

A proof-of-concept study shows the generation of an immunogenic hexavalent Coxsackie B virus vaccine that prevents disease.
Abstract
Coxsackievirus B (CVB) enteroviruses are common human pathogens known to cause severe diseases including myocarditis, chronic dilated cardiomyopathy, and aseptic meningitis. CVBs are also hypothesized to be a causal factor in type 1 diabetes. Vaccines against CVBs are not currently available, and here we describe the generation and preclinical testing of a novel hexavalent vaccine targeting the six known CVB serotypes. We show that the vaccine has an excellent safety profile in murine models and nonhuman primates and that it induces strong neutralizing antibody responses to the six serotypes in both species without an adjuvant. We also demonstrate that the vaccine provides immunity against acute CVB infections in mice, including CVB infections known to cause virus-induced myocarditis. In addition, it blocks CVB-induced diabetes in a genetically permissive mouse model. Our preclinical proof-of-concept studies demonstrate the successful generation of a promising hexavalent CVB vaccine with high immunogenicity capable of preventing CVB-induced diseases.
INTRODUCTION
Coxsackievirus Bs (CVBs) are common human single-stranded RNA viruses that belong to the enterovirus genus of Picornaviridae. In the United States, CVB infections accounted for close to 10% of reported enterovirus cases between 2014 and 2016 (1). The CVBs consist of six serotypes, CVB1–6, and in most circumstances, infections with these viruses are asymptomatic or result in mild symptoms that present in a cold- or flu-like manner. However, CVBs are also known to cause potentially life-threatening diseases, including encephalitis and aseptic meningitis (2, 3), myocarditis and chronic dilated cardiomyopathy (DCM) (4–6), pancreatitis (7), and hand-foot-and-mouth disease [HFMD; (8, 9)]. Acute infections of neonates can be lethal (10). DCM is the most frequent reason for heart transplantation in children in the United States (6) and is the second leading cause of heart transplantation worldwide (11). In addition to this, there are strong associations between CVB infections and the chronic autoimmune disease type 1 diabetes [T1D; (9, 12–14)], suggesting that these viruses are an important causal factor in an increasing number of diseases with high clinical and economic relevance. Specific CVB serotypes have been associated with these different diseases. For instance, it has been well documented that CVB3 causes viral myocarditis and DCM (4–6), CVB5 infections can result in encephalitis and aseptic meningitis outbreaks (2, 8), and CVB1 was associated with the induction of β-cell autoimmunity, which can lead to T1D (15). Thus, a vaccine that targets CVBs would be highly desirable for the prevention of CVB-induced diseases, which place a heavy burden upon health care systems and could also serve to clarify the role of these viruses in initiating other debilitating diseases, including T1D.
Despite the common nature of CVB infections and the possibility of severe manifestations, there are currently no commercially available vaccines that target CVBs. The only enterovirus vaccines in clinical use are those against polioviruses and two recently approved enterovirus 71 vaccines that target HFMD (8). A few reports have described the production and preclinical testing of CVB vaccines. These have been limited to studies addressing the efficacy of a single valence CVB vaccine in mouse models (16–21). Given that all of the CVB serotypes are capable of causing severe diseases, a vaccine targeting a single serotype is unlikely to be of high benefit, and it is improbable that motivation would exist to initiate general vaccination strategies with such a vaccine.
The objective of our study was to explore the possibility of developing a hexavalent vaccine that covers the six CVB serotypes and to test its safety, immunogenicity, and functionality. For this purpose, we used a nonhuman primate model and clinically relevant experimental murine models for CVB-induced diseases. We demonstrate that an inactivated hexavalent CVB vaccine is immunogenic in three commonly used mouse strains and in rhesus macaques, and that it induces strong neutralizing antibody (nAB) responses against all six CVB serotypes. The vaccine protected against acute CVB infections. It also prevented infection of the heart in an experimental murine model for CVB3-induced myocarditis and provided immunity against virus-induced diabetes in a genetically susceptible mouse strain.
RESULTS
Production and characterization of the hexavalent CVB1–6 vaccine
In this study, our goal was to create a hexavalent CVB vaccine based on formalin-inactivated CVB1–6 viruses and to test its immunogenicity in preclinical animal models. The individual CVB virus components of the vaccine were produced in Vero cells and inactivated using an optimized protocol. Briefly, the viruses were inactivated in 0.01% (v/v) formalin for 5 days at 37°C (as described in Materials and Methods). After inactivation and before the mixing of the serotypes, the concentration and integrity of the inactivated CVB1–6 serotypes were assessed. The concentrations of inactivated CVB1–5 were not altered when compared to the original CVB virus components, but there was some decrease in the protein concentration of CVB6 (loss compared to samples before inactivation in percent: CVB1–5, 0 to 2.2%; CVB6, 50%). SDS–polyacrylamide gel electrophoresis (SDS-PAGE) analysis with a stain-free detection system (Fig. 1A) and Western blot analysis with the in-house produced antibody 3A6 (Fig. 1B) (22) directed to VP1 revealed correctly sized bands for the viral capsid protein VP1 in the inactivated CVB1–6 serotypes. VP1 was the predominant band in Fig. 1A, and the other bands are likely to represent other viral proteins and contaminants from the virus propagation in Vero cells. The quality of the inactivated CVB1–6 viruses was determined with dynamic light scattering (DLS) analysis. DLS analysis demonstrated that all CVB1–6 serotypes contained intact virus particles (Fig. 1, C and D). The average particle sizes were between 29 and 47 nm (Fig. 1C), and the proportion of the most prominent particle sizes in the vaccine preparations are shown in Fig. 1D. Inactivated CVB1–6 serotypes were also stored at −80°C for 27 months and analyzed using the same methods, with similar results as shown in fig. S1. Transmission electron microscopy (TEM) analysis of the individual inactivated CVB1–6 viruses showed the presence of intact virus particles that had the correct morphology and were of the expected size (Fig. 1E). The quantities of DNA from the Vero cell line used to produce the viruses were very low in the vaccine preparations (1.5 ng per 6 μg vaccine dose). To further confirm that the virus was inactivated and unable to replicate, enterovirus-specific reverse transcription quantitative polymerase chain reaction (RT-qPCR) was carried out using primers that target the highly conserved 5′ non-coding region (NCR). Green monkey kidney (GMK) cell cultures were treated with vaccine (i.e., formalin-inactivated CVB1–6 vaccine, 200 μl per well) and cultured for 5 days. As a control for the RT-qPCR, additional GMK cells were infected in parallel with CVB1 (multiplicity of infection, 0.001). After this, the cells were lysed, RNA was extracted, and RT-qPCR was performed. The amounts of viral RNA in the vaccine before culture and in the GMK cells treated with CVB1–6 vaccine after culture were compared (fig. S2). No differences in the quantities of viral RNA were detected by enterovirus-specific RT-qPCR, confirming the inactivation of the CVB1–6 serotypes (fig. S2). Collectively, these studies reveal the integrity of the individual CVB1–6 vaccine components and confirm that the infectious viruses were inactivated. Therefore, the hexavalent CVB1–6 vaccine was created by mixing the individually inactivated CVB serotypes to achieve a final dose of 1 μg per serotype in 150-μl vaccine volume. TEM analysis of the mixed CVB1–6 vaccine is shown in Fig. 1E.
Fig. 1. Characterization of inactivated CVB1–6 viruses, experimental setup, and vaccine safety and immunogenicity in C57BL/6J mice.
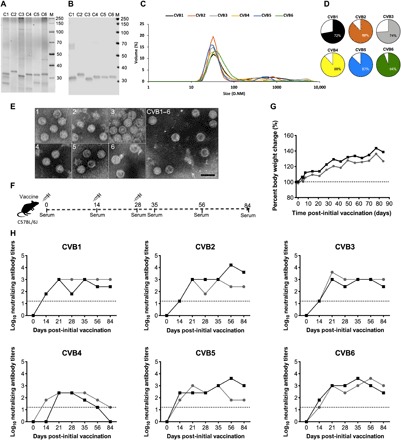
CVB1–6 viruses were propagated in Vero cells, purified, inactivated with formalin, and then characterized as described in Materials and Methods. (A) Analysis of CVB1–6 total protein and virus protein content (2 μg of virus per well) by SDS-PAGE followed by (B) Western blot analysis using the in-house produced rat monoclonal antibody 3A6, which binds to the CVB1–6 viral capsid protein VP1. (C) DLS analysis of the inactivated CVB1–6 serotypes. D.NM is the diameter in nm of the particles. (D) Volume distribution (percentages) of the most prominent particle populations in the vaccine preparations as measured by DLS analysis. (E) TEM analysis of individual inactivated CVB1–6 vaccine components (serotypes are indicated by the number in the top left-hand corner of each image) and the vaccine mix (CVB1–6). Scale bar, 50 nm. (F) Experimental setup for the C57BL/6J CVB1–6 vaccine studies. (G and H) Two female C57BL/6J mice (age 7.8 weeks), represented by individual lines, were vaccinated on days 0, 14, and 28 with CVB1–6 vaccine (1-μg dose of each serotype, 150 μl, interscapularly). (G) Percentage body weight change from the first vaccination (day 0); the dotted line indicates the weight on day 0. (H) CVB nAB titers in the serum of mice after vaccination. The dotted lines show the positivity cutoff for the method. C1, CVB1; C2, CVB2; C3, CVB3; C4, CVB4; C5, CVB5; C6, CVB6.
CVB1–6 vaccine has a good safety profile in mice and is immunogenic
Initially, the safety of the hexavalent CVB1–6 vaccine was examined in mice. It was assumed that the vaccine should be immunogenic in any mouse strain, and therefore, initial studies were performed using mice on the C57BL/6J background. Mice were vaccinated on days 0, 14, and 28 (the study setup is shown in Fig. 1F), and no changes in weight (Fig. 1G) or other clinically relevant features (as described in Materials and Methods) were seen. nAB titers against the six CVB serotypes were measured at different time points after the prime vaccination (Fig. 1H). Neutralizing capacity of the sera against all CVB1–6 serotypes was absent before the prime vaccination (day 0) in all experiments. Two weeks after the prime vaccination, nABs were present for all of the CVBs apart from CVB4 in one mouse. nAB titers against CVB1–6 were augmented after the first boost vaccination (on day 14) and were generally, and with only one exception (CVB4) in one mouse, maintained up until 84 days after the prime vaccination when the animals were sacrificed (Fig. 1H).
The immunogenicity and safety of the CVB1–6 vaccine were also tested in mice with a different background, namely non-obese diabetic (NOD) mice (23) (the experimental setup is shown in Fig. 2A). NOD mice are the most common mouse strain used in research addressing T1D (23), a disease highly associated with enterovirus infections (9, 12). The CVB1–6 vaccine had no adverse effects on either weight (Fig. 2B) or blood glucose levels (Fig. 2C) of vaccinated mice when compared to control animals. Strong nAB responses against the six CVB serotypes were detected after the first boost vaccination, and these were further augmented after the second boost vaccination (Fig. 2D). The nABs titers remained high in most animals (between 1/64 and 1/16,384) throughout the time period after the final vaccination until the experimental end point (day 41/42). To gain additional insight into the duration and magnitude of the nAB response, three additional NOD mice were vaccinated and nAB titers were measured on days 0 and 42 and between 70 and 113 days after the initial vaccination. This experiment showed that all animals maintained nAB titers ≥1/256 for CVB1, and CVB3-5 at the terminal time point, and two of three mice had nAB titers ≥1/64 for CVB2 and CVB6 at the same time point (fig. S3). Collectively, these data show that the CVB1–6 vaccine was safe in two murine strains and induced strong CVB nAB responses against all six CVB serotypes in the vast majority of animals.
Fig. 2. Safety and immunogenicity of CVB1–6 vaccine in NOD mice.
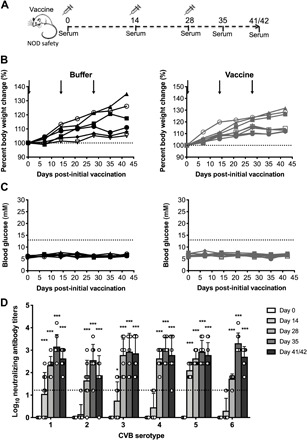
Male and female NOD mice (5 to 9 weeks old) were mock-vaccinated with vaccine buffer (150 μl, interscapularly; n = 6) or vaccinated with CVB1–6 vaccine (1 μg of each serotype in 150 μl, interscapularly; n = 8) on days 0, 14, and 28, as demonstrated in the schematic shown in (A). (B) Percentage body weight change compared to the weight on day 0 (indicated by dotted line). Buffer-treated animals are shown in black, and CVB1–6–vaccinated mice are indicated in gray. The black arrows show the vaccination time points. No statistically significant differences in percentage weight change were detected between the buffer-treated and buffer-vaccinated mice at each time point (Mann-Whitney U test). (C) Blood glucose readings of buffer-treated (black) or CVB1–6–vaccinated (gray) mice. The dotted line indicates the cutoff for diabetes. (D) Average CVB1–6 nAB titers in the serum of CVB1–6–vaccinated mice at the indicated time points. Sera from mock-vaccinated animals had no neutralizing capacity. nAB values for each individual mouse are shown by single symbols, and the bars represent means ± SD. The dotted line indicates the positivity cutoff for serum samples. *P < 0.05, ***P < 0.001 compared to day 0 for each respective serotype by two-way analysis of variance (ANOVA) with Bonferroni multiple comparisons.
CVB1–6 hexavalent vaccine protects against acute CVB infections in mice
To test the efficacy of the CVB1–6 vaccine against a selection of CVBs in vivo, female NOD mice were buffer-treated or immunized three times and then challenged with either CVB1 or CVB4 at a dose known to cause systemic infections. We also included CVB1 and CVB4 monovalent vaccines in these studies. CVB1 was included as a positive control, as we have previously documented that monovalent CVB1 vaccines are capable of preventing CVB1-induced viremia (16, 19, 20). The CVB4 monovalent vaccine was tested, as the CVB4 nAB titers were low at later time points in some (Fig. 1H), but not all, of the previous studies (Fig. 2D and fig. S3). The CVB1–6 vaccine, as well as the CVB1 and CVB4 monovalent vaccines, had no adverse effects on weight (figs. S4A and S5A) or on blood glucose values (figs. S4B and S5B). Moreover, strong nAB responses were detected against CVB1 (monovalent CVB1 vaccine; fig. S4C), CVB4 (monovalent CVB4 vaccine; fig. S5C), and the six CVB serotypes (figs. S4D and S5D). Of the mock-vaccinated (buffer-treated) mice, 80% of those challenged with CVB1 were viremic on day 3 after infection (Fig. 3B) and 100% of the equivalent CVB4-challenged buffer-treated animals had replicating virus in the blood at the same time point (Fig. 3F). Furthermore, replicating virus was present in the pancreas of CVB1- and CVB4-challenged buffer-treated mice (Fig. 3, C and G), and formalin-fixed paraffin-embedded (FFPE) pancreas showed signs of pancreatitis and was positive for viral VP1 protein in 75% of CVB1-infected (Fig. 3, D and E) and 100% of CVB4-infected buffer-treated animals (Fig. 3, H and I). In contrast, all of the CVB1–6–vaccinated mice were protected from acute CVB1 and CVB4 infections. None of the vaccinated mice had replicating virus in the blood on day 3 after infection (Fig. 3, B and F), and CVB1 and CVB4 were not detected in the pancreas at the same time point by standard plaque assay (Fig. 3, C and G, respectively). Pancreatitis and VP1 staining were completely absent in the pancreas of 75% of CVB1–6–vaccinated mice challenged with CVB1 (Fig. 3D), as represented by the image in Fig. 3E and in all of the CVB1–6–vaccinated CVB4-challenged mice (Fig. 3, H and I). Similar results were seen for the CVB1 and CVB4 monovalent vaccines, where both vaccines prevented either CVB1- or CVB4-induced viremia, respectively (figs. S4E and S5E). Moreover, the CVB1 vaccine prevented the systemic spread of CVB1 to the pancreas in 75% of CVB1-infected mice (fig. S4, F to H), while 100% of animals vaccinated with the CVB4 monovalent vaccine were protected from CVB4 infection of the pancreas (fig. S5, F to H).
Fig. 3. CVB1–6 vaccine protects mice from acute CVB1 and CVB4 infections.
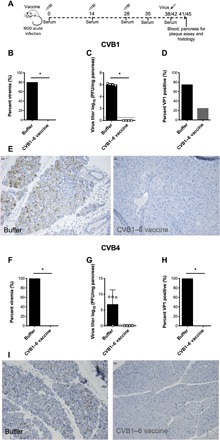
Female NOD mice (4 to 6 weeks old) were mock-vaccinated (vaccine buffer; 150 μl, interscapularly) or CVB1–6–vaccinated (1 μg of each serotype, interscapularly) on days 0, 14, and 28 and then challenged with CVB1 [106 plaque-forming units (PFU) per mouse, intraperitoneally] or CVB4 (105 PFU per mouse, intraperitoneally) on days 42 and 38, respectively, as shown in the experimental schematic in (A). (B and F) Percentage of mock-vaccinated (CVB1, n = 5; CVB4, n = 4) and CVB1–6–vaccinated (CVB1, n = 4; CVB4, n = 4) mice with viremia on day 3 after infection (measured by standard plaque assay). *P < 0.05, one-way Fisher’s exact test. (C and G) Replicating virus in the pancreas on day 3 after infection of CVB1-infected (C) or CVB4-infected (G) mice (measured by standard plaque assay). Virus titers for each mouse are shown individually. Bars represent means ± SD. The dotted line shows the limit of detection for the assay. *P < 0.05 comparing the two groups by Mann-Whitney U test. (D and H) Percentage of pancreas sections positive for VP1 by immunohistochemical staining on day 3 after infection in CVB1-infected (D) and CVB4-infected (H) mice. *P < 0.05, one-way Fisher’s exact test. (E and I) Representative VP1 staining in pancreas of mice infected with CVB1 (E) or CVB4 (I). Magnification, ×16; scale bars, 20 μm. In (C) and (D), n = 4 in the buffer-treated group, as one NOD mouse was kept for longer to ensure that SOCS-1-tg animals were not single-housed.
Vaccination may temporarily lead to nonspecific activation of innate antiviral immunity, which, in turn, can have a protective effect against infection. These effects are mainly seen with adjuvanted vaccines and vaccines based on attenuated viruses (24). Therefore, to assess whether the CVB1–6 vaccine led to activation of the host innate immune response, which, in turn, could have affected the outcomes of the acute CVB infections performed, we examined the expression of genes that are known to modulate antiviral responses to CVBs in pancreas and spleen collected 2 weeks after the final vaccination. The genes examined were interferon-α (IFN-α), IFN-β, EIF2AK2/protein kinase R (PKR), 2′-5′ oligoadenylate synthetase 1 (OAS1), and nitric oxide synthase (NOS2) (25–27). No differences were seen when comparing the buffer-treated and CVB1–6–vaccinated mice (fig. S6), suggesting that, at the time of virus challenge, nonspecific activation of antiviral immunity was negligible. Together, these results demonstrate the in vivo protective capacity of the hexavalent CVB1–6 vaccine, as well as CVB1 and CVB4 monovalent vaccines, against acute CVB infections.
CVB1–6 hexavalent vaccine is immunogenic in rhesus macaques
Rhesus macaques (nonhuman primates) share up to 93% of their genes with humans and provide one of the best human-like models for the preclinical testing of vaccine immunogenicity and safety (28). As far as we are aware, this was the first study performed in nonhuman primates with any CVB vaccine. As such, some of the animals received CVB1–6 vaccine with alum adjuvant to maximize the chances of the vaccine inducing immunity. Animals were immunized on two occasions (prime immunization on day 0 and boost immunization on day 28) with nonadjuvanted (n = 2) or alum-adjuvanted (n = 3) CVB1–6 vaccine (as shown in Fig. 4A). The weight of the animals remained stable throughout the study (Fig. 4B), as did their body temperatures (Fig. 4C) and blood glucose values (Fig. 4D).
Fig. 4. CVB1–6 vaccine is safe in rhesus macaques and induces strong immunity against all six CVB serotypes.
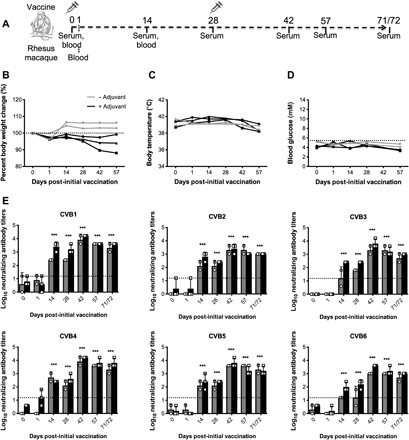
Rhesus macaques were immunized with CVB1–6 vaccine (5 μg of each CVB serotype) without adjuvant (n = 2, gray lines/bars) or CVB1–6 vaccine (5 μg of each CVB serotype) + alum adjuvant (0.2% final concentration; n = 3, black lines/bars) on days 0 and 28 by intramuscular injection, as shown in the schematic in (A). (B) Percentage weight changes compared to day 0 (prime vaccination), (C) body temperature, and (D) blood glucose values of animals [in (B), the dotted line shows the % weight at day 0, and in (C), the dotted line indicates the blood glucose concentration threshold for overt diabetes in rhesus macaques after fasting]. No statistically significant differences in the weights, temperatures, or blood glucose values were found when comparing the different time points to day 0 (two-way ANOVA with Bonferroni correction). (E) CVB1–6 nAB titers in the serum as measured by standard neutralization assay. The dotted line shows the limit of detection. nAB values for each individual animal are shown by single symbols, and the bars represent means ± SD. Samples without an error bar are all equal in value. ***P < 0.001 comparing the average nAB titer for both groups at each time point to day 0 by two-way ANOVA with Bonferroni correction.
The safety of the vaccine was further emphasized by results from liver function tests performed with samples collected on day 0 and on days 1 and 14 after the prime vaccination. No consistent alterations were seen in the levels of various liver enzymes (fig. S7, A to D) on days 1 and 14 compared with day 0, and all were in the normal range for rhesus macaques (29, 30), indicating that no liver toxicity was induced by the vaccine.
nAB titers were assessed on days 0, 1, 14, 28, 42, 56, and 71 or 72 after the prime immunization (Fig. 4E). At the early time points (days 0 and 1), the serum from all animals lacked CVB1–6 neutralizing capacity. By day 14 after the prime vaccination, serum from animals immunized with the vaccine and adjuvant showed higher nABs titers for all CVB1–6 serotypes than those from the nonadjuvanted vaccine group (apart from CVB4, where the levels were similar between the two groups; Fig. 4E). A similar trend was seen on day 28; however, the nAB titers appeared to be more comparable between the two groups after the boost vaccination (days 42, 56, and 71/72). Moreover, the nAB titers remained high up to 10 weeks after the prime vaccination in both groups when monitoring of the animals was discontinued. The data from these studies show that the CVB1–6 vaccine is safe and highly immunogenic in nonhuman primates, and adjuvant is not required for a strong and sustained nAB response after prime and boost immunizations.
CVB1–6 immunization prevents CVB infection of the heart in a model for viral myocarditis
CVB infections have been associated with viruses-induced myocarditis (4–6), and an important goal in the clinical use of CVB vaccines would be the prevention of infections that disseminate to the heart and could lead to myocarditis and DCM. CVB3 infection of young (less than 6 weeks of age) male Balb/c mice provides a murine model for CVB-induced heart disease (4, 5, 31). While our vaccine schedule precluded infection of young Balb/c mice (the last immunizations are conducted when animals are 8 to 9 weeks of age), we instead examined whether the CVB1–6 vaccine can prevent CVB3 infection of the heart (a prerequisite for myocarditis development) in Balb/c mice. Due to the aforementioned reasons, and since previous studies have shown that strong nAB responses develop following the first boost immunization, animals as young as possible were mock-immunized or immunized with CVB1–6 vaccine on two occasions (days 0 and 21) and then challenged with CVB3 on day 35 (Fig. 5A).
Fig. 5. CVB1–6 vaccine protects against acute CVB3 infection in Balb/c mice.
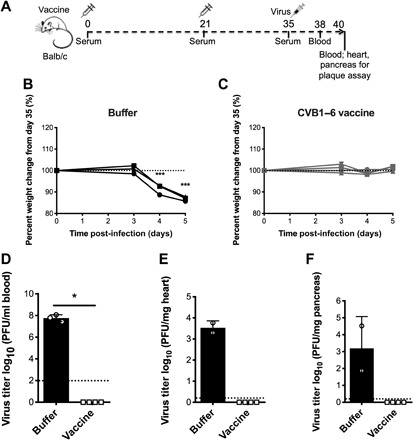
Male and female mice Balb/c mice were buffer-treated (150 μl, interscapularly; n = 3; black lines/bars) or vaccinated with CVB1–6 vaccine (1 μg of each serotype; 150 μl, interscapularly; n = 4; gray lines/bars) on days 0 and 21 and challenged with CVB3 (5 × 104 PFU per mouse, intraperitoneally) on day 35 as shown in the schematic in (A). (B and C) Percentage weight change for individual animals compared to day 35 in buffer-treated (B) or CVB1–6–vaccinated mice (C). Dotted line indicates day 35 weight. ***P < 0.001 compared to day 35 weight as measured by one-way ANOVA with Dunnett’s multiple comparison test. Replicating virus in the blood (D) on day 3 post-CVB3 infection or either the pancreas (E) or heart (F) on day 5 after infection, as determined by standard plaque assay [n = 2 for buffer-treated group in (E) and (F), as one animal was found dead on day 5 after infection]. Virus titers for individual mice have individual symbols, and the bars show means ± SD. The dotted lines show the limit of detection for the assays. *P < 0.05 comparing the two groups by Mann-Whitney U test. Insufficient n numbers in the buffer group prevented statistical analysis of (E) and (F).
Immunization did not alter the weight of mice after the prime and boost vaccinations (fig. S8, A and B), and nABs were present against all six CVB serotypes on day 35 (fig. S8C). Mock-immunized mice displayed drastic weight loss from day 3 after CVB3 challenge (Fig. 5B), while animals immunized with CVB1–6 remained stable in weight (Fig. 5C). Immunized mice also showed no signs of viremia on day 3 after infection, whereas all buffer-treated mice had high CVB3 titers in the blood at the same time point (Fig. 5D). Blood samples from all mice collected on day 5 after CVB3 infection were free of replicating virus (no plaques were detected by standard plaque assay). One buffer-treated mouse had succumbed to infection before the day 5 samples could be collected. At this same time point, replicating virus was detected in both the heart (Fig. 5E) and the pancreas (Fig. 5F) of the remaining (n = 2) buffer-treated animals. The absence of viremia on day 5 after infection confirmed that the replicating virus measured in the heart at the same time point was present due to the systemic spread of CVB3 to heart tissue rather than the presence of virus in the blood. In contrast, the CVB1–6–immunized mice were completely protected from infection of the heart and pancreas (Fig. 5, E and F). These studies demonstrate that the CVB1–6 vaccine is capable of protecting mice from acute CVB3 infections in the heart, which can portend to CVB-induced myocarditis.
CVB-induced diabetes is prevented by immunization with the CVB1–6 hexavalent vaccine
It has been postulated that CVBs may be a causative agent in T1D (9, 12, 13). To address whether the CVB vaccine also provides protection from enterovirus-induced diabetes, we used the SOCS-1-tg mouse model. SOCS-1-tg mice are a murine model for virus-induced T1D, where the β-cells are highly susceptible to CVB destruction, as the expression of the suppressor of cytokine signaling-1 (SOCS-1) under the control of the insulin promoter renders them unable to respond to IFNs and mount a successful antiviral response (20, 27, 32).
Two vaccination experiments with either CVB1 or CVB3 challenge were performed (experimental setup is shown in Fig. 6A), and the weights of the animals are shown in fig. S9 (A, B, D, and E). In both cases, most of the mock-immunized mice were viremic by day 3 after infection, whereas no replicating virus was detected in the blood of CVB1–6–vaccinated mice (Fig. 6, B and G). Mock-immunized (buffer-treated) SOCS-1-tg mice challenged with either CVB1 (Fig. 6C) or CVB3 (Fig. 6H) developed hyperglycemia as shown by sharp increases in blood glucose values in 50 or 100% of animals, respectively, whereas the CVB1- or CVB3-challenged CVB1–6–vaccinated SOCS-1-tg mice were completely protected from virus-induced diabetes (Fig. 6, D and I, respectively). This is further illustrated by the diabetes incidence curves in Fig. 6 (E and J).
Fig. 6. CVB1–6 vaccine prevents CVB1- and CVB3-induced diabetes in SOCS-1-tg mice.
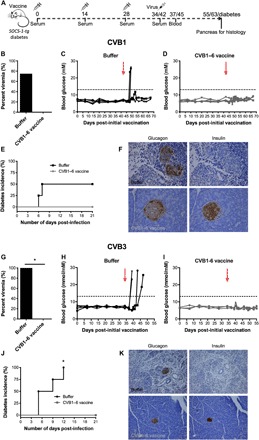
Female SOCS-1-tg mice (4 to 6 weeks old) were mock-immunized (vaccine buffer; n = 4 for each study; 150 μl, interscapularly; black bars/lines) or CVB1–6 vaccine–immunized (1 μg of each serotype; n = 4 for CVB1 and n = 3 for CVB3; i.s.; gray bars/lines) on days 0, 14, and 28 and then challenged with CVB1 (106 PFU per mouse, intraperitoneally; top) or CVB3 (106 PFU per mouse, intraperitoneally; bottom) on days 42 and 34, respectively, as indicated in the schematic shown in (A). (B and G) Percentage of mock and vaccinated mice with viremia on day 3 after infection. *P < 0.05 comparing the two groups by Fisher’s exact test. (C, D, H, and I) Blood glucose values in buffer-vaccinated (C and H; both n = 4) and CVB1–6–vaccinated (D and I; n = 4 and n = 3, respectively) mice. Arrows indicate CVB infection, and dotted lines show diabetes limit. (E and J) Diabetes incidence curves in mock- and CVB1–6–vaccinated mice infected with CVB1 (E) or CVB3 (J). *P < 0.05 comparing the two groups by Fisher’s exact test. (F and K) Representative immunohistochemical glucagon (left) and insulin (right) staining in pancreas from mock (top panels) and CVB1–6–vaccinated (bottom panels) mice infected with CVB1 (F) or CVB3 (K). Pancreas was collected at diabetes onset or on day 21 after infection. Magnification, ×40 (F) and ×16 (K). Scale bars, 20 μm.
The exocrine pancreas tissue was also destroyed in the buffer-treated CVB-challenged SOCS-1-tg mice, and this was accompanied by a loss of insulin positivity in the islets, as shown in the representative images in Fig. 6F (CVB1) and Fig. 6K (CVB3). Glucagon staining remained strong in all animals (CVB1, Fig. 6F; CVB3, Fig. 6K). Contrastingly, there was strong insulin and glucagon staining in the islets of the CVB1–6–immunized animals and the exocrine tissue remained healthy [representative images shown in Fig. 6F (CVB1-challenged) and Fig. 6K (CVB3-challenged)]. These data were supported by the nAB titers seen in the CVB1–6–immunized mice against all CVB serotypes (fig. S9, C and F). In summary, CVB1–6 immunization protected SOCS-1-tg mice against CVB1- and CVB3-induced diabetes.
DISCUSSION
Our studies document the excellent immunogenicity of a novel hexavalent CVB1–6 vaccine in several mouse strains and a nonhuman primate model. We also report that this vaccine has the ability, in relevant preclinical models, to protect against acute CVB infections, block CVB infection of the heart, and prevent CVB-induced pancreatitis and diabetes.
Historically, there have been very few attempts to create a polyvalent CVB vaccine. In 1994, See and Tilles (33) described the efficacy of a CVB1–6 vaccine in mice; however, their vaccine did not completely protect against challenge from different CVB serotypes, only one mouse strain was used, and protection against CVB-induced diseases was not explored. In our study, a strong nAB response to all CVB1–6 viruses was induced both in mice and in rhesus macaques. Also, no unwanted side effects were seen in any of the species used. Moreover, several different mouse strains immunized with the hexavalent vaccine were protected from infection with the tested serotypes (CVB1, CVB3, and CVB4) and the vaccine did not affect the expression of genes involved in innate immunity 2 weeks after the final vaccination, suggesting that protection is mediated via the nAB response rather than residual activation of the innate immune system. Collectively, this demonstrates the excellent safety and strong efficacy of this newly developed experimental vaccine.
Our CVB1–6 vaccine shares a number of similarities with the inactivated poliovirus vaccine (IPV) that is being used worldwide (alongside the oral poliovirus vaccine) in an attempt to eradicate poliomyelitis. For instance, both vaccines are produced in Vero cells and are inactivated by formalin treatment. According to the World Health Organization (WHO) Practical Guide to IPV (2014), the preferred delivery of IPV is via the intramuscular route, and two to three basic immunizations are required early in life with a number of boost vaccinations later on for maximum immunity (over 90% of cases immunized after 8 weeks of age have sufficient immunological responses against all three poliovirus serotypes present in the vaccine after two immunizations; WHO Immunological Basis for Immunization Series: Module 6 Poliomyelitis). The results obtained with the CVB1–6 vaccine in rhesus macaques mirror the IPV vaccine, where intramuscular immunization of the CVB1–6 vaccine (with a boost dose after 28 days) resulted in a strong serum immunity against all six CVB serotypes, which lasted up to 10 weeks after the prime dose, when monitoring of the animals finished. These data were further supported by the murine studies, where the vast majority of mice immunized with the CVB1–6 vaccine had high nAB titers at the terminal time point (which varied between studies). In the initial studies using C57BL/6J mice, the CVB4 neutralizing capacity was lost by day 84 after the prime vaccination. This was not, however, a consistent finding, and strong CVB4 nAB titers were seen in both NOD and Balb/c mice. In NOD mice, CVB4 nAB titers were equal to 1:256 up to a period of 70 to 113 days after the prime vaccination (fig. S3).
The coadministration of an adjuvant at the time of immunization was not necessary in either mice or rhesus macaques for strong serum CVB immunity after CVB1–6 vaccination. Based on the strategies used for IPV, the similarities between the two vaccines, and the aforementioned results we have obtained (particularly in the rhesus macaques), it would be reasonable to assume that an immunization strategy with the CVB1–6 vaccine that followed a similar approach to that used for IPV would induce sufficient serum immunity in humans to protect against CVB infections. Although other enterovirus vaccines are also known to provide long-term and effective protection against infection (9), this must of course be established in clinical trials before the ability of the vaccine to prevent CVB-induced diseases can be established. Together, the CVB1–6 vaccine shows excellent promise with regard to the induction of nABs and the duration of the response.
One of the most well-documented diseases caused by CVBs is myocarditis, which can lead to DCM. Approximately 4 to 20% of sudden cardiac deaths in young adults are attributed to myocarditis and it also causes around 9% of DCM cases (4, 5). Moreover, in children, more than 40% of DCMs are estimated to result from myocarditis (34). In the United States alone, it was estimated that cardiomyopathies place a burden on the U.S. health care system of between $4 and $10 billion annually (6). Although CVBs are not associated with every case, they are the most commonly identified cause of the disease in developed countries (4) and are thought to account for around 25% of viral myocarditis cases [approximately 70% of clinical myocarditis and/or pericarditis cases are associated with specific viral infections (5)]. Various studies describe murine models for CVB3 myocarditis, including the initial description by Woodruff and Woodruff (35), and young male Balb/c mice are a commonly used model (4, 35). Here, we show that our hexavalent vaccine prevents acute CVB3 infections and the dissemination of virus to the heart (and pancreas) in Balb/c mice, highlighting the clinical relevance of such a vaccine for use in preventing CVB-induced diseases such as myocarditis in humans.
T1D is another disease associated with enteroviruses, but there is no definitive proof confirming the causal involvement of these viruses, despite a large variety of existing data that implicates enteroviruses, and more specifically CVBs, in the pathogenesis of the disease [as reviewed in (9, 10, 12, 13)]. Here we have demonstrated in a proof-of-concept study that our hexavalent CVB vaccine is capable of protecting SOCS-1-tg mice from virus-induced diabetes that occurs due to the direct infection and destruction of the pancreatic β-cells. Vaccination of children with genetic profiles that increase their T1D disease susceptibility risk and the subsequent monitoring of disease incidence would provide empirical evidence for a possible role of these viruses in T1D. Similar randomized clinical trials, like TRIGR (36) that examined the effects of dietary interventions on T1D, provide an example of such an intervention study performed to explore the causality of T1D-associated exposures. If T1D cases dropped in number in immunized children, the causal role of CVBs in T1D would be proven and the vaccine would provide a viable preventative treatment. This would, however, require immunization early in life or potentially immunity gained via the immunization of expectant mothers.
Further research could look to increase the valency of the vaccine to include, for instance, enterovirus A serotypes such as EV71 and CVA16, which are known to cause HFMD (8), and EV-D68, an enterovirus D serotype, that can result in severe respiratory illness (37) and is associated with acute flaccid paralysis in many studies (38). To this end, a study from 2016 documented the success of a 25-valent rhinovirus vaccine in mice and a 50-valent rhinovirus vaccine in rhesus macaques (39), suggesting the feasibility of such a vaccine. In this study, the vaccines were adjuvanted, which was not required for a strong immune response with our hexavalent vaccine. As such, this could be used if necessary when the valency of the vaccine is increased to include further enteroviruses. Virus-like particles (18, 21) could also be considered as an option, especially in the case of viruses that propagate poorly in cell culture.
A limitation of the study is that we have performed studies to show that the vaccine protects against three, namely, CVB1, CVB3, and CVB4, but not all six CVB serotypes in relevant preclinical murine disease models. Another limitation of the study is that a single strain of each CVB1–6 serotype was used to produce the individual vaccine components of the CVB1–6 hexavalent vaccine. At any given time, different CVB1–6 strains will be in circulation, which poses the challenge of producing a vaccine with the ability to protect against as many different strains as possible. It will be important in the future to properly assess the extent of the CVB strain coverage provided by the vaccine when it is produced for and tested in humans. Further to this, it would be of relevance to perform studies that examine whether and how formalin inactivation alters the antigenicity of the virus particles, as this may modify the ability of the vaccine to elicit protective nABs in humans.
To summarize, we have described the development and preclinical testing of a novel experimental hexavalent CVB vaccine with a high capacity to induce serum nABs in a number of animal models. Moreover, we show in proof-of-concept studies that the vaccine prevents acute CVB infections and CVB infections that can lead to CVB-induced diseases such as myocarditis and potentially T1D. Currently, there are no commercially available preventative measures that provide protection against CVB-induced diseases such as myocarditis, meningitis, and pancreatitis or for elucidating the role of these viruses in diseases like T1D. The vaccine we describe here provides a viable option for tackling CVB infections and associated diseases. As such, it is a prime candidate for human clinical trial and efforts to develop this type of polyvalent CVB vaccine for human use have recently been initiated (14).
MATERIALS AND METHODS
Experimental design
The objectives of this study were to produce a new hexavalent CVB vaccine candidate and to test its immunogenicity in mice and nonhuman primates, and thereafter assess its ability to protect against acute infections and clinically relevant CVB-induced diseases. We hypothesized that the CVB1–6 vaccine would be immunogenic in different murine models and nonhuman primates. Moreover, we hypothesized that the vaccine would be able to prevent acute CVB infections and infections that cause CVB-induced diseases in clinically relevant murine models. The CVB1–5 strains used for the vaccine were derived from recent clinical isolates from Finland, and the CVB6 reference strain from the American Type Culture Collection (ATCC) was used, as recent clinical isolates were not as readily available for this serotype. Inactivated CVB serotypes were assessed for integrity and inactivation. All animal studies were performed in accordance with national and institutional guidelines. Three murine strains were used in the study (NOD, Balb/c, and C57BL/6J) along with the SOCS-1-tg mouse model (NOD background) and the nonhuman primate species rhesus macaques. Vaccination strategies were based on previous studies (16, 20). Mice and rhesus macaques were randomly assigned to treatment groups, and serum nAB responses were assessed by standard neutralization assay. Replicating virus in the blood, pancreas, and heart were measured by plaque assay. Pancreatitis was assessed by histological analysis. Virus protein and islet hormones were examined in the pancreas by immunohistochemical staining. A health monitoring system was followed, and if mice lost more than 15% of their highest body weight or scored four or more points after their health was assessed, they had to be removed for ethical reasons. Blood glucose levels were regularly monitored in NOD and SOCS-1-tg mice, and any animals with a single blood glucose reading of 18 mM or over, or two consecutive readings between 13 and 18 mM were deemed diabetic and removed. The number of animals included in each experimental group is indicated in the figure legends and for mice in table S1.
Animal husbandry and monitoring of animal health
C57BL/6J mice, NOD mice, SOCS-1-tg mice, and Balb/c mice, all obtained from in-house breeding, were housed in a specific pathogen–free environment in the PKL animal facility at the Karolinska University Hospital Huddinge, Sweden. All animal experiments performed were approved by the Institutional Animal Care and Use Committee and Linköpings Ethical Committee on Animal Experiments and conducted in accordance with the National Institutes of Health principles of laboratory animal care and national laws in Sweden. Extended health monitoring of mice was performed throughout the studies, and the following were monitored: general condition, movement and posture, piloerection, skin, weight, porphyrin staining, respiration, and appetite. Points were awarded for the severity of the symptoms, and if an animal reached four points or higher, they were euthanized. No mice were single-housed, and a maximum of five mice were kept in the same ventilated cage. Food and water were provided ad libitum.
SOCS-1-tg mice are generated on a NOD background, and their generation and breeding have been described previously (20, 27, 32). Briefly, SOCS-1-tg mice express the suppressor of cytokine signaling (Socs1) under the control of the insulin promoter. As such, the β-cells are unable to mount a successful antiviral IFN response, thereby leaving the animals susceptible to CVB infection and virus-induced diabetes. This occurs around 5 to 12 days after infection with CVB1, CVB3, or CVB4 (20, 27, 32).
Indian rhesus macaques were housed in the Astrid Fagraeus laboratory at the Karolinska Institute, Stockholm, Sweden according to guidelines provided by the Association for Assessment and Accreditation of Laboratory Animal Care. Procedures were performed according the Swedish Animal Welfare Agency’s provisions and general guidelines. The study was approved by the Stockholm Ethical Committee on Animal Experiments. Animal health was monitored during the study.
Virus production, purification, and characterization
Wild CVB1–5 field isolates from Finland [provided by Vactech Ltd.; (15)] and CVB6 ATCC strain Schmitt were propagated in Vero cells (Vero, ECACC no. 84113001, mycoplasma negative) as described previously (16). Viruses were recovered from the clarified Vero cell culture supernatants by 30% sucrose cushion pelleting (175,000g, 6 to 16 hours at 4°C). The pellets were resuspended in phosphate-buffered saline (PBS)–0.1% Tween 80 and further purified with gelatin affinity chromatography resin (GE Healthcare), and the virus was recovered from the flow-through. Last, samples were pelleted through discontinuous 30/50% sucrose cushion ultracentrifugation (285,000g, 14 hours at 4°C), and the pellet was dissolved in PBS–0.1% Tween 80.
Purified viruses were characterized for both their total protein content by Pierce BCA Protein Assay kit (Thermo Fisher Scientific) and their purity (virus protein content) by running the virus samples on mini-protean TGX stain-free precast gradient gels (4 to 20%) (Bio-Rad) and then visualization of the tryptophan-containing proteins in SDS-PAGE gels by ultraviolet-induced fluorescence. Subsequently, the CVB1–6 proteins were electroblotted on nitrocellulose membranes, and the viral capsid protein VP1 was detected using an in-house produced rat polyclonal antibody 3A6 (22). Hydrodynamic diameter of the purified viruses, Vero cell residual DNA content, and infective titers were measured as described previously (16).
Vaccine formulation and characterization
CVB1–6 viruses were inactivated in 0.01% (v/v) formalin for 5 days at 37°C, and inactivation was confirmed by the lack of infective virus (after culturing the inactivated viruses in GMK cells) (National Institute for Health and Welfare, Finland, mycoplasma negative) in TCID50 (median tissue culture infectious dose) end-point dilution assays [detection limit, 34 plaque-forming units (PFU)/ml] and in enterovirus-specific real-time PCR (detection limit, 0.00067 PFU/ml), as described earlier (16, 20). The vaccine was formulated in M199 medium (Gibco) containing 0.1% Tween 80 by mixing 1 μg of each CVB serotype per vaccine dose. Individual CVB1 and CVB4 monovalent vaccines were also formulated in M199 Medium containing 0.1% Tween 80 (1 μg of protein in each vaccine). The individual CVB1–6 vaccine components and the CVB1–6 vaccine were characterized by TEM. Butwar-coated copper grids were made hydrophilic via glow discharging using ESM/SC7620 Mini Sputter coater (Quorum). Inactivated virus samples were added to the grid and incubated for 15 s, after which excess virus was removed by blotting with Whatman 3MM paper. The inactivated viruses were then negatively stained through the addition of 1% phosphotungstic acid (in water, pH adjusted to 7.4) to the grid for 1 min, before blotting to remove the excess. Samples were dried overnight and imaged with a JEM-1400 (JEOL) TEM.
Murine immunizations
Male and female age-matched C57BL/6J mice, NOD mice, SOCS-1-tg mice, and Balb/c mice (4 to 9 weeks old) were assigned to treatment groups at random. Mice were vaccinated two to three times with nonadjuvanted hexavalent CVB1–6 vaccine containing 1 μg of each CVB1–6 serotype, CVB1 monovalent vaccine (1 μg), and CVB4 monovalent vaccine (1 μg) or mock-vaccinated with vaccine buffer alone (M199–0.1% Tween, v/v) in a total volume of 150 μl by interscapular injection. Before each vaccination and before virus challenge, a serum sample was collected from blood taken from the tail vein. See Figs. 1F, 2A, 3A, 5A, and 6A for the experimental setups and see table S1 for a summary of the mouse experiments (n numbers, immunization and challenge dates, ages at start, challenge CVB serotypes and concentrations, and respective figures).
Virus challenge
Mice were challenged with CVB3-Nancy, CVB4-E2 (both received from G. Frisk, University of Uppsala, Sweden), or CVB1-10796 (40) by intraperitoneal injection. Table S1 summarizes the CVB infection doses and time of infection in the different experiments. The viruses were diluted in serum-free RPMI to a final volume of 200 μl, and the doses required to yield a productive infection had been optimized previously. A blood sample was collected from the tail vein on day 3 after infection from all infected mice (1:1 ratio with 12 mmol EDTA in PBS). NOD mice were removed on day 3 after infection, and the pancreas was saved for histological analysis and measurements of replicating virus. SOCS-1-tg mice were kept until day 21 after infection or until diabetes development and the pancreas were saved for histological analysis. Balb/c mice were euthanized on day 5 after infection, blood was collected on days 3 and 5 after infection, and the pancreas and heart were saved for histological analysis and for measuring replicating virus.
Rhesus macaque immunizations
Indian rhesus macaques (4 years of age, male) were allocated to two groups and received hexavalent CVB1–6 vaccine alone (n = 2) or hexavalent CVB1–6 vaccine + alum adjuvant (n = 3). Alhydrogel (alum) adjuvant (2%) was purchased from InvivoGen (Toulouse, France). The vaccines contained 5 μg of each CVB serotype (30 μg of protein in total) in 150-μl vaccine buffer. Before immunization, the vaccine was mixed with PBS (nonadjuvant group: 150 μl of CVB1–6 vaccine +350 μl of PBS) or PBS and adjuvant (adjuvant group: 150 μl of CVB1–6 vaccine + 50 μl of alum adjuvant + 300 μl of PBS; 0.2% final concentration of alum adjuvant). Alum formulation was designed according to the manufacturers’ instructions and a previously published study (39). Animals were immunized with a final volume of 500 μl by intramuscular injection in the arm on days 0 and 28. Peripheral venous blood was collected at selected time points before or after vaccination and processed within 1 hour as previously described (30). Liver function tests using whole-blood samples were performed on days 0, 1, and 14 after the first immunization by Adlego Biomedical. Figure 4A shows the experimental setup in rhesus macaques.
Monitoring of blood glucose and diabetes development
Blood glucose was monitored from blood obtained from the tail vein of mice or from blood taken from rhesus macaques, with a Bayer Contour XT blood glucose meter. Mice with a blood glucose reading of 18 mM or over or two measurements on consecutive days between 13 and 18 mM were deemed diabetic and euthanized. Rhesus macaques were fasted before anesthetization, and a fasting blood glucose reading of more than 5.6 mM was the cutoff for overt diabetes in these animals (41).
nAB assays
Titers of CVB1–6 nABs were measured by standard virus plaque reduction assay in GMK cells (15, 20, 42). Briefly, serial 1:4 dilutions of sera were incubated with equal volumes of suspensions containing 50 PFU of the respective homotypic CVB serotypes that were used to produce the vaccine for 1 hour at 37°C and then overnight at room temperature. The virus-serum mixture was added to GMK cells grown to 95% confluency in a 12-well plate for 1 hour at 37°C, then removed and replaced with a semisolid medium (minimum essential medium supplemented with 0.67% carboxymethylcellulose), and incubated for 2 days at 37°C. Cells were then fixed and stained with crystal violet, and plaque numbers were quantified. An 80% or more reduction in plaque number compared to untreated virus suspensions was deemed positive. The detection limit of the assay was a fourfold dilution (1:4), and the positivity of serum samples was set to ≥1:16 dilution.
RNA isolation, reverse transcription, and real-time quantitative PCR
Pancreas and spleen were submerged in RNAlater immediately after harvesting and then stored at −20°C. For RNA isolation, approximately 30 mg of tissue was transferred to RLT buffer and homogenized with a TissueLyser II system (5-mm metal beads; Qiagen) according to the kit instructions. RNA was isolated using an RNeasy kit (Qiagen) with deoxyribonuclease (DNase) I treatment and quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific). RNA was reverse-transcribed using a High-Capacity cDNA Reverse Transcription kit (Applied Biosystems). Real-time quantitative PCR for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), IFN-α, IFN-β, EIF2AK2 (PKR), OAS1, and inducible NOS (NOS2) was performed with TaqMan Gene Expression Assays (Applied Biosystems) using the QuantStudio 5 System (Thermo Fisher Scientific). mRNA expression for each gene was normalized to GAPDH using the 2−(ΔCt) method.
Blood and pancreas virus titration
Pancreas tissue was homogenized with sterile ceramic beads (6×, ø2.8 mm; Qiagen) and the PowerLyzer 24 Bench Top Homogenizer (MBio Laboratories, Qiagen). Blood samples or pancreas homogenates were serially diluted 1:10, and lytic virus was measured by standard plaque assay in GMK cells (NOD and SOCS-1-tg mice, assays performed at the University of Tampere) or assessed by plaque titration in HeLa cells (Balb/c mice, assays performed at the Karolinska Institutet) as described in (27). Viral titers are expressed as PFU/ml of blood or PFU/mg tissue. In samples where no plaques were detected, the samples were assumed to be negative and given a value of 0. There is a detection limit of 3.3 PFU/mg for virus detection from tissue homogenates.
Histological analysis and immunohistochemistry
FFPE murine pancreas tissue cut into 5-μm sections was used for histological and immunohistochemical analysis. VP1 was detected with a biotinylated anti-VP1 antibody (5D8/1, Dako; biotinylated by Capra Bioscience, Ängelholm, Sweden; final concentration, 0.07 μg/ml) and tris-EDTA (pH 6) antigen retrieval. Biotin was then detected using the Vectastain ABC Elite Biotin System (Vector Laboratories) and stained using DAB (Vector Laboratories; both according to the manufacturer’s instructions). As previously described, the antibody was validated in mouse pancreas sections (20, 27, 32). The islet hormones insulin and glucagon were detected with anti-insulin (1:10,000; A0564, Dako) and anti-glucagon (1:12,000; AB92518, Abcam) antibodies. Secondary antibodies alone were used as negative controls in the hormone staining (insulin: goat anti-guinea pig, Vector Laboratories; glucagon: goat anti-rabbit, Dako) or 2% normal goat serum the case of the VP1 staining. The images were acquired on a Leica DM4000B light microscope using QWIN software.
Statistical analysis
Statistical analyses were performed using Prism 5 software (GraphPad, La Jolla, CA, USA). Percentage body weight change of vaccinated and buffer-treated mice at the same time points was compared by Mann-Whitney U test, as were the weights, temperature, and blood glucose of rhesus macaques (compared to day 0). nAB titers were assessed by two-way analysis of variance (ANOVA) with Bonferroni correction (CVB1–6 vaccine) or by one-way ANOVA with Bonferroni correction (CVB1 and CVB4 vaccines). The percentage of animals with viremia on day 3 after infection, the percentage of animals with VP1 positivity in the pancreas, and diabetes incidence were compared by one-tailed χ2 Fisher’s exact test. *P < 0.05, **P < 0.01, ***P < 0.001. nAB titers, viremia, and virus titers in the pancreas and heart are shown in log10.
Supplementary Material
Acknowledgments
We would like to acknowledge the animal staff at PKL and the Astrid Fagraeus facilities at the Karolinska Institutet for their assistance with the animal studies, E. Henriksson for her help with processing histological samples. L. Radler for laboratory management and M. Ekström for administrative support. A. Laaksonen, M. Jokela, U. Kiiskinen, N. Kähkönen, A. Karjalainen, M. Kekäläinen, M. Ovaskainen, E. Jalonen, S. Oikarinen, J. Ilomäki, and T. Kuusela from Faculty of Medicine and Health Technology (Tampere University, Finland) are acknowledged for their assistance with the vaccine production and characterization and the nAB analyses. A. Thorpe is acknowledged for help with linguistics. Funding: We would like to acknowledge the financial support from the Business Finland [formerly TEKES (THERDIAB project, diary no. 1843/31/2014)]; the Academy of Finland (grant nos. 288671 and 1309455); the Swedish Child Diabetes Foundation; and Karolinska Institutet including the Strategic Research Program in Diabetes. Author contributions: Conceptualization: M.F.-T., V.P.H., O.H.L., H.H., K.L., V.M.S., and M.M.H.; formal analysis: V.M.S., M.M.H., A.B.S.-K., V.P.H., and M.F.-T.; investigation: V.M.S., M.M.H., A.B.S.-K., A.L., M.A.M., I.M.D.L., V.M., V.P.H., and M.F.-T.; resources: M.F.-T., V.P.H., H.H., and K.L.; data curation: V.M.S. and M.F.-T.; writing (original draft): V.M.S.; writing (review and editing): V.M.S., M.M.H., O.H.L., A.B.S.-K., A.L., M.A.M., I.M.D.L., V.M., K.L., H.H., V.P.H., and M.F.-T.; visualization: V.M.S. and M.M.H.; supervision; M.F.-T., O.H.L., V.P.H., and K.L.; funding acquisition: M.F.-T., V.P.H., O.H.L., H.H., M.M.H., and V.M.S. Competing interests: H.H. is inventor on a patent related to this work filed by Vactech Oy (no. FI20075861A, filed on 30 November 2007). H.H., M.F.-T., O.H.L., and M.M.H. (former Pulkki) are inventors on a second patent related to this work filed by Vactech Oy (no. EP2768527B1, filed on 18 October 2012). H.H. is inventor on a third patent related to this work filed by Vactech Oy (no. FI20075861A, filed on 17 March 2000). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The virus strains used to produce the vaccines and the vaccines used in this study can be provided by Vactech Oy, at the sole discretion of Vactech’s exclusive licensee, pending scientific and operational review, and a completed material transfer agreement. Requests for the virus strains or vaccines should be submitted to: info@vactech.fi.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/19/eaaz2433/DC1
REFERENCES AND NOTES
- 1.Abedi G. R., Watson J. T., Nix W. A., Oberste M. S., Gerber S. I., Enterovirus and parechovirus surveillance—United States, 2014–2016. MMWR Morb. Mortal. Wkly. Rep. 67, 515–518 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tavakoli N. P., Wang H., Nattanmai S., Dupuis M., Fusco H., Hull R., Detection and typing of enteroviruses from CSF specimens from patients diagnosed with meningitis/encephalitis. J. Clin. Virol. 43, 207–211 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Hong J., Kang B., Yeo S., Jee Y., Park J.-H., Pathogenesis of coxsackievirus B2 in mice: Characterization of clinical isolates of the coxsackievirus B2 from patients with myocarditis and aseptic meningitis in Korea. J. Vet. Sci. 18, 457–464 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fairweather D., Stafford K. A., Sung Y. K., Update on coxsackievirus B3 myocarditis. Curr. Opin. Rheumatol. 24, 401–407 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pollack A., Kontorovich A. R., Fuster V., Dec G. W., Viral myocarditis—Diagnosis, treatment options, and current controversies. Nat. Rev. Cardiol. 12, 670–680 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Towbin J. A., Lowe A. M., Colan S. D., Sleeper L. A., Orav E. J., Clunie S., Messere J., Cox G. F., Lurie P. R., Hsu D., Canter C., Wilkinson J. D., Lipshultz S. E., Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 296, 1867–1876 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Huber S., Ramsingh A. I., Coxsackievirus-induced pancreatitis. Viral Immunol. 17, 358–369 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Mao Q. Y., Wang Y. P., Bian L. L., Xu M., Liang Z. L., EV-A71 vaccine licensure: A first step for multivalent enterovirus vaccine to control HFMD and other severe diseases. Emerg. Microbes Infect. 5, e75 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hyöty H., Leon F., Knip M., Developing a vaccine for type 1 diabetes by targeting coxsackievirus B. Expert Rev. Vaccines 17, 1071–1083 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Lugo D., Krogstad P., Enteroviruses in the early 21st century: New manifestations and challenges. Curr. Opin. Pediatr. 28, 107–113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dennert R., Crijns H. J., Heymans S., Acute viral myocarditis. Eur. Heart J. 29, 2073–2082 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richardson S. J., Morgan N. G., Enteroviral infections in the pathogenesis of type 1 diabetes: New insights for therapeutic intervention. Curr. Opin. Pharmacol. 43, 11–19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Principi N., Berioli M. G., Bianchini S., Esposito S., Type 1 diabetes and viral infections: What is the relationship? J. Clin. Virol. 96, 26–31 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Dunne J. L., Richardson S. J., Atkinson M. A., Craig M. E., Dahl-Jørgensen K., Flodström-Tullberg M., Hyöty H., Insel R. A., Lernmark Å., Lloyd R. E., Morgan N. G., Pugliese A., Rationale for enteroviral vaccination and antiviral therapies in human type 1 diabetes. Diabetologia 62, 744–753 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laitinen O. H., Honkanen H., Pakkanen O., Oikarinen S., Hankaniemi M. M., Huhtala H., Ruokoranta T., Lecouturier V., André P., Harju R., Virtanen S. M., Lehtonen J., Almond J. W., Simell T., Simell O., Ilonen J., Veijola R., Knip M., Hyöty H., Coxsackievirus B1 is associated with induction of β-Cell autoimmunity that portends type 1 diabetes. Diabetes 63, 446–455 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Hankaniemi M. M., Laitinen O. H., Stone V. M., Sioofy-Khojine A., Määttä J. A. E., Larsson P. G., Marjomäki V., Hyöty H., Flodström-Tullberg M., Hytönen V. P., Optimized production and purification of Coxsackievirus B1 vaccine and its preclinical evaluation in a mouse model. Vaccine 35, 3718–3725 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Hunziker I. P., Harkins S., Feuer R., Cornell C. T., Whitton J. L., Generation and analysis of an RNA vaccine that protects against coxsackievirus B3 challenge. Virology 330, 196–208 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Koho T., Koivunen M. R. L., Oikarinen S., Kummola L., Mäkinen S., Mähönen A. J., Sioofy-Khojine A., Marjomäki V., Kazmertsuk A., Junttila I., Kulomaa M. S., Hyöty H., Hytönen V. P., Laitinen O. H., Coxsackievirus B3 VLPs purified by ion exchange chromatography elicit strong immune responses in mice. Antiviral Res. 104, 93–101 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Larsson P. G., Lakshmikanth T., Laitinen O. H., Utorova R., Jacobson S., Oikarinen M., Domsgen E., Koivunen M. R. L., Chaux P., Devard N., Lecouturier V., Almond J., Knip M., Hyöty H., Flodström-Tullberg M., A preclinical study on the efficacy and safety of a new vaccine against Coxsackievirus B1 reveals no risk for accelerated diabetes development in mouse models. Diabetologia 58, 346–354 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Stone V. M., Hankaniemi M. M., Svedin E., Sioofy-Khojine A., Oikarinen S., Hyöty H., Laitinen O. H., Hytönen V. P., Flodström-Tullberg M., A Coxsackievirus B vaccine protects against virus-induced diabetes in an experimental mouse model of type 1 diabetes. Diabetologia 61, 476–481 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L., Parham N. J., Zhang F., Aasa-Chapman M., Gould E. A., Zhang H., Vaccination with coxsackievirus B3 virus-like particles elicits humoral immune response and protects mice against myocarditis. Vaccine 30, 2301–2308 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Saarinen N. V. V., Laiho J. E., Richardson S. J., Zeissler M., Stone V. M., Marjomäki V., Kantoluoto T., Horwitz M. S., Sioofy-Khojine A., Honkimaa A., Hankaniemi M. M., Flodström-Tullberg M., Hyöty H., Hytönen V. P., Laitinen O. H., A novel rat CVB1-VP1 monoclonal antibody 3A6 detects a broad range of enteroviruses. Sci. Rep. 8, 33 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buschard K., What causes type 1 diabetes? Lessons from animal models. APMIS Suppl. 119, 1–19 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Lee Y. J., Lee J. Y., Jang Y. H., Seo S.-U., Chang J., Seong B. L., Non-specific effect of vaccines: Immediate protection against respiratory syncytial virus infection by a live attenuated influenza vaccine. Front. Microbiol. 9, 83 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flodström M., Horwitz M. S., Maday A., Balakrishna D., Rodriguez E., Sarvetnick N., A critical role for inducible nitric oxide synthase in host survival following coxsackievirus B4 infection. Virology 281, 205–215 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Flodström-Tullberg M., Hultcrantz M., Stotland A., Maday A., Tsai D., Fine C., Williams B., Silverman R., Sarvetnick N., RNase L and double-stranded RNA-dependent protein kinase exert complementary roles in islet cell defense during coxsackievirus infection. J. Immunol. 174, 1171–1177 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Flodström M., Maday A., Balakrishna D., Cleary M. M., Yoshimura A., Sarvetnick N., Target cell defense prevents the development of diabetes after viral infection. Nat. Immunol. 3, 373–382 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Rhesus Macaque Genome Sequencing and Analysis Consortium, Gibbs R. A., Rogers J., Katze M. G., Bumgarner R., Weinstock G. M., Mardis E. R., Remington K. A., Strausberg R. L., Venter J. C., Wilson R. K., Batzer M. A., Bustamante C. D., Eichler E. E., Hahn M. W., Hardison R. C., Makova K. D., Miller W., Milosavljevic A., Palermo R. E., Siepel A., Sikela J. M., Attaway T., Bell S., Bernard K. E., Buhay C. J., Chandrabose M. N., Dao M., Davis C., Delehaunty K. D., Ding Y., Dinh H. H., Dugan-Rocha S., Fulton L. A., Gabisi R. A., Garner T. T., Godfrey J., Hawes A. C., Hernandez J., Hines S., Holder M., Hume J., Jhangiani S. N., Joshi V., Khan Z. M., Kirkness E. F., Cree A., Fowler R. G., Lee S., Lewis L. R., Li Z., Liu Y.-s., Moore S. M., Muzny D., Nazareth L. V., Ngo D. N., Okwuonu G. O., Pai G., Parker D., Paul H. A., Pfannkoch C., Pohl C. S., Rogers Y. H., Ruiz S. J., Sabo A., Santibanez J., Schneider B. W., Smith S. M., Sodergren E., Svatek A. F., Utterback T. R., Vattathil S., Warren W., White C. S., Chinwalla A. T., Feng Y., Halpern A. L., Hillier L. W., Huang X., Minx P., Nelson J. O., Pepin K. H., Qin X., Sutton G. G., Venter E., Walenz B. P., Wallis J. W., Worley K. C., Yang S. P., Jones S. M., Marra M. A., Rocchi M., Schein J. E., Baertsch R., Clarke L., Csürös M., Glasscock J., Harris R. A., Havlak P., Jackson A. R., Jiang H., Liu Y., Messina D. N., Shen Y., Song H. X., Wylie T., Zhang L., Birney E., Han K., Konkel M. K., Lee J., Smit A. F., Ullmer B., Wang H., Xing J., Burhans R., Cheng Z., Karro J. E., Ma J., Raney B., She X., Cox M. J., Demuth J. P., Dumas L. J., Han S. G., Hopkins J., Karimpour-Fard A., Kim Y. H., Pollack J. R., Vinar T., Addo-Quaye C., Degenhardt J., Denby A., Hubisz M. J., Indap A., Kosiol C., Lahn B. T., Lawson H. A., Marklein A., Nielsen R., Vallender E. J., Clark A. G., Ferguson B., Hernandez R. D., Hirani K., Kehrer-Sawatzki H., Kolb J., Patil S., Pu L. L., Ren Y., Smith D. G., Wheeler D. A., Schenck I., Ball E. V., Chen R., Cooper D. N., Giardine B., Hsu F., Kent W. J., Lesk A., Nelson D. L., O'brien W. E., Prüfer K., Stenson P. D., Wallace J. C., Ke H., Liu X. M., Wang P., Xiang A. P., Yang F., Barber G. P., Haussler D., Karolchik D., Kern A. D., Kuhn R. M., Smith K. E., Zwieg A. S., Evolutionary and biomedical insights from the rhesus macaque genome. Science 316, 222–234 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Chen Y., Qin S., Ding Y., Wei L., Zhang J., Li H., Bu H., Lu Y., Cheng J., Reference values of clinical chemistry and hematology parameters in rhesus monkeys (Macaca mulatta). Xenotransplantation 16, 496–501 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Thompson E. A., Ols S., Miura K., Rausch K., Narum D. L., Spångberg M., Juraska M., Wille-Reece U., Weiner A., Howard R. F., Long C. A., Duffy P. E., Johnston L., O’Neil C. P., Loré K., TLR-adjuvanted nanoparticle vaccines differentially influence the quality and longevity of responses to malaria antigen Pfs25. JCI Insight 3, 1206982 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qi X. M., Xiong S. D., Intein-mediated backbone cyclization of VP1 protein enhanced protection of CVB3-induced viral myocarditis. Sci. Rep. 7, 41485 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larsson P. G., Lakshmikanth T., Svedin E., King C., Flodström-Tullberg M., Previous maternal infection protects offspring from enterovirus infection and prevents experimental diabetes development in mice. Diabetologia 56, 867–874 (2013). [DOI] [PubMed] [Google Scholar]
- 33.See D. M., Tilles J. G., Efficacy of a polyvalent inactivated-virus vaccine in protecting mice from infection with clinical strains of group B coxsackieviruses. Scand. J. Infect. Dis. 26, 739–747 (1994). [DOI] [PubMed] [Google Scholar]
- 34.Nandi D., Rossano J. W., Epidemiology and cost of heart failure in children. Cardiol. Young 25, 1460–1468 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woodruff J. F., Woodruff J. J., Involvement of T lymphocytes in the pathogenesis of coxsackie virus B3 heart disease. J. Immunol. 113, 1726–1734 (1974). [PubMed] [Google Scholar]
- 36.Writing Group for the TRIGR Study Group, Knip M., Åkerblom H. K., Al Taji E., Becker D., Bruining J., Castano L., Danne T., de Beaufort C., Dosch H. M., Dupre J., Fraser W. D., Howard N., Ilonen J., Konrad D., Kordonouri O., Krischer J. P., Lawson M. L., Ludvigsson J., Madacsy L., Mahon J. L., Ormisson A., Palmer J. P., Pozzilli P., Savilahti E., Serrano-Rios M., Songini M., Taback S., Vaarala O., White N. H., Virtanen S. M., Wasikowa R., Effect of hydrolyzed infant formula vs conventional formula on risk of type 1 diabetes: The TRIGR randomized clinical trial. JAMA 319, 38–48 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiang Z. C., Wang J. W., Enterovirus D68 and human respiratory infections. Semin. Respir. Crit. Care Med. 37, 578–585 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suresh S., Forgie S., Robinson J., Non-polio enterovirusdetection with acute flaccid paralysis: A systematic review. J. Med. Virol. 90, 3–7 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Lee S., Nguyen M. T., Currier M. G., Jenkins J. B., Strobert E. A., Kajon A. E., Madan-Lala R., Bochkov Y. A., Gern J. E., Roy K., Lu X., Erdman D. D., Spearman P., Moore M. L., A polyvalent inactivated rhinovirus vaccine is broadly immunogenic in rhesus macaques. Nat. Commun. 7, 12838 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hämäläinen S., Nurminen N., Ahlfors H., Oikarinen S., Sioofy-Khojine A. B., Frisk G., Oberste M. S., Lahesmaa R., Pesu M., Hyöty H., Coxsackievirus B1 reveals strain specific differences in plasmacytoid dendritic cell mediated immunogenicity. J. Med. Virol. 86, 1412–1420 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Wagner J. E., Kavanagh K., Ward G. M., Auerbach B. J., Harwood H. J. Jr., Kaplan J. R., Old world nonhuman primate models of type 2 diabetes mellitus. ILAR J. 47, 259–271 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Sioofy-Khojine A. B., Lehtonen J., Nurminen N., Laitinen O. H., Oikarinen S., Huhtala H., Pakkanen O., Ruokoranta T., Hankaniemi M. M., Toppari J., Vähä-Mäkilä M., Ilonen J., Veijola R., Knip M., Hyöty H., Coxsackievirus B1 infections are associated with the initiation of insulin-driven autoimmunity that progresses to type 1 diabetes. Diabetologia 61, 1193–1202 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/19/eaaz2433/DC1