

Chemogenetic suppression of a neuronal ensemble that encodes a lasting alcohol cue memory attenuates cue-induced relapse.
Abstract
Alcohol use disorder is characterized by a high risk of relapse during periods of abstinence. Relapse is often triggered by retrieval of persistent alcohol memories upon exposure to alcohol-associated environmental cues, but little is known about the neuronal circuitry that supports the long-term storage of alcohol cue associations. We found that a small ensemble of neurons in the medial prefrontal cortex (mPFC) of mice was activated during cue-paired alcohol self-administration (SA) and that selective suppression of these neurons 1 month later attenuated cue-induced relapse to alcohol seeking. Inhibition of alcohol seeking was specific to these neurons as suppression of a non–alcohol-related or sucrose SA–activated mPFC ensemble did not affect relapse behavior. Hence, the mPFC neuronal ensemble activated during cue-paired alcohol consumption functions as a lasting memory trace that mediates cue-evoked relapse long after cessation of alcohol intake, thereby providing a potential target for treatment of alcohol relapse vulnerability.
INTRODUCTION
Alcohol use disorder (AUD) is one of the most common substance use disorders and is considered a chronic psychopathology, resulting in serious health problems for affected individuals and placing a large socioeconomic burden on societies worldwide (1, 2). A key problem in the treatment of AUD is relapse, which can occur even after long periods of abstinence (3). Alcohol-related environmental cues, such as liquor bottles or locations where alcohol is frequently consumed (e.g., bars), become associated with the rewarding effect of alcohol (4). Exposure to these cues during periods of abstinence can evoke intense feelings of craving, thereby fueling the urge to resume alcohol consumption (5, 6). Given the frequent incidence and lasting potential of alcohol-related cues to drive relapse, identification of the neuronal circuitry that supports the storage of persistent alcohol cue memories is critical for the design of improved medical intervention to reduce relapse susceptibility (7, 8).
Clinical and preclinical studies strongly implicate the medial prefrontal cortex (mPFC) in relapse to alcohol and drug seeking (9–11). For instance, neuroimaging studies performed in patients with AUD demonstrate increased activation of the mPFC in response to alcohol-associated cues, and this response correlates with increased risk of relapse (6, 11, 12). In preclinical animal models, reexposure to alcohol-paired cues evokes alcohol seeking and enhances neuronal activity in the mPFC (13, 14), as measured by expression of the immediate early gene Fos, an established molecular marker of neuronal activity (15). In line with these observations, pharmacological inhibition of the prelimbic region of the mPFC reduces conditioned alcohol seeking (16, 17), indicating that the mPFC is involved in the processing of alcohol cue associations.
Mounting evidence indicates that the expression of learned behaviors, including alcohol-related behavior, is mediated by activity of sparsely distributed neurons, so-called neuronal ensembles (15, 18–20). However, whether neuronal ensembles function as a long-term physical memory trace supporting the storage of alcohol cue associations that drive relapse has remained elusive. To address this, we investigated whether neurons in the mPFC that are activated during alcohol self-administration (SA) in the presence of alcohol-paired cues are subsequently required for cue-induced alcohol seeking weeks after cessation of alcohol intake. Furthermore, we questioned whether mPFC ensembles have a distinct role in encoding of an alcohol cue association and a learned association between a cue and a natural reinforcer, such as sucrose.
To study this, we used a mouse alcohol SA paradigm and a targeted intervention with a recently developed viral-TRAP (targeted recombination in activated populations) technique (21), which enables permanent expression of a DREADD (designer receptor exclusively activated by designer drug) in neurons that are activated during a defined time window. Using this approach, we found that mPFC neurons that are activated during cue-paired alcohol SA are subsequently required for cue-induced relapse 1 month after cessation of alcohol intake. Chemogenetic suppression had no effect when alcohol seeking was evoked in the absence of a discrete alcohol-paired cue and when mPFC neurons activated during alcohol consumption in the absence of a discrete cue were subsequently suppressed during cue-induced relapse. Furthermore, this mechanism did not generalize to cue-induced sucrose seeking. Together, our data indicate that a specific neuronal ensemble in the mPFC functions as a critical storage mechanism for alcohol cue associations, and selective chemogenetic suppression of these neurons reduced cue-induced relapse long after the last exposure to alcohol.
RESULTS
Viral-TRAP enables lasting DREADD expression in alcohol SA–activated mPFC neurons
A viral-TRAP approach was used to express a molecular tag in mPFC neurons that are activated upon alcohol SA with the aim of suppressing these specific neurons during a cue-induced alcohol seeking test performed several weeks later. This approach consists of two adeno-associated virus (AAV) vectors (Fig. 1A) (21); the first encodes an inducible Cre recombinase (CreERT2) under control of the Fos promotor, and the second encodes a Cre-dependent coding sequence of a molecular tag [e.g., hM4Di, an inhibitory DREADD (22)]. Functional expression of the molecular tag is restricted to neurons that are activated during a specific time window, as it is coupled to the Fos promoter [activated by strong neuronal activity (23)] and controlled by a systemic injection of 4-hydroxytamoxifen (4TM) (21).
Fig. 1. Viral-TRAP enables molecular tagging of alcohol SA–activated mPFC neurons.
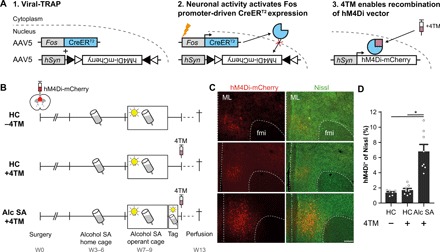
(A) Schematic overview of viral-TRAP. A mixture of AAV-Fos::CreERT2 and Cre-dependent AAV-hSyn::DIO-hM4Di-mCherry was injected bilaterally into the mPFC to enable 4TM-controlled irreversible expression of hM4Di-mCherry. (B) Experimental design for validation of viral-TRAP in the alcohol SA paradigm. All mice underwent acquisition of alcohol SA but were divided into three groups on the “Tag” day. Two groups remained in their home cage (HC) in the absence or presence of systemic 4TM treatment (HC −4TM, N = 6 and HC +4TM, N = 8, respectively). The third group underwent an additional alcohol SA session and received 4TM 2 hours later (Alc SA +4TM; N = 8). Animals were euthanized 4 weeks (W) after the Tag session. (C) Representative images of hM4Di-mCherry expression in the mPFC. ML, midline; fmi, forceps minor of the corpus callosum. Scale bar, 250 μm. (D) Percentage of hM4Di+ cells in the mPFC. Alcohol SA–tagged mice showed increased hM4Di-mCherry expression compared with controls. *P < 0.001. Bar graph, means + SEM.
We microinjected a mixture of AAV-Fos::CreERT2 and Cre-dependent hM4Di fused to mCherry (fluorescent reporter; AAV-hSyn::DIO-hM4Di-mCherry) in the prelimbic cortex of the mPFC of mice. Three weeks later, animals were first habituated to alcohol SA in their home cage (fig. S1), followed by acquisition of operant alcohol SA (Fig. 1B). During the operant phase, we trained mice in daily 60-min sessions to respond on an active lever, which resulted in presentation of a cue light and delivery of a 10-μl 8% alcohol reward. Responses on an inactive lever had no programmed consequences. Notably, we previously established that mice are more motivated to lever press for alcohol than water using this protocol (fig. S2). Two hours after a last operant alcohol SA session, mice received 4TM (25 mg/kg) to induce hM4Di-mCherry expression in mPFC neurons that were activated during alcohol SA (Alc SA +4TM). To confirm that viral-TRAP allowed tagging of mPFC neurons that were activated during operant alcohol SA, control mice remained in their home cage without or with 4TM treatment (HC −4TM and HC +4TM, respectively). Four weeks later, we euthanized the mice to examine hM4Di-mCherry expression. We detected hM4Di-mCherry in a subset of mPFC neurons along the entire anterior-posterior axis of the prelimbic cortex and to some extent in the anterior cingulate cortex (Fig. 1C and fig. S3). Alcohol SA evoked hM4Di-mCherry expression in 6.8 ± 0.92% of mPFC neurons, whereas the HC −4TM and HC +4TM groups showed significantly less hM4Di-mCherry+ cells [1.4 ± 0.14 and 1.7 ± 0.22%, respectively; one-way analysis of variance (ANOVA): F2,21 = 26.58, P < 0.001; post hoc Bonferroni test: Alc SA +4TM versus HC −4TM, P < 0.001 and versus HC +4TM, P < 0.001]. Together, this confirms that viral-TRAP allowed molecular tagging of alcohol SA–activated mPFC neurons and that hM4Di-mCherry expression persisted for at least 4 weeks after the tag session.
The alcohol SA–tagged ensemble is not required for context-induced alcohol seeking
Next, we assessed whether mPFC neuronal ensembles that are activated during cue-paired alcohol SA mediate alcohol seeking after prolonged abstinence and whether these cells are differentially involved in conditioned alcohol seeking in the absence and presence of the discrete alcohol-associated cue. To address this, we used viral-TRAP to tag alcohol SA–activated mPFC neurons with hM4Di-mCherry or mCherry alone (control). To assess potential nonspecific effects of clozapine N-oxide (CNO) used for chemogenetic suppression of tagged mPFC neurons, hM4Di-expressing animals were divided into groups that received vehicle (VEH) or CNO treatment before the relapse test. We previously showed that the excitability of mPFC neurons tagged with hM4Di using viral-TRAP is reduced by CNO, whereas neurons tagged with mCherry alone did not respond to CNO (21). Mice were first habituated to alcohol drinking in their home cage (fig. S4) and then acquired operant alcohol SA (Fig. 2A). To assess motivation of the animals to self-administer alcohol, we increased the number of lever presses required to obtain one alcohol reward gradually from fixed ratio 1 (FR1) to FR5. All groups developed a stable preference for the active lever and increased active lever pressing, but not inactive lever pressing, over sessions (Fig. 2B). ANOVA revealed a significant Session x Lever (F6.08,97.19 = 23.05, P < 0.001) but no Group x Session x Lever (F12.15,97.19 = 0.91, P = 0.55) interaction. We injected mice with 4TM after the last SA (Tag) session, and then, they underwent 3 weeks of forced abstinence in their home cage (Fig. 2A). Lever pressing did not differ between groups during the Tag session (active lever: F2,18 = 0.02, P = 0.98; inactive lever: F2,18 = 0.01, P = 0.99; Fig. 2C). We then assessed whether activity of the alcohol SA–tagged ensemble was necessary for alcohol seeking in the absence of the alcohol-paired cue light. We refer to this as context-induced alcohol seeking, as all contextual stimuli were present but active lever pressing did not result in presentation of the cue light nor alcohol reward. To selectively suppress the activity of the hM4Di-tagged mPFC ensemble, we treated mice with CNO 30 min before the test. All groups showed a preference for the active (previously alcohol-paired) lever (Fig. 2D), but no effect of chemogenetic suppression of the alcohol SA–tagged ensemble was found under these conditions [active lever: F2,18 = 0.11, P = 0.90; inactive lever: χ2(2) = 0.04, P = 0.98]. During the test, all groups showed within-session extinction of active lever pressing (fig. S5A). No differences were observed between VEH- and CNO-treated animals, confirming that CNO treatment itself did not affect lever pressing. Therefore, we did not include a VEH control group in subsequent experiments.
Fig. 2. Suppression of the alcohol SA–activated mPFC ensemble does not affect context-induced alcohol seeking.
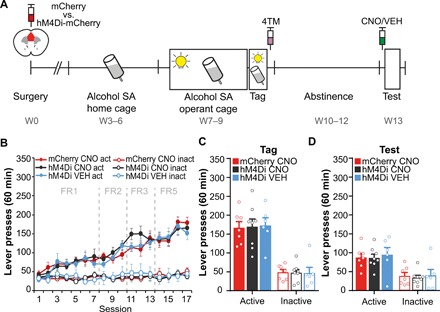
(A) Experimental design. Activated mPFC neurons were tagged after the last alcohol SA session with mCherry (N = 7) or hM4Di-mCherry [CNO (N = 8); VEH (N = 6)]. (B) All groups developed a stable preference for the active lever, and active lever presses increased over sessions, while inactive lever presses remained stable. Acquisition of alcohol SA was similar in all groups. (C) Lever pressing did not differ between groups during the Tag session (last alcohol SA session). (D) Following 3 weeks of forced abstinence, mice received CNO or VEH 30 min before the context-induced alcohol seeking test to selectively suppress the alcohol SA–tagged mPFC ensemble. During this test, all contextual stimuli were present but not the discrete cue light that was previously paired with each alcohol reward. Suppression of the tagged neuronal ensemble did not affect alcohol seeking in the absence of the cue light. All graphs, means + SEM.
The alcohol SA–tagged ensemble is required for cue-induced alcohol seeking
We next examined whether chemogenetic suppression of the alcohol SA–activated mPFC ensemble reduces alcohol seeking induced by the discrete alcohol cue. To assess this, mice were subjected to the same alcohol SA procedure, and activated mPFC neurons were again tagged during the last operant SA session (Fig. 3A). Responses of both groups were similar during the Tag session [active lever: t(23) = −0.08, P = 0.94; inactive lever: t(23) = 0.27, P = 0.79; Fig. 3B]. To investigate alcohol seeking specifically induced by the discrete alcohol cue, we first extinguished the salience of the contextual stimuli by reexposing mice to the operant chamber in the absence of alcohol reward and the cue light (Fig. 3C). This resulted in a significant reduction of active responses in both groups over sessions (F3.69, 84.96 = 11.05, P < 0.001). Then, all mice received CNO and, 30 min later, underwent a cue-induced reinstatement (i.e., relapse) test. We observed robust relapse (active lever pressing) in the control group, but this was significantly attenuated in the hM4Di group (Fig. 3D). ANOVA revealed a Session (Extinction versus Reinstatement) x Group (F1,23 = 7.95, P = 0.010) interaction for active lever presses. Post hoc analysis confirmed a significant difference between control and hM4Di mice during the reinstatement test [t(23) = 3.14, P = 0.005]. The effect of CNO was specific for the active lever, as responses on the inactive lever remained stable and did not differ between the groups and sessions (Session x Group: F1,23 = 0.89, P = 0.36). Notably, we did not find a correlation between the size of the tagged mPFC ensemble and the number of active lever presses during the Tag session and cue-induced reinstatement test (fig. S6), suggesting that the level of operant alcohol intake and seeking is not determined by the size of the activated ensemble in the mPFC. Whereas the mCherry group increased active lever pressing during the reinstatement test, responses of the hM4Di group did not change (fig. S5B). This suggests that chemogenetic suppression prevented a cue-driven increase in alcohol seeking during the relapse session, rather than facilitating extinction of lever pressing in the hM4Di group. Thus, mPFC neuronal ensembles that are activated during cue-paired alcohol SA have a critical role in cue-induced relapse to alcohol seeking 1 month later.
Fig. 3. Suppression of the alcohol SA–activated mPFC ensemble attenuates cue-induced alcohol seeking.
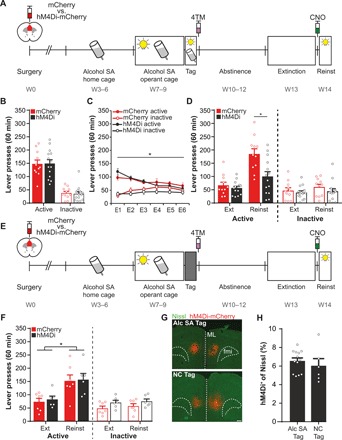
(A) Experimental design. Reinst, reinstatement test. Groups: mCherry (N = 11) and hM4Di-mCherry (N = 14). (B) Tag session. Responding did not differ between groups. (C) Extinction training reduced active lever presses in both groups. E, extinction session. *P < 0.001. (D) Reinstatement. Reexposure to the alcohol-associated cue light reinstated alcohol seeking in control mice, but active lever pressing in the hM4Di group was significantly attenuated. *P = 0.010. (E) Experimental design. Mice acquired alcohol SA and were then exposed to a novel context (NC) to tag activated mPFC neurons. Groups: mCherry (N = 7) and hM4Di-mCherry (N = 6). (F) Reinstatement. Both groups showed reinstatement (*P = 0.017), and CNO did not affect active or inactive lever pressing. (G) Representative images of hM4Di-mCherry expression in the alcohol (Alc) SA Tag and NC Tag group. Scale bar, 250 μm. (H) Quantification of hM4Di+ cells revealed no difference in the percentage of tagged neurons between both groups. All graphs, means + SEM.
To determine whether the inhibitory effect on alcohol seeking was specific to the alcohol SA–activated ensemble, another group of animals fully acquired alcohol SA, but we now tagged activated mPFC ensembles with hM4Di-mCherry or mCherry after mice explored a novel context (NC) (Fig. 3E). Subsequently, animals underwent forced abstinence and extinction, and then, we examined the effect of CNO-mediated suppression of the NC-tagged ensemble on cue-induced relapse. Both groups showed reinstatement of active lever pressing, and suppression of the NC-tagged ensemble did not affect this behavior (Fig. 3F). ANOVA confirmed a significant effect of Session (F1,14 = 7.30, P = 0.017) but not Session x Group interaction (F1,14 = 0.25, P = 0.62) nor Group effect (F1,14 = 0.06, P = 0.82). The size of the alcohol SA–tagged (Fig. 3, A to D) and NC-tagged mPFC ensemble (Fig. 3, E and F) did not differ (Fig. 3, G and H) and can therefore not explain the differential effect of CNO on cue-induced alcohol seeking.
We then assessed whether chemogenetic suppression of mPFC neurons that are activated by alcohol consumption alone (in the absence of specific cues) affects cue-induced relapse (fig. S7A). Three days after the last operant SA session, we allowed mice to consume 8% alcohol from a bottle in their home cage for 60 min (fig. S7B), and 2 hours later, we treated the animals with 4TM. Following forced abstinence and extinction sessions, suppression of the alcohol consumption–tagged ensemble did not affect cue-induced reinstatement (fig. S7C) despite the observation that the size of the ensemble (6.9 ± 0.45% neurons; fig. S7, D and E) was similar to the cue-paired alcohol SA–tagged ensemble (Fig. 3).
The alcohol SA–activated mPFC ensemble is preferentially reactivated during relapse
The observation that mPFC neurons activated during alcohol SA are subsequently required for cue-induced alcohol seeking several weeks later suggests that, specifically, these neurons are reactivated during cue-induced alcohol seeking under endogenous conditions (without chemogenetic intervention). To investigate this, we tagged mPFC ensembles after the last alcohol SA session (Alc SA Tag) or NC exploration (NC Tag; Fig. 4A). Subsequently, animals underwent abstinence and extinction and were euthanized 90 min after a cue-induced reinstatement test. Reinstatement evoked Fos expression (reflecting neuronal activity during relapse) in a similar percentage of mPFC neurons in the NC-tagged (8.2 ± 0.8%) and alcohol SA–tagged (8.9 ± 1.6%) group (Fig. 4, B and C). Next, we determined reactivation by comparing expression of Fos in the tagged (mCherry+) and nontagged (mCherry−) population in both groups (Fig. 4D). ANOVA revealed an interaction between Population (Fos+/mCherry− versus Fos+/mCherry+) and Group (F1,8 = 5.68, P = 0.044). Post hoc analysis revealed a significant difference between the Fos+/mCherry+ and Fos+/mCherry− population of the alcohol SA group [t(8) = 4.64, P = 0.003]. Hence, this confirms that alcohol SA–activated mPFC neurons are preferentially reactivated during cue-induced relapse 1 month later and further indicates that a distinct population of mPFC neurons is activated during alcohol SA and NC exploration.
Fig. 4. The alcohol SA–activated mPFC ensemble is preferentially reactivated during cue-induced alcohol seeking.
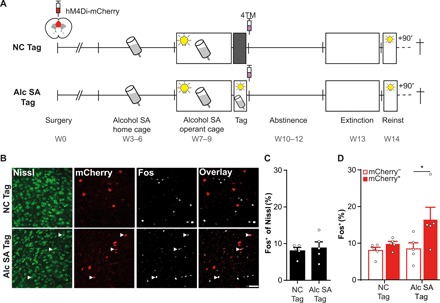
(A) Experimental design. Activated mPFC ensembles were tagged after a last alcohol SA session (Alc SA Tag; N = 5) or after exposure to an NC (NC Tag; N = 5). Animals were perfused 90 min after a cue-induced reinstatement test to investigate colocalization of Fos expression (induced by the test) and hM4Di-mCherry in the NC-tagged and alcohol SA–tagged ensemble. (B) Example images of Fos+ and mCherry+ cells and colocalization of both markers (white arrowheads). Scale bar, 50 μm. (C) Fos expression in the total neuronal population (Nissl+) in the mPFC did not differ between the NC-tagged group (8.2 ± 0.8%) and alcohol SA–tagged group (8.9 ± 1.6%). (D) Percentage of Fos+ within the mCherry− and mCherry+ population demonstrates enhanced colocalization of Fos and the alcohol SA–tagged ensemble. *P = 0.044. All bar graphs, means + SEM.
Sucrose SA–activated mPFC neurons are not involved in cue-induced sucrose seeking
We next questioned whether storage of a cue association by mPFC ensembles generalizes to sucrose reward, a natural reinforcer. To answer this, we trained animals to lever press for a 10% sucrose solution under conditions identical to the alcohol experiments (Fig. 5A). Mice readily acquired a robust preference for the active lever, and active lever responses increased over sessions [Fig. 5B; Session x Lever: F14,168 = 25.25, P < 0.001(ANOVA)]. We tagged neurons with hM4Di-mCherry or mCherry after the last sucrose SA session (Fig. 5C), during which responding did not differ between groups [active lever: t(12) = −0.31, P = 0.76; inactive lever: t(12) = −0.982, P = 0.35]. Similar to cue-induced alcohol seeking, mice first underwent 3 weeks of forced abstinence, after which active lever pressing was successfully extinguished in both groups [active lever: F1.42, 17.05 = 35.42, P < 0.001 (ANOVA); Fig. 5D]. Subsequently, we investigated the effect of CNO-mediated suppression of sucrose SA–tagged ensembles during a cue-induced reinstatement test. Both groups showed increased active lever pressing compared with the last extinction session, and responding did not differ between groups (Fig. 5E). ANOVA confirmed an effect of Session (F1,12 = 6.42, P = 0.026) but not Session x Group (F1,12 = 0.02, P = 0.88) nor Group (F1,12 = 0.25, P = 0.63).
Fig. 5. A sucrose SA–activated mPFC ensemble is not involved in cue-induced sucrose seeking.
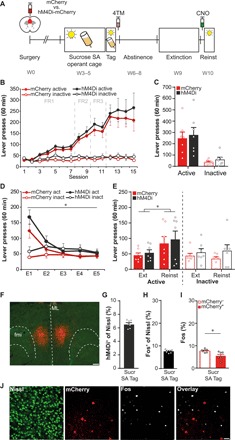
(A) Experimental design. Groups: mCherry (N = 7) and hM4Di-mCherry (N = 7). (B) Acquisition of sucrose SA was similar in both groups. (C) Tag session. Lever pressing did not differ between the groups. (D) Active lever pressing declined over extinction sessions. *P < 0.001. (E) Cue reexposure reinstated active lever pressing (*P = 0.026), but CNO-mediated suppression of the sucrose SA–tagged ensemble did not affect sucrose seeking. Inactive lever presses did not differ between sessions and groups. (F) Example of mPFC sucrose SA–tagged hM4Di-mCherry+ expression. Scale bar, 250 μm. (G) The sucrose SA–tagged ensemble comprised 6.6 ± 0.2% of the total neuronal mPFC population. (H) A second reinstatement test evoked Fos expression in 7.6 ± 0.3% of mPFC neurons. (I) Percentage of Fos+ within the mCherry− and mCherry+ populations. Colocalization of Fos with mCherry+ population was lower than colocalization with mCherry− population. *P = 0.020. (J) Example images of Fos+ and mCherry+ cells in the mPFC. Scale bar, 50 μm. All bar graphs, means + SEM.
Similar to alcohol SA, the hM4Di-tagged ensemble after sucrose SA comprised 6.6 ± 0.2% of the total neuronal population in the mPFC (Fig. 5, F and G). On the basis of the lack of effect of CNO on reinstatement, we hypothesized that the sucrose SA–tagged ensemble was not preferentially reactivated during cue-induced sucrose seeking. To address this, we used the mCherry control group to perform a second cue-induced reinstatement test 24 hours after the first test and euthanized mice 90 min later. Active lever presses was not extinguished over both tests (fig. S8). Cue-induced sucrose seeking evoked Fos expression in 7.6 ± 0.3% of mPFC neurons (Fig. 5H), similar to alcohol seeking (Fig. 4C). We found that colocalization of Fos with the mCherry+ population was significantly lower than with the mCherry− population [t(6) = −3.15, P = 0.020; Fig. 5, I and J], indicating that the sucrose SA–tagged ensemble was preferentially not reactivated upon reexposure to the sucrose cue.
Alcohol SA– and sucrose SA–tagged mPFC ensembles differ in composition and efferent projections
Given the functional difference between operant cue-paired alcohol SA– and sucrose SA–activated mPFC ensembles, we next investigated whether these neuronal ensembles differ in cellular composition and axonal projections. To examine the cellular composition, we quantified the percentage of GABAergic [glutamic acid decarboxylase 67–positive (GAD67+)] neurons within the tagged (mCherry+) and nontagged (mCherry−) mPFC population in both groups (Fig. 6A). As expected, the overall percentage of GABAergic neurons in the mPFC did not differ between groups (Fig. 6B). However, ANOVA revealed a Group (Alc SA versus Sucr SA Tag) x Population interaction (F1,13 = 7.46, P = 0.017; Fig. 6C). Post hoc analyses showed that GAD67+ cells were overrepresented in the mCherry+ population in both groups [Alc SA Tag: t(13) = 3.04, P = 0.019; Sucr SA Tag: t(13) = 6.59, P < 0.001], but the sucrose SA–tagged ensemble comprised a higher percentage of GAD67+ cells compared with the alcohol SA–tagged ensemble [t(26) = 3.52, P = 0.003; Fig. 6C].
Fig. 6. Alcohol SA– and sucrose SA–tagged ensembles differ in composition and efferent projections.
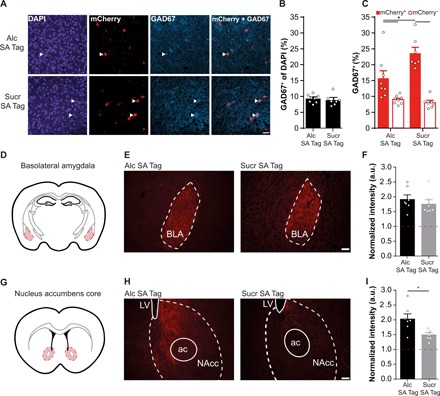
(A) Representative images of nuclei [4′,6-diamidino-2-phenylindole (DAPI)], ensemble neurons (mCherry+), and GABAergic (GAD67+) cells in the mPFC of alcohol SA–tagged (Alc SA Tag) and sucrose SA–tagged (Sucr SA Tag) mice. White arrowheads indicate colocalization of GAD67 and mCherry. Scale bar, 50 μm. (B) Percentage of GAD67+ cells in the mPFC was similar between alcohol SA (N = 8) and sucrose SA (N = 7) groups (9.2 ± 0.5 and 8.8 ± 0.7% of DAPI, respectively). (C) Percentage of GAD67+ within the mCherry− (DAPI+) and mCherry+ populations. Colocalization of GAD67+ with the mCherry+ population was higher in the sucrose SA–tagged than the alcohol SA–tagged group. *P = 0.015. (D) Schematic of the BLA region (BLA; shaded red) that was imaged for analysis of projections of mPFC ensembles. (E) Representative images of axonal fibers (mCherry+) of alcohol SA– and sucrose SA–tagged mPFC ensembles. Scale bar, 100 μm. (F) Fiber intensity in the BLA did not differ between the alcohol SA– and sucrose SA–tagged ensemble. (G) Schematic of the NAcc region (shaded red) that was imaged for analysis of projections of mPFC ensembles. (H) Representative images of axonal fibers (mCherry+) of the alcohol SA– and sucrose SA–tagged mPFC ensemble. Scale bar, 100 μm. LV, lateral ventricle; ac, anterior commissure, olfactory limb. (I) Intensity of the alcohol SA–tagged ensemble fibers was higher than those of the sucrose SA–tagged ensemble in the NAcc (P = 0.012). *P < 0.012. a.u., arbitrary units.
Next, we investigated whether the density of axonal fibers (mCherry+) differed between the alcohol SA– and sucrose SA–tagged groups in the basolateral amygdala (BLA; Fig. 6D) and nucleus accumbens core (NAcc; Fig. 6G), two well-established mPFC target regions that are known to play a role in alcohol-related behavior (24). In the BLA, mCherry+ intensity did not differ between tagged ensembles (U = 17, P = 0.38; Fig. 6, E and F), whereas in the NAcc, a higher intensity of mCherry+ fibers was detected in the alcohol SA–tagged group [t(12) = 2.96, P = 0.012; Fig. 6, H and I]. Together, this confirms that alcohol SA– and sucrose SA–activated mPFC ensembles have different cellular properties.
DISCUSSION
Our study in mice reveals that mPFC neurons that are activated during cue-paired alcohol SA are preferentially reactivated upon cue exposure and that selective suppression of this neuronal ensemble reduces relapse to alcohol seeking 1 month after the last alcohol exposure. The specificity of the alcohol cue–encoding mPFC ensemble is supported by the observation that (i) alcohol seeking in the absence of the alcohol cue was not affected by suppression of the operant alcohol SA–tagged ensemble, (ii) suppression of a similar-sized non–alcohol-related ensemble or an ensemble activated by alcohol consumption in the absence of the cue had no effect on cue-induced alcohol seeking, and (iii) the inhibitory effect on cue reactivity did not generalize to mPFC neurons activated during operant SA of a natural reinforcer. On the basis of this, we conclude that a learned alcohol cue association is encoded by a specific neuronal ensemble in the mPFC and that this ensemble functions as a lasting physical trace that mediates cue-evoked alcohol seeking after cessation of alcohol intake.
It is well established that the mPFC has an important role in learning and memory associated with exposure to illicit drugs and alcohol (6, 11–14, 25). The mPFC ensemble that is activated during alcohol SA likely functions as a critical circuitry hub in a larger network of ensembles together comprising the persistent alcohol cue memory trace (18). The nucleus accumbens and amygdala have also been implicated in cue-induced alcohol seeking (26), as well as excessive alcohol drinking (20, 27), and these regions receive dense projections from the mPFC. Selective ablation of mPFC neurons that project to the nucleus accumbens attenuates cue-induced alcohol seeking, whereas ablation of mPFC neurons that project to the BLA do not affect relapse (24). In line with this, we found that the alcohol SA–activated mPFC ensemble more densely innervates the NAcc than a sucrose SA–activated mPFC ensemble, whereas this difference is not present in the BLA. Hence, neurons of the alcohol SA–activated mPFC ensemble may project to an alcohol cue–encoding ensemble in the NAcc to promote responding to alcohol cues.
Suppression of the mPFC ensemble did not affect relapse when the discrete cue was not present, i.e., when alcohol seeking was evoked by contextual stimuli only. This points to a difference in the neuronal circuitry that mediates the processing of contextual and discrete cues, in line with what has been described by others for alcohol and other drugs of abuse (28, 29). Moreover, chemogenetic suppression of an ensemble that was tagged after alcohol SA in the home cage (i.e., in the absence of all cues that were present in the operant cage) did not affect cue-induced reinstatement. This suggests that alcohol consumption in the presence and absence of cues that signal the availability of alcohol activates a different subset of mPFC neurons.
In contrast to cue-induced alcohol seeking, we found that mPFC neurons activated during cue-paired sucrose SA were not required for subsequent cue-induced sucrose seeking, indicating that responding to alcohol and sucrose cues is regulated through a different neuronal substrate. This has also been reported for heroin- and cocaine-paired cues versus sucrose-paired cues (30–32). Moreover, we found that sucrose SA–tagged mPFC neurons were less likely to be activated during cue-induced sucrose seeking than neighboring nontagged neurons, which notably differs with the preferential reactivation of the alcohol SA–activated ensemble. In addition, we found that a higher number of GABAergic neurons were recruited in the sucrose SA–tagged ensemble compared to the alcohol SA–tagged ensemble. On the basis of the difference in reactivation of these ensembles, we speculate that GABAergic neurons that are activated during operant SA serve to suppress activity of non-ensemble neurons in the mPFC during cue-induced alcohol seeking, whereas during sucrose seeking, they may suppress activity of other neurons in the ensemble. Alternatively, cue-induced seeking of a natural reinforcer depends on neuronal activity in other subregions of the mPFC, such as the infralimbic cortex (33), or other brain regions. Furthermore, we speculate that an alcohol cue memory might be more persistent than a learned association between a natural reinforcer and an environmental cue. This is supported by the difference in active lever responses evoked by reexposure to the alcohol- and sucrose-associated cue and in line with the observation that rats trained to self-administer alcohol and saccharin (an artificial sweetener) show stronger cue-induced relapse for alcohol than saccharin (34).
In our study, we investigated the persistency of a neuronal ensemble that encodes an alcohol cue association, and therefore, relapse to alcohol seeking was assessed after a prolonged period of abstinence, i.e., at a remote time point after learning the alcohol cue association. For a memory to persist, it is thought to undergo a process of systems consolidation, involving gradual maturation of the memory in cortical networks (35). Recently, we and others discovered that expression of a remote, but not recent, contextual fear memory depends on activity of a learning-activated neuronal ensemble in the mPFC (21, 36). Therefore, the alcohol SA–activated mPFC ensemble may not be involved in cue-induced alcohol seeking at a recent time point after acquisition of cue-paired alcohol SA. However, as Pavlovian fear conditioning and incremental instrumental conditioning involve different forms of associative learning, consolidation of these memories may occur through alternate neurobiological mechanisms and at different time scales. Thus, it remains to be determined whether cue-evoked alcohol seeking after recent cessation of alcohol SA is mediated by the same mPFC ensemble that mediates relapse at a remote time point.
The viral-TRAP technique that we used to visualize and intervene with neurons that are activated during alcohol SA depends on activation of the Fos promoter. This is thought to primarily occur through stimulation of the mitogen-activated protein kinase pathway following sustained calcium influx through N-methyl-d-aspartate receptors and voltage-sensitive calcium channels (37). However, not all activated neurons may reach intracellular calcium levels that are sufficient to activate the Fos promoter. Therefore, a limitation of our approach is that we may have intervened with a subset of activated neurons in the mPFC. This may have resulted in incomplete suppression of the alcohol SA–tagged ensemble and cue-induced alcohol seeking. Yet, we demonstrate that suppression of the small population (~6 to 7%) of mPFC neurons that we tagged on the basis of activation of the Fos promoter clearly is sufficient to substantially reduce cue-induced relapse to alcohol seeking. Whether suppression of the alcohol SA–tagged mPFC ensemble equally affects mice that acquire different levels of operant alcohol SA remains a topic of future research. We found that the percentage of tagged mPFC neurons did not correlate with the number of active lever presses during the tag session or the reinstatement test, suggesting that operant performance is not determined by the size of the activated mPFC ensemble. Thus, the number of activated (Fos-expressing) neurons may not represent the strength of a learned alcohol cue association.
We found that the alcohol SA–activated ensemble was partially reactivated upon reexposure to the alcohol cue. This observation may point to an overestimation of the number of tagged mPFC neurons that actually encode the alcohol cue memory, as probably not all neurons that are activated during alcohol SA are involved in the processing of the acquired cue association. Moreover, it is possible that we did not visualize, and thus quantify, all neurons that were activated during the relapse test because of a detection threshold of the Fos immunostaining. Alternatively, it is possible that cue-induced alcohol seeking involves reactivation of only a fraction of the total neuronal ensemble that encodes the alcohol cue memory and that upon each exposure to alcohol-associated cues, a different combination of neurons that belong to the memory ensemble can be used to drive alcohol seeking.
To conclude, we demonstrate causality between activity of the mPFC neuronal ensemble that is activated during alcohol SA and cue-induced relapse to alcohol seeking, thereby providing a rationale for design of therapeutic intervention to silence this ensemble with the aim to reduce cue reactivity in patients with AUD. Note that we used only male mice in our study. Although AUD is worldwide more prevalent in men than women (1), it remains to be determined whether our findings can be generalized to females. In addition, it will be crucial to understand how an alcohol cue memory is stored by mPFC ensembles. Memory-encoding ensembles have been shown to undergo specific physiological and molecular changes (38, 39). Elucidation and reversal of the physical changes that occur in the mPFC ensemble that stores the alcohol cue association hold promise for disruption of persistent alcohol memories that contribute to the risk of relapse after prolonged abstinence.
MATERIALS AND METHODS
Experimental design
With our study, we aimed to determine whether mPFC neurons that are activated during cue-paired alcohol consumption function as a lasting physical trace for alcohol cue associations that maintain cue reactivity after prolonged abstinence. For this, we applied a viral-TRAP method to selectively tag and manipulate mPFC neurons that are activated during alcohol SA in mice. In this study, a total of N = 162 C57BL/6J mice were trained to self-administer alcohol (N = 144) or sucrose (N = 18) in operant chambers, and activated mPFC neurons were tagged with an inhibitory DREADD during a last SA session or after exposure to non–alcohol-related context. Relapse tests were performed 3 to 4 weeks after cessation of alcohol/sucrose intake, and the effect of chemogenetic suppression of tagged mPFC ensembles on alcohol/sucrose seeking (measured by resumption of active lever pressing) in the absence and presence of an alcohol-paired cue was examined. At the end of behavioral experiments, mice were euthanized and their brains were isolated. After immunohistochemical stainings, confocal microscopy was used to quantify the number of tagged neurons (mCherry+), GABAergic neurons (GAD67+), activated neurons (Fos+), and overlap between markers, as well as for analysis of ensemble-derived axonal fibers (mCherry+) in the BLA and NAcc.
Animals
Male wild-type C57BL/6J mice aged 6 to 8 weeks at the start of the experiments were housed individually 1 day before surgery (see below). One week after surgery, animals were transferred to a 12-hour reversed light/dark cycle (lights off at 07:00 a.m.) until the end of the experiment. Mice had ad libitum access to regular laboratory chow food and water throughout the experiment, except for the limited drinking phase during habituation (see below). Behavioral experiments were conducted during the animal’s dark phase. This study was approved by The Netherlands Central Committee for Animal Experiments (Centrale Commissie Dierproeven) and the Animal Ethical Care Committee Dierexperimentencommissie of the Vrije Universiteit Amsterdam. We excluded 16 mice because of misplaced virus injections and 42 mice because they failed to acquire stable alcohol SA (see below). The latter animals were used in control groups to validate the viral-TRAP technique.
AAV vectors and stereotactic microinjections
The AAV-Fos-CreERT2 plasmid was generated as previously described (21). AAV-Fos::CreERT2 (titer, 1.2 × 1013) and Cre-dependent AAVs AAV-hSyn::DIO-hM4Di-mCherry and AAV-hSyn::DIO-mCherry (titers, 5.0 × 1012 to 6.0 × 1012) were packaged as serotype 5 virus. A virus mixture of AAV5-Fos::CreERT2 and Cre-dependent AAV was injected at a ratio of 1:500 (final titer AAV5-Fos::CreERT2, 2.4 × 1010). Animals received Temgesic (0.05 mg/kg; RB Pharmaceuticals, UK) and carprofen (5 mg/kg; Rimadyl, Zoetis B.V., Netherlands) 30 min before surgery. Subsequently, animals were anesthetized with isoflurane (2.5% induction and 1.5% maintenance) and mounted into a stereotactic frame. To provide local analgesia, lidocaine (2%; Sigma-Aldrich Chemie N.V., Netherlands) was topically applied to the skull before incision. The virus mixture was bilaterally (0.5 μl per hemisphere) infused into the mPFC (+1.8 mm anteroposterior; ±0.45 mm mediolateral; −2.1 mm dorsoventral; relative to bregma) using microinjection glass needles. Virus was infused at a flow rate of 0.1 μl/min, followed by 5 min of diffusion time and stepwise retraction of the needle. Analgesia was provided at 24 and 48 hours after surgery (carprofen; 5 mg/kg). Animals recovered for 1 week and were then transferred to a different housing room on a reversed day-night cycle, where they remained in their home cage for two additional weeks until the start of the behavioral procedures.
4TM treatment
4TM (H6278, Sigma-Aldrich Chemie N.V., Netherlands) was prepared as described previously (21). First, 10 mg of 4TM was first dissolved in 200 μl of 100% dimethyl sulfoxide (DMSO) (D8418, Sigma-Aldrich Chemie N.V., Netherlands). This stock solution was stepwise-diluted in 1900 μl of saline containing 2%Tween 80 (P1754, Sigma-Aldrich Chemie N.V., Netherlands) and then once more in the same volume of saline. This resulted in a final solution of 4TM (2.5 mg/ml), 5% DMSO, and 1% Tween 80 in saline. Animals received 4TM [25 mg/kg, intraperitoneally (ip)] 2 hours after a tag session (see below).
Chemogenetic manipulation
CNO (Hello Bio Limited, Bristol, UK) was dissolved in saline at a concentration of 0.5 mg/ml. Animals received an injection of CNO (5 mg/kg, ip) 30 min before a test. VEH-injected animals received the same volume of saline.
Behavioral procedures
Habituation: Two-choice phase
To habituate mice to alcohol, mice were first subjected to alcohol drinking in their home cage. During the first two habituation weeks, mice had unlimited access to both water and alcohol with increasing concentrations [days 1 to 2, 4% (w/v); days 3 to 8, 6% (w/v); days 9 to 14, 8% (w/v) in water]. Bottles were refreshed and weighed three times a week to measure water and alcohol intake. Two bottles were placed on an empty cage to control for leakiness. To avoid side preference, the position of the bottles on the cage was alternated after each time that they were weighed.
Habituation: Limited drinking phase
To motivate animals to drink during their dark phase, mice had access exclusively to a bottle containing 8% alcohol (w/v) for 8 hours starting 2 hours after the beginning of the dark phase. The water bottle was available during the other 16 hours of the day. Bottles were weighed daily, and solutions were replaced three times a week. In this phase, mice were handled for three consecutive days.
Operant SA
Mice were trained in operant conditioning chambers in sound-attenuating cubicles (TSE Systems, Bad Homburg, Germany) for 5 to 7 days a week (60 min per session throughout the experiment). During training sessions, a red house light centered above the operant chamber was illuminated. Active and inactive levers were located at opposite sides of the chamber. A predefined number of responses on the active lever resulted in the delivery of 10 μl of reward solution (8% alcohol or 10% sucrose solution) in the receptacle and illumination of a yellow cue light (discrete cue) located above the receptacle for 2 s. This also initiated a time-out period of 8 s, during which the house light was dimmed and lever presses were recorded but had no consequences. Responses on the inactive lever were recorded but had not programmed consequences. Because of a technical failure, one data point of inactive lever presses is missing in session FR3-3 and another in session FR5-2 in Fig. 3B.
Mice were initially trained to respond on an FR1 schedule of reinforcement for seven sessions, followed by an FR2 schedule for two to four sessions, an FR3 schedule for two to four sessions, and an FR5 schedule for four sessions. Alcohol SA and sucrose SA tag sessions were performed at FR5. Insufficient acquisition of alcohol SA was based on the following criteria: (i) active lever press average below 80 during FR5 sessions, (ii) no FR5 session with more than 100 active lever presses, and (iii) active lever preference (active lever presses per active + inactive lever presses) lower than 0.65. Mice that met two or more criteria (N = 42) were a priori excluded from alcohol seeking tests. Active and inactive lever responses (excluding time-out responses) are reported in graphs and used for statistical analyses of SA sessions.
Tag sessions
Alcohol and sucrose Tag sessions were equivalent to an operant SA session at FR5 with availability of the reward and presentation of the cue light. The NC tag was performed by a different experimenter 72 hours after the last operant SA session by exposing mice to an NC (square white box with black stripes) in a different room for 60 min. The alcohol consumption tag was performed 72 hours after the last operant SA session by giving mice access to a bottle containing 8% alcohol (which replaced a water bottle) in their home cage for 60 min.
Abstinence
After the tag session, animals underwent 3 weeks of forced abstinence in their home cage without access to alcohol.
Context-induced alcohol seeking
Animals were placed back into the operant chamber for 60 min under extinction conditions. Lever presses were recorded but did not result in reward delivery or presentation of the cue light. The red house light was illuminated throughout the session. As there was no time-out period during this session, the total number of active lever presses is reported.
Extinction
Extinction sessions were equivalent to the context-induced alcohol seeking test described above. Alcohol seeking was considered extinguished after a minimum of five extinction sessions and when active responses were stably reduced to the level of inactive responses.
Cue-induced alcohol seeking
Animals were placed in the operant chamber for 60 min, and lever pressing resulted in presentation of the discrete cue light on an FR5 schedule, with the exception that the first lever press also resulted in presentation of the cue light. Alcohol was not available during this session. In addition, when the FR was reached, this triggered a time-out period and activation of the pump that previously delivered the reward, similar to SA sessions. The total number of active and inactive lever presses are reported in graphs and used for statistical analyses.
Immunohistochemistry
At the end of experiments, all mice were transcardially perfused with ice-cold phosphate-buffered saline (PBS) (pH 7.4), followed by ice-cold 4% paraformaldehyde (PFA) in PBS. Brains were extracted, postfixed overnight in 4% PFA, and then transferred to 30% sucrose in PBS with 0.02% NaN3. After at least 2 days of immersion in sucrose, brains were sliced in 35-μm coronal sections using a cryostat and stored in PBS with 0.02% NaN3 and stored at 4°C until further use. Subsequent immunohistochemical stainings were performed as described previously (40) using the following antibodies: rabbit anti-Fos (1:300; sc52, Santa Cruz Biotechnology, USA), rabbit anti–red fluorescent protein (1:500; Rockland, USA), and mouse anti-GAD67 (1:1000; Merck Millipore, Netherlands). NeuroTrace 500/525 Green Fluorescent Nissl Stain was used to visualize neuronal cell bodies or DAPI to visualize cell nuclei. A confocal microscope was used to generate four to six z-stacks per animal by an experimenter blinded to treatment conditions. Fiji software (https://fiji.sc/) was used to determine the regions of interest (ROIs) of Nissl+ cells (Gaussian filter, Huang threshold, and watershed), and cells with a size of 80 to 2000 square units and circularity of 0.5 to 1.0 were included. MATLAB (MathWorks) was used to group ROIs belonging to the same Nissl+ cell and then to count the total number of Nissl+ cells in a z-stack. Cells expressing hM4Di-mCherry, mCherry, GAD67, or Fos were counted manually by an experimenter blinded to the conditions. Representative images were generated using a wide-field fluorescent microscope (DMi8, Leica Microsystems, Germany). One alcohol SA–tagged mouse was used for patch-clamp recording to confirm that CNO-reduced excitability of hM4Di-expressing cells. Therefore, this mouse was not included in the quantification of the percentage of hM4Di-expressing neurons in Fig. 3H. For analysis of the density of axonal fibers, six single-plane images of the NAcc and BLA were obtained at ×10 magnification per animal. The intensity of fluorescent signal in manually drawn ROIs (based on Nissl) was measured using Fiji and normalized to the background intensity of the section per animal (background was determined by defining an ROI in a nontarget region that was devoid of mCherry+ fibers).
Statistics
Data in graphs represent means ± SEM, and the number of animals per group is provided in the figure legends. SPSS software (version 25; IBM Corp.) was used for statistical analysis. We determined whether the data were modeled by a normal distribution using a Kolmogorov-Smirnov test. A one-way ANOVA with post hoc Bonferroni correction was used to investigate the number of tagged cells between multiple conditions. To compare drinking behavior in the home cage, a three-factor repeated measures ANOVA was used (Group x Time point x Solution). Acquisition of operant SA was investigated by a three-factor repeated measures ANOVA (Group x Sessions x Lever), followed by a separate two-factor repeated measures ANOVA (Group x Sessions) for the active and inactive lever. Active and inactive lever presses during the Tag session were separately compared between groups using an unpaired Student’s t test or by a Mann-Whitney U test in case data were not normally distributed. For the context-induced alcohol seeking experiment involving three independent groups, data were analyzed using a one-way ANOVA or a Kruskal-Wallis test in case of not-normally distributed data. Extinction was examined using a two-factor repeated measures ANOVA (Group x Sessions) for active lever pressing. Cue-induced reinstatement was compared with the last extinction session and analyzed separately for active and inactive responses using a two-factor repeated measures ANOVA (Group x Sessions). Reactivation of the tagged ensemble was investigated by a two-factor repeated measures ANOVA [Group x Population (Fos+/mCherry− versus Fos+/mCherry+)]. The proportion of GABAergic cells in the tagged ensembles was investigated using a repeated measures ANOVA [Tag x Population (GAD67+/mCherry− versus GAD67+/mCherry+)]. The fiber intensity of ensemble projections in target areas was compared using an unpaired Student’s t test or by a Mann-Whitney U test when data were not normally distributed. In case the assumption of sphericity was violated, a Greenhouse-Geisser correction was applied. Significance was set at P < 0.05.
Supplementary Material
Acknowledgments
We would like to thank Y. Gouwenberg for AAV packaging and K. A. Jonkman, I. Koutlas, and N. Milićević for help with behavioral experiments and immunohistochemical stainings. Funding: This study was funded by an NWO VIDI grant (016.168.313) to E.V. and M.C.v.d.O., a VIDI grant (016.Vidi.188.022) to N.J.M., and an Amsterdam Neuroscience PoC grant (8-722 PoC-CIA-2017) to M.C.v.d.O. and N.J.M. Author contributions: Conceptualization: E.V., N.J.M., T.J.d.V., A.B.S., and M.C.v.d.O. Methodology: E.V., M.R.M., and M.C.v.d.O. Formal analysis: E.V. and M.C.v.d.O. Investigation: E.V., M.R.M., and R.J.v.d.L. Writing: E.V., A.B.S., and M.C.v.d.O. Supervision: T.J.d.V., A.B.S., and M.C.v.d.O. Funding acquisition: N.J.M., A.B.S., and M.C.v.d.O. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions of the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper and requests for resources and reagents may be requested from the corresponding author M.C.v.d.O.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/19/eaax7060/DC1
REFERENCES AND NOTES
- 1.World Health Organization, Global Status Report On Alcohol And Health 2018 (World Health Organization, S.l., 2019).
- 2.Whiteford H. A., Degenhardt L., Rehm J., Baxter A. J., Ferrari A. J., Erskine H. E., Charlson F. J., Norman R. E., Flaxman A. D., Johns N., Burstein R., Murray C. J., Vos T., Global burden of disease attributable to mental and substance use disorders: Findings from the Global Burden of Disease Study 2010. Lancet. 382, 1575–1586 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Moos R. H., Moos B. S., Rates and predictors of relapse after natural and treated remission from alcohol use disorders. Addiction 101, 212–222 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heinz A., Beck A., Grüsser S. M., Grace A. A., Wrase J., Identifying the neural circuitry of alcohol craving and relapse vulnerability. Addict. Biol. 14, 108–118 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Monti P. M., Binkoff J. A., Abrams D. B., Zwick W. R., Nirenberg T. D., Liepman M. R., Reactivity of alcoholics and nonalcoholics to drinking cues. J. Abnorm. Psychol. 96, 122–126 (1987). [DOI] [PubMed] [Google Scholar]
- 6.Schacht J. P., Anton R. F., Myrick H., Functional neuroimaging studies of alcohol cue reactivity: A quantitative meta-analysis and systematic review. Addict. Biol. 18, 121–133 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sussman S., Horn J. L., Gilewski M. J., Cue-exposure interventions for alcohol relapse prevention: Need for a memory modification component. Int. J. Addict. 25, 921–929 (1990). [DOI] [PubMed] [Google Scholar]
- 8.Whitaker L. R., Hope B. T., Chasing the addicted engram: Identifying functional alterations in Fos-expressing neuronal ensembles that mediate drug-related learned behavior. Learn. Mem. 25, 455–460 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldstein R. Z., Volkow N. D., Dysfunction of the prefrontal cortex in addiction: Neuroimaging findings and clinical implications. Nat. Rev. Neurosci. 12, 652–669 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van den Oever M. C., Spijker S., Smit A. B., De Vries T. J., Prefrontal cortex plasticity mechanisms in drug seeking and relapse. Neurosci. Biobehav. Rev. 35, 276–284 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Grüsser S. M., Wrase J., Klein S., Hermann D., Smolka M. N., Ruf M., Weber-Fahr W., Flor H., Mann K., Braus D. F., Heinz A., Cue-induced activation of the striatum and medial prefrontal cortex is associated with subsequent relapse in abstinent alcoholics. Psychopharmacology (Berl) 175, 296–302 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Beck A., Wüstenberg T., Genauck A., Wrase J., Schlagenhauf F., Smolka M. N., Mann K., Heinz A., Effect of brain structure, brain function, and brain connectivity on relapse in alcohol-dependent patients. Arch. Gen. Psychiatry 69, 842–852 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Dayas C. V., Liu X., Simms J. A., Weiss F., Distinct patterns of neural activation associated with ethanol seeking: Effects of naltrexone. Biol. Psychiatry 61, 979–989 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jupp B., Krstew E., Dezsi G., Lawrence A. J., Discrete cue-conditioned alcohol-seeking after protracted abstinence: Pattern of neural activation and involvement of orexin1 receptors. Br. J. Pharmacol. 162, 880–889 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cruz F. C., Koya E., Guez-Barber D. H., Bossert J. M., Lupica C. R., Shaham Y., Hope B. T., New technologies for examining the role of neuronal ensembles in drug addiction and fear. Nat. Rev. Neurosci. 14, 743–754 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palombo P., Leao R. M., Bianchi P. C., de Oliveira P. E. C., Planeta C. D. S., Cruz F. C., Inactivation of the prelimbic cortex impairs the context-induced reinstatement of ethanol seeking. Front. Pharmacol. 8, 725 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Willcocks A. L., McNally G. P., The role of medial prefrontal cortex in extinction and reinstatement of alcohol-seeking in rats. Eur. J. Neurosci. 37, 259–268 (2013). [DOI] [PubMed] [Google Scholar]
- 18.George O., Hope B. T., Cortical and amygdalar neuronal ensembles in alcohol seeking, drinking and withdrawal. Neuropharmacology 122, 107–114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pfarr S., Meinhardt M. W., Klee M. L., Hansson A. C., Vengeliene V., Schönig K., Bartsch D., Hope B. T., Spanagel R., Sommer W. H., Losing control: Excessive alcohol seeking after selective inactivation of cue-responsive neurons in the infralimbic cortex. J. Neurosci. 35, 10750–10761 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Guglielmo G., Crawford E., Kim S., Vendruscolo L. F., Hope B. T., Brennan M., Cole M., Koob G. F., George O., Recruitment of a neuronal ensemble in the central nucleus of the amygdala is required for alcohol dependence. J. Neurosci. 36, 9446–9453 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matos M. R., Visser E., Kramvis I., van der Loo R. J., Gebuis T., Zalm R., Rao-Ruiz P., Mansvelder H. D., Smit A. B., Van den Oever M. C., Memory strength gates the involvement of a CREB-dependent cortical fear engram in remote memory. Nat. Commun. 10, 2315 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dong S., Allen J. A., Farrell M., Roth B. L., A chemical-genetic approach for precise spatio-temporal control of cellular signaling. Mol. Biosyst. 6, 1376–1380 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Cruz F. C., Javier Rubio F., Hope B. T., Using c-fos to study neuronal ensembles in corticostriatal circuitry of addiction. Brain Res. 1628, 157–173 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keistler C. R., Hammarlund E., Barker J. M., Bond C. W., DiLeone R. J., Pittenger C., Taylor J. R., Regulation of alcohol extinction and cue-induced reinstatement by specific projections among medial prefrontal cortex, nucleus accumbens, and basolateral amygdala. J. Neurosci. 37, 4462–4471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalivas P. W., Volkow N. D., The Neural Basis of Addiction: A Pathology of Motivation and Choice. Am. J. Psychiatry 162, 1403–1413 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Millan E. Z., Reese R. M., Grossman C. D., Chaudhri N., Janak P. H., Nucleus accumbens and posterior amygdala mediate cue-triggered alcohol seeking and suppress behavior during the omission of alcohol-predictive cues. Neuropsychopharmacology 40, 2555–2565 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Augier E., Barbier E., Dulman R. S., Licheri V., Augier G., Domi E., Barchiesi R., Farris S., Nätt D., Mayfield R. D., Adermark L., Heilig M., A molecular mechanism for choosing alcohol over an alternative reward. Science 360, 1321–1326 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Bossert J. M., Marchant N. J., Calu D. J., Shaham Y., The reinstatement model of drug relapse: Recent neurobiological findings, emerging research topics, and translational research. Psychopharmacology (Berl) 229, 453–476 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chaudhri N., Sahuque L. L., Schairer W. W., Janak P. H., Separable roles of the nucleus accumbens core and shell in context- and cue-induced alcohol-seeking. Neuropsychopharmacology 35, 783–791 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van den Oever M. C., Goriounova N. A., Wan Li K., Van der Schors R. C., Binnekade R., Schoffelmeer A. N. M., Mansvelder H. D., Smit A. B., Spijker S., De Vries T. J., Prefrontal cortex AMPA receptor plasticity is crucial for cue-induced relapse to heroin-seeking. Nat. Neurosci. 11, 1053–1058 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Schmidt E. D., Voorn P., Binnekade R., Schoffelmeer A. N. M., De Vries T. J., Differential involvement of the prelimbic cortex and striatum in conditioned heroin and sucrose seeking following long-term extinction. Eur. J. Neurosci. 22, 2347–2356 (2005). [DOI] [PubMed] [Google Scholar]
- 32.McGlinchey E. M., James M. H., Mahler S. V., Pantazis C., Aston-Jones G., Prelimbic to accumbens core pathway is recruited in a dopamine-dependent manner to drive cued reinstatement of cocaine seeking. J. Neurosci. 36, 8700–8711 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Warren B. L., Mendoza M. P., Cruz F. C., Leao R. M., Caprioli D., Rubio F. J., Whitaker L. R., McPherson K. B., Bossert J. M., Shaham Y., Hope B. T., Distinct fos-expressing neuronal ensembles in the ventromedial prefrontal cortex mediate food reward and extinction memories. J. Neurosci. 36, 6691–6703 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pfarr S., Schaaf L., Reinert J. K., Paul E., Herrmannsdörfer F., Roßmanith M., Kuner T., Hansson A. C., Spanagel R., Körber C., Sommer W. H., Choice for drug or natural reward engages largely overlapping neuronal ensembles in the infralimbic prefrontal cortex. J. Neurosci. 38, 3507–3519 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wiltgen B. J., Brown R. A. M., Talton L. E., Silva A. J., New circuits for old memoreis: The role of the neocortex in consolidation. Neuron 44, 101–108 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Kitamura T., Ogawa S. K., Roy D. S., Okuyama T., Morrissey M. D., Smith L. M., Redondo R. L., Tonegawa S., Engrams and circuits crucial for systems consolidation of a memory. Science 356, 73–78 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomas G. M., Huganir R. L., MAPK cascade signalling and synaptic plasticity. Nat. Rev. Neurosci. 5, 173–183 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Whitaker L. R., Warren B. L., Venniro M., Harte T. C., McPherson K. B., Beidel J., Bossert J. M., Shaham Y., Bonci A., Hope B. T., Bidirectional modulation of intrinsic excitability in rat prelimbic cortex neuronal ensembles and non-ensembles after operant learning. J. Neurosci. 37, 8845–8856 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rao-Ruiz P., Couey J. J., Marcelo I. M., Bouwkamp C. G., Slump D. E., Matos M. R., van der Loo R. J., Martins G. J., van den Hout M., van IJcken W. F., Costa R. M., Van den Oever M. C., Kushner S. A., Engram-specific transcriptome profiling of contextual memory consolidation. Nat. Commun. 10, 2232 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van den Oever M. C., Rotaru D. C., Heinsbroek J. A., Gouwenberg Y., Deisseroth K., Stuber G. D., Mansvelder H. D., Smit A. B., Ventromedial prefrontal cortex pyramidal cells have a temporal dynamic role in recall and extinction of cocaine-associated memory. J. Neurosci. 33, 18225–18233 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/19/eaax7060/DC1