cells could reverse the protective effects of RAG1 deficiency, suggesting that IFN-γ-producing CD4 T cells mediate components of adverse LV remodeling following ischemic myocardial injury. This possibility was explored further in the setting of dectin 2 deficiency, which is a pattern recognition receptor that recognizes components of the fungal cell wall. Dectin 2 deficiency led to a reduction in the number of IFN-γ-producing CD4+ T cells in the infarcted heart, increased survival, and reduced cardiac rupture (104). Interestingly, T cell activation was found to rely on antigen recognition, as mice with transgenic T cell receptors (TCRs) on CD4+ T cells that only recognize ovalbumin exhibit poor T cell proliferation following myocardial infarction (99).
IFN-γ-producing CD4 T cells also play important roles in models of nonischemic heart failure. Absence of this T cell subset preserved LV systolic function following pressure overload (97). Adoptive transfer of IFN-γ-producing CD4 T cells into T cell–deficient mice partially reversed the protective effects seen in T cell–deficient mice following TAC (96). Finally, transgenic mice bearing TCRs that recognize ovalbumin exhibit diminished LV remodeling and systolic dysfunction in the TAC model, implicating antigen recognition in T cell activation (97). To the best of our knowledge, the importance of T cell antigen recognition has not been studied in the context of Ang II infusion. Interestingly, it was recently found that T cell activation during Ang II infusion requires contact with macrophages (105). A feed-forward loop was evident, where T cell–derived IFN-γ stimulated CCL2 production by macrophages leading to further monocyte recruitment and T cell activation (105).
T cell activation also impacts inflammatory resolution through activation of regulatory T cells (Tregs). Depletion of Tregs via Foxp3-DTR mice or treatment with anti-CD25 antibody led to reductions in LV systolic function following myocardial infarction. Conversely, Treg activation using a superagonist CD28 antibody led to improved survival, preserved LV systolic function, and increased the number of CD206+ macrophages following myocardial infarction (98). These findings are consistent with other studies reporting beneficial effects of Treg adoptive transfer in both ischemic and nonischemic heart failure models (106–108). Treg activation may also depend on antigen recognition, as transgenic mice bearing TCRs that only recognize ovalbumin exhibit increased incidence of death and cardiac rupture following myocardial infarction, which was attributed to the loss of Foxp3+ Tregs (99). Treg activation and/or trafficking may also be regulated by interactions with macrophages. Deletion of CCR5 (necessary for macrophage recruitment and differentiation) leads to reduced Treg infiltration (109, 110). The exact mechanisms by which different monocyte and macrophage populations interact with T cells remain an area of intense research that is only likely to broaden as investigators embrace the extent and importance of immune cell diversity within the infarcted heart.
TARGETING MONOCYTES AND MACROPHAGES IN HEART FAILURE
Preclinical and human pathological studies have implicated CCR2+ macrophages as a potential cellular target to mitigate damaging inflammatory responses following myocardial injury. In mice and humans, CCR2+ macrophages represent an inflammatory population that contributes to heart failure progression. Mechanistically, CCR2+ macrophages are activated by MYD88 signaling and generate inflammatory chemokines, cytokines, and oxidative products that orchestrate neutrophil and monocyte recruitment, selection of inflammatory fates, and collateral myocardial injury (32, 33, 46).
The possible benefit of targeting this population was first demonstrated using CCR2-targeted siRNAs. To selectively target phagocytic cells, the CCR2 siRNA was encapsulated in a lipid nanoparticle, which readily distributes between the blood, spleen, and bone marrow following intravenous injection. Application of this agent in mouse models of myocardial infarction and atherosclerosis confirmed reductions in infarct size and reduced atherosclerotic lesion size. Molecular imaging using a myeloperoxidase-activatable gadolinium-based MRI agent (MPO-Gd) to track inflammation revealed that the CCR2 siRNA decreased myeloperoxidase activity. This correlated with decreased expression of CCR2, MCP-1, NF-κB, MPO, IL-6, IL-1β, and TNF-α and an increased expression of IL-10 and arginase. Finally, these findings correlated with improved LV systolic function and decreased LV remodeling (111). Importantly, this study provided evidence that phagocytic cells can be targeted without attenuating tissue repair processes.
An important limitation to modalities that either suppress or modulate the immune system is the potential to increase the risk of opportunistic or life-threatening infections. This was clearly demonstrated in the recent CANTOS trial (10). As such, it is essential to devise diagnostic strategies to identify individuals that are most likely to benefit from the therapy in question. Furthermore, it is equally important to have the capacity to monitor on-target responses to ensure appropriate dosing of the therapeutic agent. Molecular imaging represents one such approach. Recent advances have allowed the direct imaging of CCR2+ cells within the heart. A peptide-based PET tracer was developed that allosterically binds to an extracellular loop of CCR2 (ECL1i-DOTA). The imaging agent can then be visualized using either radioactive gallium or copper. ECL1i-DOTA specifically identified CCR2+ monocytes and macrophages in mouse models of cardiomyocyte ablation and myocardial infarction. Importantly, ECL1i-DOTA uptake on day 4 following myocardial infarction was predictive of infarct size and LV systolic function 4 weeks later. Finally, this agent may also be appropriate for human imaging, as it readily identified CCR2+ macrophages in human myocardial tissue (112).
Imaging agents may also be used to deliver therapies. This concept is referred to as theranostics, modalities used to simultaneously diagnose, deliver therapy, and monitor treatment effects (113). An example of this approach is a prodrug–fluorophore conjugate designed to release both its drug cargo (doxorubicin) and fluorescent marker (BODIPY) when taken up by macrophages. Incubation with cultured macrophages derived from human monocytes resulted in release of the fluorescent signal and cell death when macrophages were stimulated with LPS. In vivo application of this reagent in zebrafish demonstrated selectivity for inflammatory macrophages after tailfin amputation (113). This concept could certainly be applied to CCR2+ macrophages using a radionucleotide tracer.
An alternative strategy is to selectively deliver pharmaceuticals to CCR2+ macrophages that inhibit their activation or differentiation. Indeed, monocytes express CCR2 prior to differentiating into monocyte-derived macrophages (32, 33, 46). MYD88 and NF-κB represent potential targets to inhibit CCR2+ macrophage activation. MicroRNAs may be delivered using this approach (114). In principle, these compounds may be delivered to CCR2+ macrophages using ECL1i-targeted nanoparticles or nanoclusters. Such nanotherapeutics may be radiolabeled to provide information regarding target availability. Currently, we know little regarding how monocytes select inflammatory versus reparative fates once infiltrating the injured or diseased heart. Once the molecular mechanisms by which these fate decisions are elucidated, it may be possible to directly manipulate recruited monocytes in a manner that favors acquisition of reparative phenotypes. This approach has tremendous clinical potential given the abundance of monocytes that are recruited to the injured heart.
Lastly, it is plausible to target the effector pathways by which inflammatory populations of macrophages exert their effects. Cytokines such as IL-1β and IL-6 are robustly expressed by CCR2+ macrophages, and neutralizing antibodies are clinically available. It is intriguing to postulate that delivery of neutralizing IL-1β or IL-1 receptor antibodies to patients with abundant numbers of CCR2+ macrophages in the heart may be a tractable and effective approach. Other possibilities include inhibition of chemokines that potentiate inflammatory monocyte and T cell recruitment and/or activation.
CONCLUSION AND FUTURE DIRECTIONS
Despite mounting evidence implicating inflammation in the progression from cardiac injury to heart failure, therapeutic translation has proven difficult. While recent clinical trials have shown mixed results, targeting of IL-1β in atherosclerosis has provided renewed hope and confirmed that inflammation is a viable therapeutic target in cardiovascular diseases (10, 11). Here, we have reviewed recent preclinical data on the roles of various subsets of macrophages in cardiac homeostasis, with a focus on resident and recruited macrophage diversity. Among the identified macrophage populations within the heart, CCR2+ monocytes and macrophages represent an attractive target for therapies that inhibit immune cell activation, impact monocyte differentiation, or neutralize effector cytokines and chemokines. Although our understanding of the immune system is largely incomplete (115), selective targeting of macrophage subsets represents an important and exciting opportunity for the emerging field of cardiac immunology.
ACKNOWLEDGMENTS
K.J.L. is supported by the US National Institutes of Health (NIH) grants K08 HL123519, R01 HL138466, and R01 HL139714; the Burroughs Wellcome Fund (grant 1014782); Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (grants CH-II-2015-462, CH-II-2017-628); and the Foundation for Barnes-Jewish Hospital (grant 8038-88). All figures created with BioRender software (https://www.biorender.com).
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Dick SA, Epelman S. 2016. Chronic heart failure and inflammation: What do we really know? Circ. Res 119:159–76 [DOI] [PubMed] [Google Scholar]
- 2.Moore KJ, Koplev S, Fisher EA, Tabas I, Björkegren JLM, et al. 2018. Macrophage trafficking, inflammatory resolution, and genomics in atherosclerosis: JACC macrophage in CVD series (part 2). J. Am. Coll. Cardiol 72:2181–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ueland T, Gullestad L, Nymo SH, Yndestad A, Aukrust P, Askevold ET. 2015. Inflammatory cytokines as biomarkers in heart failure. Clin. Chim. Acta 443:71–77 [DOI] [PubMed] [Google Scholar]
- 4.DuBrock HM, AbouEzzeddine OF, Redfield MM. 2018. High-sensitivity C-reactive protein in heart failure with preserved ejection fraction. PLOS ONE 13:e0201836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalogeropoulos A, Georgiopoulou V, Psaty BM, Rodondi N, Smith AL, et al. 2010. Inflammatory markers and incident heart failure risk in older adults: the Health ABC (Health, Aging, and Body Composition) study. J. Am. Coll. Cardiol 55:2129–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nymo SH, Hulthe J, Ueland T, McMurray J, Wikstrand J, et al. 2014. Inflammatory cytokines in chronic heart failure: interleukin-8 is associated with adverse outcome. Results from CORONA. Eur. J. Heart Fail 16:68–75 [DOI] [PubMed] [Google Scholar]
- 7.Panahi M, Papanikolaou A, Torabi A, Zhang JG, Khan H, et al. 2018. Immunomodulatory interventions in myocardial infarction and heart failure: a systematic review of clinical trials and meta-analysis of IL-1 inhibition. Cardiovasc. Res 114:1445–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mann DL, McMurray JJ, Packer M, Swedberg K, Borer JS, et al. 2004. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation 109:1594–602 [DOI] [PubMed] [Google Scholar]
- 9.Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT, et al. 2003. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-α, in patients with moderate-to-severe heart failure: results of the Anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 107:3133–40 [DOI] [PubMed] [Google Scholar]
- 10.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, et al. 2017. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med 377:1119–31 [DOI] [PubMed] [Google Scholar]
- 11.Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ. 2018. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet 391:319–28 [DOI] [PubMed] [Google Scholar]
- 12.Tardif JC, Tanguay JF, Wright SR, Duchatelle V, Petroni T, et al. 2013. Effects of the P-selectin antagonist inclacumab on myocardial damage after percutaneous coronary intervention for non-ST-segment elevation myocardial infarction: results of the SELECT-ACS trial. J. Am. Coll. Cardiol 61:2048–55 [DOI] [PubMed] [Google Scholar]
- 13.Stähli BE, Gebhard C, Duchatelle V, Cournoyer D, Petroni T, et al. 2016. Effects of the P-selectin antagonist inclacumab on myocardial damage after percutaneous coronary intervention according to timing of infusion: insights from the SELECT-ACS trial. J. Am. Heart Assoc 5:e004255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramos GC, van den Berg A, Nunes-Silva V, Weirather J, Peters L, et al. 2017. Myocardial aging as a T-cell-mediated phenomenon. PNAS 114:E2420.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, et al. 2014. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40:91–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pinto AR, Paolicelli R, Salimova E, Gospocic J, Slonimsky E, et al. 2012. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLOS ONE 7:e36814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, et al. 2014. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ. Res 114:1611–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nahrendorf M, Swirski FK. 2016. Abandoning M1/M2 for a network model of macrophage function. Circ. Res 119:414–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walter W, Alonso-Herranz L, Trappetti V, Crespo I, Ibberson M, et al. 2018. Deciphering the dynamic transcriptional and post-transcriptional networks of macrophages in the healthy heart and after myocardial injury. Cell Rep 23:622–36 [DOI] [PubMed] [Google Scholar]
- 20.Varga T, Mounier R, Horvath A, Cuvellier S, Dumont F, et al. 2016. Highly dynamic transcriptional signature of distinct macrophage subsets during sterile inflammation, resolution, and tissue repair. J. Immunol 196:4771–82 [DOI] [PubMed] [Google Scholar]
- 21.Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, et al. 2014. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159:1312–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lavine KJ, Pinto AR, Epelman S, Kopecky BJ, Clemente-Casares X, et al. 2018. The macrophage in cardiac homeostasis and disease: JACC macrophage in CVD series (part 4). J. Am. Coll. Cardiol 72:2213–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ensan S, Li A, Besla R, Degousee N, Cosme J, et al. 2016. Self-renewing resident arterial macrophages arise from embryonic CX3CR1+ precursors and circulating monocytes immediately after birth. Nat. Immunol 17:159–68 [DOI] [PubMed] [Google Scholar]
- 24.Röszer T 2018. Understanding the biology of self-renewing macrophages. Cells 7:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gentek R, Molawi K, Sieweke MH. 2014. Tissue macrophage identity and self-renewal. Immunol. Rev 262:56–73 [DOI] [PubMed] [Google Scholar]
- 26.Williams JW, Giannarelli C, Rahman A, Randolph GJ, Kovacic JC. 2018. Macrophage biology, classification, and phenotype in cardiovascular disease: JACC macrophage in CVD series (part 1). J. Am. Coll. Cardiol 72:2166–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, et al. 2017. Macrophages facilitate electrical conduction in the heart. Cell 169:510–22.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heidt T, Courties G, Dutta P, Sager HB, Sebas M, et al. 2014. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res 115:284–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sager HB, Hulsmans M, Lavine KJ, Moreira MB, Heidt T, et al. 2016. Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ. Res 119:853–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, et al. 2019. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol 20:29–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stevens SM, von Gise A, VanDusen N, Zhou B, Pu WT. 2016. Epicardium is required for cardiac seeding by yolk sac macrophages, precursors of resident macrophages of the adult heart. Dev. Biol 413:153–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, et al. 2019. Tissue resident CCR2− and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ. Res 124:263–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, et al. 2014. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. PNAS 111:16029–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leid J, Carrelha J, Boukarabila H, Epelman S, Jacobsen SE, Lavine KJ. 2016. Primitive embryonic macrophages are required for coronary development and maturation. Circ. Res 118:1498–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun Z, Zhou D, Xie X, Wang S, Wang Z, et al. 2016. Cross-talk between macrophages and atrial myocytes in atrial fibrillation. Basic Res. Cardiol 111:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, et al. 2018. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med 24:1234–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalogeris T, Baines CP, Krenz M, Korthuis RJ. 2012. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol 298:229–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glezeva N, Voon V, Watson C, Horgan S, McDonald K, et al. 2015. Exaggerated inflammation and monocytosis associate with diastolic dysfunction in heart failure with preserved ejection fraction: evidence of M2 macrophage activation in disease pathogenesis. J. Card. Fail 21:167–77 [DOI] [PubMed] [Google Scholar]
- 39.Nakayama H, Otsu K. 2013. Translation of hemodynamic stress to sterile inflammation in the heart. Trends Endocrinol. Metab 24:546–53 [DOI] [PubMed] [Google Scholar]
- 40.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, et al. 2012. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485:251–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nishida K, Otsu K. 2017. Sterile inflammation and degradation systems in heart failure. Circ. J 81:622–28 [DOI] [PubMed] [Google Scholar]
- 42.Nakayama H, Otsu K. 2018. Mitochondrial DNA as an inflammatory mediator in cardiovascular diseases. Biochem. J 475:839–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krychtiuk KA, Wurm R, Ruhittel S, Lenz M, Huber K, et al. 2019. Release of mitochondrial DNA is associated with mortality in severe acute heart failure. Eur. Heart J. Acute Cardiovasc. Care 10.1177/2048872618823405 [DOI] [PubMed] [Google Scholar]
- 44.Dhondup Y, Ueland T, Dahl CP, Askevold ET, Sandanger O, et al. 2016. Low circulating levels of mitochondrial and high levels of nuclear DNA predict mortality in chronic heart failure. J. Card. Fail 22:823–28 [DOI] [PubMed] [Google Scholar]
- 45.Cao DJ, Schiattarella GG, Villalobos E, Jiang N, May HI, et al. 2018. Cytosolic DNA sensing promotes macrophage transformation and governs myocardial ischemic injury. Circulation 137:2613–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li W, Hsiao HM, Higashikubo R, Saunders BT, Bharat A, et al. 2016. Heart-resident CCR2+ macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight 1:e87315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.King KR, Aguirre AD, Ye YX, Sun Y, Roh JD, et al. 2017. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med 23:1481–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang L, Cai X, Liu J, Jia Z, Jiao J, et al. 2013. CpG-ODN attenuates pathological cardiac hypertrophy and heart failure by activation of PI3Kα-Akt signaling. PLOS ONE 8:e62373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Velten M, Duerr GD, Pessies T, Schild J, Lohner R, et al. 2012. Priming with synthetic oligonucleotides attenuates pressure overload-induced inflammation and cardiac hypertrophy in mice. Cardiovasc. Res 96:422–32 [DOI] [PubMed] [Google Scholar]
- 50.Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. 2007. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat. Med 13:851–56 [DOI] [PubMed] [Google Scholar]
- 51.Ehrentraut H, Ehrentraut SF, Boehm O, El Aissati S, Foltz F, et al. 2015. Tlr4 deficiency protects against cardiac pressure overload induced hyperinflammation. PLOS ONE 10:e0142921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ehrentraut H, Weber C, Ehrentraut S, Schwederski M, Boehm O, et al. 2011. The Toll-like receptor 4-antagonist eritoran reduces murine cardiac hypertrophy. Eur. J. Heart Fail 13:602–10 [DOI] [PubMed] [Google Scholar]
- 53.Higashikuni Y, Tanaka K, Kato M, Nureki O, Hirata Y, et al. 2013. Toll-like receptor-2 mediates adaptive cardiac hypertrophy in response to pressure overload through interleukin-1β upregulation via nuclear factor κB activation. J. Am. Heart Assoc 2:e000267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matsuda S, Umemoto S, Yoshimura K, Itoh S, Murata T, et al. 2015. Angiotensin activates MCP-1 and induces cardiac hypertrophy and dysfunction via Toll-like receptor 4. J. Atheroscler. Thromb 22:833–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang L, Li YL, Zhang CC, Cui W, Wang X, et al. 2014. Inhibition of Toll-like receptor 2 reduces cardiac fibrosis by attenuating macrophage-mediated inflammation. Cardiovasc. Res 101:383–92 [DOI] [PubMed] [Google Scholar]
- 56.Han J, Zou C, Mei L, Zhang Y, Qian Y, et al. 2017. MD2 mediates angiotensin II-induced cardiac inflammation and remodeling via directly binding to Ang II and activating TLR4/NF-κB signaling pathway. Basic Res. Cardiol 112:9. [DOI] [PubMed] [Google Scholar]
- 57.Strand ME, Herum KM, Rana ZA, Skrbic B, Askevold ET, et al. 2013. Innate immune signaling induces expression and shedding of the heparan sulfate proteoglycan syndecan-4 in cardiac fibroblasts and myocytes, affecting inflammation in the pressure-overloaded heart. FEBS J. 280:2228–47 [DOI] [PubMed] [Google Scholar]
- 58.Honsho S, Nishikawa S, Amano K, Zen K, Adachi Y, et al. 2009. Pressure-mediated hypertrophy and mechanical stretch induces IL-1 release and subsequent IGF-1 generation to maintain compensative hypertrophy by affecting Akt and JNK pathways. Circ. Res 105:1149–58 [DOI] [PubMed] [Google Scholar]
- 59.Leychenko A, Konorev E, Jijiwa M, Matter ML. 2011. Stretch-induced hypertrophy activates NFκB-mediated VEGF secretion in adult cardiomyocytes. PLOS ONE 6:e29055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, et al. 2005. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J. Clin. Investig 115:2108–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oka T, Akazawa H, Naito AT, Komuro I. 2014. Angiogenesis and cardiac hypertrophy: maintenance of cardiac function and causative roles in heart failure. Circ. Res 114:565–71 [DOI] [PubMed] [Google Scholar]
- 62.Kehat I, Molkentin JD. 2010. Molecular pathways underlying cardiac remodeling during pathophysio-logical stimulation. Circulation 122:2727–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Volz HC, Laohachewin D, Seidel C, Lasitschka F, Keilbach K, et al. 2012. S100A8/A9 aggravates post-ischemic heart failure through activation of RAGE-dependent NF-κB signaling. Basic Res. Cardiol 107:250. [DOI] [PubMed] [Google Scholar]
- 64.Ramasamy R, Schmidt AM. 2012. Receptor for advanced glycation end products (RAGE) and implications for the pathophysiology of heart failure. Curr. Heart Fail. Rep 9:107–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Uderhardt S, Martins AJ, Tsang JS, Lammermann T, Germain RN. 2019. Resident macrophages cloak tissue microlesions to prevent neutrophil-driven inflammatory damage. Cell 177:541–55.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, et al. 2014. Macrophages are required for neonatal heart regeneration. J. Clin. Investig 124:1382–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liao X, Shen Y, Zhang R, Sugi K, Vasudevan NT, et al. 2018. Distinct roles of resident and nonresident macrophages in nonischemic cardiomyopathy. PNAS 115:E4661–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Clemente-Casares X, Hosseinzadeh S, Barbu I, Dick SA, Macklin JA, et al. 2017. A CD103+ conventional dendritic cell surveillance system prevents development of overt heart failure during subclinical viral myocarditis. Immunity 47:974–89.e8 [DOI] [PubMed] [Google Scholar]
- 69.Li W, Feng G, Gauthier JM, Lokshina I, Higashikubo R, et al. 2019. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J. Clin. Investig 129:2293–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dutta P, Sager HB, Stengel KR, Naxerova K, Courties G, et al. 2015. Myocardial infarction activates CCR2+ hematopoietic stem and progenitor cells. Cell Stem Cell 16:477–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang NP, Erskine J, Zhang WW, Zheng RH, Zhang LH, et al. 2017. Recruitment of macrophages from the spleen contributes to myocardial fibrosis and hypertension induced by angiotensin II. J. Renin-Angiotensin Aldosterone Syst 18 10.1177/1470320317706653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Prabhu SD. 2018. The cardiosplenic axis is essential for the pathogenesis of ischemic heart failure. Trans. Am. Clin. Climatol. Assoc 129:202–14 [PMC free article] [PubMed] [Google Scholar]
- 73.Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD. 2014. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ. Res 114:266–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Emami H, Singh P, MacNabb M, Vucic E, Lavender Z, et al. 2015. Splenic metabolic activity predicts risk of future cardiovascular events: demonstration of a cardiosplenic axis in humans. JACC Cardiovasc. Imaging 8:121–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Burns J, Sivananthan MU, Ball SG, Mackintosh AF, Mary DA, Greenwood JP. 2007. Relationship between central sympathetic drive and magnetic resonance imaging-determined left ventricular mass in essential hypertension. Circulation 115:1999–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kiriazis H, Wang K, Xu Q, Gao XM, Ming Z, et al. 2008. Knockout of β1- and β2-adrenoceptors attenuates pressure overload-induced cardiac hypertrophy and fibrosis. Br. J. Pharmacol 153:684–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Verloop WL, Beeftink MM, Santema BT, Bots ML, Blankestijn PJ, et al. 2015. A systematic review concerning the relation between the sympathetic nervous system and heart failure with preserved left ventricular ejection fraction. PLOS ONE 10:e0117332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, et al. 2017. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J 38:187–97 [DOI] [PubMed] [Google Scholar]
- 79.Yang W, Tao Y, Wu Y, Zhao X, Ye W, et al. 2019. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat. Commun 10:1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Puhl SL, Steffens S. 2019. Neutrophils in post-myocardial infarction inflammation: Damage versus resolution? Front. Cardiovasc. Med 6:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Geng S, Zhang Y, Lee C, Li L. 2019. Novel reprogramming of neutrophils modulates inflammation resolution during atherosclerosis. Sci. Adv 5:eaav2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mouton AJ, DeLeon-Pennell KY, Rivera Gonzalez OJ, Flynn ER, Freeman TC, et al. 2018. Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res. Cardiol 113:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, et al. 2007. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med 204:3037–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hayasaki T, Kaikita K, Okuma T, Yamamoto E, Kuziel WA, et al. 2006. CC chemokine receptor-2 deficiency attenuates oxidative stress and infarct size caused by myocardial ischemia-reperfusion in mice. Circ. J 70:342–51 [DOI] [PubMed] [Google Scholar]
- 85.Kaikita K, Hayasaki T, Okuma T, Kuziel WA, Ogawa H, Takeya M. 2004. Targeted deletion of CC chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction. Am. J. Pathol 165:439–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Struthers M, Pasternak A. 2010. CCR2 antagonists. Curr. Top. Med. Chem 10:1278–98 [DOI] [PubMed] [Google Scholar]
- 87.Weisheit C, Zhang Y, Faron A, Kopke O, Weisheit G, et al. 2014. Ly6Clow and not Ly6Chigh macrophages accumulate first in the heart in a model of murine pressure-overload. PLOS ONE 9:e112710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Patel B, Ismahil MA, Hamid T, Bansal SS, Prabhu SD. 2017. Mononuclear phagocytes are dispensable for cardiac remodeling in established pressure-overload heart failure. PLOS ONE 12:e0170781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Patel B, Bansal SS, Ismahil MA, Hamid T, Rokosh G, et al. 2018. CCR2+ monocyte-derived infiltrating macrophages are required for adverse cardiac remodeling during pressure overload. JACC Basic Transl. Sci 3:230–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Howangyin KY, Zlatanova I, Pinto C, Ngkelo A, Cochain C, et al. 2016. Myeloid-epithelial-reproductive receptor tyrosine kinase and milk fat globule epidermal growth factor 8 coordinately improve remodeling after myocardial infarction via local delivery of vascular endothelial growth factor. Circulation 133:826–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Deng KQ, Li J, She ZG, Gong J, Cheng WL, et al. 2017. Restoration of circulating MFGE8 (milk fat globule-EGF factor 8) attenuates cardiac hypertrophy through inhibition of Akt pathway. Hypertension 70:770–79 [DOI] [PubMed] [Google Scholar]
- 92.Anzai A, Choi JL, He S, Fenn AM, Nairz M, et al. 2017. The infarcted myocardium solicits GM-CSF for the detrimental oversupply of inflammatory leukocytes. J. Exp. Med 214:3293–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee JS, Jeong SJ, Kim S, Chalifour L, Yun TJ, et al. 2018. Conventional dendritic cells impair recovery after myocardial infarction. J. Immunol 201:1784–98 [DOI] [PubMed] [Google Scholar]
- 94.Nagai T, Honda S, Sugano Y, Matsuyama TA, Ohta-Ogo K, et al. 2014. Decreased myocardial dendritic cells is associated with impaired reparative fibrosis and development of cardiac rupture after myocardial infarction in humans. J. Am. Heart Assoc 3:e000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Anzai A, Anzai T, Nagai S, Maekawa Y, Naito K, et al. 2012. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation 125:1234–45 [DOI] [PubMed] [Google Scholar]
- 96.Nevers T, Salvador AM, Grodecki-Pena A, Knapp A, Velazquez F, et al. 2015. Left ventricular T-cell recruitment contributes to the pathogenesis of heart failure. Circ. Heart Fail 8:776–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Laroumanie F, Douin-Echinard V, Pozzo J, Lairez O, Tortosa F, et al. 2014. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation 129:2111–24 [DOI] [PubMed] [Google Scholar]
- 98.Weirather J, Hofmann UD, Beyersdorf N, Ramos GC, Vogel B, et al. 2014. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ. Res 115:55–67 [DOI] [PubMed] [Google Scholar]
- 99.Hofmann U, Beyersdorf N, Weirather J, Podolskaya A, Bauersachs J, et al. 2012. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation 125:1652–63 [DOI] [PubMed] [Google Scholar]
- 100.Gröschel C, Sasse A, Röhrborn C, Monecke S, Didié M, et al. 2017. T helper cells with specificity for an antigen in cardiomyocytes promote pressure overload-induced progression from hypertrophy to heart failure. Sci. Rep 7:15998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Choo EH, Lee JH, Park EH, Park HE, Jung NC, et al. 2017. Infarcted myocardium-primed dendritic cells improve remodeling and cardiac function after myocardial infarction by modulating the regulatory T cell and macrophage polarization. Circulation 135:1444–57 [DOI] [PubMed] [Google Scholar]
- 102.Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, et al. 2018. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med 24:1234–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yang Z, Day YJ, Toufektsian MC, Xu Y, Ramos SI, et al. 2006. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation 114:2056–64 [DOI] [PubMed] [Google Scholar]
- 104.Yan X, Zhang H, Fan Q, Hu J, Tao R, et al. 2017. Dectin-2 deficiency modulates Th1 differentiation and improves wound healing after myocardial infarction. Circ. Res 120:1116–29 [DOI] [PubMed] [Google Scholar]
- 105.Han YL, Li YL, Jia LX, Cheng JZ, Qi YF, et al. 2012. Reciprocal interaction between macrophages and T cells stimulates IFN-γ and MCP-1 production in Ang II-induced cardiac inflammation and fibrosis. PLOS ONE 7:e35506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Matsumoto K, Ogawa M, Suzuki J, Hirata Y, Nagai R, Isobe M. 2011. Regulatory T lymphocytes attenuate myocardial infarction-induced ventricular remodeling in mice. Int. Heart J 52:382–87 [DOI] [PubMed] [Google Scholar]
- 107.Kanellakis P, Dinh TN, Agrotis A, Bobik A. 2011. CD4+CD25+Foxp3+ regulatory T cells suppress cardiac fibrosis in the hypertensive heart. J. Hypertens 29:1820–28 [DOI] [PubMed] [Google Scholar]
- 108.Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, et al. 2009. Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation 119:2904–12 [DOI] [PubMed] [Google Scholar]
- 109.Dobaczewski M, Xia Y, Bujak M, Gonzalez-Quesada C, Frangogiannis NG. 2010. CCR5 signaling sup-presses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am. J. Pathol 176:2177–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zamilpa R, Kanakia R, Cigarroa J, Dai Q, Escobar GP, et al. 2011. CC chemokine receptor 5 deletion impairs macrophage activation and induces adverse remodeling following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol 300:H1418–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Majmudar MD, Keliher EJ, Heidt T, Leuschner F, Truelove J, et al. 2013. Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation 127:2038–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Heo GS, Kopecky B, Sultan D, Ou M, Feng G, et al. 2019. Molecular imaging visualizes recruitment of inflammatory monocytes and macrophages to the injured heart. Circ. Res 124:881–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fernandez A, Vermeren M, Humphries D, Subiros-Funosas R, Barth N, et al. 2017. Chemical modulation of in vivo macrophage function with subpopulation-specific fluorescent prodrug conjugates. ACS Cent. Sci 3:995–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Amici SA, Dong J, Guerau-de-Arellano M. 2017. Molecular mechanisms modulating the phenotype of macrophages and microglia. Front. Immunol 8:1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, et al. 2006. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med 355:1018–28 [DOI] [PubMed] [Google Scholar]