

Abstract
Secondary diversification of the immunoglobulin (Ig) repertoire occurs through somatic hypermutation (SHM), gene conversion (GCV), and class switch recombination (CSR)—three processes that are initiated by activation-induced cytidine deaminase (AID). AID targets Ig genes at rates orders of magnitude higher than the rest of the genome, but the basis for this specificity is poorly understood. We have previously demonstrated that enhancers and enhancer-like sequences from Ig genes are capable of stimulating SHM of neighboring genes in a capacity distinct from their roles in increasing transcription. Here, we use an in vitro proteomics approach to identify E-box, MEF2, Ets, and Ikaros transcription factor family members as potential binders of these enhancers. Chromatin immunoprecipitation (ChIP) assays in the hypermutating Ramos B cell line confirmed that many of these factors bound the endogenous Igλ enhancer and/or the IgH intronic enhancer (Eμ) in vivo. Further investigation using SHM reporter assays identified binding sites for E2A and MEF2B in Eμ and demonstrated an association between loss of factor binding and decreases in the SHM stimulating activity of Eμ mutants. Our results provide novel insights into trans-acting factors that dictate SHM targeting and link their activity to specific DNA binding sites within Ig enhancers.
Keywords: somatic hypermutation, AID, E2A, MEF2B, Ramos B cell line
Graphical Abstract
An in vitro mass spectrometry based approach uncovers transcription factors that bind to Ig enhancers capable of targeting SHM known as Diversification Activator (DIVAC) sequences. Binding of transcription factors to these sequences is shown to be linked to their capability to target somatic hypermutation.
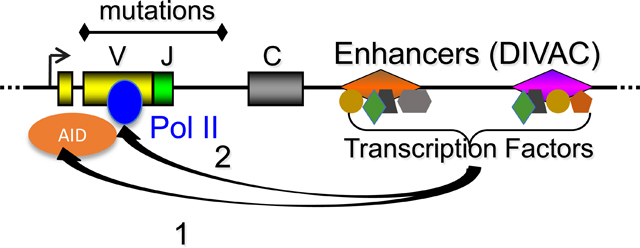
INTRODUCTION
The primary immunoglobulin (Ig) repertoire is generated by V(D)J recombination in developing B cells. Additional diversification occurs in antigen activated B cells through the processes of somatic hypermutation (SHM), gene conversion (GCV), and class switch recombination (CSR), and is initiated by the enzymatic activity of activation-induced cytidine deaminase (AID), which deaminates cytosines to uracils in the Ig loci [1–5].
During SHM, AID is targeted to the rearranged variable region, and processing of uracils by error-prone base excision repair (BER) and mismatch repair (MMR) pathways leads to the accumulation of mutations [6]. AID similarly acts on V regions in GCV, but deamination results in the formation of single and double-stranded DNA breaks that initiate a “copy and paste” mechanism whereby pseudo V (ψV) genes serve as donor templates to be transferred into the rearranged V gene [7, 8]. B cell clones with alterations to their rearranged V-regions that increase the ability of the antibody to bind antigen are selected for and expanded through a cellular process occurring in the germinal center (GC) called affinity maturation [9]. Finally, in CSR, AID is targeted to GC-rich switch regions where processing of uracils leads to double-stranded breaks and recombination that allows for the expression of different Ig isotypes [5].
SHM occurs in V regions over an approximately 1.5 kb region starting approximately 150 bp downstream of the transcription start site (TSS) and extending into the intronic region 3’ of the assembled V exon [10]. Deamination requires transcription which is thought to produce the single-stranded DNA that serves as a substrate for AID activity [11–13]. Levels of transcription, however, are inadequate to explain targeting, as the Ig genes are targeted at orders of magnitude higher rates than the rest of the genome [14, 15]. Recent work has suggested that the qualitative aspects of transcription, such as premature pausing or stalling of the polymerase in the rearranged V region, might potentiate AID activity. Current models for AID targeting have noted its association with RNA Polymerase II (PolII) [16] and other PolII associated factors such as the RNA exosome [17–19] and Spt5 [20, 21], and have thus proposed that AID attaches to a transcriptional elongation complex as it travels through the gene body to initiate its activity when the complex becomes paused or stalled [22, 23].
Though AID targets Ig loci preferentially, it has also been shown to deaminate non-Ig loci, leading to off-target mutations and chromosomal translocations that can subsequently contribute to B cell tumorigenesis [15, 24–26]. Expression of AID in a number of hematological malignancies correlates with poor prognosis [27, 28], and experiments in mice have shown that physiological AID expression can drive the development or progression of B cell lymphomas[29–32]. Studies on off-targeting have suggested a number of potential mechanisms by which AID might be drawn to deaminate non-Ig loci, such as presence of convergent transcription [33], divergent transcription [18], and topologically complex, highly transcribed super-enhancers [34]. However, it is not clear if the mechanisms that mediate off-targeting are similar or distinct from those that govern targeting at the Ig loci.
One potential unifying model is that cis-elements in Ig loci and in targeted non-Ig loci, provide the basis for AID’s targeting. Studies of Ig genes, however, have shown that neither the endogenous assembled V region [35, 36] nor Ig promoters [37–40] are necessary for SHM. Initial studies on Ig enhancers were inconclusive as deletion of these elements had different effects in different contexts and often resulted in reductions in transcription [10, 41, 42].
More recently, work using the chicken DT40 cell line, which constitutively undergoes high rates of GCV and SHM, has shown that cis-elements downstream of the Ig light chain (IgL) constant region are responsible for targeting AID-mediated deamination [43–50]. These elements, which we have termed DIVAC (for diversification activator), are capable of directing hypermutation to a neighboring reporter gene even when placed outside of the Ig loci, indicating their sufficiency for targeting SHM [48].
Using a more sensitive reporter system that allowed for the examination of SHM activity of small cis-elements, we have shown that AID is targeted by Ig enhancer and enhancer-like elements from chicken, human, and mouse [51]. Mutation analysis revealed that the activity of these DIVAC sequences was dependent on a recurrent set of transcription factor motifs—including E-box, NF-κB, MEF2, and Ets—that were conserved across species [51]. Most strikingly, DIVACs from human and mouse showed strong SHM stimulating activity in DT40 cells, indicating a high level of evolutionary conservation [51]. In sum, these studies from our lab and others confirmed a role for DIVACs in SHM beyond their activity as traditional transcriptional enhancers.
While our previous work implicated the binding sites for a number of transcription factor families in DIVAC activity, the identity of the specific factors binding to these sites remained a mystery. To address the potential role of DIVAC-binding factors in targeting SHM, we undertook a proteomics approach where proteins that remained bound to immobilized DIVAC DNA sequences following incubation with nuclear lysate from hypermutating cell lines were identified by mass spectrometry. This work implicated factors that were members of Ikaros, E-box, MEF2, and Ets transcription factor families as DIVAC binders. We then confirmed that a number of the proteomic hits were in fact bound to the endogenous IgH intronic enhancer (Eμ) or Igλ enhancer in vivo using chromatin immunoprecipitation (ChIP). To further dissect the roles of these trans-acting factors in SHM, we performed an extensive binding site mutation analysis of Eμ and found that perturbation of E-box and MEF2 binding sites had the most deleterious effects on Eμ’s SHM stimulating activity. ChIP analysis of the binding site mutants revealed that loss of MEF2B and E2A binding at Eμ mutants in our reporter construct correlated with loss of their SHM stimulating activity. Taken together, our work here provides more evidence for the critical role of enhancers in SHM targeting and suggests that specific classes of transcription factors act in concert to recruit the SHM machinery to Ig loci.
RESULTS
Immobilized template analysis linked to mass spectrometry reveals possible DIVAC-interacting factors
Studies from our lab and others had demonstrated that the Ig enhancers were the critical cis-elements that targeted the SHM machinery. Most recently, we have shown that putative transcription factor (TF) binding sites were necessary for the SHM stimulation activity of these cis-elements. However, any number of TFs from a TF family can potentially bind a single motif, complicating the search for relevant factors. This is best illustrated by the case of E-boxes, which can potentially be bound—either directly or indirectly—by over a hundred basic helix-loop-helix (bHLH) proteins [52]. In other cases, binding sites for one family of TFs in one context can serve as bindings sites for a different set of TFs in an alternate context. For instance, our previous work has implicated NF-κB binding sites in the DIVAC activity of certain Ig cis-elements, but NF-κB motifs have been shown to be competed for and bound by Sp1 under certain circumstances [53, 54].
To overcome these issues and create a manageable set of targets for downstream investigation, we coupled an immobilized template assay to liquid chromatography tandem mass-spectrometry to identify potential DIVAC interacting factors (DIFs) (Figure 1A). We selected the human IgH intronic enhancer (Eμ), the human Igλ enhancer, and the IgL enhancer (cIgLE, also called the 5’ core) and 3’ core from chicken IgL as bait sequences since they were four of the most highly active DIVAC elements in our previous assays in DT40 cells [49, 51]. We paired the human DIVAC elements together to create a single bait sequence and labeled this sequence IgH+L (Supplementary Table 1H). Likewise, the chicken DIVAC elements were combined and named 5+3 (Supplementary Table 1I). As controls, we synthesized mutant versions of the human (mIgH+L) and chicken (5m+3m) bait sequences where all potential minimal TF motifs (based on a TRANSFAC and JASPAR search) were scrambled (Supplementary Tables 1H–I).
Figure 1. Identification of Trans-Factors binding to DIVAC sequences in vitro.

A) Strategy for immobilized template analysis to identify DIVAC interacting trans-factors. Nuclear extract is made from cell lines constitutively undergoing SHM—such as DT40 or Ramos—and then incubated with biotinylated DIVAC sequences and competitor DNA. Following a series of wash steps, DNA-bound proteins are eluted for analysis by SDS-PAGE and western blotting or LC-MS/MS.
B) Representative Sypro Orange-stained SDS-PAGE gels from two independent experiments of eluates from DT40 or Ramos cell extract incubated with chicken DIVAC (5+3) or mutated counterpart (5m+3m) sequences. Red boxes highlight regions of differential banding.
C) Western blot analysis of E2A levels in eluates from chicken bait sequences incubated with Ramos extract from one independent experiment. The same nuclear lysate was incubated with each bait sequence, eluted, and equal volumes loaded.
D) Plot of Log10 values of Peptide Spectral Matches (PSMs) vs abundance ratio of WT/control human and chicken DIVAC sequences incubated with Ramos extract. Data is from three independent experiments with one sample for each group per experiment, and derived from Supplementary Tables 1A and 1B, respectively. Transcription factor families recurrently represented across different samples are highlighted in different colors, as indicated. Some PSMs align with multiple isoforms of the same protein.
Since our previous work had established that DIVAC function was conserved across species [51], our preliminary experiments paired either DT40 and Ramos nuclear extract with the well characterized chicken bait sequences. Eluates from either pairing demonstrated different patterns of bands between WT (5+3) and control (5m+3 m) DIVACs by SDS-PAGE (Figure 1B). As E2A has been implicated by a number of studies as a potential trans-factor critical to SHM[55, 56], we blotted Ramos eluates from chicken DIVACs using an E2A antibody and found that WT 5+3 bound higher levels of E2A protein compared to the control 5m+3m sequence (Figure 1C). Taken together these results suggested that our approach has the potential to uncover novel DIFs.
We performed mass spectrometry analysis on Ramos and DT40 nuclear lysates paired with either chicken or human DIVAC baits and their matched controls. The results showed an enrichment of hits from a variety of TF families—including Ikaros, Ets, and bHLH—when comparing the proteins bound to WT versus mutant DIVAC sequences (Supplementary Tables 1A–D). As most databases and commercial reagents are centered around mouse and human, we focused on hits from the two conditions using Ramos extract. We narrowed the list of potential candidates by looking at factors that had at least 10 peptide spectral matches, 2-fold enrichment for peptides in WT versus control, and were members of recurrently represented transcription factor families (Figure 1D).
The abundance of bHLH factors (Supplementary Tables 1A–D) was notable given the numerous studies implicating roles for E-boxes in SHM targeting [50, 57, 58]. E2A and TFAP4 were the most well represented bHLH family members in the two Ramos extract conditions (Supplementary Tables 1A–B). Other bHLH proteins included—but were not limited to—MGA, MLX, and MAX (Supplementary Tables 1A–B).
Ikaros family members, which have been shown to play critical roles in lymphocyte development [59], were the most abundantly represented family (Figure 1D). Three members of the family, Ikaros (IKZF1), Helios (IKZF2), and Aiolos (IKZF3) were hits in both Ramos extract conditions, with Ikaros and Aiolos being the most strongly represented (Supplementary Tables 1A–B). ELF1 and ELF2 were the only factors from the Ets family found in either of the four conditions (Supplementary Tables 1A–D), with ELF1 found only in the Ramos extract/Chicken DIVAC conditions and ELF2 found in all conditions, but at much lower abundance (Supplementary Tables 1A–D). PU.1 binding motifs had been implicated by work from our lab [51] and others [47] in the DIVAC activity of chicken IgL sequences. Surprisingly, we found no evidence of PU.1 binding from any of the four combinations of extract and bait (Supplementary Tables 1A–D). Other notable hits included members of the Sp, MEF2, and ying yang (YY) family of factors (Supplementary Tables 1A–D).
Several potential DIFs bind endogenous human DIVACs in vivo
To start validation of our proteomics data, we first wanted to determine if putative DIFs were bound to DIVACs in vivo. The Ramos cell line is an Igλ expressing cell line [60] and the functionally rearranged variable regions at both IGH and IGL have been shown to undergo constitutive SHM [60, 61]. Hence, we examined the binding of various putative DIFs at endogenous Eμ and the Igλ enhancer by ChIP-qPCR.
We demonstrated strong binding of E2A to both Eμ and the Igλ enhancer (Figure 2A). MEF2B, the only MEF2 family member to be identified in our proteomics work, likewise bound strongly at both endogenous DIVACs (Figure 2B). We observed modest binding of Aiolos at the Igλ enhancer (Figure 2C) but were not able to detect a signal for either Ikaros or Helios above that seen with the control antibody at either enhancer region (data not shown). YY1 binding was strong at Eμ (Figure 2D), which was expected as there is a strong YY1 motif at the 5’ end of the sequence. We also tested the binding of PU.1—a factor implicated in DIVAC function by binding site mutagenesis [47, 51] but missing from our list of candidate hits—and found strong binding at the Igλ enhancer (Figure 2E). This finding indicates that negative results from our proteomics approach should by itself not be grounds for excluding a factor as a potential DIF.
Figure 2. Putative DIVAC Interacting Factors bind to DIVAC in vivo.

ChIP-qPCR analysis of binding of A) E2A (n=4), B) MEF2B (n=4), C ) Aiolos (n=2) D) YY1 (n=2), and E) PU.1 (n=2) at the endogenous Igλ enhancer (Igλ enh.) and Eμ in WT Ramos cells (n represents the number of independent chromatin samples analyzed for each transcription factor, with each sample subject to ChIP with the specific Ab and control Ig). Data are from 2–3 technical qPCR replicates for each independent ChIP, and represent the mean with S.E.M.
F) Diagram of GFP7 vector to assay SHM targeting capacity of DIVAC elements. CMV - cytomegalovirus promoter, HM7 - Hypermutation Targeting Sequence 7, 2A – 2A self-cleaving peptide, IRES – internal ribosome entry site, BSR – blasticidin resistance gene. Arrow indicates site where DIVAC elements are inserted.
G) Diagram of Chicken IgL locus indicating the portion of cIgLE<->5’core that was cloned into the GFP7 vector. CR1 – chicken repeat region.
ChIP-qPCR analysis of binding of H) E2A (n=2), I) MEF2B (n=2), and J) YY1 (n=2) binding at exogenous cIgLE and 5’core sequences in the GFP7 vector of bulk transduced Ramos cells (n defined as for panels A-E; 2 technical qPCR replicates for each ChIP). Data plotted represent the mean with S.E.M.
For all ChIP analyses, statistical analysis uses unpaired two-tailed T-Test with Welch’s correction. ns = not significant, * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001.
Our lab has recently developed GFP7 [62] a lentiviral SHM reporter capable of assaying DIVAC elements in Ramos cells (Figure 2F), to take advantage of the expanded set of reagents and tools available for use in human cell lines. GFP7 allows for potential DIVAC sequences to be cloned upstream of a truncated CMV promoter driving expression of a hypermutation target sequence (HTS)-T2A-GFP fusion gene (Figure 2F). The HTS is composed of SHM hotspots that upon mutation of cytidine yield stop codons, and the T2A peptide functions through a ribosome skipping mechanism to separate HTS from GFP and prevent decreases in GFP fluorescence intensity. Following a brief, high dose treatment with blasticidin that removes cells with a silenced reporter, SHM at the vector due to the DIVAC element can be read out as loss of GFP expression by FACS—a finding we have confirmed by sequencing the GFP gene after sorting [62].
We cloned an ~1.6 kb fragment from chicken IgL spanning the cIgλE to 3’ core sequences (Figure 2G) into GFP7 before transducing Ramos cells with the vector. We performed ChIP-qPCR at the cIgλE and 3’ core in bulk infected cells and demonstrated binding of E2A at both sequences (Figure 2H). However, binding of MEF2B at chicken sequences (Figure 2I)—while detectable—was weaker than at the endogenous human DIVACs (Figure 2B). YY1 appeared to bind modestly to both cIgλE and 3’ core sequences in the lentivirus (Figure 2J).
Our ability to validate binding of a number of other promising DIFs, such as TFAP4, YY2, Sp3, and BEND3, was limited by lack of availability of suitable ChIP-grade antibodies. Nevertheless, we were able to confirm binding of E2A, MEF2B, Aiolos, and YY1, providing validation for our approach and suggesting a role in SHM targeting for a subset of DIFs identified by proteomics.
Disruption of individual DIF-encoding genes in Ramos has only a modest effect on SHM
To test the functional significance of the DIFs discovered through our proteomics approach, we used transiently expressed CRISPR-Cas9 and sgRNA to inactivate the genes encoding representatives of some of the most abundant and highly enriched TF families in the Ramos proteomics data. Since most of the factors we were ablating have a role in regulation of gene expression, we wanted to mitigate the effects of a potential alteration in the regulation of expression of endogenous AICDA (encoding AID). To this end, we used a Ramos subclone, named A23, that was stably transduced with an MSCV retrovirus over-expressing AID [63] for our gene targeting experiments.
We were able to verify homozygous gene disruption and loss of protein expression for Ikaros, Aiolos, TFAP4 by western blotting (Figure 3A) and through detection of nonsense and frameshift mutations by Sanger sequencing of PCR products (Supplementary Table 1J). Homozygous disruption of BEND3 was validated only by Sanger sequencing (Supplementary Table 1J) due to a lack of a suitable immunoblotting antibody.
Figure 3. Single factor perturbations of DIVAC-interacting factors have modest effects on SHM.

A) Representative western blot analysis of CRISPR-Cas9 mediated gene disruption in Ramos A23 cells from two independent experiments by probing with antibodies against Aiolos, Ikaros, AP4, E2A, and βactin.
B) IgM loss data collected 3 weeks post-sort for IgM+ cells. Each data point represents an independent experiment with bar height equal to the mean of the data for each cell line, plotted +/− S.E.M. All statistical comparisons are to the WT (A23) sample. Statistical analysis uses unpaired two-tailed T-Test with Welch’s correction. * = p<.05, ** = p<.01, *** = p<.001. Statistical analysis using the nonparametric Mann Whitney U Test confirmed that data that were significantly different by T-test were also significantly different (p<.05) using Mann Whitney U Test.
C) Proliferation data for WT (A23) and TCF3hyp cells. Cells were split to 0.2 × 106 cells/mL at d0 and the cell density measured at days 2 and 3. Statistical analysis uses unpaired two-tailed T-Test with Welch’s correction. * = p<.05, ** = p<.01.
D) Representative western blot analysis from two independent experiments of AID and βactin expression in A23 and gene disrupted A23 cells.
E) RT-qPCR expression analysis for endogenous, retroviral, or total AID expression normalized to the RAB7A housekeeping gene. All statistical comparisons are to A23. Data for each bar derive from four technical RT-qPCR replicates for each of two independent RNA preparations for the indicated cell line. Bar height represents the mean of the 8 values and is plotted +/− S.E.M. All statistical analysis uses unpaired two-tailed T-Test with Welch’s correction. * = p<.05, ** = p<.01, *** = p<.001. Bars without an asterisk are not significantly different from A23.
We initially appeared to have also generated an E2A knockout, but further analysis by immunoblotting revealed that this clone was a hypomorph expressing very low levels of an anti-E2A antibody-reactive species (Figure 3A). Our attempts to create a full E2A knockout were likely unsuccessful because Burkitt Lymphoma cell lines, such as Ramos, are addicted to E2A [64]. Our attempts to knockout MEF2B were likewise unsuccessful, leaving us only with heterozygous mutants (data not shown). This might be due to MEF2B being a critical lineage defining TF in germinal center B cells and their derivative tumors, necessary for both survival and proliferation [65, 66].
To determine the functional effects on SHM, we subjected the parental A23 cell line and the knockout subclones to an IgM loss assay in which loss of IgM expression, detected by flow cytometry, serves a read-out for levels of SHM at the Ig loci [60, 67, 68]. We first confirmed that IgM loss was AID-dependent by assaying AID KO cells (Supplementary Figure 1A) and finding they accumulated IgM negative cells at background levels (Figure 3B). Compared to WT A23 cells, knockout subclones exhibited only modest decreases in SHM, with no line showing a more than a two-fold reduction in levels of SHM (Figure 3B). Knockout of IKZF3 and hypomorphic levels of E2A caused the largest reductions in SHM levels with decreases of 44% and 36%, respectively (Figure 3B). However, the E2A hypomorph grew noticeably more slowly than the parental A23 cells, which we confirmed by measuring proliferation over the course of 72 hours (Figure 3C). This defect was not surprising as E2A has been shown to be critical for both the growth of several Burkitt Lymphoma lines [64] as well as a critical TF in germinal center B cells [69, 70]. Since the accumulation of mutations might be linked to cell division, it is possible that some or all of the SHM defect in the clone is explained by decreases in proliferation.
Additionally, to rule out perturbations due to changes in levels of AID expression, we measured total AID protein levels by western blot (Figure 3D) and total AID transcript levels as well as AID transcripts from the endogenous locus and the retroviral vector (Figure 3E). The IKZF3 knockout clone had a 20% decrease in AID transcript levels and decreased levels of AID protein (Figure 3D and 3E), which could partially explain the defects in SHM relative to WT A23.
Surprisingly, of the two IKZF1 KO clones examined, one exhibited a decrease in IgM loss levels (~30%), while the other displayed an increase (~23%) (Figure 3B). The reason for this difference is not known, but could potentially be due to a secondary mutation acquired by one of the two clones. Lastly, loss of BEND3, a protein thought to maintain heterochromatin through its interaction with HP1, increased SHM levels ~1.3-fold (Figure 3B). This phenotype potentially implicates BEND3 in repressing DIVAC function, but could also be due to global increases in chromatin openness and accessibility.
In sum, the data from our gene targeting experiments in the A23 line suggested that loss of any single DIF was likely to have only modest effects on SHM in Ramos cells. We hypothesized that this could be due to redundancy between different factors in the same TF family allowing for compensatory binding. In our initial attempt to address this possibility, we transduced Cas9-expressing Ramos cells with lentiviruses expressing gRNAs targeting IKZF1 and IKZF3, two of the strongest hits from our proteomics approach. Though we were able to recover single knockouts of IKZF1 and IKZF3, we found that the Cas9+ Ramos cells that received gRNAs targeting both genes did not recover from selection (data not shown). As many of the hits from our proteomics analysis are TFs that have roles in global gene regulation and lineage determination, it is unsurprising that perturbing factors with redundant functions might compromise cell survival.
Eμ binding site mutants reveal roles for putative DIFs in stimulating SHM
As genetic ablation of DIFs to determine their necessity in SHM yielded limited insight, we decided instead to look at their potential sufficiency in AID targeting. First, we focused our efforts on an extensive dissection of putative TF binding sites in Eμ, as the element was one of the least extensively characterized in our previous study despite being the most potent small DIVAC element described to date [51].
To create a sensitive SHM assay able to detect changes in activity due to alterations in individual TF binding sites, we engineered a Ramos cell line with high levels of AID expression and activity. CRISPR-Cas9 was used to create an AID-deficient Ramos cell line (Supplementary Figure 1A) into which we introduced a single copy of an all-in-one doxycycline (dox) inducible AID cassette into one allele of AAVS1 (Supplementary Figure 1B–E), to generate the AID7.3in cell line. The cassette expresses AID7.3, an AID mutant with an ~3-fold increase in catalytic activity [71].
We used the JASPAR and TRANSFAC databases to search for potential TF binding motifs in Eμ and compared that to our proteomics data to identify ten binding sites for mutagenesis that included E-boxes and sites for Ets, IRF, YY1 and MEF2 (Figure 4A). WT and mutant Eμ elements were cloned into the GFP7 reporter vector, and the resulting constructs were introduced into AID7.3in cells, sorted, and analyzed for levels of GFP loss 14 days after dox induction. Our analysis showed that mutation of either E-box (M1 or M4) had a substantial effect on SHM of the reporter, with mutation of M1 dropping SHM levels 3-fold (Figure 4B). Notably, mutation of both E-boxes (M1/M4) crippled hypermutation, yielding levels of GFP loss nearly as low as the empty “no DIVAC” (ND) vector control (Figure 4B). With the exception of M7 and M3, all other single binding site mutants showed statistically significant reductions in GFP loss activity compared to WT Eμ using either parametric or non-parametric statistical analysis, but in no case did the reduction exceed 40% (Figure 4B). A putative MEF2 binding site mutant, M9, showed close to a 40% reduction as did a putative IRF binding site mutant, M5 (Figure 4B). Double mutants showed decreased GFP loss relative to single mutants in a largely additive fashion (Figure 4B). Of note, the M9 (MEF2) and M8 (Ets/ELF1) binding sites overlap by 3 bp (Figure 4A), suggesting that cooperative binding of two adjacently spaced factors was unlikely, and only one of the two binding sites was occupied by a DIF.
Figure 4. Eμ binding site mutants have reduced SHM stimulating activity.

A) Diagram of Eμ and the mutations made to binding sites for potential DIVAC interacting factors.
B) GFP loss analysis in AID7.3in Ramos cells transduced with SHM reporter lentiviral vectors carrying empty (ND), Eμ, or mutant sequences of Eμ. Cells were sorted 3–5 days after transduction and GFP loss levels measured 2 weeks following addition of 150 ng/mL of doxycycline to induce AID expression. Data from 3 (M2, M3, M5, M7, M10, M1/M4, M7/M8, M6/M9), 4 (M4, M8), 5 (M6, M9), 6 (M1), or 7 (ND, Eμ) independent transductions. All statistical comparisons are to the Eμ sample using unpaired two-tailed T-Test with Welch’s correction. Data represent the mean +/− S.E.M. ns = not significant, * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001. Statistical analysis using the nonparametric Mann Whitney U Test confirmed that data that were significantly different by T-test were also significantly different (p<.05) using Mann Whitney U Test with the exception of M3.
In sum, our binding site mutation analysis showed that numerous binding sites within Eμ contribute to its SHM stimulating properties, with two E boxes (M1, M4) and a putative MEF2 site (M9) being among the most important. This analysis builds on our previous work demonstrating the critical role of E-boxes in chicken DIVAC function [50] and confirms the importance of the cooperative contribution of TF binding sites to DIVAC function [50, 51].
Loss of MEF2B and E2A binding to Eμ correlates with reductions in SHM
E2A and MEF2B bind endogenous Eμ (Figure 2A and 2B) and mutation of E-boxes and a putative MEF2 site in Eμ substantially reduced DIVAC function (Figure 4B). We were therefore interested in determining whether E2A or MEF2B bind to the functionally relevant sites in Eμ. To investigate this, we performed ChIP-qPCR for E2A binding at Eμ and E-box mutant Eμ sequences in GFP7. Both M1 and M4 mutations compromised E2A binding, with the M4 mutation causing a larger reduction that the M1 mutation and the M1/M4 double mutant showing an additive loss of E2A binding (Figure 5A). Taken together these experiments strongly suggest that E2A binds both E-box sites, but do not rule out other E-box-binding factors as important players in Eμ DIVAC activity.
Figure 5. E2A and MEF2B bind at sites critical to the SHM stimulating activity of Eμ.

ChIP-qPCR analysis of binding of A) E2A (n=2) or B) MEF2B (n=2) at mutated WT or mutated Eμ in the context of the GFP7 vector (n defined as for panels A-E of Figure 2; 2 technical qPCR replicates for each ChIP). Data represent the mean with S.E.M. Statistical analysis only within E2A and MEF2B ChIP-qPCR data using unpaired two-tailed T-Test with Welch’s correction. * = p<0.05, ** = p<0.01.
Similarly, we conducted MEF2B ChIP-qPCR experiments using cells transduced with GFP7 containing WT Eμ or the MEF2 binding site mutants M6 and M9 singularly and in combination. The M9 mutations reduced levels of MEF2B binding significantly while the M6 mutation had no effect on binding, either alone or in combination with the M9 mutation (Figure 5B). These results demonstrate that the motif at M9 is an important MEF2B binding site in Eμ. While M6 does not disrupt MEF2B binding, it could potentially disrupt binding of another DIF either at the site of M6 or indirectly through perturbation of factor binding at adjoining M5 and M10 motifs.
Our data demonstrate that three of the most critical binding sites for Eμ DIVAC activity are bound by E2A and MEF2B and that these factors bind endogenous Eμ. These findings are fully consistent with the hypothesis that E2A and MEF2B contribute directly to the SHM stimulating activity of Eμ.
Eμ’s SHM and transcriptional enhancing properties are not separable using the GFP7 assay
Our previous work in DT40 cells demonstrated that the DIVAC activity of Ig enhancers was not a direct effect of their ability to increase the efficiency of transcription, in part by showing that some strong DIVACs were modest transcriptional enhancers and vice versa [51]. However, what remains unexamined is the degree to which SHM and transcriptional enhancing functions for a given DIVAC element are distinct. We hypothesized that if these two activities were separable, some Eμ binding site mutations would compromise SHM at GFP7 to a larger degree than they did transcription, while others would strongly compromise SHM, but would have little or no effect on transcription.
To address this possibility, we examined normalized median fluorescence intensity (MFI) as a measure of relative GFP gene transcription for Ramos cells transduced with GFP7 containing WT or mutant versions of Eμ. We found that reductions in GFP MFI due to binding site mutations (Figure 6A) was quite similar in pattern to reductions in SHM caused by the mutations (Figure 4B), and that when the data for all replicates was plotted, a strong association between the two features was observed (Figure 6B). We note, however, that relatively small changes in GFP MFI correspond to much larger changes in SHM, with, for example, insertion of Eμ in GFP7 increasing GFP loss ~6-fold over the no-DIVAC background while only increasing MFI ~60%. It remains unclear to what extent there is a causal link between these two phenomena. These data clearly indicate, however, that within the context of GFP7 in Ramos cells, the DIVAC and transcriptional enhancer activities of Eμ are not separable and could not be attributed to distinct TF binding modalities.
Figure 6. GFP loss in the SHM reporter correlates with transcription levels of the GFP gene.

A) Analysis of GFP expression levels of Eμ and Eμ mutants by dividing sample median fluorescence intensity (MFI) by MFI of the empty vector no-DIVAC (ND) sample analyzed concomitantly by flow cytometry. Data from 3 (M2, M3, M5, M7, M10, M1/M4, M7/M8, M6/M9), 4 (M4, M8), 5 (M6, M9), 6 (M1), or 7 (ND, Eμ) independent experiments with the bar representing the mean plotted +/− S.E.M.
B) Scatterplot of GFP loss vs normalized GFP expression for each sample. R2 value calculated for linear regression. P value is a measure of likelihood the slope of the line is non-zero.
DISCUSSION
Previous studies from our lab [49–51] and others [44–48, 72] have implicated cis-acting DIVAC elements from Ig loci in targeting SHM to rearranged V regions. Here, we show that transcription factor binding at these DIVAC elements correlates with targeting of SHM in the context of an SHM reporter assay. Using a proteomics approach, we uncovered numerous factors that interact biochemically with DIVAC elements—with a number of them, including E2A, MEF2B, and members of the Ikaros family reported to be involved in B cell development or germinal center B cell function [65, 66, 69, 70, 73].
Prior work on the chicken DT40 cell line had shown sizable defects in gene conversion upon genetic ablation of either Aiolos [74] or E2A [56] that was independent of AID expression. However, in Ramos, a human EBV-negative Burkitt Lymphoma cell line, single knockout of any one of four putative DIFs (TFAP4, Ikaros, Aiolos, and BEND3) or a E2A hypomorphic mutant had only modest effects on SHM as determined by the IgM loss assay (Figure 3B). What might explain this discrepancy? In an effort to avoid the effects that the knockout of DIFs might have on AID expression, we performed our gene editing experiments in an AID overexpressing line. As a result, it is possible that AID overexpression might have masked more robust impacts of DIF perturbation on SHM. However, Ramos cells express modest levels of AID and have mutation rates below those estimated for germinal center B cells[60]. While A23 cells express high levels of AID relative to WT Ramos, it is unclear the extent to which this is an overexpression relative to the physiological state.
E2A has previously been shown to be essential for the viability of Burkitt Lymphoma lines [64], suggesting that E2A deficiency in our hypomorphic clone perturbs hypermutation modestly, but residual E2A protein levels (Figure 3A) prevent SHM deficiencies to the extent seen in DT40 knockouts. Likewise, no defects in SHM were detected in the B cells of Aiolos knockout mice [73], suggesting that the SHM defect observed in Aiolos-deficient DT40 is not evolutionary conserved or might be restricted to the DT40 line.
Our proteomics analysis identified multiple hits belonging to several families of TFs, raising the possibility that members of the same family of TFs might be able to compensate functionally for one another both in the context of gene transcription and SHM. While this hypothesis is promising, our initial attempts to test it by creating Ramos cells deficient in both Aiolos and Ikaros resulted in a loss of viability. This finding indicates that redundant functions in the context of SHM are likely mirrored by redundant functions in the context of cell survival.
Our prior studies of chicken IgL provided clear evidence of functional redundancy between cis-elements [48–50]. Likewise, numerous studies involving deletions of portions of murine Ig loci have suggested a role for redundancy between enhancer elements [75–82]. An alternative—though not mutually exclusive—explanation for a lack of strong SHM defect in our Ramos TF knockout lines is that different cis-elements within the same locus (such as Eμ and hs3/hs4 of the 3’ regulatory region) bind different members of a TF family, making a lack of SHM defect in our knockouts an indirect result of the redundancy of DIVAC elements.
While our knockout approach yielded limited insight into the role of DIFs in SHM, our ChIP-qPCR studies demonstrated strong binding of E2A and MEF2B at human DIVACs. Furthermore, our binding site mutant analysis showed that three functionally important binding sites in Eμ are E-boxes or MEF2 sites. We further demonstrated an association between E2A and MEF2B binding and Eμ’s SHM stimulating activity by showing loss of binding of both these factors as a consequence of binding site mutations in our lentiviral reporter construct in vivo. Studies showing the critical importance of both E2A [69, 70] and MEF2B [65, 66] in the germinal center reaction in mice support the possibility that these factors have a role in SHM targeting. Finally, recruitment of an E47-LacI protein to a PolyLacO site inserted into the pseudogenes of the IgL locus in chicken DT40 cells increased rates of GCV [55], and a similar phenomenon could be at play at Eμ in Ramos cells.
Previous studies in mice using deletions of enhancers in Ig transgenes or at the endogenous Ig loci often had difficulty dissociating effects on SHM and transcription [38, 76, 77, 81–83]. Our previous approaches circumvented these issues by utilizing a knock-in SHM reporter construct powered by a strong RSV promoter that masked the effect of test sequences on transcription [48–51]. Our results suggested the possibility that different TF binding sites (and their accompanying TFs) mediated aspects of transcription enhancement and mutation enhancement to varying degrees. While the RSV promoter is capable of powering high levels of transcription in DT40, we found that it was both a poor transcriptional activator and highly prone to silencing in the context of GFP7 in Ramos cells (data not shown). As such, we used a truncated CMV promoter in GFP7, which in Ramos cells is of modest strength and as a result is more sensitive to test sequence-mediated effects on transcription. This system revealed a correlation between the effects of Eμ binding site mutants on SHM and on GFP transcription. At least for Eμ, DIVAC activity and transcriptional enhancer activity do not appear to be properties of different binding modalities, suggesting that there are qualitative differences in the way transcription is regulated by enhancers capable of targeting SHM and those that don’t.
Our data on E2A and MEF2B also have implications for AID off-target activity across the genome. Given that both E2A and MEF2B appear to be lineage-defining TFs for GC B cells [65, 66, 69, 70], they would be expected to bind at super-enhancers in these cell types. Given AID’s proclivity for acting at super-enhancers [33, 34], our data predict that off-target loci should be enriched in MEF2B and/or E2A binding. We have recently completed a study identifying regions of the genome that are either susceptible or resistant to AID-mediated mutation despite having similar levels of transcription [62]. As predicted, both E2A and MEF2B binding are significantly enriched in susceptible versus resistant regions of the genome.
The most pressing unanswered question in the field is the elucidation of mechanisms by which the hypermutation machinery is specifically recruited to the Ig loci. A long-standing model in the field posits that AID associates with the transcription machinery until pausing or stalling of PolII allows for AID dissociation and subsequent activity. Until recently, pausing was though to occur largely in the promoter-proximal region, but recent genome-wide approaches have demonstrated that pausing occurs throughout the gene body [84–87]. Some posited mechanisms of PolII pausing appear to be sequence dependent, but others appear to be dependent on factors such as CTCF which are thought to serve as a roadblock to PolII elongation [88, 89]. An appealing mechanism by which DIVAC elements might function is to recruit DIFs that either directly or indirectly increase pausing in the gene body. To our knowledge, there are no studies that detail how enhancers or elements distal from the promoter can act to promote pausing or stalling. However, the understanding of how pausing is directed to and occurs at gene bodies is still in its infancy [90], suggesting exciting avenues for further research.
MATERIALS AND METHODS
Cell Culture
Ramos cells and cell lines derived from Ramos cells were grown in RPMI-1640 (Gibco 11875) supplemented with 10% Fetal Bovine Serum (Sigma), 0.5 mg/mL Penicillin-Streptomycin-Glutamine (Gibco 10378016), and 0.1 mg/mL Normocin (Invivogen ant-nr-2) at 37 °C, 5% CO2. DT40 Cells were grown at 41 °C, 5% CO2 in the same media as Ramos cells with the additional supplementation of 0.1 mM ß-mercaptoethanol. 293T cells were grown at 37 °C, 5% CO2 in DMEM (Gibco 10566) supplemented in similar fashion to Ramos cells. Adherent cells were disassociated using Tryp-LE Express Enzyme (Gibco 12605010) and split every 2–3 days.
Western Blotting
Cells were pelleted, washed once with 1X PBS, and lysed using 200 μL/106 cells of RIPA Buffer. Immunoblotting was carried out as described previously[91]. The following primary antibodies were used for Western Blotting: AID Monoclonal Antibody (Invitrogen ZA001), Ikaros D10E5 Rabbit mAb (CST 9034), Aiolos D1C1E Rabbit mAb (CST 15103), TFAP4 Monoclonal Antibody clone 6B1 (Abnova H00007023-M01), E2A D2B1 Rabbit mAb (CST 12258), E2A (v-18) antibody (SCBT 349), and monoclonal Anti-β-actin (Sigma A2228).
Immobilized Template Analysis and Mass Spectrometry
In brief, nuclei from 300–600 × 106 DT40 or Ramos cells were isolated using a sucrose cushion according to a previously published protocol [92]. The nuclei were resuspended in a high salt buffer (20 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl) and incubated on a rotating wheel at 4 °C for 1 hour before being centrifuged at 20,000 xg for 20 min. The nuclear extract was aliquoted and moved to −80°C for long term storage.
Human DIVACs (Supplementary Table 1H) and chicken DIVACs (Supplementary Table 1I) or their control counterparts (Supplementary Tables 1H–I) were amplified from pIgL GFP2- backbone vectors using biotin-TEG oligos (IDT) and spin-column purified using the Nucleospin Gel and PCR Clean-up kit (Macherney and Nagel 740609). The purified DNA was then bound to M-280 Streptavidin Dynabeads (Invitrogen 11205D) according to manufacturer’s protocol and then washed with Protein Binding Buffer[93]. Four hundred μg of nuclear lysate was combined with 15 μg of either WT or control biotinylated DNA and 25 μg of PolydAdT (Sigma P0883). Protein binding buffer was added to a final volume of 600 μl and the mixture was placed on a rotation wheel at 4 °C for 90 min. Following the incubation, the beads were pelleted and washed three times with Protein Wash Buffer [93] on a magnetic rack. For SDS-PAGE and immunoblotting analysis, the beads were incubated with RIPA Buffer to elute the proteins. For mass spectrometry analysis, the beads were subjected to an on-bead digestion with trypsin (Promega V5111) as described previously [94]. The eluted peptides were flash frozen on dry ice and placed at −80 °C.
All mass spectrometry data was acquired on Thermo Scientific™ Orbitrap Fusion™ Tribrid™ mass spectrometer from 300 to 1200 m/z with a resolution of 120,000 at m/z 200, AGC target 3E5 charges, and a max inject time of 100ms. MS result files were evaluated using Thermo Scientific Proteome Discoverer Beta V2.2.0.336. HCD and CID spectra were searched using the SEQUEST algorithm, and ETD spectra using the MASCOT algorithm against either the Chicken TrEMBL database or the Human Swissprot database downloaded from Uniprot. Final result tables were filtered to remove proteins that did not appear in at least 2 biological replicates, have at least 2 peptide spectral matches, and have a mean sample to control ratio of greater than or equal to 2.0. Protein Hits with the highest number of peptide spectral matches (PSMs) and sample/control ratios were manually validated for MS2 assignments and the accuracy of the label free quantitation.
Plasmids
Combined human DIVAC (IgH intronic enhancer and Igλ enhancer), chicken DIVAC (cIgLE and IgL 3’ core), and their mutated counterparts were synthesized by Blue Heron Biotech, PCR amplified, and cloned into pIgL- GFP2 [48] digested with NheI and SpeI using the In-Fusion method (Takara Bio 638920).
pX458 [95] (Addgene plasmid 4813) and espCas9(1.1) [96] (Addgene plasmid 71814) were gifts from Feng Zhang. espCas9(1.1)-2A-GFP was cloned by replacing SpCas9 in pX458 with espCas9(1.1). espCas9(1.1)-2A-GFP Gln tRNA was subsequently derived by removing the U6 promoter and replacing it with a gln tRNA sequence upstream of the gRNA scaffold while preserving the BbsI cloning sites. Guide RNAs were cloned into these vectors using a previously published protocol [95]. A full list of oligos used to clone sgRNAs can be found in Supplementary Table 1G.
AAVS1 SA-2A-puro-pA donor [97] (Addgene 22075) was a gift from Rudolf Jaenisch. The vector was cut with HindIII to release the insert placed between the AAVS1 targeting arms, and an all-in-one dox-inducible cassette was inserted by a four-fragment In-Fusion reaction that stitched together PCR fragments containing the third-generation TRE response element, a bovine growth hormone polyA site, CMV-Tet3G, and IRES-HisD to create the AAVS1 TCIH vector. AID 7.3 was amplified from Peak8 hAIDup7.3 (a gift from Julian Sale) and cloned into AAVS1 TCIH at a SalI site downstream of the TRE element.
Eμ and Eμ mutants were cloned into the GFP7 vector[62] by insertion at a HpaI site immediately upstream of the central polypurine tract. Two-insert overlapping In-Fusion reactions were used to create binding site mutants. A complete list of primers and strategies used to create the vectors can be found in Supplementary Table 1E.
CRISPR-Cas9 Genome Editing in Ramos Cells
Guide RNAs targeting human AICDA, IKZF1, IKZF3, BEND3, TFAP4, TCF3, or AAVS1 were designed using either MIT’s CRISPR Design ( http://crispr.mit.edu) or the Broad Institute’s sgRNA designer (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). Guide RNAs were cloned into pX458 or espCas9(1.1)-2A-GFP Gln tRNA vectors. For single factor knockouts, sgRNA containing plasmids were transiently transfected into either WT or A23 Ramos cells with Gene Pulser Electroporation Buffer (BioRad 1652676) using the Gene Pulser XCell Electroporation System (BioRad 1652660) and the following settings: exponential wave, 250 V, 800 μF, and infinite resistance. The shocked cells were then placed in 20% FBS containing media and left to recover for 36–48 hours before being single cell sorted for the GFPhi population into 96 well plates containing 200 μL of 20% FBS conditioned media in every well.
Fourteen to 21 days following sorting, colonies in 96 well plates were expanded in 24 well plates. Concurrently, approximately 1,000–10,000 cells were collected and digested in 20 μL of 1X Phusion PCR Buffer (NEB B0518S) with 1 mg/mL Proteinase K (Life Technologies 25530‐049) at 65°C for 1 hour. One μL of the crude genomic DNA preparation was then used as input for PCR amplification of the targeted genomic region. Three μL of the PCR product from Cas9 transfected subclones was then combined with 3 μL of PCR product from amplified WT DNA, denatured at 95 °C in a thermal cycler, and allowed to form heteroduplex DNA by reducing temperature 2.5 °C/min to 25 °C. The heteroduplex DNA was then subjected to a T7 endonuclease assay at 37 °C for 1 hour before being visualized on an EtBr stained 2% agarose gel. Mismatches between the WT DNA and potential genome edited clones were visualized as bands smaller than the primary amplicon.
PCR reactions from clones showing evidence of genome editing were TA-Cloned using the Topo TA Cloning Kit (Invitrogen K4574) and Sanger sequenced. Clones showing evidence of large deletions or nonsense mutations were expanded before 2–5 × 106 cells were collected, washed with PBS, and lysed in RIPA Buffer. Knockout of the gene of interest was confirmed by western blotting when commercially available antibodies were available.
To create the AID7.3in cell line, AID-deficient Ramos cells were transfected with the AAVS1 TCIH AID 7.3 vector and the espCas9(1.1) Gln tRNA vector carrying gRNAs targeting AAVS1 using the conditions described above. Transfected cells were returned to 20% FBS media for four days after recovery, before histidinol (Sigma H6647) selection was added to a final concentration of 0.5 mg/mL and the cells dispensed into 96 well plates. After 14–17 days, clones were picked, expanded, and checked for targeted integration of the cassette by genomic PCR.
Lentiviral transductions
293T Cells were grown to 50–80% confluence and transfected with 3 μg of total DNA mixed with JetPrime reagent (Polyplus 114–07) in 6-well plates according to the manufacturer’s protocol. The transfection media was replaced with 2 mL of UltraCULTURE media (Lonza 12–725F) supplemented with 1X Glutamax (Gibco 35050061) and 0.1 mg/mL Normocin four hours later. Forty-eight hours after transfection the viral supernatant was collected and Ramos cells were transduced as reported previously [68].
IgM Loss Assay
For the IgM loss assay, ~1.0 × 106 cells were stained with APC Mouse Anti-Human IgM (BD 551062) diluted 1:100 in FACS Buffer (1X PBS with 2% FBS) and incubated for 20 min at RT. Cells were washed 2X with FACS Buffer and sorted for the IgM+ population in bulk. Following 3 weeks in culture, cells were again stained, and the percentage of IgM+ and IgM− cells measured in accordance with published guidelines on flow cytometry [98] (Supplementary Figure 2).
GFP Loss Assay
For the GFP loss assay, cells were transduced with GFP7 vectors and selected two days later with 5 μg/mL blasticidin (Sigma A111392). On day 5–7, cells were sorted for the GFP+ population in bulk and recovered in media without selection. Eleven days following the sort, the cells were selected with 20 μg/mL blasticidin for three days and assayed for the percentage of GFP+ and GFP− cells on day 14 post-sort in accordance with published guidelines on flow cytometry [98].
RNA preparation and Quantitative Real-time PCR (qRT-PCR)
RNA was prepared using the RNeasy Mini Kit (Qiagen 74106) according to the manufacturer’s protocol from 2–8 × 106 cells. cDNA was prepared from RNA using Superscript II (Qiagen 18080–051) and random primers (Invitrogen 48190011). iTaq Universal SYBR Green Supermix (Bio-Rad 1725120) was used to perform qPCR analysis of cDNA in duplicate or triplicate for every sample according to manufacturer’s protocol. Ct values for a GOI were normalized to RAB7A housekeeping gene [99] using the double delta Ct method [100]. Primers used for qPCR assays are detailed in Supplementary Table 1F.
Chromatin Immunoprecipitation (ChIP)-qPCR
Approximately 40 × 106 Ramos cells were pelleted and resuspended in 9 mL of RPMI supplemented with 2% FBS in a 15 mL conical tube. 600 uL of 16% formaldehyde (Thermo Fisher 28906) was added and the tube was placed on a rocker for 10 min at RT before 1 mL of 10X Glycine solution from the SimpleChIP Enzymatic Chromatin IP Kit (CST 9003) was added to quench the reaction. The rest of the procedure to isolate and purify ChIP DNA was completed using reagents from the SimpleChIP kit according to the manufacturer’s protocol. qPCR using ChIP DNA or 2% input sample was completed using iTaq Universal Probes Supermix Kit (Bio-Rad 1725130) according to the manufacturer’s protocol. Percent input was calculated with the following formula: Fraction of the Input = 2% x 2(C[T] 2%Input Sample – C[T] IP Sample). Primers and probes used for qPCR assays are detailed in Supplementary Table 1F.
The following antibodies were used for ChIP: Aiolos D1C1E Rabbit mAb (CST 15103), E2A D2B1 Rabbit mAb (CST 12258), YY1(SCBT 7341X), PU.1 9G7 Rabbit mAb (CST 2258), Ikaros (D10E5) Rabbit mAb (CST 9034), Helios D8W4X (CST 4247), and MEF2B (Abcam 33540).
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank members of the Schatz and Hunt labs for helpful discussions, and the staff of the Yale Flow Cytometry Core for assistance with cell sorting experiments. This work was supported by in part by R01 AI127642 (D.G.S.), GM 037537 (D.F.H.), a Gruber Science Fellowship and National Science Foundation Graduate Research Fellowship (R.K.D.), grant 15-24776S from the Czech Science Foundation (F.S.), and grants from Sigrid Juselius Foundation, Jane and Aatos Erkko Foundation, Jenny and Antti Wihuri Foundation, Ella and Georg Ehrnrooth Foundation, Cancer Society of South-West Finland and Emil Aaltonen Foundation (J.A.).
Footnotes
CONFLICTS OF INTEREST
The authors declare no commercial or financial conflicts of interest.
REFERENCES
- 1.Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO and Honjo T, Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem 1999. 274: 18470–18476. [DOI] [PubMed] [Google Scholar]
- 2.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y. and Honjo T, Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000. 102: 553–563. [DOI] [PubMed] [Google Scholar]
- 3.Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N. et al. , Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell 2000. 102: 565–575. [DOI] [PubMed] [Google Scholar]
- 4.Arakawa H, Hauschild J. and Buerstedde JM, Requirement of the activation-induced deaminase (AID) gene for immunoglobulin gene conversion. Science 2002. 295: 1301–1306. [DOI] [PubMed] [Google Scholar]
- 5.Di Noia JM and Neuberger MS, Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem 2007. 76: 1–22. [DOI] [PubMed] [Google Scholar]
- 6.Peled JU, Kuang FL, Iglesias-Ussel MD, Roa S, Kalis SL, Goodman MF and Scharff MD, The biochemistry of somatic hypermutation. Annu Rev Immunol 2008. 26: 481–511. [DOI] [PubMed] [Google Scholar]
- 7.Chen JM, Cooper DN, Chuzhanova N, Ferec C. and Patrinos GP, Gene conversion: mechanisms, evolution and human disease. Nat Rev Genet 2007. 8: 762–775. [DOI] [PubMed] [Google Scholar]
- 8.Arakawa H. and Buerstedde JM, Immunoglobulin gene conversion: insights from bursal B cells and the DT40 cell line. Dev Dyn 2004. 229: 458–464. [DOI] [PubMed] [Google Scholar]
- 9.Victora GD and Nussenzweig MC, Germinal centers. Annu Rev Immunol 2012. 30: 429–457. [DOI] [PubMed] [Google Scholar]
- 10.Odegard VH and Schatz DG, Targeting of somatic hypermutation. Nat Rev Immunol 2006. 6: 573–583. [DOI] [PubMed] [Google Scholar]
- 11.Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E. and Alt FW, Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature 2003. 422: 726–730. [DOI] [PubMed] [Google Scholar]
- 12.Bransteitter R, Pham P, Scharff MD and Goodman MF, Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc Natl Acad Sci U S A 2003. 100: 4102–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petersen-Mahrt SK, Harris RS and Neuberger MS, AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature 2002. 418: 99–103. [DOI] [PubMed] [Google Scholar]
- 14.Rajewsky K, Forster I. and Cumano A, Evolutionary and somatic selection of the antibody repertoire in the mouse. Science 1987. 238: 1088–1094. [DOI] [PubMed] [Google Scholar]
- 15.Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH and Schatz DG, Two levels of protection for the B cell genome during somatic hypermutation. Nature 2008. 451: 841–845. [DOI] [PubMed] [Google Scholar]
- 16.Nambu Y, Sugai M, Gonda H, Lee CG, Katakai T, Agata Y, Yokota Y. and Shimizu A, Transcription-coupled events associating with immunoglobulin switch region chromatin. Science 2003. 302: 2137–2140. [DOI] [PubMed] [Google Scholar]
- 17.Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, Zhang T. et al. , The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell 2011. 144: 353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pefanis E, Wang J, Rothschild G, Lim J, Chao J, Rabadan R, Economides AN and Basu U, Noncoding RNA transcription targets AID to divergently transcribed loci in B cells. Nature 2014. 514: 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pefanis E, Wang J, Rothschild G, Lim J, Kazadi D, Sun J, Federation A. et al. , RNA exosome-regulated long non-coding RNA transcription controls super-enhancer activity. Cell 2015. 161: 774–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, Resch W. et al. , Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell 2010. 143: 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maul RW, Cao Z, Venkataraman L, Giorgetti CA, Press JL, Denizot Y, Du H. et al. , Spt5 accumulation at variable genes distinguishes somatic hypermutation in germinal center B cells from ex vivo-activated cells. J Exp Med 2014. 211: 2297–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peters A. and Storb U, Somatic hypermutation of immunoglobulin genes is linked to transcription initiation. Immunity 1996. 4: 57–65. [DOI] [PubMed] [Google Scholar]
- 23.Sun J, Rothschild G, Pefanis E. and Basu U, Transcriptional stalling in B-lymphocytes: a mechanism for antibody diversification and maintenance of genomic integrity. Transcription 2013. 4: 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Casellas R, Resch W, Hakim O. and Nussenzweig MC, The origin of B cell recurrent chromosomal translocations: proximity versus DNA damage. Mol Cell 2013. 51: 275–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robbiani DF and Nussenzweig MC, Chromosome translocation, B cell lymphoma, and activation-induced cytidine deaminase. Annu Rev Pathol 2013. 8: 79–103. [DOI] [PubMed] [Google Scholar]
- 26.Alvarez-Prado AF, Perez-Duran P, Perez-Garcia A, Benguria A, Torroja C, de Yebenes VG and Ramiro AR, A broad atlas of somatic hypermutation allows prediction of activation-induced deaminase targets. J Exp Med 2018. 215: 761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Casellas R, Basu U, Yewdell WT, Chaudhuri J, Robbiani DF and Di Noia JM, Mutations, kataegis and translocations in B cells: understanding AID promiscuous activity. Nat Rev Immunol 2016. 16: 164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swaminathan S, Klemm L, Park E, Papaemmanuil E, Ford A, Kweon SM, Trageser D. et al. , Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nat Immunol 2015. 16: 766–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pasqualucci L, Bhagat G, Jankovic M, Compagno M, Smith P, Muramatsu M, Honjo T. et al. , AID is required for germinal center-derived lymphomagenesis. Nat Genet 2008. 40: 108–112. [DOI] [PubMed] [Google Scholar]
- 30.Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio S, Chen-Kiang S, Muramatsu M, Honjo T. et al. , AID is required for c-myc/IgH chromosome translocations in vivo. Cell 2004. 118: 431–438. [DOI] [PubMed] [Google Scholar]
- 31.Robbiani DF, Bothmer A, Callen E, Reina-San-Martin B, Dorsett Y, Difilippantonio S, Bolland DJ et al. , AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell 2008. 135: 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dorsett Y, Robbiani DF, Jankovic M, Reina-San-Martin B, Eisenreich TR and Nussenzweig MC, A role for AID in chromosome translocations between c-myc and the IgH variable region. J Exp Med 2007. 204: 2225–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meng FL, Du Z, Federation A, Hu J, Wang Q, Kieffer-Kwon KR, Meyers RM et al. , Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell 2014. 159: 1538–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qian J, Wang Q, Dose M, Pruett N, Kieffer-Kwon KR, Resch W, Liang G. et al. , B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell 2014. 159: 1524–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yelamos J, Klix N, Goyenechea B, Lozano F, Chui YL, Gonzalez Fernandez A, Pannell R. et al. , Targeting of non-Ig sequences in place of the V segment by somatic hypermutation. Nature 1995. 376: 225–229. [DOI] [PubMed] [Google Scholar]
- 36.Yeap LS, Hwang JK, Du Z, Meyers RM, Meng FL, Jakubauskaite A, Liu M. et al. , Sequence-Intrinsic Mechanisms that Target AID Mutational Outcomes on Antibody Genes. Cell 2015. 163: 1124–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fukita Y, Jacobs H. and Rajewsky K, Somatic hypermutation in the heavy chain locus correlates with transcription. Immunity 1998. 9: 105–114. [DOI] [PubMed] [Google Scholar]
- 38.Betz AG, Milstein C, Gonzalez-Fernandez A, Pannell R, Larson T. and Neuberger MS, Elements regulating somatic hypermutation of an immunoglobulin kappa gene: critical role for the intron enhancer/matrix attachment region. Cell 1994. 77: 239–248. [DOI] [PubMed] [Google Scholar]
- 39.Tumas-Brundage KM, Vora KA and Manser T, Evaluation of the role of the 3’alpha heavy chain enhancer [3’alpha E(hs1,2)] in Vh gene somatic hypermutation. Mol Immunol 1997. 34: 367–378. [DOI] [PubMed] [Google Scholar]
- 40.Yang SY, Fugmann SD and Schatz DG, Control of gene conversion and somatic hypermutation by immunoglobulin promoter and enhancer sequences. J Exp Med 2006. 203: 2919–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang SY and Schatz DG, Targeting of AID-mediated sequence diversification by cis-acting determinants. Adv Immunol 2007. 94: 109–125. [DOI] [PubMed] [Google Scholar]
- 42.Kothapalli NR and Fugmann SD, Targeting of AID-mediated sequence diversification to immunoglobulin genes. Curr Opin Immunol 2011. 23: 184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kothapalli N, Norton DD and Fugmann SD, Cutting edge: a cis-acting DNA element targets AID-mediated sequence diversification to the chicken Ig light chain gene locus. J Immunol 2008. 180: 2019–2023. [DOI] [PubMed] [Google Scholar]
- 44.Kothapalli NR, Collura KM, Norton DD and Fugmann SD, Separation of mutational and transcriptional enhancers in Ig genes. J Immunol 2011. 187: 3247–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim Y. and Tian M, The recruitment of activation induced cytidine deaminase to the immunoglobulin locus by a regulatory element. Mol Immunol 2010. 47: 1860–1865. [DOI] [PubMed] [Google Scholar]
- 46.Kim Y. and Tian M, NF-kappaB family of transcription factor facilitates gene conversion in chicken B cells. Mol Immunol 2009. 46: 3283–3291. [DOI] [PubMed] [Google Scholar]
- 47.Luo H. and Tian M, Transcription factors PU.1 and IRF4 regulate activation induced cytidine deaminase in chicken B cells. Mol Immunol 2010. 47: 1383–1395. [DOI] [PubMed] [Google Scholar]
- 48.Blagodatski A, Batrak V, Schmidl S, Schoetz U, Caldwell RB, Arakawa H. and Buerstedde JM, A cis-acting diversification activator both necessary and sufficient for AID-mediated hypermutation. PLoS Genet 2009. 5: e1000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kohler KM, McDonald JJ, Duke JL, Arakawa H, Tan S, Kleinstein SH, Buerstedde JM and Schatz DG, Identification of core DNA elements that target somatic hypermutation. J Immunol 2012. 189: 5314–5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McDonald JJ, Alinikula J, Buerstedde JM and Schatz DG, A critical context-dependent role for E boxes in the targeting of somatic hypermutation. J Immunol 2013. 191: 1556–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buerstedde JM, Alinikula J, Arakawa H, McDonald JJ and Schatz DG, Targeting of somatic hypermutation by immunoglobulin enhancer and enhancer-like sequences. PLoS Biol 2014. 12: e1001831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murre C, Helix-loop-helix proteins and the advent of cellular diversity: 30 years of discovery. Genes Dev 2019. 33: 6–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hirano F, Tanaka H, Hirano Y, Hiramoto M, Handa H, Makino I. and Scheidereit C, Functional interference of Sp1 and NF-kappaB through the same DNA binding site. Mol Cell Biol 1998. 18: 1266–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mao XR, Moerman-Herzog AM, Chen Y. and Barger SW, Unique aspects of transcriptional regulation in neurons--nuances in NFkappaB and Sp1-related factors. J Neuroinflammation 2009. 6: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yabuki M, Ordinario EC, Cummings WJ, Fujii MM and Maizels N, E2A acts in cis in G1 phase of cell cycle to promote Ig gene diversification. J Immunol 2009. 182: 408–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schoetz U, Cervelli M, Wang YD, Fiedler P. and Buerstedde JM, E2A expression stimulates Ig hypermutation. J Immunol 2006. 177: 395–400. [DOI] [PubMed] [Google Scholar]
- 57.Michael N, Shen HM, Longerich S, Kim N, Longacre A. and Storb U, The E box motif CAGGTG enhances somatic hypermutation without enhancing transcription. Immunity 2003. 19: 235–242. [DOI] [PubMed] [Google Scholar]
- 58.Tanaka A, Shen HM, Ratnam S, Kodgire P. and Storb U, Attracting AID to targets of somatic hypermutation. J Exp Med 2010. 207: 405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heizmann B, Kastner P. and Chan S, The Ikaros family in lymphocyte development. Curr Opin Immunol 2018. 51: 14–23. [DOI] [PubMed] [Google Scholar]
- 60.Sale JE and Neuberger MS, TdT-accessible breaks are scattered over the immunoglobulin V domain in a constitutively hypermutating B cell line. Immunity 1998. 9: 859–869. [DOI] [PubMed] [Google Scholar]
- 61.Papavasiliou FN and Schatz DG, Cell-cycle-regulated DNA double-stranded breaks in somatic hypermutation of immunoglobulin genes. Nature 2000. 408: 216–221. [DOI] [PubMed] [Google Scholar]
- 62.Senigl F, Maman Y, Dinesh RK, Alinikula J, Seth RB, Pecnova L, Omer AD et al. , Topologically associated domains delineate susceptibility to somatic hypermutation. Cell Rep 2019. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koduru S, Wong E, Strowig T, Sundaram R, Zhang L, Strout MP, Flavell RA et al. , Dendritic cell-mediated activation-induced cytidine deaminase (AID)-dependent induction of genomic instability in human myeloma. Blood 2012. 119: 2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, Wright G. et al. , Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012. 490: 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brescia P, Schneider C, Holmes AB, Shen Q, Hussein S, Pasqualucci L, Basso K. and Dalla-Favera R, MEF2B Instructs Germinal Center Development and Acts as an Oncogene in B Cell Lymphomagenesis. Cancer Cell 2018. 34: 453–465 e459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ying CY, Dominguez-Sola D, Fabi M, Lorenz IC, Hussein S, Bansal M, Califano A. et al. , MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat Immunol 2013. 14: 1084–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rada C, Jarvis JM and Milstein C, AID-GFP chimeric protein increases hypermutation of Ig genes with no evidence of nuclear localization. Proc Natl Acad Sci U S A 2002. 99: 7003–7008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Upton DC and Unniraman S, Assessing somatic hypermutation in Ramos B cells after overexpression or knockdown of specific genes. J Vis Exp 2011: e3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kwon K, Hutter C, Sun Q, Bilic I, Cobaleda C, Malin S. and Busslinger M, Instructive role of the transcription factor E2A in early B lymphopoiesis and germinal center B cell development. Immunity 2008. 28: 751–762. [DOI] [PubMed] [Google Scholar]
- 70.Wohner M, Tagoh H, Bilic I, Jaritz M, Poliakova DK, Fischer M. and Busslinger M, Molecular functions of the transcription factors E2A and E2–2 in controlling germinal center B cell and plasma cell development. J Exp Med 2016. 213: 1201–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang M, Yang Z, Rada C. and Neuberger MS, AID upmutants isolated using a high-throughput screen highlight the immunity/cancer balance limiting DNA deaminase activity. Nat Struct Mol Biol 2009. 16: 769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kothapalli N. and Fugmann SD, Characterizing somatic hypermutation and gene conversion in the chicken DT40 cell system. Methods Mol Biol 2011. 748: 255–271. [DOI] [PubMed] [Google Scholar]
- 73.Cortes M. and Georgopoulos K, Aiolos is required for the generation of high affinity bone marrow plasma cells responsible for long-term immunity. J Exp Med 2004. 199: 209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Narvi E, Nera KP, Terho P, Mustonen L, Granberg J. and Lassila O, Aiolos controls gene conversion and cell death in DT40 B cells. Scand J Immunol 2007. 65: 503–513. [DOI] [PubMed] [Google Scholar]
- 75.Gorman JR, van der Stoep N, Monroe R, Cogne M, Davidson L. and Alt FW, The Ig(kappa) enhancer influences the ratio of Ig(kappa) versus Ig(lambda) B lymphocytes. Immunity 1996. 5: 241–252. [DOI] [PubMed] [Google Scholar]
- 76.van der Stoep N, Gorman JR and Alt FW, Reevaluation of 3’Ekappa function in stage- and lineage-specific rearrangement and somatic hypermutation. Immunity 1998. 8: 743–750. [DOI] [PubMed] [Google Scholar]
- 77.Inlay MA, Gao HH, Odegard VH, Lin T, Schatz DG and Xu Y, Roles of the Ig kappa light chain intronic and 3’ enhancers in Igk somatic hypermutation. J Immunol 2006. 177: 1146–1151. [DOI] [PubMed] [Google Scholar]
- 78.Perlot T, Alt FW, Bassing CH, Suh H. and Pinaud E, Elucidation of IgH intronic enhancer functions via germ-line deletion. Proc Natl Acad Sci U S A 2005. 102: 14362–14367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pinaud E, Khamlichi AA, Le Morvan C, Drouet M, Nalesso V, Le Bert M. and Cogne M, Localization of the 3’ IgH locus elements that effect long-distance regulation of class switch recombination. Immunity 2001. 15: 187–199. [DOI] [PubMed] [Google Scholar]
- 80.Morvan CL, Pinaud E, Decourt C, Cuvillier A. and Cogne M, The immunoglobulin heavy-chain locus hs3b and hs4 3’ enhancers are dispensable for VDJ assembly and somatic hypermutation. Blood 2003. 102: 1421–1427. [DOI] [PubMed] [Google Scholar]
- 81.Dunnick WA, Collins JT, Shi J, Westfield G, Fontaine C, Hakimpour P. and Papavasiliou FN, Switch recombination and somatic hypermutation are controlled by the heavy chain 3’ enhancer region. J Exp Med 2009. 206: 2613–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rouaud P, Vincent-Fabert C, Saintamand A, Fiancette R, Marquet M, Robert I, Reina-San-Martin B. et al. , The IgH 3’ regulatory region controls somatic hypermutation in germinal center B cells. J Exp Med 2013. 210: 1501–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Goyenechea B, Klix N, Yelamos J, Williams GT, Riddell A, Neuberger MS and Milstein C, Cells strongly expressing Ig(kappa) transgenes show clonal recruitment of hypermutation: a role for both MAR and the enhancers. EMBO J 1997. 16: 3987–3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mayer A. and Churchman LS, Genome-wide profiling of RNA polymerase transcription at nucleotide resolution in human cells with native elongating transcript sequencing. Nat Protoc 2016. 11: 813–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kwak H, Fuda NJ, Core LJ and Lis JT, Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 2013. 339: 950–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang IX, Core LJ, Kwak H, Brady L, Bruzel A, McDaniel L, Richards AL et al. , RNA-DNA differences are generated in human cells within seconds after RNA exits polymerase II. Cell Rep 2014. 6: 906–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nojima T, Gomes T, Grosso ARF, Kimura H, Dye MJ, Dhir S, Carmo-Fonseca M. and Proudfoot NJ, Mammalian NET-Seq Reveals Genome-wide Nascent Transcription Coupled to RNA Processing. Cell 2015. 161: 526–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mayer A, di Iulio J, Maleri S, Eser U, Vierstra J, Reynolds A, Sandstrom R. et al. , Native elongating transcript sequencing reveals human transcriptional activity at nucleotide resolution. Cell 2015. 161: 541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, Oberdoerffer P. et al. , CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 2011. 479: 74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mayer A, Landry HM and Churchman LS, Pause & go: from the discovery of RNA polymerase pausing to its functional implications. Curr Opin Cell Biol 2017. 46: 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Carmona LM, Fugmann SD and Schatz DG, Collaboration of RAG2 with RAG1-like proteins during the evolution of V(D)J recombination. Genes Dev 2016. 30: 909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dyer RB and Herzog NK, Isolation of intact nuclei for nuclear extract preparation from a fragile B-lymphocyte cell line. Biotechniques 1995. 19: 192–195. [PubMed] [Google Scholar]
- 93.Spruijt CG, Baymaz HI and Vermeulen M, Identifying specific protein-DNA interactions using SILAC-based quantitative proteomics. Methods Mol Biol 2013. 977: 137–157. [DOI] [PubMed] [Google Scholar]
- 94.Baymaz HI, Spruijt CG and Vermeulen M, Identifying nuclear protein-protein interactions using GFP affinity purification and SILAC-based quantitative mass spectrometry. Methods Mol Biol 2014. 1188: 207–226. [DOI] [PubMed] [Google Scholar]
- 95.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA and Zhang F, Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013. 8: 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX and Zhang F, Rationally engineered Cas9 nucleases with improved specificity. Science 2016. 351: 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, Katibah GE et al. , Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol 2009. 27: 851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cossarizza A, Chang HD, Radbruch A, Akdis M, Andra I, Annunziato F, Bacher P. et al. , Guidelines for the use of flow cytometry and cell sorting in immunological studies. Eur J Immunol 2017. 47: 1584–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Eisenberg E. and Levanon EY, Human housekeeping genes, revisited. Trends Genet 2013. 29: 569–574. [DOI] [PubMed] [Google Scholar]
- 100.Livak KJ and Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001. 25: 402–408. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.