

Gene editing of FOXP3 ensures regulated expression and restored function in T cells, supporting clinical applicability.
Abstract
The prototypical genetic autoimmune disease is immune dysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome, a severe pediatric disease with limited treatment options. IPEX syndrome is caused by mutations in the forkhead box protein 3 (FOXP3) gene, which plays a critical role in immune regulation. As a monogenic disease, IPEX is an ideal candidate for a therapeutic approach in which autologous hematopoietic stem and progenitor (HSPC) cells or T cells are gene edited ex vivo and reinfused. Here, we describe a CRISPR-based gene correction permitting regulated expression of FOXP3 protein. We demonstrate that gene editing preserves HSPC differentiation potential, and that edited regulatory and effector T cells maintain their in vitro phenotype and function. Additionally, we show that this strategy is suitable for IPEX patient cells with diverse mutations. These results demonstrate the feasibility of gene correction, which will be instrumental for the development of therapeutic approaches for other genetic autoimmune diseases.
INTRODUCTION
Primary immunodeficiencies comprise a group of genetic immune diseases, which typically present with recurrent infections, but may instead manifest with predominant autoimmunity (1, 2). Over 350 monogenic immune diseases have been described to date, and this number has been rapidly increasing with technological advances in DNA sequencing and expanding accessibility of genetic screening (1). The prototype of genetic autoimmunity is immune dysregulation polyendocrinopathy enteropathy and X-linked (IPEX) syndrome, which is a severe X-linked disease with early onset (2, 3). The most frequent autoimmune manifestations of IPEX syndrome include type 1 diabetes, eczema, and life-threatening enteropathy (4). Other common autoimmune manifestations include cytopenia, autoimmune hepatitis, and thyroiditis. IPEX syndrome is classified as a Tregopathy, a class of diseases that selectively affect the function of regulatory T cells (Tregs) and, in the case of IPEX syndrome, CD4+CD25highFOXP3+ Tregs (5, 6). In patients with IPEX, nonfunctional Tregs are produced that are unable to prevent the development of autoimmunity or allergy because they lack the ability to suppress the function and proliferation of effector T (Teff) cells (7). In recent years, the pathophysiology of IPEX syndrome has been dissected, but few therapeutic advances have been made, and limited treatment options exist (2). Currently, patients with IPEX are treated with pharmacological immunosuppression, which has only partial efficacy in the acute phase of the disease and cannot prevent long-term disease progression (2, 4). Furthermore, administration of immunosuppressive drugs carries the risk of severe side effects associated with toxicity and susceptibility to infections. The only curative treatment available for IPEX is allogeneic hematopoietic stem cell transplantation. However, many patients do not find a suitable donor or suffer from transplant-related complications (4).
IPEX syndrome is caused by mutations in the forkhead box protein 3 (FOXP3) gene, and over 70 unique mutations throughout the FOXP3 locus have been identified (4, 8). FOXP3 is a master transcription factor required for the function of Tregs, which up-regulates Treg-associated markers, such as CD25 and CTLA-4, and represses proinflammatory cytokine production (9). While Tregs rely on constitutive FOXP3 expression, Teff cells transiently express FOXP3 following T cell receptor (TCR) activation (7). This cell type–specific regulation is a result of a complex network of promoter and enhancer elements (9, 10). The high and persistent FOXP3 expression in Tregs is due to epigenetic marks established during T cell development, including the Treg-specific demethylated region (TSDR) (10). It has previously been shown that Teff cells require transient FOXP3 expression for intrinsic regulation of proliferation, cytokine production, and TCR signaling (11, 12). According to the current understanding of IPEX, impairment of both Teff and Treg function underlies IPEX syndrome pathology (2, 9). In human Teff cells and Tregs, FOXP3 pre-mRNA is alternatively spliced, and the two predominant spliced isoforms are the full-length (FOXP3FL) isoform and a shorter version that lacks exon 2 (FOXP3dE2) (13, 14). The FOXP3FL and FOXP3dE2 isoforms each represent roughly half of the FOXP3 expressed; however, the proportion is skewed in different cell activation states and in a number of inflammatory diseases (14). Causative IPEX mutations in exon 2 have been described, with a subset of patients presenting with milder clinical phenotypes (15, 16). Because these mutations spare the FOXP3dE2 isoform, it has been suggested that FOXP3dE2 can partially compensate for FOXP3FL loss but that FOXP3FL is required for complete Treg and Teff cell function and prevention of IPEX syndrome (16).
Because IPEX syndrome is a monogenic immune disease caused by mutations in FOXP3, gene therapy could be a useful approach to treat the disease. We previously developed a FOXP3 gene delivery protocol for ex vivo generation of genetically engineered Tregs that uses lentiviral vector (LV)–mediated delivery of copy of the complementary DNA (cDNA) of the full-length isoform of FOXP3 (17). Because this vector expresses FOXP3 under a constitutive promoter, EF1α, it is able to convert IPEX patient conventional CD4+ T cells into potent Treg-like suppressor cells. We are currently optimizing the LV-FOXP3 system for use as a T cell–based therapy for patients with IPEX. However, this approach does not address other cell types, such as Teff cells, that also contribute to the IPEX pathology. An additional limitation of this approach is that it cannot be used on long-term repopulating hematopoietic stem and progenitor cells (HSPCs) because of the adverse effects of FOXP3 overexpression on stem cell proliferation and differentiation (18). For the development of a successful FOXP3 gene therapy using HSPCs, it is necessary to achieve constitutive expression of FOXP3 in the Treg compartment to restore suppressive function without having FOXP3 overexpression perturb the proliferation and function of HSPCs or Teff cells. To maintain this cell type–specific expression, an ideal approach would precisely deliver FOXP3 to its endogenous gene locus and allow regulation of FOXP3 in its own genomic context.
To deliver the FOXP3 cDNA in a site-specific manner while preserving endogenous regulation, we propose gene editing with the clustered regularly interspaced short palindromic repeat (CRISPR)–CRISPR-associated protein 9 (Cas9) system. In general, this one-size-fits-all cDNA insertion approach is designed to benefit all or the majority of patients, given that the causative mutations are located downstream of the insertion site (19, 20). We hypothesized that this site-specific approach would permit gene delivery to patient-derived HSPCs for an autologous transplant. This gene correction approach could provide a more complete and long-term treatment for IPEX syndrome and circumvent the need for matched hematopoietic stem cell donors. Here, we combine the CRISPR system with a DNA repair homology donor to insert the FOXP3 cDNA into the endogenous locus via homology-directed repair (HDR). We report that this gene editing platform can accurately and specifically target FOXP3 in HSPCs and that edited HSPCs maintain normal differentiation potential in vitro and in vivo in immunodeficient mice. We demonstrate that both Tregs and Teff cells retain their key biologic properties when the cDNA is inserted into the endogenous locus, including normal proliferation of Teff cells. We show that the FOXP3 gene can be corrected in cells from patients with IPEX with diverse mutations, which demonstrates the feasibility of a CRISPR-based FOXP3 gene correction approach for IPEX syndrome.
RESULTS
Efficient and precise editing of FOXP3 locus in human HSPCs and T cells using CRIS
To achieve gene editing at the FOXP3 locus, we designed a CRISPR system targeting the FOXP3 gene downstream of the translation start codon in exon 1 (E1) and a corresponding HDR donor containing FOXP3 cDNA (Fig. 1A). The donor construct was designed to insert a codon-diverged FOXP3 cDNA and restore wild-type (WT) FOXP3 protein expression in patient cells with diverse and scattered FOXP3 mutations. The gene replacement donor template was also designed to knock-in a marker gene, the truncated nerve growth factor receptor (tNGFR), which is used clinically as a surface marker for selection and tracking of genetically engineered cells. We placed tNGFR under the control of a constitutive promoter such that it would be expressed independently of FOXP3. Polyadenylation (pA) signals were included in the construct to ensure termination and inactivation of the downstream endogenous gene elements. In addition to the full-length FOXP3 cDNA construct (FOXP3FL), we also developed two similar experimental constructs including a FOXP3 cDNA of a naturally occurring alternatively spliced isoform of FOXP3 lacking exon 2 (FOXP3dE2) and a FOXP3 knockout (FOXP3KO) construct that disrupts the FOXP3 gene by inserting only the tNGFR maker gene flanked by pA signals (fig. S1A).
Fig. 1. The FOXP3 locus is precisely targeted using the CRISPR system in primary HSPCs and T cells.
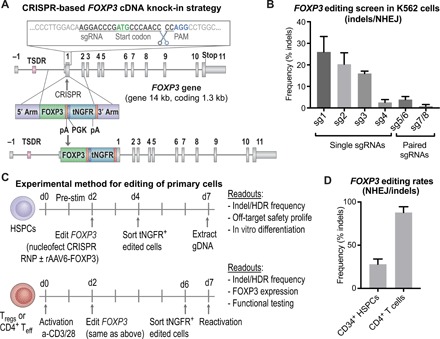
(A) Schematic representation of CRISPR-based editing of the FOXP3 gene showing the CRISPR cut site in first coding exon, E1 (exons depicted by gray boxes separated by lines representing introns; the first coding exon, E1, is preceded by the noncoding exon E-1 and the enhancer with TSDR). A zoomed-in view of the sgRNA binding site relative to the start codon, PAM site, and cleavage site is shown. Homology donor depicted below with arms of homology, codon divergent FOXP3 cDNA, BGH polyadenylation (pA) signal included to terminate the FOXP3 transcript, truncated NGFR (tNGFR) marker gene under the Phosphoglycerate Kinase (PGK) promoter to drive marker expression independent of FOXP3 expression, and a second pA. (B) Screening of sgRNAs targeting the first coding exon of the FOXP3 gene. Plasmids encoding WT Cas9 or nickase variant of Cas9 (paired sgRNAs) and FOXP3 sgRNAs nucleofected into K562 cell lines. CRISPR efficiency measured by TIDE analysis to detect insertion deletion (indel) mutations created by nonhomologous end joining (NHEJ)–mediated DNA repair. (C) Experimental method for editing of HSPCs and T cells with functional readouts listed. (D) CRISPR cutting efficiency in CD34+ HSPCs and CD4+ T cells quantified by TIDE analysis for the detection of indel mutations created by the NHEJ repair pathway.
We screened FOXP3 CRISPR single-guide RNAs (sgRNAs) for on-target cutting activity in immortalized K562 cells (Fig. 1B, fig. S1B, and table S1). The sgRNAs 1 and 2 triggered the highest on-target activity (26 ± 7% and 20 ± 5%, respectively, mean ± SD, n = 4) (Fig. 1B), as indicated by the frequency of insertion deletion (indel) mutations detected by TIDE analysis (21). Of the sgRNAs screened, sgRNA 2 was selected because of a combination of its on-target activity, safe predicted off-target profile, location in the coding sequence of the gene, and proximity to the start codon. Cutting efficiency of sgRNA 2 was 28 ± 6% in human CD34+ HSPCs and 88 ± 7% in CD4+ T cells (mean ± SD, n = 4), thus validating that the CRISPR system efficiently targets FOXP3 in the primary cells of interest (Fig. 1, C and D).
Using FOXP3 sgRNA 2, we edited the allele via the HDR-mediated pathway by transducing HSPCs with a FOXP3 DNA repair donor delivered as a recombinant adeno-associated virus of serotype 6 (rAAV6) (22, 23). Similarly, we gene edited CD4+ T cells to enable functional testing on T cells. We detected successful HDR as evidenced by an in-out polymerase chain reaction (PCR) amplification of genomic DNA (Fig. 2A and fig. S2A). Precise insertion and the FOXP3 cDNA sequence were confirmed by sequencing analysis (table S1). Rates of HDR-mediated editing detected by tNGFR expression were 14 ± 7% in primary CD4+CD25high Tregs (n = 14), 17 ± 5% in the immortalized MT-2 Treg-like cell line (n = 7) (24), 24 ± 10% in CD4+CD25−/low Teff cells (n = 21), and 29 ± 8% in HSPCs (n = 27) (Fig. 2B). Testing of the alternative constructs, FOXP3dE2 and FOXP3KO, in HSPCs revealed average HDR rates of 26 ± 9% and 23 ± 5%, respectively (fig. S2B). To confirm that tNGFR expression was an accurate measure of editing rates, we performed editing on HSPCs from three cell donors and tested editing in each donor in parallel by both tNGFR expression and a quantitative in-out PCR using the Digital Droplet PCR (ddPCR) system (19). We found concordance between the two methods of editing detection, 25 ± 2% editing for tNGFR flow cytometry and 29 ± 5% for ddPCR (n = 3), confirming the accuracy of detection (fig. S2C). Mock-treated cells were nucleofected and transduced with rAAV-donor templates in the absence of CRISPR and showed low levels of background episomal tNGFR expression (Fig. 2C and fig. S2D). Enrichment of edited cells using the tNGFR maker resulted in a population consistently above 90% purity (Fig. 2C), confirming that tNGFR could be used to isolate a pure population of edited cells.
Fig. 2. CRISPR combined with a rAAV6 homology donor enables precise HDR-mediated FOXP3 cDNA transgene insertion into the endogenous locus.
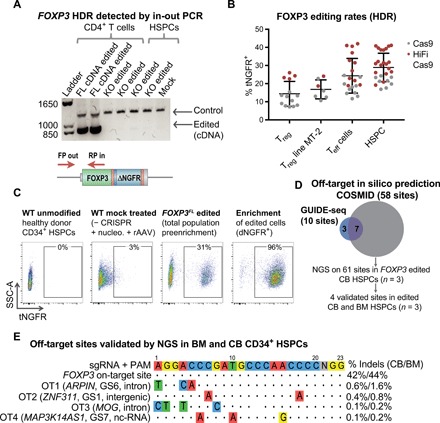
(A) Editing observed at the DNA level by an in-out PCR strategy that uses a primer inside the inserted divergent cDNA construct and a second primer outside of the 5′ arm of homology. Control band represents unmodified region in the FOXP3 gene as a positive control for the presence of genomic DNA. PCR using in-out primers resulted in band only present in samples in which the cDNA was inserted (FL cDNA) and not in FOXP3 knockout (KO) or mock-treated samples. After the ladder, the first four lanes represent CD4+ T cells, and the last three lanes represent HSPCs. (B) Rates of HDR-mediated FOXP3 editing detected by flow cytometry for tNGFR marker gene expression in primary human CD4+CD25++ Tregs, MT-2 Treg cell line, primary CD4+CD25− Teff cells, and CD34+ HSPCs. Editing rates with standard Sp Cas9 (gray) and high-fidelity (HiFi) Cas9 (red) are shown for comparison. Dots represent cells from individual donors. (C) Representative flow cytometry plots showing tNGFR marker gene expression in edited cord blood–derived CD34+ HSPCs. Negative control: Mock-treated cells nucleofected with phosphate-buffered saline in place of CRISPR and transduced with rAAV6-FOXP3 donor. Edited cells enriched using tNGFR selection and purity are shown by flow cytometry. SSC-A, side scatter area. (D) Venn diagram showing overlap in predicted off-target sites identified by COSMID in silico prediction and GUIDE-seq DSB capture. All predicted sites were tested by NGS in edited CD34+ HSPCs derived from cord blood (CB) and validated by NGS in edited bone marrow (BM)–derived HSPCs. (E) The four off-target (OT) sites validated by NGS shown with highlighted mismatches in off-target site versus FOXP3 sgRNA target sequence, closest gene name, gene region, and average frequency of indel mutations detected by NGS (mean, n = 3).
Specific FOXP3 gene editing in HSPC using CRISPR
We investigated off-target activity using three complementary methods: bioinformatic prediction, a double-strand break (DSB) capture assay, and next-generation sequencing (NGS). Initially, 58 potential off-target sites were predicted by bioinformatic in silico prediction using the CRISPR Search with Mismatches, Insertions and/or Deletions (COSMID) tool (25), of which 96% were in noncoding regions of the genome (fig. S2E and tables S2 and S3, A to E). Ten off-target sites were identified using DSB capture by genome-wide, unbiased identification of DSBs enabled by sequencing (GUIDE)–seq GUIDE-seq (26) in the U2OS cell line, of which seven were also predicted by in silico analysis (Fig. 2D and fig. S2F). The 61 sites predicted by the combination of COSMID and GUIDE-seq were then evaluated in FOXP3 edited HSPCs by NGS. Four sites were validated by NGS, three of which were identified by all three methods. The four sites identified as off-targets were ranked as 1, 3, 4, and 14 by the COSMID algorithm. When excluding a mismatch at the 5′ distal nucleotide, none of the validated sites had more than three mismatches. As compared with the NGS-identified indel rates in the FOXP3 gene of 42 and 44% in edited cord blood– and bone marrow–derived HSPCs, respectively, all off-target sites had less than 2% targeting (Fig. 2E). None of the validated sites were in coding regions of genes, and on the basis of gene annotation, none of the sites would have a clear impact on hematopoiesis or cell cycle regulation in hematopoietic cells. The results from this off-target analysis suggest that the FOXP3 CRISPR system is a relatively specific platform for gene editing in hematopoietic cells.
Persistent FOXP3 protein expression and preserved Tregs phenotype and function in FOXP3 edited Tregs
We next evaluated our FOXP3 cDNA insertion strategy in Tregs, the major cell type that expresses FOXP3 and is implicated in IPEX syndrome. We used the immortalized MT-2 Treg cell line and primary peripheral blood–derived Tregs separated by CD25 enrichment beads. The purity of the primary Tregs was confirmed by flow cytometry and epigenetic TSDR demethylation (fig. S3, A and B). We initially tested FOXP3 expression in MT-2 cells and observed that FOXP3FL edited cells expressed FOXP3 protein, but at a lower level compared with unmodified MT-2 cells (Fig. 3A and fig. S4A). In contrast, editing with the FOXP3KO construct led to complete loss of FOXP3 protein expression as predicted (fig. S4B). Insertion of the second isoform of FOXP3 (FOXP3dE2) led to approximately 50% FOXP3 expression relative to WT cells (fig. S5A), revealing that delivery of either isoform cDNA individually can support a similar level of FOXP3 expression in Tregs. To improve FOXP3 protein expression, we designed two additional constructs: a codon-optimized FOXP3 cDNA construct (FOXP3FLco) and the codon-optimized FOXP3 construct followed by a woodchuck hepatitis virus posttranscriptional regulatory element (FOXP3co + WPRE or FOXP3FLcoW), added to increase mRNA stability and protein translation (Fig. 3B) (20). The FOXP3co construct yielded similar FOXP3 protein levels to the FOXP3FL construct, whereas the FOXP3FLcoW construct resulted in moderately increased FOXP3 protein expression levels (Fig. 3C).
Fig. 3. FOXP3 edited Tregs express FOXP3 protein and display characteristic in vitro phenotype and function.
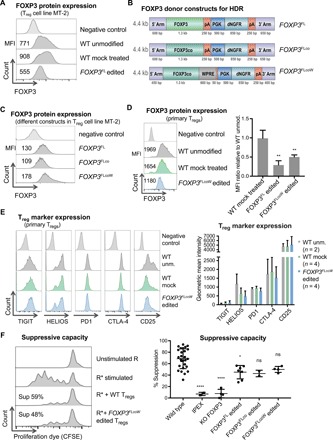
(A) Quantification of FOXP3 protein expression in the MT-2 Treg cell line by flow cytometry, showing median fluorescence intensity (MFI) values for one representative experiment out of three performed. (B) FOXP3 homology donor constructs designed to improve FOXP3 protein expression, including further codon optimization (FOXP3FLco) and addition of a WPRE element (FOXP3FLcoW). (C) Flow cytometry for FOXP3 in MT-2 cells comparing the different donor constructs. (D) FOXP3 expression in primary Tregs by flow cytometry and quantification represented as ratio of MFI in treated cells versus WT unmodified Tregs [mean ± SD, one-way analysis of variance (ANOVA), Tukey’s multiple comparisons]. Significance comparing FOXP3FL (n = 2, **P < 0.01) or FOXP3FLcoW (n = 6, **P < 0.01) to WT mock treated (n = 2). (E) Expression of signature Treg marker proteins on primary Tregs by flow cytometry and corresponding quantification (mean ± SD, n = 2 to 4). (F) Suppression assay showing unstimulated responders (R), stimulated responders (R*) alone or in coculture with Tregs, and calculated percent suppression. Responders stained with proliferation dye, carboxyfluorescein diacetate succinimidyl ester (CFSE). Average suppressive potential over several independent experiments is quantified (right). Significance comparing the WT Tregs with following Tregs: IPEX, FOXP3 KO, FOXP3FL edited, FOXP3FLco edited, and FOXP3FLcoW edited (mean ± SD, one-way ANOVA, Tukey’s multiple comparisons test, *P < 0.05; ****P < 0.0001; ns, not significant).
We next evaluated FOXP3 expression in human peripheral blood–derived CD4+CD25high Tregs. The pattern of FOXP3 expression from the different donor constructs in MT-2 cells was reflected in peripheral blood–derived Tregs. Tregs edited with the FOXP3FL cDNA expressed a lower level of FOXP3 protein, and a modest increase in FOXP3 protein expression from the FOXP3FLcoW construct was observed (Fig. 3D). While the various cDNA donors were not able to fully reach WT levels of FOXP3 protein expression in the MT-2 Treg cell line or primary Tregs, we hypothesized that this level of FOXP3 protein expression might still be sufficient to restore Treg function.
Immunophenotypic analysis for Treg cell markers, including TIGIT, HELIOS, PD1, CTLA-4, and CD25, revealed that all markers were expressed in FOXP3 edited Tregs (Fig. 3E). The average intensity of HELIOS was decreased after editing, but there was no statistically significant difference in HELIOS expression between edited and mock-treated samples. Overall, the expression levels of all Treg makers were similar in WT and edited Tregs with no statistically significant differences observed (Fig. 3E). These results suggest that the CRISPR edited Tregs maintain the expression of key proteins characteristic of WT Tregs.
We next tested the function of gene edited Tregs and their hallmark ability to suppress the proliferation of activated Teff cells in a coculture suppression assay. WT Tregs from healthy donors displayed suppression capacity at an average of 69 ± 17% (mean ± SD) (Fig. 3F). In contrast, IPEX Tregs and FOXP3 knockout Tregs displayed diminished suppression at rates of 7 ± 3% and 8 ± 7%, respectively (Fig. 3F). Tregs that were edited with the various FOXP3 cDNA knock-in constructs all displayed suppressive function, although not fully reaching the level of WT Tregs [FOXP3FL 45 ± 12%, n = 7, *P < 0.05 versus WT; FOXP3co 43 ± 16%, n = 2, P = ns (not significant); FOXP3FLcoW 50 ± 5%, n = 4, P = ns] (Fig. 3F). In addition, we tested the suppressive function of FOXP3dE2 Tregs and found them to perform comparably to FOXP3FL and FOXP3FLcoW edited Tregs (fig. S5, B and C). As a negative control, FOXP3FLcoW edited Teff cells that were cultured and edited in parallel were tested and found to lack suppressive function (fig. S5, C and D). Given the ability to suppress Teff proliferation and the relatively higher FOXP3 expression, we selected the FOXP3FLcoW construct for subsequent functional testing. Overall, FOXP3 cDNA knock-in Tregs displayed suppressive function that overlapped with lower normal range of suppressive function observed in WT Tregs from different donors, suggesting that editing might be sufficient to restore suppressive capacity to nonfunctional Tregs.
Physiologically regulated FOXP3 expression and preserved function in FOXP3 edited Teff cells
Because Teff cells transiently express FOXP3 upon TCR activation, we monitored FOXP3 protein expression in FOXP3 edited Teff cells by flow cytometry over a 2-week time course after activation. In nonactivated cells, a low level of background FOXP3 expression was observed, likely due to the pre-editing activation and culturing (Fig. 4A). Upon TCR-mediated reactivation, FOXP3 expression in both edited cells and controls was induced and nearly doubled by day 3 before gradually returning to baseline (Fig. 4A). Overall, the regulation of FOXP3 expression in Teff cells closely mirrored that in WT controls without statistically significant differences, confirming that endogenous regulation of expression was preserved.
Fig. 4. FOXP3 editing of Teff cells preserves physiological regulation of FOXP3 expression and in vitro function.
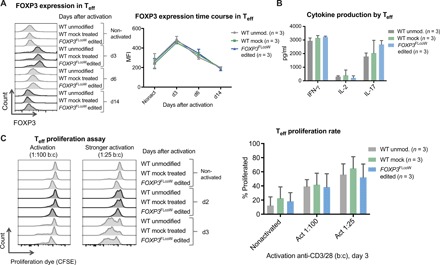
(A) Flow cytometry time course showing kinetics of FOXP3 expression in nonactivated Teff cells and activated Teff cells on subsequent days after activation [day 3 (d3), d6, and d14], comparing WT unmodified, WT mock-treated, and FOXP3FLcoW edited Teff cells. FOXP3 expression quantified in Teff cells over time course of activation, showing average MFI (mean ± SD, n = 3). (B) Cytokine production in WT and FOXP3 gene edited Teff cells determined by enzyme-linked immunosorbent assay (ELISA). Supernatants collected at 24 hours (IL-2) and 48 hours (IFN-γ and IL-17) after activation with anti-CD3/28 (mean ± SD, n = 3). (C) Teff cell proliferation in response to activation measured by the proliferation assay. Flow cytometry plots of carboxyfluorescein diacetate succinimidyl ester (CFSE) dye–stained Teff cells with progressive dilution of dye as proliferation progresses from nonactivated to day 2 and day 3 after activation with anti-CD3/28 Dynabeads are shown. Comparison of proliferation rates in response to activation with a bead:cell ratio of 1:100 and 1:25. Quantification of average proliferative response of Teff cells from proliferation assay at day 3 is shown to the right, comparing different doses of activation beads (mean ± SD, n = 3).
In addition to FOXP3 expression, we evaluated the cytokine production profile and proliferation potential of gene edited Teff cells and controls to demonstrate that the intrinsic regulatory effect of FOXP3 was maintained. High levels of interferon-γ (IFN-γ) and low levels of interleukin-2 (IL-2) were produced in both WT and FOXP3 edited Teff cells (Fig. 4B). IL-17 production was higher in FOXP3 edited Teff cells, but not statistically different as compared with the WT control (Fig. 4B). In addition, all edited and nonedited Teff cells displayed similar kinetics of activation-induced proliferation over time that was dependent on the strength of activation (Fig. 4C and fig. S6). Together, these results indicate that FOXP3 edited Teff cells maintain physiological regulation of FOXP3 expression, cytokine production, and proliferation.
Restored functional FOXP3 expression to IPEX patient Tregs and Teff cells after CRISPR-based FOXP3 editing
We obtained cells from six patients with IPEX, including two sets of brothers with identical pathologic mutations (mutation locations are depicted in Fig. 5A; see table S4 for patient information). Mutations ranged from point mutations to complete abrogation of gene expression. Gene editing of IPEX T cells using the FOXP3 CRISPR system resulted in precise HDR-mediated editing as revealed by in-out PCR of the edited locus (Fig. 5B). In addition, HDR rates determined by tNGFR expression revealed that IPEX cells could all be targeted with comparable efficiency as healthy donor cells [editing rates for IPEX patients (pt.) 24 and 64 are shown and were reproduced in pt. 37, 65, 77, and 78; Fig. 5C].
Fig. 5. CRISPR-based editing enables FOXP3 gene correction in IPEX patient cells.
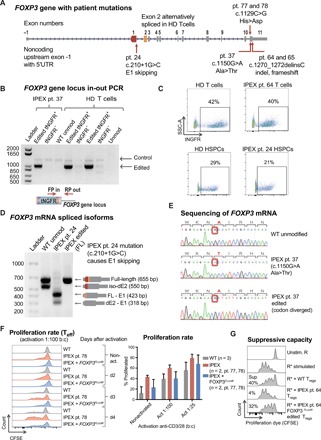
(A) Schematic of FOXP3 gene highlighting mutations of patients involved in this study. (B) Editing of IPEX and HD T cells observed at the DNA level by in-out PCR strategy. Forward primer is in the tNGFR cassette, and the reverse primer is in the FOXP3 gene locus outside the 3′ arm of homology. Positive and negative fractions after tNGFR enrichment (+/−) analyzed by PCR. (C) Flow cytometry plots of tNGFR staining. (D) Expression of FOXP3 mRNA demonstrated by RT-PCR in activated IPEX and HD T cells (schematic mRNA isoforms are shown to the right of corresponding PCR product bands). After ladder, first lane represents WT cells with two naturally occurring alternatively spliced isoforms, FL and dE2. The second lane shows aberrant skipping of E1 in IPEX pt24 mRNA that results in truncated transcripts. The third lane shows edited IPEX cells with restoration of the FL mRNA. (E) Sanger sequencing showing c.1150G>A mutation in the mRNA of pt37, resulting in an Ala>Thr change, and CRISPR-based insertion of divergent FOXP3 cDNA restoring correct amino acid sequence. (F) Proliferation of Teff cells in response to activation measured by the proliferation assay, comparing HD WT cells and IPEX pt78 cells. Quantification of average proliferation response of Teff cells from proliferation assay at day 3. (G) Functional testing of gene edited IPEX Tregs using the in vitro suppression assay. Flow cytometry plots of CFSE-stained Teff responder cells (R) cultured with or without Tregs. Calculated suppressive potential shows diminished suppressive function of IPEX pt. 64 Tregs, which was partially restored by FOXP3 gene editing.
Gene editing restored expression of FOXP3 in gene edited IPEX T cells harboring different mutations. We edited IPEX pt. 24 cells, which harbor a c.210+1G>C mutation that disrupts the exon-intron border of the first coding exon and leads to aberrant E1 skipping (27). FOXP3 mRNA isoforms in this patient are shorter due to the missing E1 as shown by reverse transcription (RT)–PCR (Fig. 5D). Gene editing of FOXP3 in IPEX pt. 24 cells with the full-length FOXP3 cDNA construct restored expression of the full-length FOXP3 mRNA isoform (Fig. 5D). We also analyzed mRNA expression after gene editing of T cells from IPEX pt. 37, carrying a c.1150G>A mutation that leads to an alanine-to-threonine amino acid change. RT-PCR and Sanger sequencing revealed that the edited pt. 37 cells expressed FOXP3 mRNA from the inserted FOXP3 codon-diverged cDNA construct, as confirmed by a restoration of a codon encoding for the correct alanine residue (Fig. 5E). In addition, the entire mRNA was sequenced to ensure that precise HDR-mediated FOXP3 cDNA insertion led to the predicted full-length mRNA encoding WT FOXP3 protein. Together, these results provide evidence that the FOXP3 CRISPR system can be used to restore expression of FOXP3 in T cells from patients with IPEX with diverse mutations.
We next evaluated proliferative responses of FOXP3 gene–edited Teff cells from IPEX pt. 77 and 78. Compared with WT healthy donor Teff cells, the Teff cells from both pt. 77 and 78 proliferated at an accelerated rate (Fig. 5F), consistent with previous findings that FOXP3 loss leads to hyperproliferation (12). FOXP3 gene editing was able to normalize proliferation rates closer to the level of healthy donor Teff cells, showing that restoration of FOXP3 expression reestablished regulation of proliferation in IPEX Teff cells (Fig. 5F). To test for restoration of Treg-suppressive activity in IPEX cells, we gene edited Tregs of IPEX pt. 64. The WT Tregs from healthy donor displayed suppressive function (40%) in coculture conditions with Teff responder cells, whereas IPEX pt. 64 Tregs showed diminished suppressive function (4%) (Fig. 5G). Gene editing of IPEX pt. 64 Tregs with the FOXP3FLcoW construct was able to increase suppressive function (32%) near the levels typical of healthy donor–edited cells (Fig. 5G), and these results were confirmed on two independent blood draws. Together, the results from functional analyses of edited IPEX Treg and Teff cells show that FOXP3 correction using the CRISPR system restores physiological regulation of WT FOXP3 expression and has the potential to provide a functional benefit to IPEX patient cells.
We then evaluated the differentiation potential of FOXP3 edited HSPCs using in vitro and in vivo approaches to confirm safety and feasibility. We performed in vitro colony-forming unit (CFU) assays to test the short-term differentiation of FOXP3 edited HSPCs into myeloid and erythroid lineages. As compared with WT controls, the FOXP3 edited HSPCs differentiated into hematopoietic progenitor colonies at similar rates with no statistically significant differences (Fig. 6A). We then tested the in vivo engraftment and multilineage reconstitution of edited HSPCs in a humanized mouse (hu-mouse) model. FOXP3FLcoW edited and control HSPCs (WT unmodified and WT mock) were injected into the liver of 3- to 4-day-old neonatal immunodeficient mice, and engraftment was monitored over a 14-week time course (Fig. 6B). The NSG-SGM3 strain of mice was selected for engraftment studies because of their expression of several human cytokines and their reported higher proportion of FOXP3+ Tregs relative to standard NSG mice (28). Before injection, edited HSPCs were phenotyped by flow cytometry for purity (%CD34+) and markers of hematopoietic progenitor subsets (fig. S7A). Gene edited and control HSPCs from three cord blood donors were injected, without prior enrichment for tNGFR, into three corresponding litters of mice. A total of 27 mice were injected, including 10 FOXP3FLcoW edited, 9 WT unmodified, and 8 WT mock conditions. The overall survival of the mice over the course of the study was comparable among conditions (fig. S7B). Human cell engraftment, determined by flow cytometric analysis of hCD45+ expression, steadily increased over time in the peripheral blood of the mice and was found to be comparable among conditions with no statistically significant differences (Fig. 6C).
Fig. 6. FOXP3 edited HSPCs undergo multilineage hematopoietic differentiation and engraftment in vitro and in vivo.
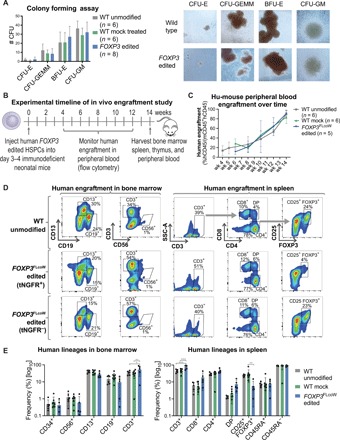
(A) Differentiation potential of edited HSPCs tested by the in vitro CFU assay. Four resulting hematopoietic progenitor colony types: CFU-E (mature erythroid progenitors), CFU-GEMM (granulocyte, erythrocyte, macrophage, and megakaryocyte), BFU-E (primitive erythroid progenitors), and CFU-GM (granulocyte and macrophage progenitors). Representative images of colonies from the CFU assay, showing similar morphology (×10 magnification). (B) Experimental timeline of hu-mouse study using NSG-SGM3 mice. (C) Human engraftment kinetics in the peripheral blood of hu-mice at corresponding weeks after injection. Engraftment was measured by flow cytometry for hCD45 marker on human cells, and frequency was quantified relative to the total of human (hCD45+) and mouse (mCD45+) cells (mean ± SD). (D) Representative flow cytometry plots of engrafted human hematopoietic subsets in the bone marrow (left) and spleen (right) of hu-mice at 14 weeks after injection. Populations gated out of human cells (hCD45+). FOXP3 edited samples were divided into tNGFR+ and tNGFR− gates for comparability. (E) Quantification of human hematopoietic lineages by flow cytometry with each symbol representing a single mouse (mean ± SD). In spleen, the CD8+, CD4+, and CD4+CD8+ double-positive (DP) populations were gated out of CD3+ T cells. The CD25+FOXP3+, naïve CD45RA+, and memory CD45RA− populations were gated out of CD4+ single-positive T cell subset (*P < 0.5, **P < 0.01, ***P < 0.001).
Multilineage human engraftment was observed in the peripheral blood, bone marrow, and spleen in all experimental mice (Fig. 6, D and E). The tNGFR+ edited cells persisted in vivo (fig. S7, C to E), although three mice were excluded for having less than 5% tNGFR+ cells. Analysis of editing rates comparing the proportion of alleles edited by HDR (cDNA insertion) and nonhomologous end joining (NHEJ) (indels) relative to WT alleles in the bone marrow of the mice revealed similar proportions to that of the edited HSPCs before injection with no apparent abnormal expansion of edited cells (fig. S7E). The hematopoietic lineages were analyzed by flow cytometry, and among the edited conditions, the cells were subgated into tNGFR+ and tNGFR− fractions for comparison (Fig. 6, D and E). We observed the presence of CD34+ HSPCs, CD56+ natural killer cells, CD13+ myeloid cells, CD19+ B cells, and CD3+ T cells in the bone marrow of FOXP3FLcoW edited (tNGFR+ and tNFGR− fraction) and control mice (Fig. 6, D and E). T cell subsets were further evaluated in the spleen, with CD3+, CD4+ single-positive, CD8+ single-positive, CD4+CD8+ double-positive, CD4+CD25+FOXP3+ Tregs, memory CD4+CD45RA−, and naïve CD4+CD45RA+ T cells all present in both FOXP3FLcoW edited (tNGFR+ and tNFGR− fraction) and control mice (Fig. 6, D and E). Some differences in the frequencies of cell subsets were observed between conditions, such as higher proportion of CD3+ cells and a lower proportion of CD25+FOXP3+ cells in the tNGFR+ fraction. However, no overt changes to immune reconstitution were observed, and each hematopoietic cell lineage was represented among the different experimental conditions (Fig. 6E).
To test the function of Tregs and Teff cells derived from in vivo differentiation of edited HSPCs, we sorted CD3+CD4+ T cells from hu-mouse spleens and separated them into CD25high (Treg) and CD25low (Teff) fractions. The Treg and Teff fractions from the edited conditions were further sorted into tNGFR+ and tNGFR− fractions. The purity of FOXP3+ Tregs within the CD25high-sorted fraction was tested by epigenetic TSDR demethylation analysis and found to be above 78% for the WT unmodified, mock, and FOXP3FLcoW edited tNGFR− and undefined for tNGFR+ fraction due to low cell count. Teff cells derived from the hu-mouse were found to proliferate at comparable rates to control human peripheral blood–derived Teff cells (fig. S7F). Among hu-mouse–derived Teff cells, all conditions proliferated at similar rates in a dose-dependent manner relative to the strength of activation (fig. S7F). In addition, the CD25high-sorted cells were found to be suppressive upon coculture with responder Teff cells, and the FOXP3FLcoW edited Tregs displayed comparable suppressive capacity to WT controls (fig. S7G). Overall, these results demonstrate that FOXP3 edited HSPCs can engraft in vivo and undergo multilineage differentiation, including the production of functional Tregs and Teff cells.
DISCUSSION
Monogenic immune and blood disorders are prime candidates for gene therapy approaches that target autologous HSPCs. Traditionally, viral vectors, such as LVs, that integrate gene cDNAs semirandomly in the genome have been used for gene therapy. While LVs have proven to facilitate efficient gene delivery and are now being widely tested in clinical trials, they are limited in their ability to preserve locus-specific endogenous regulation of gene expression. For this reason, site-specific gene editing has been emerging as a therapeutic approach for the delivery of genes that are regulated by a complex network of noncoding elements. Here, we developed a gene editing approach for IPEX syndrome, which is a prototypical genetic autoimmune disease with unmet medical need. We harnessed site-specific gene editing by the CRISPR system to insert a FOXP3 cDNA into the endogenous gene locus, preserving spatiotemporal regulation by endogenous regulatory elements.
Using the CRISPR methodology, we efficiently and specifically edited FOXP3 in HSPCs and in the two main cell types that express FOXP3, Tregs and Teff cells. FOXP3 edited HSPCs did not express FOXP3 protein, whereas edited Teff cells transiently expressed FOXP3 after activation, and edited Tregs persistently expressed FOXP3. This cell type–specific expression is consistent with endogenous expression patterns. Edited HSPCs maintained their differentiation potential and displayed a safe off-target profile. Transient FOXP3 expression in Teff cells from healthy donors and patients with IPEX allowed for maintenance of normal proliferative potential and cytokine production capacity. Persistent expression of FOXP3 in Tregs preserved their phenotype and ability to suppress T cell function. The level of FOXP3 expression in edited Teff cells closely mirrored that of WT cells, while edited Tregs displayed partial FOXP3 protein expression. This cell type–distinct result could be attributed to the fact that Tregs physiologically express a much higher level of FOXP3 than activated Teff cells. Although the level of FOXP3 expression in edited Tregs led to average suppressive function less than that of WT Tregs from normal donors, the suppressive rates of edited cells were still within the lower range of nonedited normal donor function. The range in suppressive function among healthy donor Tregs highlights the variability of in vitro regulatory function among individuals. Restoration of a similar level of FOXP3 expression in IPEX patient Tregs was sufficient to reestablish suppressive activity in coculture with Teff cell responders.
By using the CRISPR system, we avoided the delivery of FOXP3 under a constitutive promoter and subsequent FOXP3 overexpression, as in our previously developed LV-EF1α-FOXP3 vector platform (17). The CRISPR gene editing approach prevents the detrimental effects on HSPC proliferation and differentiation caused by FOXP3 constitutive overexpression (18). LV-mediated gene delivery of FOXP3 with its promoter and enhancer elements has been recently reported in mouse models (29). While on the basis of a strong rationale, the capacity to restore spatiotemporally regulated expression of FOXP3 in human IPEX cells using this vector remains to be determined. In general, limitations of LV-mediated delivery include the effect of vector copy number on transgene expression and the potential for position effect variegation. For example, the therapeutic gene could be silenced if positioned into a nonexpressed locus or overexpressed if integrated into a site proximal to a strong promoter or enhancer. In addition, the semirandom insertion of the transgene promoter and enhancer elements into the genome may raise concerns of inadvertently activating proto-oncogenes or genes that would be detrimental to hematopoiesis. These concerns underscore the benefits of site-specific gene editing as a more precise method for therapeutic gene delivery.
In this study, the CRISPR system enabled efficient HDR-mediated editing of FOXP3 in HSPCs, the cell type used for autologous HSPC transplantation and long-term reconstitution of the immune system. In HSPCs, the overall targeted integration frequency was 29 ± 8% when using tNGFR marker to identify targeted cells. In prior studies, we observed that targeting frequency increased to 50% or greater when the HDR donor was shortened by removal of the tNGFR marker gene (19). While the tNGFR marker facilitates enrichment of edited cells for functional testing, it may not always be necessary in clinical settings. The marker gene could potentially be removed to improve editing rates if it becomes apparent that selection and tracking of tNGFR-expressing cells are not essential.
While we envision HSPCs being used for autologous transplant, the CRISPR system could be applied to adoptive cell therapy using differentiated T cells or T cell precursor cells. In addition, the efficient CRISPR-based editing of T cells facilitated functional testing of edited Tregs and Teff cells. The T cell functional assays performed in this study used the codon-optimized divergent FOXP3 cDNA sequence followed by a WPRE, which was expected to provide optimal protein expression. To further improve FOXP3 expression and optimize the construct, several avenues may be explored. For example, different codon-diverged sequences could be evaluated in Tregs, and modifications to the CG content of the cDNA could be examined. It is also possible that expression from the endogenous FOXP3 gene and translation of FOXP3 protein are normally enhanced by endogenous intron-exon splice sites and 3′ untranslated region (3′UTR) elements. We did not include endogenous FOXP3 introns or 3′UTR elements in the cDNA knock-in constructs because of the potential for causing premature crossover events during homologous recombination. Thus, additional future modifications to the construct aimed at improving expression could include incorporation of short exogenous intronic sequences or 3′UTRs from genes highly expressed in Tregs.
On the basis of the knowledge that two major spliced isoforms of FOXP3 are expressed in human T cells, FOXP3FL and FOXP3dE2 (13, 14), we speculate that both isoforms could be required to reach WT levels of FOXP3 expression in Tregs. We observed that CRISPR-based knock-in of either of the two isoforms leads to partial FOXP3 expression and Treg cell–suppressive function. It has been previously shown that overexpression of either isoform is sufficient to confer Treg function to conventional CD4+ T cells (13, 30, 31). However, these results could have been confounded by the artificially high level of forced FOXP3 overexpression and the background genomic expression of both isoforms. By comparison, CRISPR-mediated knock-in uses endogenous FOXP3 promoters and enhancers and allows each isoform to be expressed individually under physiological conditions. Although expression of each isoform was only ~50 to 60% of WT levels, each isoform alone at that level was able to support suppressor function within the lower range of healthy donor cells. Nonetheless, the ability to deliver individual isoforms into the endogenous locus allows the FOXP3 CRISPR system to be used as a tool to investigate the FOXP3FL and FOXP3dE2 isoforms individually. For therapeutic purposes, it may be necessary to use a gene delivery platform that preserves expression of both major FOXP3 isoforms. In support of this hypothesis, we have previously shown that LV-mediated codelivery of both FOXP3FL and FOXP3dE2 cDNAs and subsequent simultaneous overexpression of both isoforms in CD4+ T cells were able to increase FOXP3 protein expression and confer more potent Treg properties than either of the isoforms individually (13). Therefore, it is possible that the two isoforms work in concert to regulate a network of Treg-related genes that drive Treg cell function, and the CRISPR system could be used to delineate the distinct and complementary roles of the FOXP3 isoforms.
The FOXP3 CRISPR system was similarly used as a tool to study the effects of complete FOXP3KO. We observed that FOXP3 loss in Tregs ablated suppressive function. The tNGFR marker was used to isolate a pure population of FOXP3KO cells for functional analysis. The ability to purify FOXP3-null cells makes this approach superior to an incomplete knockdown or heterogeneous indel-mediated knockout of FOXP3 as previously used to investigate the effects of FOXP3 loss (11). Thus, the FOXP3 CRISPR system allows for the creation of IPEX-like cell models in more readily available healthy donor cells. Similarly, the CRISPR system could be used to knock-in FOXP3 cDNAs harboring patient-specific mutations, which would provide insight into the molecular mechanisms underlying the heterogeneity of clinical presentation in IPEX syndrome.
While FOXP3 loss in both FOXP3KO and IPEX Tregs abrogated suppressive function, FOXP3 cDNA knock-in Tregs displayed in vitro function and maintained characteristic Treg phenotypic markers. Whether restoring IPEX Tregs to the lower range of normal function could be sufficient to provide a stable therapeutic benefit in patients with IPEX remains to be addressed. For example, in the context of certain disease settings such as hemophilia, it has been shown that as little as 5 to 10% gene function can provide therapeutic benefit (32–35). Moreover, transplanted patients with IPEX with low overall chimerism of donor cells can still undergo tremendous disease regression, especially due to the fact that the Treg compartment shows a selective advantage toward donor cells (36–38). Similarly, carrier mothers display a selective advantage in the Treg compartment such that the mutated FOXP3 allele is predominantly located in the inactivated X chromosome, while the WT allele of FOXP3 is expressed from the active X chromosome in the Treg compartment (38). These observations highlight the selective advantage of cells harboring a WT copy of FOXP3 and demonstrate that a small subset of WT Tregs can exert a strong protective effect against autoimmunity. We observed that FOXP3 CRISPR gene editing of IPEX cells led to several improvements, including restoration of WT FOXP3 expression, increased Treg-suppressive function, and normalization of Teff cell proliferation rates.
We evaluated the differentiation potential of FOXP3 edited HSPCs using a combination of in vitro and in vivo testing. The differentiation of edited HSPCs into myeloid and erythroid lineages was confirmed in vitro. The in vivo studies demonstrated engraftment and multilineage hematopoietic reconstitution and allowed us to observe lymphocyte lineages in more depth. The gene edited cells (tNGFR+ population) persisted in vivo, although in some mice, the percentage of tNGFR+ cells was lower than that in the edited HSPC population before injection. This decrease could be due to several factors, including the previously observed preferential editing of short-term progenitor cells over long-term HSCs and the decrease in cell viability after gene editing (22, 39). However, the lack of expansion of edited cells alleviates any general concerns of clonal expansion of genetically modified cells. This notion is further underscored by the comparable survival rates of hu-mice engrafted with edited and nonedited HSPCs over the course of the study. While there were some differences in cell population frequencies among conditions, there were no major changes or skewing in immune reconstitution. A decrease in the proportion of FOXP3+CD25+ cells in the tNGFR+ edited condition was found, possibly due to lower FOXP3 protein expression in edited cells. However, edited FOXP3+ Tregs isolated from the hu-mouse spleen were found to have similar suppressive capacity to WT counterparts. In addition, hu-mouse–derived FOXP3 edited Teff cells were found to be functional in vitro and proliferate at comparable rates to WT Teff cells. These functional assays demonstrate that FOXP3 edited HSPCs retain the capacity to give rise to functional Tregs and Teff cells in vivo.
These results lay the foundation for using the FOXP3 CRISPR system to gene correct autologous HSPCs, T cell precursors, or Treg cells directly for clinical purposes. Ultimately, we envision the CRISPR system being used for FOXP3 gene repair in IPEX HSPCs for autologous transplantation, circumventing the need to find immunologically matched HSPC donors and lowering the risks associated with transplantation. More broadly, the translation of a CRISPR-based therapy for IPEX syndrome will help pave the way for similar therapeutic approaches in other primary immunodeficiencies with autoimmunity.
MATERIALS AND METHODS
Cell isolation and culture
All research on cells from patients with IPEX and healthy donor controls was approved by the Stanford University Institutional Review Board (IRB) (in accordance with the IRB-approved protocol, IRB-34131) or by the IRB of the Ospedale San Raffaele (protocol TIGET06). Written informed consent was obtained from all patients or patient families. Additional healthy donor HSPCs were isolated from umbilical cord blood donors provided by the Binns Program for Cord Blood Research at the Stanford University or purchased commercially from AllCells or StemCell Technologies. For HSPC isolation from cord blood and bone marrow, mononuclear cells were first obtained by Ficoll-Paque (GE Healthcare) density gradient separation followed by ammonium chloride red blood cell lysis (STEMCELL Technologies). HSPCs were next purified by magnetic cell isolation using the CD34 MicroBead UltraPure Kit (Miltenyi Biotec) according to the manufacturer’s recommendations and plated at a cell concentration of 2.5 × 105 cells/ml. The HSPCs were cultured at 37°C with 5% CO2 and low oxygen (5% O2) in StemSpan SFEM II medium (STEMCELL Technologies) supplemented with stem cell factor (100 ng/ml; PeproTech), IL-6 (100 ng/ml; PeproTech), thrombopoietin (100 ng/ml; PeproTech), Flt3L (100 ng/ml; PeproTech), 750 nM StemRegenin1 (STEMCELL Technologies), and 35 nM UM171 (STEMCELL Technologies). Additional healthy donor Treg and Teff cells were obtained from the Stanford Blood Center, and peripheral blood mononuclear cells were isolated by Ficoll-Paque density gradient separation. All edited cells were from male donor origin to match male IPEX patient samples, with the exception of a few female mock controls. Tregs and Teff cells were separated by magnetic bead isolation using the CD4+CD25+ Regulatory T Cell Isolation Kit (Miltenyi Biotec) according to the manufacturer’s protocol. Both cell fractions were activated with immobilized plate-bound anti-CD3 [10 μg/ml; OKT3 monoclonal antibody (mAb), Miltenyi] with soluble anti-CD28 mAb (1 μg/ml; NA/LE, BD) for 2 to 3 days before editing and then switched to nonactivation conditions. Tregs were cultured in X-VIVO 15 (Lonza) with 5% human serum from male AB plasma (Sigma-Aldrich), IL-2 (300 U/ml; PeproTech), and 100 nmol rapamycin (STEMCELL Technologies; only added for certain experiments). Teff cells were cultured in X-VIVO 15, 5% human serum, and IL-2 (50 U/ml). The Tregs and Teff cells were cultured at 37°C with 5% CO2 and ambient oxygen levels. Treg-like MT-2 cells were cultured in X-VIVO 15 with 5% human serum and 1% penicillin/streptomycin. K562 cells (American Type Culture Collection) were cultured in RPMI 1640 medium (Thermo Fisher Scientific) with 10% fetal bovine serum (Thermo Fisher Scientific) at 37°C with 5% CO2 and ambient oxygen levels. For all cells, fresh medium was added every 2 to 3 days.
Screening of sgRNA efficiency by TIDE analysis
CRISPR chimeric sgRNA was designed using the Desktop Genetics web-based tool (Massachusetts Institute of Technology) and cloned into expression vectors using the px330 plasmid backbone (Addgene). The sgRNAs were placed under the human U6 promoter in the px330 plasmid, which also contained an expression cassette for human codon–optimized SpCas9. For paired sgRNA nickase experiments, the sgRNAs were cloned into the px335 plasmid (Addgene) containing the Cas9 nickase expression cassette and were codelivered as paired plasmids. For the initial screen, 2 μg of sgRNA/Cas9 plasmid DNA was nucleofected into 1 million K562 cells using the Lonza Nucleofector 2b (program T-016). For each reaction, 100 μl of nucleofection solution was used [100 mM KH2PO4, 15 mM NaHCO3, 12 mM MgCl2.6H2O, 8 mM adenosine triphosphate, and 2 mM glucose (pH 7.4)]. The cells were cultured for 2 to 4 days, and genomic DNA was extracted using QuickExtract DNA Extraction Solution (Epicentre) according to the manufacturer’s recommendations. The site of DNA cleavage was PCR amplified using Herculase II Fusion Polymerase (Agilent Technologies) and primers flanking the region 5′-CTAGAGCTGGGGTGCAACTATGG-3′ and 5′-GACTACAATACGGCCTCCTCCTCTC-3′. The PCR amplicons were gel purified (Qiagen) and sequenced by Sanger sequencing using the forward primer 5′-CTAGAGCTGGGGTGCAACTATGG-3′. The resulting sequences were used to calculate indel frequencies using the TIDE analysis web-based software (https://tide.nki.nl/). A list of all sgRNA and primer sequences is provided in table S1.
FOXP3 homology donor design
The FOXP3 cDNA sequence was modified, or diverged, to incorporate synonymous mutations at the nucleotide level according to the redundant codon usage system to prevent premature recombination while still encoding for the WT FOXP3 protein. A constitutive Phosphoglycerate Kinase (PGK) promoter was positioned upstream of the tNGFR gene such that the marker would be expressed in all edited cells independent of FOXP3 expression. A strong Bovine Growth Hormone (BGH) pA signal was positioned after the FOXP3 cDNA, and another pA was included after the tNGFR marker gene to allow independent expression of the FOXP3 cDNA and tNGFR and to ensure inactivation of the remaining endogenous FOXP3 locus. The homology arms were centered on the cut site of the sgRNA 2. The 3′ arm (right arm) started at the cut site and reached approximately 650 base pairs (bp) downstream of the cut site, whereas the 5′ arm (left arm) included a region approximately 600 bp upstream of the cut site. The FOXP3FLcoW construct contained a shorter synthetic pA site in place of the BGH pA and shorter arms of homology to accommodate the addition of a WPRE element while maintaining an overall similar donor length.
Production of rAAV-FOXP3 homology donors
All FOXP3 homology donors were cloned into pAAV-MCS AAV vectors (Agilent Technologies) containing AAV inverted terminal repeats. Cloning was performed using Not I restriction digest [New England Biolabs (NEB)] followed by ligation with T4 DNA ligase (NEB). Plasmid preparation was performed by transforming plasmids into Stbl3 Escherichia coli (Life Technologies) and extracting plasmid DNA with Endotoxin-Free Maxi Prep kits (Qiagen). For rAAV production, pAAV-FOXP3 plasmids were cotransfected with rAAV6 helper plasmid DNA into the 293FT Cell Line (Life Technologies). After 72 hours, rAAV6-FOXP3 viral particles were extracted using the AAVpro kit (Clontech, Takara) according to the manufacturer’s instructions. The viral stocks were titered using quantitative PCR (qPCR) with primers and probe annealing to the inverted terminal repeats (ITRs). Briefly, the rAAV genomic DNA was isolated using QIAamp MinElute Virus Spin Kit (Qiagen), qPCR was performed on the Roche LightCycler 480, and viral titer (vector genomes per microliter) was calculated using a standard curve generated from a circular pAAV-MCS-donor plasmid of known concentration.
Gene editing by nucleofection and rAAV transduction
Gene editing of primary human HSPCs, Tregs, and Teff cells was performed using synthetic sgRNA 2 (5′-AGGACCCGATGCCCAACCCC-3′) complexed to SpCas9 protein [Integrated DNA Technologies (IDT)] as an ribonucleoprotein (RNP) system. The sgRNA was synthesized as a 100-mer RNA molecule with 2′-O-methyl 3′phosphorothioate (MS) chemical modifications at the three terminal nucleotides on the 5′ and 3′ ends [5′-2'OMe(A(ps)G(ps)G(ps))ACC CGA UGC CCA ACC CCG UUU UAG AGC UAG AAA UAG CAA GUU AAA AUA AGG CUA GUC CGU UAU CAA CUU GAA AAA GUG GCA CCG AGU CGG UGC UUU 2'OMe(U(ps) U(ps)U)-3′; ps indicates phosphorothioate, and 2'OMe indicates 2′-O-methyl]. The sgRNAs were purified by reversed-phase high-performance liquid chromatography (HPLC) and quantified by mass spectrometry. The sgRNAs were purchased from TriLink BioTechnologies and, in later experiments, from Synthego (not HPLC-purified), and editing rates triggered by sgRNAs from the two respective companies were comparable when tested in parallel. The sgRNA was complexed with Cas9 for 10 min at 25°C at an approximate Cas9:sgRNA molar ratio of 1:2.5, using 8 μg of sgRNA and 15 μg of Cas9 per 100 μl of nucleofection solution containing 2.5 × 105 to 1 × 106 cells. After switching to high-fidelity (HiFi) Cas9 (IDT) that showed slightly lower efficiency in parallel experiments, the amount of HiFi Cas9 was increased, and the molar ratio was adjusted to 1:1.8, using 8 μg of sgRNA with 22 μg of HiFi Cas9 per 100 μl of nucleofection solution. The sgRNA/Cas9 complexes were nucleofected into Tregs and Teff cells after 2 to 3 days of activation using the P3 Primary Cell Nucleofection Kit (Lonza) and the Lonza Nucleofector 4D (program E-0115). On the day of nucleofection, additional antibiotic (penicillin/streptomycin) was removed from the medium, and rapamycin was removed from the Treg medium. The following day, medium was changed, and antibiotic/rapamycin was replaced. For HSPC editing, the cells were nucleofected using the P3 Primary Cell Nucleofection Kit (Lonza) and the Lonza Nucleofector 4D (program DZ-100). For analysis of indel frequencies, genomic DNA was extracted using QuickExtract DNA Extraction Solution (Epicentre), and TIDE analysis was performed as described above. For HDR experiments, rAAV6-FOXP3 donor transduction was performed following nucleofection at a multiplicity of infection (MOI) of 1 × 105 to 1 × 106 viral genomes per cell. After 24 hours of transduction, the medium was changed to remove excess viral particles.
Assessment of HDR-mediated targeted integration by flow cytometry and in-out PCR
To assess HDR at the genomic level, we performed in-out PCR 2 to 4 days after editing using a forward primer outside of the 5′ arm of homology (5′-ATGTCAGCTCGGTCCTTCCA-3′) and a reverse primer inside the inserted divergent cDNA construct (5′-TGGCATAGGATTAAGGGAACTG-3′). A second in-out PCR strategy targeting the 3′ end used a forward primer inside the inserted tNGFR region (5′-AGCCTTCAAGAGGTGGAACA-3′) of the construct and a reverse primer in the endogenous FOXP3 locus outside of the 3′ arm of homology (5′-AGGCCATCCTGATCCTCAC-3′). As a control for the presence of genomic DNA, a PCR strategy targeting a downstream, unmodified region of FOXP3 was performed (forward primer: 5′-TGCCTCCTCTTCTTCCTTGA-3′; reverse primer: 5′-GAGCCTCGAAAACCCTGACT-3′). Herculase II fusion polymerase (Agilent Technologies) was used for all PCR amplification steps. The resulting PCR products were resolved by agarose gel electrophoresis. For absolute quantification of genomic integration events at the DNA level, an in-out PCR strategy quantified using the ddPCR (Bio-Rad) system was used. For ddPCR, we used two primer/probe sets: the first for the edited region of the FOXP3 locus (forward primer: 5′-GGGAGGATTGGGAAGACAAT-3′; FAM-labeled probe: 5′-TCAGAGATTGGAGGCTCTCC-3′; reverse primer: 5′-ACAATACGGCCTCCTCCTCT-3′) and a second primer/probe control set targeting a nonmodified region of the FOXP3 gene used as a reference (forward primer: 5′-CACCGAAATCGGTATTAGTTTG-3′; HEX-labeled probe: 5′-CAGTTCTGGAGGCCAGAGTC-3′; reverse: 5′-CCCGGGGGAGTATAGAAGG-3′). The two regions were amplified and quantified as previously described with modifications (19). The PCR used 100 ng of genomic DNA digested with Bam HI-HF (10 U) and an annealing temperature of 62°C. The percentage targeting was calculated as the ratio of FAM (targeted allele) to HEX signal.
Enrichment of tNGFR+ edited cells
Edited tNFGR+ cells were enriched by fluorescence-activated cell sorting (FACS) 2 to 4 days after editing on a FACSAria II SORP (BD Biosciences). Cells were stained with anti-NGFR/CD271 mAb [BioLegend, clone ME20.4, phycoerythrin (PE)–Cy7 conjugated or allophycocyanin (APC) conjugated]. When edited cells were present at low cell numbers, magnetic bead cell isolation was used to increase yield and avoid cell loss associated with FACS. Positive selection of tNGFR+ cells was performed using CD271 (tNGFR) Microbead Kit (Miltenyi Biotec) according to the manufacturer’s instructions. Briefly, cells were magnetically labeled with anti-tNGFR microbeads and separated on an MS or LS column in the magnetic field of a MACS separator. Columns were washed and eluted, and the cells were passed through a second column to increase purity.
Off-target analysis
Off-target sites were bioinformatically predicted using the in silico COSMID prediction tool (25). For in vitro off-target analysis, GUIDE-seq was performed as previously described (26). In brief, the px330-FOXP3-sgRNA2-Cas9 plasmid was electroporated into U2OS cells along with a double-stranded oligodeoxynucleotide (dsODN; 5′-GTTTAATTGAGTTGTCATATGTTAATAACGGTAT-3′). The T7 and restriction fragment length polymorphism (RFLP) assays were performed to confirm editing and tag integration (using TIDE primers 5′-CTAGAGCTGGGGTGCAACTATGG and 5′-GACTACAATACGGCCTCCTCCTCTC-3′). Genomic DNA was extracted, sequencing libraries were prepared, and NGS was performed to identify all sites in which the dsODN was integrated into DNA breaks. GUIDE-seq reads were filtered using a cutoff of eight mismatches according to previous studies on the CRISPR tolerance of mismatches (25, 40). A total of 62 sites identified by the combination of COSMID and GUIDE-seq analyses were then tested by NGS (Illumina MiSeq) in edited HSPCs. HSPCs derived from male healthy donor cord blood (n = 3) and bone marrow (n = 3) were edited with CRISPR sgRNA2/HiFi Cas9 combined with rAAV6-FOXP3 FL donor, and total cells (tNGFR+/−) were used for genomic DNA extraction. NGS reads that were identified at similar rates in edited cells and mock-treated samples were eliminated from the analysis. High background in the mock-treated samples was attributed to the proximity of the sequencing primer to the polynucleotide sequence 5′-CCCC-3′ in the sgRNA target site, as polyN sequences commonly cause errors in NGS and can lead to false-positive indel identification. Only sites with indel mutation rates above mock control background were selected for NGS validation in edited bone marrow–derived HSPCs. Four validated sites were identified in edited bone marrow–derived HSPC, and these sites were analyzed by bioinformatic gene annotation to predict expression in hematopoietic lineages and contribution to hematopoiesis or cell cycle regulation using the UCSC (University of California, Santa Cruz) genome browser, UniProt, GeneCards, and publically archived microarray and RNA sequencing data.
Determination of FOXP3 expression by RT-PCR and flow cytometry
FOXP3 mRNA expression was detected by RT-PCR in FOXP3FL edited CD4+ T cells and controls after 3 days of reactivation with Human T-Activator anti-CD3/28 Dynabeads (Life Technologies, 1:25 bead:cell ratio). RNA was extracted with TRI Reagent (Sigma-Aldrich), and polyA+ mRNA was reverse transcribed into cDNA using SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific). PCR amplification of FOXP3 cDNA was performed using Herculase II fusion polymerase (Agilent Technologies), and primers are listed in table S1. For assessing FOXP3 protein expression by flow cytometry, cells were fixed and permeabilized using FOXP3 staining solutions (eBioscience) and stained with anti-FOXP3 mAb (clone 259D/C7) conjugated to either AF647 (BD Biosciences) or AF488 (BioLegend) following the manufacturer’s instructions. Fluorescence was detected on a FACSAria II SORP (BD Biosciences), analyzed using FlowJo software v4 10.5.0, and median florescence intensity (MFI) was recorded.
Treg phenotyping and suppression assay
For Treg phenotyping, cells were stained for flow cytometry using the following antibodies: CD25-BV605 (clone 2A3, BD Biosciences), CTLA-4-PerCPCy5.5 (L3D10, BioLegend), FOXP3-AF647 (259D/C7, BD), HELIOS-PE (22F6, BioLegend), NGFR-BV421 (cME20.4, BioLegend), PD1–fluorescein isothiocyanate (FITC) (MIH4, BD), and TIGIT-PE-Cy7 (MBSA43, eBioscience). Intracellular staining for FOXP3, CTLA-4, and HELIOS was performed after fixing and permeabilizing with FOXP3 staining solutions (eBioscience). Expression was detected on a FACSAria II SORP (BD Biosciences), and geometric mean intensity was analyzed using FlowJo software v4 10.5.0. The function of gene edited Tregs was tested by the suppression assay using allogenic CD4+ T cell responders that were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE) proliferation dye (CellTrace CFSE Cell Proliferation Kit, Life Technologies). Responders were plated at a concentration of 2 × 104 cells per well and cocultured with Tregs at a 1:1 or 1:0.5 ratio of responders:suppressors. The cells were activated with a 1:25 ratio of beads:cells using Human T-Activator anti-CD3/28 Dynabeads (Life Technologies). As a reference control, responders were cocultured with an equal number of unstained Teff cells. The cells were cultured in 96-well round well plates and analyzed for CFSE staining on days 3 to 5 using a FACSAria II SORP (BD Biosciences). Nonactivated responders were used for gating, and the percentage of proliferated cells was analyzed using FlowJo software v4 10.5.0. Percent suppression was calculated using the following equation: % suppression = ((%proliferated R*) − (%proliferated R* + Treg))/((%proliferated R*) × 100), where R* represents stimulated CFSE-stained responder Teff. Treg purity was performed by flow cytometric analysis using the following antibodies: CD4-APC-Cy7 (RPA-T4, BioLegend), CD25-PE (4E3, Miltenyi), CD127-PerCP–Cy5.5 (A019D5, BioLegend), and FOXP3-AF647 (clone 259D/C7, BD). The frequency of demethylated TSDR Tregs was quantified by epigenetic bisulfite qPCR in collaboration with Epimune/Epiontis GmbH (Berlin, Germany) as previously described (41).
Teff cytokine quantification and proliferation assay
Teff cytokine production was quantified using enzyme-linked immunosorbent assay (ELISA) for IL-2 (BD Biosciences), IFN-γ (BD Biosciences), and IL-17 (R&D Systems). Teff cells were activated using Human T-Activator anti-CD3/28 Dynabeads (Life Technologies) at a 1:25 ratio of beads:cells in 96-well round well plates at 2 × 105 cells per 200 μl. Supernatants were collected at 24 hours (IL-2) and 48 hours (IFN-γ and IL-17) after activation. For the proliferation assay, Teff cells were stained using the CellTrace CFSE Cell Proliferation Kit (Life Technologies) and cultured at 5 × 104 cells per well in 96-well round well plates. The stained cells were activated with a 1:25 ratio of anti-CD3/28 Dynabeads and analyzed for CFSE staining on days 2 to 4 after activation on a FACSAria II SORP (BD Biosciences). The percentage of proliferated cells was determined using FlowJo software v4 10.5.0 and gated using nonactivated responders as a reference.
CFU assay to assess in vitro HSPC differentiation
Gene edited cord blood–derived HSPCs were FACS sorted 2 to 4 days after editing and differentiated in vitro using the CFU assay. For each condition, 500 cells were plated in 1.1 ml of semisolid methylcellulose medium (MethoCult H4434, STEMCELL Technologies) and performed in duplicate or triplicate. The cells suspended in MethoCult were incubated at 37°C with 5% CO2 and ambient oxygen levels, and the resulting progenitor colonies were counted and scored after 14 days [BFU-E (primitive erythroid progenitors), CFU-E (mature erythroid progenitors), CFU-GM (granulocyte and macrophage progenitors), and CFU-GEMM (granulocyte, erythrocyte, macrophage, and megakaryocyte)].
Engraftment studies in immunodeficient mice
All animal experiments were conducted in accordance with the protocols approved by the Stanford University’s Administrative Panel on Laboratory Animal Care Research Committee under the Division of Laboratory Medicine. Human cord blood–derived CD34+ HSPCs were gene edited as described above and injected 2 days after editing (without enriching for tNGFR+ cells). Cultured HSPCs were phenotyped by flow cytometry to ensure purity using the antibodies CD34-PE-Cy7 (4H11, eBioscience), CD38-Percp-Cy5.5 (HIT2, BioLegend), CD45RA-FITC (HI100, BD Biosciences), CD90-APC-Cy7 (5E10, BioLegend), CD49f-PE (GoH3, BioLegend), and combined lineage markers (Lin) on APC as follows: CD45-APC (30-F11, BioLegend), CD19-APC (HIB19, BioLegend), CD14-APC (HCD14, BioLegend), CD235a-APC (HIR2, BioLegend), CD20-APC (2H7, BioLegend), CD16-APC (3G8, BioLegend), CD2-APC (RPA-2.10, BioLegend), CD3-APC (SK7, BioLegend), CD4-APC (SK3, BioLegend), CD8-APC (SK1, BioLegend), and CD13-APC (WM15, BioLegend). On the day of injection, 3- to 4-day-old NSG-SGM3 (the Jackson laboratory, JAX:013062) neonatal pups were irradiated with 100 centigray and rested for 6 hours before injection. HSPCs (between 1.5 × 105 and 1.0 × 106) were resuspended in 30 μl of expansion media, and cells were injected intrahepatically using a 28.5-gauge insulin syringe. Starting at week 6, mice were checked for peripheral engraftment of human CD45+ cells via biweekly retro-orbital bleed. The mice were euthanized between 11 and 14 weeks, and blood, spleen, bone marrow, and thymus were harvested. For blood and spleen samples, red blood cells were lysed following a 5-min incubation on ice with 1× red blood cell lysis buffer (eBioscience) and were resuspended in staining buffer (phosphate-buffered saline, 0.25% bovine serum albumin, and 1 mM EDTA). Cells purified from tissues were stained using the following antibodies: hCD45 BV510 (HI30, BD Biosciences), mCD45-APC (30-F11, BioLegend), CD3-Percp-Cy5.5 (OKT3, BioLegend), CD56-PE (5.1H11, BioLegend), CD13-APC-Cy7 (WM15, BioLegend), and CD19-FITC (HIB19, BD Biosciences). An additional antibody panel for T cell subsets included hCD45 BV510 (HI30, BD Biosciences), mCD45 PE (30-F11, BioLegend), CD4 APC-Cy7 (RPA-T4, BioLegend), CD8 BV650 (SK1, BioLegend), CD25 BV605 (2A3, BD Biosciences), CD45RA FITC (HI100, BD Biosciences), and FOXP3 AF647 (259D, BioLegend). Cells were either analyzed by flow cytometric analysis (CytoFLEX BD) or sorted (BD, FACSAria). Sorted CD25high and CD25low populations were analyzed by suppression and proliferation assays, respectively, as described above.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software v7.0c (GraphPad Inc.). Averages were represented as mean ± SD, and the number of replicates was indicated in respective figures and figure legends. For comparison of two datasets, two-tailed Student’s t test was performed (α = 5.0%). For paired donor samples, a paired Student’s t test was performed (α = 5.0%). Differences between multiple groups were identified using one- or two-way analysis of variance (ANOVA) and Tukey’s multiple comparisons test (α = 5.0%). Significances were indicated as *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Supplementary Material
Acknowledgments
We would like to thank all members of the Bacchetta, Roncarolo, and Porteus laboratories for feedback and supportive roles, especially L. Nguyen, BS; A. Cepika, MD, PhD; J. Schulze, PhD; D. Dever, PhD; R. Bak, PhD; and I. Aversa, BS. We appreciate the useful discussions and advice from R. Parkman, MD, and K. Weinberg, MD, and feedback on the manuscript from R. Perriman, PhD. We are grateful for patient participation and the excellent patient care by clinicians and medical staff, including R. Agarwal, MD; K. Weinacht, MD; J. Chu, MD; S. Stone, MSN; S. Loeb, RN, BSN; B. Hines, MD; and S. Wadera, MD. We acknowledge F. Romana Santoni de Sio, PhD, for contribution to experimental protocols. We also thank the Binns Program for Cord Blood Research at the Stanford University, particularly S. Mantri, BS, and L. Ren, BS, who provided umbilical cord blood and HSPC samples. We thank Synthego for help with the chemically modified sgRNAs. We thank patients and their families for their trust in our research. Funding: We gratefully acknowledge the support from several grants and awards, including the NIH NIAID (R21 AI123896), the CIRM (DISC2-09526), the Stanford SPARK (1110504-308-DHBTC), and the Sutardja Foundation (1198779-101-GHFBB), and the Cancer Prevention and Research Institute of Texas to G.B. (RR140081 and RR170721), in addition to a donation from an anonymous donor to the Stanford Center for Genetic Immune Diseases (CGID) for immunological studies on the patient samples. M.G. thanks the Stanford University for the tremendous support, including the Stanford Maternal and Child Health Research Institute (MCHRI) Postdoctoral Award (Stanford NIH-NCATS-CTSA, UL1 TR001085) and the Stanford University Office of the Dean Postdoctoral Award (1182084-100-DHDEZ). E.L. received funding support from the Agency for Science, Technology and Research, Singapore and the Stanford Masters of Science in Medicine scholarship.Author contributions: R.B., M.G., and M.P. designed the study and CRISPR strategy. M.G. and E.L. performed or supervised the experiments, and M.G., E.L., R.B., and M.G.R. interpreted the results and analyzed the data. U.L., S.S., L.F., M.N., A. She., M.P.-D., and A.H. helped with protocol design, performing experiments, and/or data collection. C.M.L. and G.B. performed off-target analysis. M.G. wrote the manuscript, which was edited by R.B., M.P., and M.G.R. Patient samples were provided by the dedicated clinicians, including F.B., L.P., C.S.B., H.K.M., M.G.-L., M.J.B., A.B., and A. Sha. Competing interests: M.P. is on the Scientific Advisory Board and has equity in CRISPR Therapeutics and Allogene Therapeutics. The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/19/eaaz0571/DC1
REFERENCES AND NOTES
- 1.Notarangelo L. D., Primary immunodeficiencies. J. Allergy Clin. Immunol. 125, S182–S194 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Bacchetta R., Barzaghi F., Roncarolo M.-G., From IPEX syndrome to FOXP3 mutation: A lesson on immune dysregulation. Ann. N. Y. Acad. Sci. 1417, 5–22 (2018). [DOI] [PubMed] [Google Scholar]
- 3.d'Hennezel E., Ben-Shoshan M., Ochs H. D., Torgerson T. R., Russell L. J., Lejtenyi C., Noya F. J., Jabado N., Mazer B., Piccirillo C. A., FOXP3 forkhead domain mutation and regulatory T cells in the IPEX syndrome. N. Engl. J. Med. 361, 1710–1713 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Barzaghi F., Amaya Hernandez L. C., Neven B., Ricci S., Kucuk Z. Y., Bleesing J. J., Nademi Z., Slatter M. A., Ulloa E. R., Shcherbina A., Roppelt A., Worth A., Silva J., Aiuti A., Murguia-Favela L., Speckmann C., Carneiro-Sampaio M., Fernandes J. F., Baris S., Ozen A., Karakoc-Aydiner E., Kiykim A., Schulz A., Steinmann S., Notarangelo L. D., Gambineri E., Lionetti P., Shearer W. T., Forbes L. R., Martinez C., Moshous D., Blanche S., Fisher A., Ruemmele F. M., Tissandier C., Ouachee-Chardin M., Rieux-Laucat F., Cavazzana M., Qasim W., Lucarelli B., Albert M. H., Kobayashi I., Alonso L., Diaz de Heredia C., Kanegane H., Lawitschka A., Seo J. J., Gonzalez-Vicent M., Diaz M. A., Goyal R. K., Sauer M. G., Yesilipek A., Kim M., Yilmaz-Demirdag Y., Bhatia M., Khlevner J., Richmond Padilla E. J., Martino S., Montin D., Neth O., Molinos-Quintana A., Valverde-Fernandez J., Broides A., Pinsk V., Ballauf A., Haerynck F., Bordon V., Dhooge C., Garcia-Lloret M. L., Bredius R. G., Kałwak K., Haddad E., Seidel M. G., Duckers G., Pai S. Y., Dvorak C. C., Ehl S., Locatelli F., Goldman F., Gennery A. R., Cowan M. J., Roncarolo M. G., Bacchetta R.; Primary Immune Deficiency Treatment Consortium (PIDTC) and the Inborn Errors Working Party (IEWP) of the European Society for Blood and Marrow Transplantation (EBMT) , Long-term follow-up of IPEX syndrome patients after different therapeutic strategies: An international multicenter retrospective study. J. Allergy Clin. Immunol. 141, 1036–1049.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cepika A. M., Sato Y., Liu J. M., Uyeda M. J., Bacchetta R., Roncarolo M. G., Tregopathies: Monogenic diseases resulting in regulatory T-cell deficiency. J. Allergy Clin. Immunol. 142, 1679–1695 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Ziegler S. F., FOXP3: Of mice and men. Annu. Rev. Immunol. 24, 209–226 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Bacchetta R., Passerini L., Gambineri E., Dai M., Allan S. E., Perroni L., Dagna-Bricarelli F., Sartirana C., Matthes-Martin S., Lawitschka A., Azzari C., Ziegler S. F., Levings M. K., Roncarolo M. G., Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J. Clin. Invest. 116, 1713–1722 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.d'Hennezel E., Bin Dhuban K., Torgerson T., Piccirillo C. A., The immunogenetics of immune dysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J. Med. Genet. 49, 291–302 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Passerini L., Santoni de Sio F. R., Roncarolo M. G., Bacchetta R., Forkhead box P3: The peacekeeper of the immune system. Int. Rev. Immunol. 33, 129–145 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Huehn J., Polansky J. K., Hamann A., Epigenetic control of FOXP3 expression: The key to a stable regulatory T-cell lineage? Nat. Rev. Immunol. 9, 83–89 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Santoni de Sio F. R., Passerini L., Restelli S., Valente M. M., Pramov A., Maccari M. E., Sanvito F., Roncarolo M. G., Porteus M., Bacchetta R., Role of human forkhead box P3 in early thymic maturation and peripheral T-cell homeostasis. J. Allergy Clin. Immunol. 142, 1909–1921.e9 (2018). [DOI] [PubMed] [Google Scholar]
- 12.McMurchy A. N., Gillies J., Gizzi M. C., Riba M., Garcia-Manteiga J. M., Cittaro D., Lazarevic D., Nunzio S. D., Piras I. S., Bulfone A., Roncarolo M. G., Stupka E., Bacchetta R., Levings M. K., A novel function for FOXP3 in humans: Intrinsic regulation of conventional T cells. Blood 121, 1265–1275 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Allan S. E., Passerini L., Bacchetta R., Crellin N., Dai M., Orban P. C., Ziegler S. F., Roncarolo M. G., Levings M. K., The role of 2 FOXP3 isoforms in the generation of human CD4+ Tregs. J. Clin. Invest. 115, 3276–3284 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mailer R. K. W., Alternative splicing of FOXP3—Virtue and vice. Front. Immunol. 9, 530 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gambineri E., Mannurita S. C., Hagin D., Vignoli M., Anover-Sombke S., De Boer S., Segundo G. R. S., Allenspach E. J., Favre C., Ochs H. D., Torgerson T. R., Clinical, immunological, and molecular heterogeneity of 173 patients with the phenotype of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Front. Immunol. 9, 2411 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frith K., Joly A. L., Ma C. S., Tangye S. G., Lohse Z., Seitz C., Verge C. F., Andersson J., Gray P., The FOXP3Δ2 isoform supports Treg cell development and protects against severe IPEX syndrome. J. Allergy Clin. Immunol. 144, 317–320.e8 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Passerini L., Mel E. R., Sartirana C., Fousteri G., Bondanza A., Naldini L., Roncarolo M. G., Bacchetta R., CD4+ T cells from IPEX patients convert into functional and stable regulatory T cells by FOXP3 gene transfer. Sci. Transl. Med. 5, 215ra174 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Santoni de Sio F. R., Passerini L., Valente M. M., Russo F., Naldini L., Roncarolo M. G., Bacchetta R., Ectopic FOXP3 expression preserves primitive features of human hematopoietic stem cells while impairing functional T cell differentiation. Sci. Rep. 7, 15820 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pavel-Dinu M., Lee C., Bao G., Kildebeck E. J., Punjya N., Sindhu C., Inlay M. A., Saxena N., De Ravin S. S., Malech H., Roncarolo M. G., Weinberg K. I., Porteus M. H., Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat. Commun. 10, 1634 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hubbard N., Hagin D., Sommer K., Song Y., Khan I., Clough C., Ochs H. D., Rawlings D. J., Scharenberg A. M., Torgerson T. R., Targeted gene editing restores regulated CD40L expression and function in X-HIGM T cells. Blood 127, 2513–2522 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Brinkman E. K., Chen T., Amendola M., van Steensel B., Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bak R. O., Dever D. P., Porteus M. H., CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat. Protoc. 13, 358–376 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J., Exline C. M., De Clercq J. J., Llewellyn G. N., Hayward S. B., Li P. W.-L., Shivak D. A., Surosky R. T., Gregory P. D., Holmes M. C., Cannon P. M., Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat. Biotechnol. 33, 1256–1263 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamano R., Wu X., Wang Y., Oppenheim J. J., Chen X., Characterization of MT-2 cells as a human regulatory T cell-like cell line. Cell. Mol. Immunol. 12, 780–782 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cradick T. J., Qiu P., Lee C. M., Fine E. J., Bao G., COSMID: A web-based tool for identifying and validating CRISPR/Cas off-target sites. Mol. Ther. Nucleic Acids 3, e214 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsai S. Q., Zheng Z., Nguyen N. T., Liebers M., Topkar V. V., Thapar V., Wyvekens N., Khayter C., Iafrate A. J., Le L. P., Aryee M. J., Joung J. K., GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Passerini L., Barzaghi F., Curto R., Sartirana C., Barera G., Tucci F., Albarello L., Mariani A., Testoni P. A., Bazzigaluppi E., Bosi E., Lampasona V., Neth O., Zama D., Hoenig M., Schulz A., Seidel M. G., Rabbone I., Olek S., Roncarolo M. G., Cicalese M. P., Aiuti A., Bacchetta R., Treatment with rapamycin can restore regulatory T cell function in IPEX patients. J. Allergy Clin. Immunol., S0091-6749(19)32592-8 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Billerbeck E., Barry W. T., Mu K., Dorner M., Rice C. M., Ploss A., Development of human CD4+FoxP3+ regulatory T cells in human stem cell factor-, granulocyte-macrophage colony-stimulating factor-, and interleukin-3-expressing NOD-SCID IL2Rgammanull humanized mice. Blood 117, 3076–3086 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masiuk K. E., Laborada J., Roncarolo M. G., Hollis R. P., Kohn D. B., Lentiviral gene therapy in HSCs restores lineage-specific Foxp3 expression and suppresses autoimmunity in a mouse model of IPEX syndrome. Cell Stem Cell 24, 309–317.e7 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Allan S. E., Alstad A. N., Merindol N., Crellin N. K., Amendola M., Bacchetta R., Naldini L., Roncarolo M. G., Soudeyns H., Levings M. K., Generation of potent and stable human CD4+ T regulatory cells by activation-independent expression of FOXP3. Mol. Ther. 16, 194–202 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Aarts-Riemens T., Emmelot M. E., Verdonck L. F., Mutis T., Forced overexpression of either of the two common human Foxp3 isoforms can induce regulatory T cells from CD4(+)CD25(−) cells. Eur. J. Immunol. 38, 1381–1390 (2008). [DOI] [PubMed] [Google Scholar]
- 32.De Ravin S. S., Wu X., Moir S., Kardava L., Anaya-O’Brien S., Kwatemaa N., Littel P., Theobald N., Choi U., Su L., Marquesen M., Hilligoss D., Lee J., Buckner C. M., Zarember K. A., O’Connor G., Vicar D. M., Kuhns D., Throm R. E., Zhou S., Notarangelo L. D., Hanson I. C., Cowan M. J., Kang E., Hadigan C., Meagher M., Gray J. T., Sorrentino B. P., Malech H. L., Lentiviral hematopoietic stem cell gene therapy for X-linked severe combined immunodeficiency. Sci. Transl. Med. 8, 335ra57 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nathwani A. C., Reiss U. M., Tuddenham E. G., Rosales C., Chowdary P., McIntosh J., Della Peruta M., Lheriteau E., Patel N., Raj D., Riddell A., Pie J., Rangarajan S., Bevan D., Recht M., Shen Y. M., Halka K. G., Basner-Tschakarjan E., Mingozzi F., High K. A., Allay J., Kay M. A., Ng C. Y., Zhou J., Cancio M., Morton C. L., Gray J. T., Srivastava D., Nienhuis A. W., Davidoff A. M., Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 371, 1994–2004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nathwani A. C., Tuddenham E. G., Rangarajan S., Rosales C., McIntosh J., Linch D. C., Chowdary P., Riddell A., Pie A. J., Harrington C., O'Beirne J., Smith K., Pasi J., Glader B., Rustagi P., Ng C. Y., Kay M. A., Zhou J., Spence Y., Morton C. L., Allay J., Coleman J., Sleep S., Cunningham J. M., Srivastava D., Basner-Tschakarjan E., Mingozzi F., High K. A., Gray J. T., Reiss U. M., Nienhuis A. W., Davidoff A. M., Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 365, 2357–2365 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stephan V., Wahn V., Le Deist F., Dirksen U., Broker B., Müller-Fleckenstein I., Horneff G., Schroten H., Fischer A., de Saint Basile G., Atypical X-linked severe combined immunodeficiency due to possible spontaneous reversion of the genetic defect in T cells. N. Engl. J. Med. 335, 1563–1567 (1996). [DOI] [PubMed] [Google Scholar]
- 36.Seidel M. G., Fritsch G., Lion T., Jürgens B., Heitger A., Bacchetta R., Lawitschka A., Peters C., Gadner H., Matthes-Martin S., Selective engraftment of donor CD4+25high FOXP3-positive T cells in IPEX syndrome after nonmyeloablative hematopoietic stem cell transplantation. Blood 113, 5689–5691 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Kasow K. A., Morales-Tirado V. M., Wichlan D., Shurtleff S. A., Abraham A., Persons D. A., Riberdy J. M., Therapeutic in vivo selection of thymic-derived natural T regulatory cells following non-myeloablative hematopoietic stem cell transplant for IPEX. Clin. Immunol. 141, 169–176 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Di Nunzio S., Cecconi M., Passerini L., McMurchy A. N., Baron U., Turbachova I., Vignola S., Valencic E., Tommasini A., Junker A., Cazzola G., Olek S., Levings M. K., Perroni L., Roncarolo M. G., Bacchetta R., Wild-type FOXP3 is selectively active in CD4+CD25hi regulatory T cells of healthy female carriers of different FOXP3 mutations. Blood 114, 4138–4141 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Dever D. P., Bak R. O., Reinisch A., Camarena J., Washington G., Nicolas C. E., Pavel-Dinu M., Saxena N., Wilkens A. B., Mantri S., Uchida N., Hendel A., Narla A., Majeti R., Weinberg K. I., Porteus M. H., CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature 539, 384–389 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anderson E. M., Haupt A., Schiel J. A., Chou E., Machado H. B., Strezoska Ž., Lenger S., McClelland S., Birmingham A., Vermeulen A., van Brabant Smith A., Systematic analysis of CRISPR–Cas9 mismatch tolerance reveals low levels of off-target activity. J. Biotechnol. 211, 56–65 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Baron U., Werner J., Schildknecht K., Schulze J. J., Mulu A., Liebert U.-G., Sack U., Speckmann C., Gossen M., Wong R. J., Stevenson D. K., Babel N., Schürmann D., Baldinger T., Bacchetta R., Grützkau A., Borte S., Olek S., Epigenetic immune cell counting in human blood samples for immunodiagnostics. Sci. Transl. Med. 10, eaan3508 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/19/eaaz0571/DC1