

Abstract
An adolescent male with a history of autoimmune enteropathy, autoimmune hypothyroidism, aphthous stomatitis and recurrent oral Candida infections only in the setting of curative antibiotic courses presented with cryptococcal pneumonia and perihilar adenitis, which was successfully treated with antifungal therapy. The patient had a complex history with several immunological anomalies. Whole exome sequencing revealed a known STAT1 pathogenic variant, associated with gain of function (GOF). This case expands our understanding of the broad clinical phenotype manifested by STAT1 GOF and emphasises the importance of consideration of this diagnosis in patients presenting with opportunistic infections and autoimmunity.
Keywords: Cryptococcus, infections, immunological products and vaccines, radiology, drugs: respiratory system
Background
Signal transducer and activator of transcription 1 (STAT1) is a transcription factor which plays a key role in mediating cellular responses to interferon (IFN) signalling. In 2011, gain-of-function (GOF) mutations in STAT1 were found to be the cause of nearly half of all cases of chronic mucocutaneous candidiasis (CMC).1 2 Since then, our understanding of the clinical spectrum of STAT1 GOF has broadened; in addition to CMC in 98% of patients, STAT1 GOF has also been found to be associated with autoimmune conditions, recurrent bacterial and viral infections, and rarely, fungal infections.3
Cryptococcus is an opportunistic fungal pathogen that is found ubiquitously in the environment manifests as pneumonia or meningitis in individuals with impaired T cell function. To the best of our knowledge, just 10 cases of Cryptococcus, including 5 with cryptococcal pneumonia have been reported in individuals with STAT1 GOF to date.3–7 Herein, we report a case of a patient with a known STAT1 GOF variant presenting with cryptococcal pneumonia and perihilar adenitis and a history of autoimmune enteropathy, autoimmune hypothyroidism, aphthous stomatitis and recurrent oral Candida infections occurring only in the setting of curative antibiotic courses.
Case presentation
The patient is a 17-year-old young man with a history of recurrent otitis media and viral and bacterial pneumonia since 4 months of age, leading to development of progressive bronchiectasis. He had six episodes of otitis media by 9 months of age, and averaged two to three episodes of pneumonia per year, with infectious agents identified in respiratory isolates including Pneumocystis jiroveci, Pseudomonas aeruginosa and methicillin-resistant Staphylococcus aureus, in addition to common bacterial organisms.
The patient’s course was further complicated by seven episodes of oral candidiasis and one episode of candidal esophagitis, each of which occurred in the setting of recent curative antibiotic courses and responded to treatment with nystatin or fluconazole. The patient had onset of autoimmune enteropathy at age 4 years, associated with chronic diarrhoea and complicated by separate episodes of cytomegalovirus (CMV) colitis and Clostridium difficile infection at age 7 years. He was diagnosed with autoimmune hypothyroidism at age 6 years, based on positive thyroid peroxidase antibodies and manifesting primarily with a lack of linear growth (tracking at the second percentile). His medical history was otherwise complicated by sinusitis, myositis, several episodes of aphthous stomatitis, varicella zoster infection, recurrent impetigo and chronic nasal congestion.
The patient presented to our institution with a 1-week history of a cough and worsening haemoptysis, prompting further workup. At the time of presentation, the patient had been receiving intravenous immunoglobulin therapy and weekly prophylactic azithromycin and trimethoprim–sulfamethoxazole.
Investigations
The patient’s chest CT revealed infiltrate in the lingula suggestive of infection, in addition to bilateral hilar adenopathy (figures 1–3). Bronchoscopy with bronchoalveolar fluid culture revealed Cryptococcus neoformans on fungal culture. Left hilar mediastinal lymph node biopsy was performed, revealing caseating necrosis, granulomatous inflammation and the presence of fungal yeast forms highlighted on mucicarmine stain within the lymph node (figures 4 and 5). Serum cryptococcal antigen titre by lateral flow assay was ≥1:2560. Cerebrospinal fluid cryptococcal antigen was negative. Based on characteristic microscopic and serological findings, the patient was diagnosed with cryptococcal pneumonia. In light of the patient’s history of recurrent infections, immunological investigations were undertaken at 3 years of age, suggesting a primary immunodeficiency of unclear aetiology, with findings significant for an IgA level of 2 mg/dL (reference range 27–246 mg/dL) consistent with complete IgA deficiency, and functional antibody deficiency with abnormally low tetanus, diphtheria and Streptococcus pneumonia titres despite vaccination. Investigations for adenosine deaminase deficiency, alpha-1-antitrypsin deficiency, hyper-IgM syndrome, leucocyte adhesion deficiency, complement deficiencies, cystic fibrosis and coeliac disease were all negative. The patient also had normal T cell function by cellular proliferation studies, and normal neutrophil oxidative burst ruling out chronic granulomatous disease. Flow cytometric analysis was notable for natural killer (NK) cell lymphopenia (35 cells/μL; reference range 69–691 cells/μL), a reduction in total memory B cells (1.3% of B cells, reference range 4.6%–49% of B cells) related to substantially reduced class-switched memory B cells (0.3% of B cells; reference range 1.9%–30.4%), complete absence of IgM-only memory B cells (reference range 0.3%–13.1%), normal total B cells (reference range 92–792 cells/μL) and mildly reduced CD3+ (857 cells/μL; reference range 865–3618 cells/μL) and CD4+ T cells (432 cells/μL; reference range 497–2267 cells/μL).
Figure 1.

Coronal chest CT demonstrating bilateral hilar adenopathy, worse on the left, causing compression of central airways.
Figure 2.
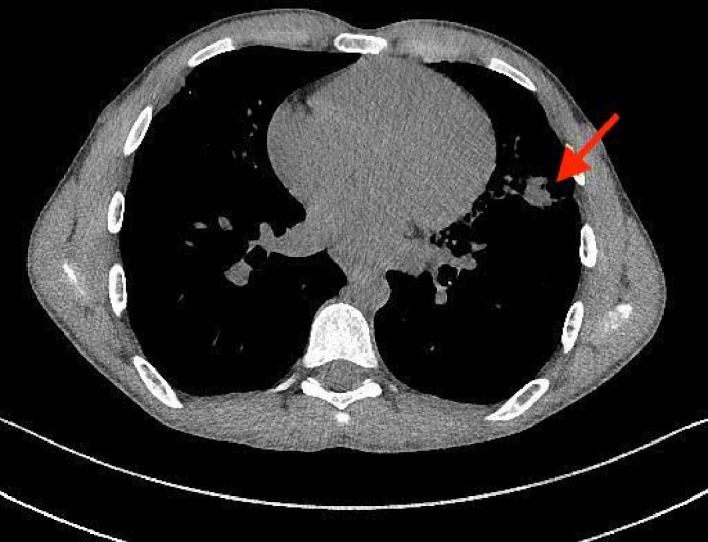
Axial chest CT demonstrating consolidation and volume loss within the lingula (arrow).
Figure 3.
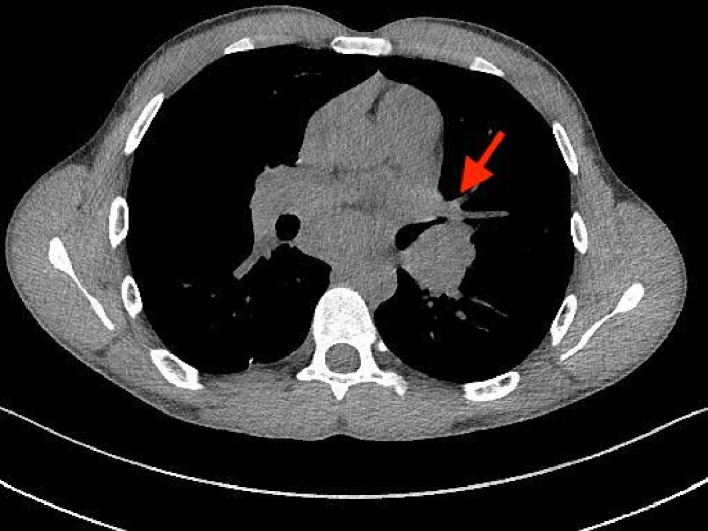
Axial chest CT demonstrating bilateral hilar adenopathy, worse on the left (arrow), causing compression of central airways.
Figure 4.

Perihilar lymph node fine needle aspiration (H&E stain, ×400) depicting numerous cryptococcal yeast forms (arrows) and caseating necrosis.
Figure 5.

Perihilar lymph node fine needle aspiration (mucicarmine stain, ×400) depicting numerous cryptococcal yeast forms (arrows).
At the time of presentation with cryptococcal pneumonia, a research-based whole exome analysis was performed, identifying a previously reported heterozygous pathogenic variant in STAT1 (c.1154C>pThr385 Met; Lal et al, manuscript in preparation), which was confirmed by Sanger sequencing.8–12 Though functional studies were not performed, the patient was presumed to have STAT1 GOF based on the identification of a genetic variant previously shown to be associated with GOF and a compatible clinical phenotype.
Differential diagnosis
Phenotypic presentations associated with STAT1 GOF variants are heterogeneous and have significant overlap with common variable immune deficiency (CVID) or combined immunodeficiency (CID). At the time initial immunological investigations were undertaken, GOF of STAT1 was not yet a known cause of disease. CID is defined by the European Society for Immunodeficiencies (ESID) by the fulfilment of at least one of the following criteria: at least one severe infection, manifestation of immune dysregulation, malignancy and having an affected family member. In addition, two of the following criteria must be met: reduced CD3, CD4 or CD8 T cells, reduced naïve CD4 and/or CD8 T cells, elevated g/d T cells and reduced proliferation to mitogen or T-cell receptor (TCR) stimulation.13 This patient had a CID phenotype with multiple severe infections requiring hospitalisation, autoimmune enteropathy and hypothyroidism, reduced CD4 T cells, and relatively low naïve CD4 T cells. CVID is defined by the ESID by the presence of a marked reduction in IgG and IgA levels, exclusion of secondary causes of hypogammaglobulinaemia, either poor antibody response to vaccines or low memory B cells, and fulfilment of at least one of the following criteria: increased susceptibility to infection, autoimmune conditions, granulomatous disease, unexplained polyclonal lymphoproliferation and the presence of affected family members with antibody deficiency.13 Though this patient had complete IgA deficiency, evidence of functional antibody deficiency and T cell lymphopenia, he did not meet diagnostic criteria for CVID because of his normal total IgG.14
Since the discovery of the significance of GOF of STAT1 in 2011, several studies characterised the clinical and immunological features associated with this disease. The largest cohort described to date reported CMC in 98% of cases, bacterial infections in 74% and autoimmune manifestations in 37%, with the most common being thyroid disease. About 1% of cases have been reported with autoimmune gastrointestinal involvement.3 While our patient had recurrent bacterial infections, autoimmune hypothyroidism and recurrent oral candidiasis as observed in other STAT1 GOF patients, he also had several atypical manifestations, including occurrence of Candida infections only in the setting of curative antibiotic courses, in addition to the presence of cryptococcal pneumonia and autoimmune enteropathy.
Despite this atypical presentation, genetic investigation ultimately facilitated the diagnosis of STAT1 GOF after a diagnostic odyssey of approximately 14 years. This case suggests that STAT1 GOF variants may underlie the aetiology of a subset of patients with a diagnosis of CVID, CID or unknown primary immunodeficiency, and in this era of easier accessibility to genetic testing, may have shorter time to diagnosis and effective and personalised therapy.
Treatment
On day 10 of a planned 14-day course of amphotericin B liposomal and flucytosine, the patient developed acute kidney injury and was therefore transitioned earlier than anticipated to a 2-month course of high-dose fluconazole at 800 mg once daily (12 mg/kg/dose). One month into therapy, his serum cryptococcal antigen titre had dropped to 1:640. His cough and haemoptysis resolved. He was then continued on prophylactic fluconazole 200 mg daily indefinitely. Eleven months later, repeat chest CT showed near-complete resolution of mediastinal and hilar adenopathy, as well as decrease in nodular consolidation in the lingula.
Because of the patient’s recurrent infections and quantitative and functional immunological anomalies, he was initiated as a child on long-term intravenous immunoglobulin therapy and weekly prophylactic azithromycin and trimethoprim–sulfamethoxazole. With the new diagnosis of STAT1 GOF, the patient will be initiated on a trial of ruxolotinib, a Janus kinase inhibitor which has been shown to restore immune defects and address autoimmune conditions in some patients in whom immunosuppressive regimens have failed.6 15–17
Outcome and follow-up
The patient continues on prophylactic fluconazole therapy and at 6-month follow-up is clinically doing well.
We are still fighting for insurance authorisation for ruxolotinib.
Discussion
This case expands our understanding of the broad clinical phenotype manifested by STAT1 GOF and emphasises the importance of consideration of this diagnosis in patients presenting with opportunistic infections and autoimmunity. The clinical course of cryptococcal pneumonia in a patient with STAT1 GOF has not been previously discussed in the literature.
Over 70 STAT1 GOF variants have been identified to date, with sites spanning both the DNA binding and coiled coil domains.3 All reported cases with a T385M variant, as found in our patient, were associated with CMC and recurrent upper and/or lower respiratory tract infections. The first report of a T385M variant involved two Japanese children, the first of which additionally presented with autoimmune thyroiditis, and the second of which proceeded to develop haemophagocytic lymphohistiocytosis and Evans syndrome.9 Subsequent reports include a patient from Ukraine with a history of oesophageal candidiasis causing strictures and recurrent severe herpes simplex virus (HSV) infections, a patient with eosinophilic oesophagitis and recurrent diarrhoea, a patient with enteropathy, cerebral aneurysms and type 1 diabetes mellitus, and a patient with hypothyroidism.8–12 The present case adds to the small cohort of reported T385M STAT1 variant patients and corroborates previous findings that no clear correlation between mutation site and clinical phenotype is currently identifiable.
Although most commonly observed in association with advanced HIV infection, cryptococcal infection has also been reported in several primary immunodeficiencies, to include idiopathic CD4 T cell lymphopenia, autosomal dominant GATA2 deficiency, X-linked CD40L deficiency, patients with inhibitory autoantibodies against IFN-γ or granulocyte-macrophage-colony stimulating factor (GM-CSF), and STAT1 GOF.8–11 18–24 In mouse models, STAT1 signalling has been found to play an important role in cryptococcal immunity, with loss of function causing a shift from TH1-type and TH17-type cytokine responses activating M1-type macrophages to TH2-type cytokine responses activating M2-type macrophages. The M2 macrophages in this study had higher pH and lower nitric oxide production, resulting in inefficient control of fungal proliferation.25 GOF of STAT1 causes a paradoxical increased susceptibility to Cryptococcus as seen in our patient and seven previously reported patients. Increased STAT1 phosphorylation results in enhanced activity in cytokines such as IFNs-α, β and γ, and interleukin (IL)-27, in addition to impaired IL-17 production and TH17 cell development.3
Limited data exist regarding management strategies for non-HIV-infected, immunosuppressed children with pulmonary cryptococcosis. The Infectious Disease Society of America formulated 2010 management guidelines are based on reported data from treatment of central nervous system (CNS) and pulmonary cryptococcosis primarily in HIV-infected individuals. Current recommendations state that children with disseminated cryptococcosis should be treated with an initial induction therapy consisting of amphotericin B deoxycholate and flucytosine for 2 weeks followed by consolidation with fluconazole (10–12 mg/kg per day orally) for 8 weeks, and maintenance fluconazole (6–12 mg/kg per day orally).26 More recently, however, the 2018 WHO guidelines for management of cryptococcal meningitis in patients infected with HIV recommended a shortened induction therapy period of 1 week, based on evidence that it reduces mortality and side effects and improves efficacy in comparison with the previously recommended 2-week regimen.27–29
Treatment of STAT1 GOF remains challenging. Supportive therapy, to include long-term antifungal and antimicrobial therapy, in addition to immune globulin replacement therapy in deficient patients, is important, though frequently insufficient on its own. Immunosuppressive therapy has been used to control autoimmune manifestations of STAT1 GOF with limited success and frequently resulting in significant side effects.6 More recently, ruxolotinib has been shown in case reports and series to be effective in restoring immune defects and address autoimmune conditions in some patients in whom immunosuppression has failed.6 15–17 More research is needed, however, to investigate the safety and efficacy of this drug in the treatment of STAT1 GOF. Haematopoietic stem cell transplantation (HSCT) has been used as well, though poor survival and high rates of graft failure and rejection limit its widespread use.30 Moving forward, it has been suggested that ruxolotinib may provide an effective bridge therapy until HSCT, provided this therapy is further optimised.31
In conclusion, because STAT1 GOF mutations can present with a wide range of clinical phenotypes, consideration of this defect should be given in patients with opportunistic infections or recurrent bacterial infections along with autoimmune conditions, even in the absence of conventional CMC. This is especially important in light of recent reports suggesting that targeted therapy with ruxolotinib may be beneficial in some patients failing to respond to other therapies.
Learning points.
STAT1 gain of function (GOF) presents with a broad clinical phenotype and should be considered in the differential diagnosis in patients presenting with chronic mucocutaneous candidiasis, recurrent infections and/or autoimmunity.
While recurrent bacterial, viral and candidal infections are most common, patients may also rarely present with other fungal infections such as Cryptococcus, Aspergillus, Pneumocystis jirovecci and Aspergillus.
Guidelines for the management of pulmonary cryptococcal infection in immunosuppressed individuals are based predominantly on studies in HIV-infected individuals. Further research is needed to optimise management in non-HIV-infected immunosuppressed individuals.
Patients on antifungal therapy for cryptococcal infection should be monitored for side effects, to include nephrotoxicity, electrolyte abnormalities and anaemia in association with amphotericin, hepatotoxicity, drug–drug interactions, gastrointestinal symptoms and rash in association with fluconazole, and bone marrow suppression, hepatotoxicity and gastrointestinal symptoms in association with flucytosine.
While treatment of STAT1 GOF remains challenging, recent case reports and series suggest that ruxolotinib may be effective in restoring immune defects and addressing autoimmune conditions in some patients.
Footnotes
Twitter: @ERistagno
Contributors: All authors have contributed equally to the manuscript. AJ detailed contribution statement: LM: planning, conduct, design, data gathering and manuscript writing. ER, PF and RA: conduct, design and manuscript writing. AJ: conduct, design, interpretation and manuscript writing.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Patient consent for publication: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Liu L, Okada S, Kong X-F, et al. Gain-Of-Function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med 2011;208:1635–48. 10.1084/jem.20110958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van de Veerdonk FL, Plantinga TS, Hoischen A, et al. STAT1 Mutations in Autosomal Dominant Chronic Mucocutaneous Candidiasis. N Engl J Med 2011;365:54–61. 10.1056/NEJMoa1100102 [DOI] [PubMed] [Google Scholar]
- 3.Toubiana J, Okada S, Hiller J, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood 2016;127:3154–64. 10.1182/blood-2015-11-679902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernasconi AR, Yancoski J, Villa M, et al. Increased STAT1 amounts correlate with the Phospho-STAT1 level in STAT1 gain-of-function defects. J Clin Immunol 2018;38:745–7. 10.1007/s10875-018-0557-0 [DOI] [PubMed] [Google Scholar]
- 5.Leiding JW, Okada S, Hagin D, et al. Hematopoietic stem cell transplantation in patients with gain-of-function signal transducer and activator of transcription 1 mutations. J Allergy Clin Immunol 2018;141:704–17. 10.1016/j.jaci.2017.03.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forbes LR, Vogel TP, Cooper MA, et al. Jakinibs for the treatment of immune dysregulation in patients with gain-of-function signal transducer and activator of transcription 1 (STAT1) or STAT3 mutations. J Allergy Clin Immunol 2018;142:1665–9. 10.1016/j.jaci.2018.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mizoguchi Y, Tsumura M, Okada S, et al. Simple diagnosis of STAT1 gain-of-function alleles in patients with chronic mucocutaneous candidiasis. J Leukoc Biol 2014;95:667–76. 10.1189/jlb.0513250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eren Akarcan S, Ulusoy Severcan E, Edeer Karaca N, et al. Gain-of-Function Mutations in STAT1: A Recently Defined Cause for Chronic Mucocutaneous Candidiasis Disease Mimicking Combined Immunodeficiencies. Case Reports Immunol 2017;2017:1–6. 10.1155/2017/2846928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takezaki S, Yamada M, Kato M, et al. Chronic mucocutaneous candidiasis caused by a gain-of-function mutation in the STAT1 DNA-binding domain. J Immunol 2012;189:1521–6. 10.4049/jimmunol.1200926 [DOI] [PubMed] [Google Scholar]
- 10.Soltész B, Tóth B, Shabashova N, et al. New and recurrent gain-of-function STAT1 mutations in patients with chronic mucocutaneous candidiasis from Eastern and Central Europe. J Med Genet 2013;50:567–78. 10.1136/jmedgenet-2013-101570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uzel G, Sampaio EP, Lawrence MG, et al. Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like syndrome. J Allergy Clin Immunol 2013;131:1611–23. 10.1016/j.jaci.2012.11.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Depner M, Fuchs S, Raabe J, et al. The extended clinical phenotype of 26 patients with chronic mucocutaneous candidiasis due to gain-of-function mutations in STAT1. J Clin Immunol 2016;36:73–84. 10.1007/s10875-015-0214-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.ESID - European Society for Immunodeficiencies. Available: https://esid.org/Working-Parties/Registry-Working-Party/Diagnosis-criteria [Accessed 25 Feb 2020].
- 14.ESID - European Society for Immunodeficiencies. Available: https://esid.org/Working-Parties/Clinical-Working-Party/Resources/Diagnostic-criteria-for-PID2 [Accessed 30 Nov 2019].
- 15.Mössner R, Diering N, Bader O, et al. Ruxolitinib induces interleukin 17 and ameliorates chronic mucocutaneous candidiasis caused by STAT1 gain-of-function mutation. Clin Infect Dis 2016;62:951.2–3. 10.1093/cid/ciw020 [DOI] [PubMed] [Google Scholar]
- 16.Higgins E, Al Shehri T, McAleer MA, et al. Use of ruxolitinib to successfully treat chronic mucocutaneous candidiasis caused by gain-of-function signal transducer and activator of transcription 1 (STAT1) mutation. J Allergy Clin Immunol 2015;135:551–3. 10.1016/j.jaci.2014.12.1867 [DOI] [PubMed] [Google Scholar]
- 17.Weinacht KG, Charbonnier L-M, Alroqi F, et al. Ruxolitinib reverses dysregulated T helper cell responses and controls autoimmunity caused by a novel signal transducer and activator of transcription 1 (STAT1) gain-of-function mutation. J Allergy Clin Immunol 2017;139:1629–40. 10.1016/j.jaci.2016.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zonios DI, Falloon J, Huang C-Y, et al. Cryptococcosis and idiopathic CD4 lymphocytopenia. Medicine 2007;86:78–92. 10.1097/md.0b013e31803b52f5 [DOI] [PubMed] [Google Scholar]
- 19.Scott-Algara D, Balabanian K, Chakrabarti LA, et al. Idiopathic CD4+ T-cell lymphocytopenia is associated with impaired membrane expression of the chemokine receptor CXCR4. Blood 2010;115:3708–17. 10.1182/blood-2009-02-202796 [DOI] [PubMed] [Google Scholar]
- 20.Vinh DC, Patel SY, Uzel G, et al. Autosomal dominant and sporadic Monocytopenia with susceptibility to mycobacteria, fungi, papillomaviruses, and myelodysplasia. Blood 2010;115:1519–29. 10.1182/blood-2009-03-208629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iseki M, Anzo M, Yamashita N, et al. Hyper-Igm immunodeficiency with disseminated cryptococcosis. Acta Paediatr 1994;83:780–2. 10.1111/j.1651-2227.1994.tb13140.x [DOI] [PubMed] [Google Scholar]
- 22.Winkelstein JA, Marino MC, Ochs H, et al. The X-linked hyper-IgM syndrome: clinical and immunologic features of 79 patients. Medicine 2003;82:373–84. [DOI] [PubMed] [Google Scholar]
- 23.Browne SK, Burbelo PD, Chetchotisakd P, et al. Adult-Onset immunodeficiency in Thailand and Taiwan. N Engl J Med 2012;367:725–34. 10.1056/NEJMoa1111160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosen LB, Freeman AF, Yang LM, et al. Anti-GM-CSF autoantibodies in patients with cryptococcal meningitis. J Immunol 2013;190:3959–66. 10.4049/jimmunol.1202526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leopold Wager CM, Hole CR, Wozniak KL, et al. Stat1 signaling is essential for protection against Cryptococcus neoformans infection in mice. J Immunol 2014;193:4060–71. 10.4049/jimmunol.1400318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perfect JR, Dismukes WE, Dromer F, et al. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the infectious diseases Society of America. Clin Infect Dis 2010;50:291–322. 10.1086/649858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.WHO Who | guidelines for the diagnosis, prevention and management of cryptococcal disease in HIV-infected adults, adolescents and children 2019. [PubMed]
- 28.Tenforde MW, Shapiro AE, Rouse B, et al. Treatment for HIV-associated cryptococcal meningitis. Cochrane Database of Systematic Reviews 2018;374 10.1002/14651858.CD005647.pub3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Molloy SF, Kanyama C, Heyderman RS, et al. Antifungal combinations for treatment of cryptococcal meningitis in Africa. N Engl J Med 2018;378:1004–17. 10.1056/NEJMoa1710922 [DOI] [PubMed] [Google Scholar]
- 30.Leiding JW, Okada S, Hagin D, et al. Hematopoietic stem cell transplantation in patients with gain of function STAT1 mutation. J Allergy Clin Immunol 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olbrich P, Freeman AF. Stat1 and STAT3 mutations: important lessons for clinical immunologists. Expert Rev Clin Immunol 2018;14:1029–41. 10.1080/1744666X.2018.1531704 [DOI] [PubMed] [Google Scholar]