

In vitro 3D human brain–like cultures infected with herpes virus develop Alzheimer’s disease characteristics.
Abstract
Alzheimer’s disease (AD) is a neurodegenerative disorder that causes cognitive decline, memory loss, and inability to perform everyday functions. Hallmark features of AD—including generation of amyloid plaques, neurofibrillary tangles, gliosis, and inflammation in the brain—are well defined; however, the cause of the disease remains elusive. Growing evidence implicates pathogens in AD development, with herpes simplex virus type I (HSV-1) gaining increasing attention as a potential causative agent. Here, we describe a multidisciplinary approach to produce physiologically relevant human tissues to study AD using human-induced neural stem cells (hiNSCs) and HSV-1 infection in a 3D bioengineered brain model. We report a herpes-induced tissue model of AD that mimics human disease with multicellular amyloid plaque–like formations, gliosis, neuroinflammation, and decreased functionality, completely in the absence of any exogenous mediators of AD. This model will allow for future studies to identify potential downstream drug targets for treating this devastating disease.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that causes severe cognitive decline, short-term memory loss, impaired speech, and ultimately an inability to function in day-to-day activities (1). AD is the sixth leading cause of death in the United States, currently afflicting nearly 5.8 million Americans with an expected doubling as the population ages in the absence of medical advances to cure or at least halt the progression of this devastating disease (1). Early-onset AD (EOAD) describes patients developing the disease at age 60 or younger. However, this population represents approximately 1 to 6% of all AD cases, most of which are classified as familial EOAD in that multiple relatives are also diagnosed with AD and, in certain cases, even demonstrate an autosomal pattern of inheritance associated with several specific gene mutations (2). Sporadic or late-onset AD accounts for approximately 95% of all AD cases and has a complex multifactorial etiology that, despite years of research, still remains quite elusive.
The hallmark features of AD are the formation of neurofibrillary tangles (NFTs) and deleterious senile plaques that develop in the brain. NFTs are aggregates of the microtubule-associated protein Tau that result from aberrant hyperphosphorylation and misfolding. Senile plaques consist of extracellular accumulation of β-amyloid (Aβ) peptide, which is a cleavage product of the integral transmembrane protein amyloid precursor protein (APP) found in neurons. Full-length APP has been shown to be involved in neuron growth, guidance, and synapse formation (3); however, APP also undergoes sequential cleavage events to generate various isoforms with different functions. During the first cleavage step of the amyloidogenic pathway, the extracellular domain of APP is cleaved by the enzyme β-secretase 1 (BACE1), which releases the soluble APPβ fragment into the extracellular space. A second enzyme, γ-secretase composed of multiple catalytic subunits including presenilins 1 and 2 (PSEN1/2) (4), subsequently cleaves the remaining intramembranous fragment C99 to form APP intracellular domain and Aβ (5). These Aβ peptides vary in size from 38 to 43 amino acids; however, the most common isoforms are Aβ1–40, which makes up approximately 90% of the resulting Aβ fragments, and Aβ1–42, which accounts for approximately 10%. This Aβ1–42 isoform can exist as monomers or oligomers and has the capacity to generate fibrils that aggregate and contribute to senile plaque formation (5). Other features of AD include cell death (6), hyperactivation of glia (7), neuroinflammation (8), and synaptic dysfunction and disruption of neural networks (9). While these diagnostic criteria of AD are well defined, the cause of this disease is not well understood.
The autosomal dominant form of EOAD has been attributed to the overproduction of Aβ as the result of mutations in one of three genes: APP, PSEN1, or PSEN2 (2). Most in vitro human models of AD rely on cells derived from patients with familial EOAD, which allow for these specific gene mutations to initiate onset of disease (10). While these models allow for the investigation of certain downstream pathways involved in the pathogenesis of this inherited type of AD, which only accounts for approximately 5% or less of all afflicted patients, they often provide little mechanistic insight into causality and potential environmental risk factors involved in most AD cases.
Growing evidence implicates pathogen involvement in the development of sporadic AD (11–15). The formation of Aβ1–42 monomers has actually been shown to be neuroprotective, supporting the survival of neurons against trophic deprivation and excitotoxic death (16). It is the aggregation of these monomers into oligomers, fibrils, and ultimately senile plaques that ultimately correlates with the neurotoxicity seen in AD. The AD pathogen hypothesis suggests that various pathogens can act as triggers to induce and/or lead to the accumulation of these Aβ1–42 monomers (14). Many of these pathogens, such as Chlamydophila pneumoniae (11), Borrelia burgdorferi (17), and Candida glabrata (18), are able to evade the host immune response, particularly in the immunocompromised and/or the elderly, and to infiltrate the brain, forming latent and potentially chronic infections. In turn, these pathogens also go on to induce reactive gliosis (19) and a pro-inflammatory response (8), ultimately leading to progressive neurodegeneration and dementia.
Herpes simplex virus type I (HSV-1) is a pathogen that is gaining increasing attention as a potential causative agent in sporadic AD pathogenesis (15, 20, 21). HSV-1 is a neurotropic double-stranded DNA virus that typically resides throughout the peripheral nervous system in a latent state. HSV-1 reactivation can be asymptomatic, manifest as cold sores, or, in rare cases, results in herpes simplex encephalitis (HSE), indicating the ability of the virus to penetrate the blood-brain barrier (22). Some of the earliest indications that HSV-1 might be linked to AD were the observations that acute HSV-1 infection resulting in HSE often targets the regions of the brain most often affected by AD, with patients demonstrating cognitive and behavioral symptoms typical of patients with AD (23). Furthermore, pioneering studies from the Itzhaki laboratory reported detection of high levels of Herpesviridae DNA from amyloid plaques harvested from postmortem brain tissue of patients with sporadic AD using polymerase chain reaction (PCR) (24–26). Even more recently, an epidemiological study in Taiwan reported that HSV-1 infection and seropositivity led to a significant risk of later development of AD and that antiherpetic treatment at the time of infection markedly reduced that risk by a factor of 10 (27). Given the mounting evidence that HSV-1 has potential causality in the development of AD, we aimed to generate a three-dimensional (3D) human brain model of herpes-induced AD.
Our laboratory has developed a technique to generate expandable and rapidly differentiating human-induced neural stem cell (hiNSC) lines through the direct reprogramming of somatic starting cells such as dermal fibroblasts (28). These hiNSC lines are ideally suited for this brain tissue model for a variety of reasons. Specifically, hiNSCs spontaneously and rapidly differentiate into multiple neuronal and glial subtypes in as few as 4 days without the requirement of lengthy differentiation protocols or media requirements and are stable in long-term cultures. Furthermore, we have demonstrated that hiNSCs are highly responsive to various growth factors, guidance cues, and pharmaceuticals and that they are also highly infectable by neurotropic viruses such as Zika (29). We have also demonstrated that hiNSCs grow well in our previously established 3D brain model (30). This 3D brain-like tissue model was developed using a “top-down bioengineering approach” that involves seeding cells into a biomaterial-based scaffold to recapitulate structural features representative of native brain tissue. Porous silk protein sponges were formed into concentrically shaped donuts recapitulating the neuron-rich gray matter of the brain, which were subsequently infused with type I collagen gel, to support the growth of neurite extensions, thereby mimicking the axon-dense white matter. This 3D brain tissue model system allows for visualization and quantification of neurite patterning and networks and electrophysiological readouts in real time.
While previous reports have suggested a potential link between HSV-1 and up-regulation of certain AD mediators (31), to date, no direct causality of AD by herpes virus in a 3D human brain model has been demonstrated. The aim of this study was to examine the effects of HSV-1 infection in our established 3D human brain–like tissue model (28, 30) to assay for correlations to AD pathology. Here, we demonstrate that relatively high levels of HSV-1 infection in our hiNSC model resulted in cell death, similar to what has been previously reported using other cell types (32), while low levels of the virus induced a remarkable AD-like phenotype not previously reported in vitro. After as few as 3 days of HSV-1 infection [multiplicity of infection (MOI) of 0.0001], we demonstrated that hiNSCs develop large, multicellular, dense Aβ+ fibrillar plaque-like formations (PLFs) upon HSV-1 infection, reminiscent of amyloid plaques found in patients with sporadic AD. HSV-1 also led to endogenous up-regulation of known AD mediators PSEN1 and PSEN2. Furthermore, we demonstrated that HSV-1 infection also induced reactive gliosis and high levels of neuroinflammation frequently observed in neurodegenerative disorders like AD. We also report that antiviral medication worked to diminish this AD-like phenotype, suggesting that this type of intervention might be useful for clinical treatment of AD.
Last, ours is the first to report a 3D human brain–like tissue model of AD that encompasses so many facets of physiological human disease, including Aβ plaque formation, neuronal loss, reactive gliosis, neuroinflammation, and diminished neural network functionality. Our tissue model achieves this robust AD phenotype in a short time relative to other 3D model systems of AD (33) and using unmodified wild-type cells completely in the absence of any exogenous factors specifically known to regulate or induce AD (33–35). Together, we describe a 3D human brain–like tissue model of herpes-induced AD that demonstrates rapid, robust, and physiologically relevant phenotypes in a short period of time. This model will allow for future studies to further elucidate mechanisms of HSV-1–mediated AD pathogenesis and to identify potential downstream targets for treating this devastating disease.
MATERIALS AND METHODS
Generation of hiNSCs
hiNSCs were generated as previously described (28). Briefly, human foreskin fibroblasts were plated at a concentration of 105 cells in one gelatin-coated well of a six-well plate and cultured in fibroblast media [Dulbecco’s modified Eagle’s medium (DMEM), 10% fetal bovine serum, and 1% antibiotic-antimycotic]. Concentrated aliquots of a polycistronic lentivirus expressing OCT4, KLF4, SOX2, and cMYC (Addgene, no. 24603, a gift from J. Cibelli) were used to infect cells in fibroblast medium using polybrene (Millipore) at a MOI of 1 to 2. Media was changed to hiNSC media: knockout (KO) DMEM supplemented with 20% KO xeno-free serum replacement, recombinant basic fibroblast growth factor (20 ng/ml), 1% glutamax, 1% antibiotic-antimycotic, and 0.1 mM β-mercaptoethanol, which also contained 1% KO growth factor cocktail (GFC) (Invitrogen). Four days later, cells were trypsinized and replated onto mouse embryonic fibroblast (MEF) feeder layers that had been previously inactivated by mitomycin C. hiNSC media (without KO-GFC) was subsequently changed every 1 to 3 days. At day 30 or later, colonies were mechanically picked and passaged onto fresh feeder MEF plates. Each picked colony was expanded to generate one hiNSC line. hiNSCs were enzymatically passaged as colonies using trypsin-like enzyme TrypLE (Invitrogen), expanded, and subsequently frozen to make stocks. All generated lines tested negative for mycoplasma contamination.
HSV-1 production and titration
HSV-1 McIntyre strain VR-539 was purchased from American Type Culture Collection (Manassas, VA). Monolayers of Vero cells were infected with HSV-1 at a MOI of 0.01. After 48 hours in culture, infected cells were harvested with three freeze-and-thaw cycles to disrupt cell membranes and release virions. Cells were subjected to low-speed centrifugation, and virus titers were subsequently measured by standard plaque assay. The resulting titer of concentrated HSV-1 was 2 × 107 plaque-forming units (PFU)/ml.
hiNSC differentiation, HSV-1 infection, and valacyclovir HCl treatment
hiNSC colonies were trypsinized off of MEF feeder layers using TrypLE (Invitrogen) and then dissociated by manual pipetting. Cell suspensions were passaged through a 40- to 70-μM cell strainer to remove larger aggregates. Dissociated hiNSCs were cultured on gelatin-coated plates in neurobasal media supplemented with 2% B27 (Invitrogen), 1% glutamax, and 1% antibiotic-antimycotic. For HSV infection, we used purified HSV-1 to directly infect hiNSCs at a range of MOIs (0.00001 to 1), which was calculated based on the starting virus concentration (2 × 107 PFU/ml) and initial seeding density.
Once added to media, inoculum was allowed to remain in cell cultures for the duration of the experiment to mimic low-level brain infection. For mock infections, an equal volume of control culture medium from uninfected Vero cells was used. All virus work was approved by the Tufts Institutional Biosafety Committee. Valacyclovir (VCV) HCl was reconstituted in dH2O and used at a concentration of 200 μM for all subsequent experiments. VCV was added at day 0 (D0; concurrently with HSV-1 infection) or day 1 (D1; 24 hours after infection).
3D cortical brain tissue model
A 3D cortical brain tissue model was generated as previously described (28, 30). Briefly, silk sponges with pore sizes of 500 to 600 μm were prepared from 6% (w/v) Bombyx mori–derived silk solution. Sponges were biopsy-punched into 6-mm discs, with 2-mm holes punched in the center to form donut-shaped scaffolds. Scaffolds were autoclaved and coated with laminin (0.5 mg/ml; Roche, Indianapolis, IN). Dissociated hiNSCs were seeded into the silk porous scaffolds at a density of 106 and allowed to adhere overnight. The following day, collagen gels were made using type I rat tail collagen (Corning, Bedford, MA, USA) as previously described. 3D human brain–like tissue constructs were then cultured in neurobasal media (Invitrogen, Carlsbad, CA) supplemented with 2% B27 (Invitrogen, Carlsbad, CA), 0.5 mM glutamax, and 1% antibiotic-antimycotic (Invitrogen, Carlsbad, CA) for 4 weeks to allow for mature network formation before subsequent HSV-1 infection, with media changes every 3 days.
Quantitative reverse transcription PCR
Total RNA was isolated using the RNeasy Mini Kit (Qiagen). Complementary DNA was generated using iScript (Bio-Rad) according to the manufacturer’s protocols. Quantitative reverse transcription PCR (qRT-PCR) was performed using SYBR Green and the CFX96 Real-Time PCR Detection System (Bio-Rad) and normalized against the housekeeping gene glyceraldehyde phosphate dehydrogenase. All primer sequences are listed in table S1. The qPCR-based Qiagen RT Profiler Array for AD (catalog no. PAHS-057Z) was performed according to the manufacturer’s instructions.
Immunofluorescence
Cells grown in tissue culture plates or in 3D scaffolds were fixed in 4% paraformaldehyde and washed with 1X phosphate-buffered saline (PBS). Samples were incubated with a blocking buffer consisting of PBS, 10% goat serum, and 0.1% Triton X-100. Primary antibodies were added to the blocking buffer and incubated with samples overnight at 4°C. The next day, samples were washed several times with PBS and incubated with a corresponding fluorescently conjugated secondary antibody in a blocking buffer for 1 hour at room temperature. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (Invitrogen). All antibodies used in this study are listed in table S2.
Aß ELISA kit
Aβ40 and Aβ42 Human enzyme-linked immunosorbent assay (ELISA) kits were purchased from Thermo Fisher Scientific. Conditioned media (CM) harvested from mock- and HSV-1–infected hiNSCs were filtered and then subjected to ELISA assay according to the manufacturer’s instructions.
Microscopy
Fluorescent images were obtained using a Keyence BZ-X700 microscope and associated software. Bright-field and fluorescent images were obtained using a Keyence BZ-X700 microscope and associated software. Fluorescent images of 3D samples were taken using a Leica TCS FLIM SP8 (Wetzlar, Germany) confocal microscope. Scanning electron microscopy (SEM) was performed using a Zeiss Evo MA10 (Carl Zeiss Microscopy, Germany) to visualize the morphological properties of 3D brain-like tissue samples. Constructs were thoroughly dried and cross-sectioned and then gold-sputter–coated to prepare for subsequent SEM analysis.
Electrophysiology
Function was assessed by sampling spontaneous electrical activity within mock- and HSV-1–infected samples. Local field potentials (LFPs) were collected using methods consistent with our previous electrophysiological experiments. Briefly, samples were placed into 35-mm plastic petri dishes containing 2 ml of extracellular solution: 130 mM NaCl, 1.25 mM NaH2PO4, 1.8 mM MgSO4, 1.6 mM CaCl2, 3 mM KCl, 10 mM Hepes-NaOH, and 5.5 mM glucose (pH 7.4). Dishes were placed onto a WP-16 Warmed Platform (with a TC-134A Handheld Temperature Controller), which maintained physiological temperature (37°C). Next, samples were secured to the bottom of the dish with a fixed depressor to eliminate signal artifacts associated with the movement of the scaffold. A recording electrode was then positioned within the collagen hydrogel along silk fibers to measure LFPs. Electrodes were fabricated using borosilicate glass pipettes (length, 5 cm; inner diameter, 0.86 mm; outer diameter, 1.5 mm), which were pulled with a Sutter P-97 (Navato, CA, USA). Each glass pipette was pulled to the following specifications: <1 μm in tip diameter, 5- to 8-mm taper, and 40- to 80-megaohm resistance. Before each recording, impedance was measured where 100 megaohm was considered suitable for measurement. LFPs (mV) were collected (Axon Instruments DAC; Intan digital amplifier) with a sampling frequency of 2500 Hz for 2 min to assess the presence of spontaneous electrical activity. Data were collected using Clampex 10.7 (Axon Instruments) and exported to Clampfit 10.7, and traces were processed to extract spontaneous spikes (~1 ms, ±0.35 mV). Spikes per minute were then computed and compared across groups.
Statistics
All data are expressed as means ± SD, and at least three biological replicates were analyzed per experiment. Independent experiments were repeated three times. Data with statistically significant differences were determined by one-factor analysis of variance (ANOVA) with post hoc Tukey test using the statistics software SYSTAT12 (Systat). P < 0.05 was considered significant.
RESULTS
hiNSCs are highly infectable by HSV-1
We first aimed to determine whether hiNSCs were infectable by HSV-1 and to establish an infection regimen for subsequent experiments. hiNSCs were exposed to mock or HSV-1 (MOI of 0.01 to 1) for 24 hours and then subsequently immunostained to identify HSV-1–infected cells (Fig. 1, A and B). At an MOI of 1, nearly 100% of cells were infected after 24 hours. We also determined that expression of HSV-1–UL29 increased over time in culture (Fig. 1C). Furthermore, we demonstrate that virus-infected hiNSCs secrete HSV-1 that is capable of infecting new cells (fig. S1). CM were harvested and filtered from mock- or HSV-1–infected hiNSCs that had been cultured for 2 days. Mock or fresh HSV-1 virus was added to CM, and new hiNSCs were cultured for 4 days. In virus CM samples without additional virus, there were HSV-1–positive cells, suggesting that infected hiNSCs secrete active HSV-1. Together, these data demonstrate how low-level viral inoculations lead to high levels of infection over time, which is reminiscent of chronically reactivating HSV-1 infections in patients.
Fig. 1. hiNSCs are highly infectable and responsive to HSV-1 exposure.
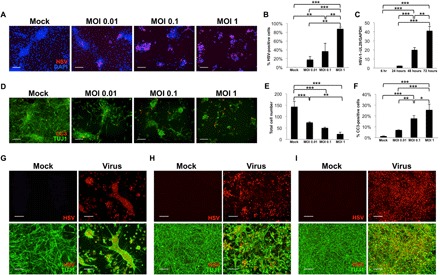
hiNSCs were cultured with varying MOIs for 24 hours and assayed for virus expression by immunostaining (A and B) and quantitative polymerase chain reaction (qPCR) showing viral HSV-1–UL29 expression over time (C). Results from β-III tubulin (TUJ1) and cleaved Caspase3 (CC3) immunostaining (D) indicate that relatively high MOI (~1 or greater) results in rapid and robust cell death in 24 hours, with corresponding quantification of cell number (E) and percentage of CC3-positive cells in response to increasing MOI in hiNSCs (F). Low-level HSV-1 infection causes morphological changes. hiNSCs were cultured 1 day (G), 4 days (H), or 7 days (I) before HSV-1 exposure (MOI of 0.0001) for 2 to 3 days. Immature hiNSCs form HSV-1–positive cell conglomerates in response to infection. Scale bars, 100 μm. Asterisks indicate statistically significant differences with error bars showing means ± SD (*P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001). DAPI, 4′,6-diamidino-2-phenylindole; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
High level of HSV-1 infection results in cell death, while low level infection induces profound morphological changes in hiNSCs
Having established that hiNSCs are highly infectable even at very low MOIs, we also wanted to discern the effect of HSV-1 infection on apoptosis in hiNSCs. hiNSCs were exposed to mock or HSV-1 (MOI of 0.01 to 1) for 24 hours and then subjected to immunostaining against apoptosis marker, cleaved Caspase3 (CC3) (Fig. 1D). We found that increasing MOI resulted in fewer total cells (Fig. 1E) and increased apoptosis (Fig. 1F). Given that even a very low MOI of 0.01 caused substantial cell death in 24 hours, we performed subsequent HSV-1 infections at an MOI of 0.0001. We cultured hiNSCs for 1, 4, or 7 days before HSV-1 exposure for 2 to 3 days and subsequent immunostaining against HSV-1 and pan-neuronal marker β-III tubulin (TUJ1) (Fig. 1, G to I). We found that low-level HSV-1 infection caused profound morphological changes in hiNSCs. Briefly, infected hiNSCs formed large multicellular structures that retained neurite extensions, reminiscent of previously described syncytia present in the extracellular space of AD brains (36). This type of cell fusion has also been reported in a monkey kidney epithelial cell line in response to HSV-1 infection (37). The more immature the neurons at the time of infection, the larger the conglomerate structure formed. This result suggests that relatively high HSV-1 infection results in cell death, while low-level infection formed large, multicellular structures.
hiNSCs produce Aβ+ PLFs and specifically regulate AD mediators in response to HSV-1 infection
Next, we wanted to characterize the structures that formed in response to low-level HSV-1 infection in hiNSCs. Thioflavins are histological dyes used for visualizing protein aggregation. Thioflavin T (ThT) is a benzothiazole dye that exhibits enhanced fluorescence (emission, 445 to 482 nm) upon binding to amyloid fibrils and is considered the “gold standard” for identifying these structures in clinical samples (38). We cultured hiNSCs for 4 days and then subjected them to mock or low-level HSV-1 infection (MOI of 0.0001) for 3 days before subsequent fixation and ThT histological staining (Fig. 2A). These multicellular structures formed in HSV-1–infected hiNSCs stained strongly positive for ThT, suggesting the presence of amyloid fibrils within these structures. We confirmed HSV-1 infection by qPCR (Fig. 2B) and went on to further characterize the virally induced protein aggregates by performing an ELISA against the Aβ isoforms Aβ1–40 and Aβ1–42 using CM from mock- or HSV-1–infected hiNSCs (Fig. 2C). We found a statistically significant increase in Aβ1–42, but not Aβ1–40, in HSV-1–infected CM, which is consistent with AD-like pathology.
Fig. 2. HSV infection causes Aβ+ PLFs and specific regulation of AD mediators in hiNSCs.
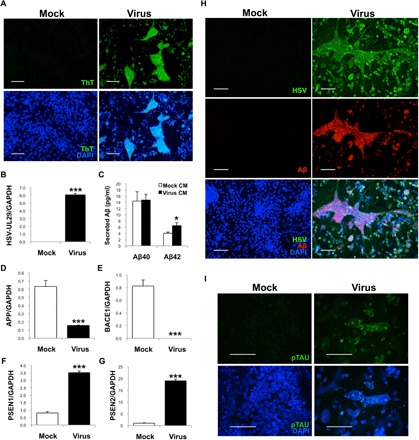
(A) hiNSCs were cultured for 4 days then subjected to HSV-1 infection for 3 days. HSV-1–infected hiNSCs demonstrated large areas of thioflavin T (ThT)–positive regions. (B) qPCR confirms HSV-1 infection in hiNSCs. (C) Results from an ELISA against Aβ isoforms Aβ1–40 and Aβ1–42, using CM from mock- or HSV-1–infected hiNSCs, reveals a statistically significant increase in Aβ1–42, but not Aβ1–40, in HSV-1–infected CM. qPCR analysis reveals significant down-regulation of APP and BACE1 (D and E) and robust up-regulation of γ-secretase subunits PSEN1/2 (F and G). (H) Further immunostaining reveals large multicellular conglomerates of Aβ+ PLFs that overlaps with HSV staining. (I) Those PLFs also stain positive for hyperphosphorylated Tau. Scale bars, 100 μm. Asterisks indicate statistically significant differences with error bars showing means ± SD (*P ≤ 0.05 and ***P ≤ 0.001).
We performed additional qPCR to ascertain whether known mediators involved in AD were also involved in the HSV-1–induced generation of PLFs in hiNSCs. qPCR analysis demonstrated that HSV-1 caused down-regulation of APP and BACE1 (Fig. 2, D and E), as well as up-regulation of PSEN1/2 (Fig. 2, F and G) in infected hiNSCs when compared to uninfected controls.
To further characterize the PLFs that formed in response to HSV-1 infection in hiNSCs, we immunostained against HSV and Aβ fibrils (Fig. 2H) to determine whether infected cells specifically expressed Aβ protein. After 3 days of low-level HSV-1 infection (MOI of 0.0001), nearly all hiNSCs were infected, with a portion of those infected cells expressing Aβ almost exclusively in regions of PLFs. The average number of nuclei within each individual PLF was 19, suggesting that these PLFs were composed of multiple cells.
As Aβ plaques are not the only pathological features of AD, we also assayed for the presence of NFTs, which are formed by the hyperphosphorylation and subsequent aggregation of a microtubule-associated protein known as tau. We immunostained mock- and virus-infected samples using a phospho-Tau antibody that specifically recognizes paired helical filament–Tau with phosphorylated residues Ser202/Thr205 (Fig. 2I). In HSV-1–treated hiNSCs, strongly positive Tau staining is visible on or near PLFs.
HSV-1 infection results in increased gliosis and neuroinflammation in hiNSCs
We have previously demonstrated that hiNSCs spontaneously differentiate into neurons and glial fibrillary acidic protein (GFAP)–positive glia (28). Given that HSV-1 infection resulted in induction of PLFs similar to those found in patients with AD, we also aimed to discern whether other phenotypic characteristics of AD were induced in HSV-1–infected hiNSCs, such as reactive gliosis and up-regulation of pro-inflammatory cytokines. We cultured hiNSCs for 4 days, before subjecting to mock or HSV-1 (MOI of 0.0001) for 3 days, and then performed subsequent analyses.
We first stained using an antibody against GFAP (Fig. 3A). In mock-infected hiNSCs, ~9% of cells demonstrated GFAP-positive staining after 1 week in culture (arrows), consistent with previous reports (28). HSV-1–infected hiNSCs expressed ~71% GFAP positivity (arrowheads), which was consistent with qPCR results (Fig. 3B). Furthermore, these GFAP-positive cells induced by HSV-1 have a swollen, globular morphology similar to that of gemistocytic astrocytes (39). The altered morphology is reminiscent of what is observed in postmortem tissue from AD patients with numerous clusters of GFAP-positive astrocytes that become more confluent as the disease progresses (7). We also tested other known markers of reactive astrocytes, vimentin, Lipocalin2 (LCN2), and Serpina3 (SERP3) (Fig. 3, C to E) and found that these markers were all highly up-regulated in response to HSV-1 infection in hiNSCs.
Fig. 3. HSV-1 causes reactive gliosis and inflammation reminiscent of neurodegeneration in hiNSCs.
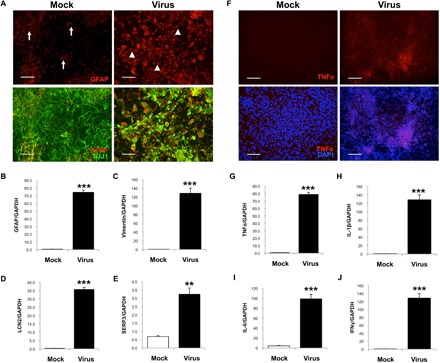
(A and B) HSV-1–infected hiNSCs highly express glia marker GFAP. Immunostaining results reveal increased number of GFAP+ cells and an altered morphology suggestive of gliosis. Other known markers of reactive astrocytes were also up-regulated in HSV-1–infected hiNSCs: (C) vimentin, (D) LCN2, and (E) SERP3. (F and G) Pro-inflammatory marker TNFα was also up-regulated. In HSV-1–infected hiNSCs, qPCR analysis revealed similarly high expression of other inflammatory markers known to be involved in AD: (H) IL-1β, (I) IL-6, and (J) IFNγ. Scale bars, 100 μm. Asterisks indicate statistically significant differences with error bars showing means ± SD (**P ≤ 0.01 and ***P ≤ 0.001).
We also aimed to assay markers of neuroinflammation. Tumor necrosis factor–α (TNFα) is one of the major pro-inflammatory cytokines known to play a role in neurodegenerative diseases (40). We found that TNFα was up-regulated in HSV-1–infected hiNSCs as indicated by immunostaining and qPCR (Fig. 3, F and G). Other known inflammatory mediators involved in AD are interleukin-1β (IL1β), IL-6, and interferon γ (IFNγ) (8). We assayed for expression of these markers and observed high levels of expression in virus-infected hiNSCs compared to uninfected controls.
VCV treatment results in decreased HSV infection, decreased Aβ+ plaque-like formation, and normalized expression of AD mediators in infected hiNSCs
Given the data to suggest that HSV-1 directly caused an AD-like phenotype in hiNSCs, we wanted to determine whether inhibiting the virus using established antiviral medication would prevent or reduce the resulting AD phenotype. One of the most commonly used antiviral agents for treating HSV-1 infections is VCV, which specifically targets viral DNA replication in infected cells. We cultured hiNSCs for 4 days before subjecting to either mock or HSV-1 infection (MOI 0.0001). In addition, we added either vehicle or 200 μM VCV at D0 or D1 to observe potential changes with regard to timing of drug administration in the acquisition of an AD-like phenotype. hiNSCs treated with vehicle or VCV alone in the absence of virus had no discernible effect (data not shown). We first immunostained against HSV and Aβ (Fig. 4A) to determine any differences in the rate of HSV-1 infection and PLF generation in response to drug treatment. As expected, VCV caused a significant decrease in HSV-1 infection using both drug regimens as indicated by immunostaining (Fig. 4B) and qPCR (Fig. 4C); however, VCV treatment at D0 almost completely abolished infection. VCV treatment also significantly reduced the average number and size of PLFs in HSV-1–infected hiNSCs (Fig. 4, D and E). We went on to assay the expression of previously characterized AD mediators—such as APP, BACE1, PSEN1, and PSEN2 (Fig. 4, F to I)–and found that VCV treatment at D0 nearly rescued all markers back to control values, while D1 VCV treatment showed partial rescue.
Fig. 4. VCV treatment results in reduced AD-like phenotype induced by HSV-1 infection in hiNSCs.
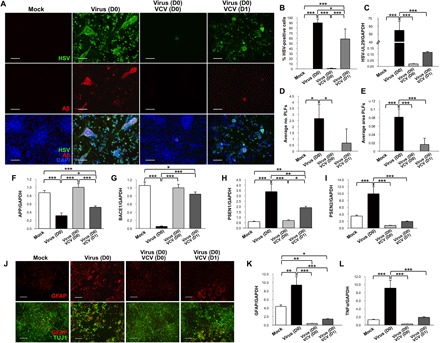
(A) Treatment with VCV reduces HSV-1 infection as well as generation of Aβ+ PLFs. (B) Quantification of infection and (C) qPCR for HSV-UL29. VCV causes a decrease in (D) total number and (E) average area of PLFs. VCV helps to normalize expression of AD mediators back to control levels in HSV-1–infected hiNSCs: (F) APP, (G) BACE1, (H) PSEN1, and (I) PSEN2. (J) VCV treatment rescues gliosis phenotype in HSV-1–infected hiNSCs. qPCR analysis reveals restoration of (K) GFAP and (L) TNFα expression. Scale bars, 100 μm. Asterisks indicate statistically significant differences with error bars showing means ± SD (*P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001).
VCV treatment of HSV-1–infected hiNSCs results in reduced gliosis and neuroinflammation
We also aimed to determine whether VCV treatment had a similar effect in reducing HSV-1–induced gliosis and neuroinflammation in hiNSCs. We performed parallel experiments using the previously described dosing regimens and then immunostained for glial marker GFAP as well as TUJ1 to visualize neurons. As observed in previous experiments, HSV-1 induced robust glial formation (Fig. 4J) in hiNSCs compared to controls. VCV treatment at D0 almost completely restored GFAP expression to mock-treated samples as indicated by both immunostaining and qPCR (Fig. 4K). We also found that VCV treatment in HSV-1–infected hiNSCs significantly reduced the pro-inflammatory mediator TNFα at both D0 and D1 dosing regimens.
Moreover, we aimed to understand whether HSV-1–secreted factors could cause gliosis even in the absence of infection (fig. S2). CM were harvested and filtered from mock- or HSV-1–infected hiNSCs that had been cultured for 2 days. Fresh HSV-1 virus was added to CM in the absence or presence of antiviral treatment VCV, and new hiNSCs were cultured for 4 days. β-III tubulin (TUJ1) and GFAP immunostaining was performed to visualize neurons and glia, respectively. Preventing HSV-1 infection using VCV in virus CM resulted in increased reactive glia formation, suggesting that HSV-1–induced factors play a role in astrogliosis even when no virus is active. This suggests that an initial low-level infection is sufficient to initiate a cascade of neurodegenerative events that is independent of the initial HSV-1 infection.
hiNSCs cultured in 3D brain model develop AD-like phenotype in response to low-level HSV-1 infection
With the observation of robust AD-like phenotypes in our monolayer cultures, we pursued our goal of generating a 3D human brain model of herpes-induced AD. To achieve this goal, we used our established silk protein scaffold–based system, which was previously described (28, 30). We first developed a seeding and infection protocol (Fig. 5A). Briefly, silk porous laminin-coated scaffolds (height, 2 mm; outer diameter, 6 mm; inner diameter, 2 mm) were seeded with 106 hiNSCs per sample, which were allowed to adhere overnight. The following day, the porous donuts were infused with type I collagen and allowed to stabilize over the course of 4 weeks in culture. These in vitro brain tissues were then subjected to mock or low-level HSV-1 infection (MOI of 0.0001) for 1 week before subsequent analyses.
Fig. 5. hiNSCs cultured in a 3D brain model develop an AD-like phenotype in response to low-level HSV-1 infection.
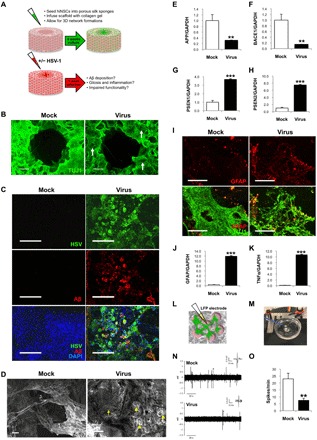
(A) Model of 3D human brain–like model. hiNSCs were cultured in the donut model for 4 weeks before HSV-1 infection for 1 week. (B) Low-magnification images of brain model showing β-III tubulin (TUJI1) and beta amyloid (Aβ) immunostaining. Arrows point to regions of neuronal loss in HSV-1–infected tissues. (C) Confocal images showing HSV and Aβ immunostaining in both mock- and virus-infected constructs. (D) Scanning electron micrographs reveal morphological changes in HSV-1–infected tissue constructs. Arrowheads and arrow indicate the presence of both small and relatively larger PLFs, respectively. Scale bars, 10 μm. (E) APP and (F) BACE1 were down-regulated, and (G and H) PSEN1/2 were up-regulated, similar to results in monolayer cultures. HSV-1 induces gliosis and neuroinflammation in 3D cultures. Glial marker GFAP is up-regulated as evidenced by (I) immunostaining and (J) qPCR, as well as (K) pro-inflammatory marker TNFα. LFP recordings were performed to assess functionality in 3D cultures. Schematic representation of microelectrode placement for LFP assessments (L) and an image of constructs placed on electrophysiology rigs for recordings (M) (photo credit: Dana M Cairns, Tufts University). (N and O) Representative traces of mock versus infected brain models shows that HSV-1–treated donuts show significantly less activity reminiscent of impaired functionality in patients with AD. Asterisks indicate statistically significant differences with error bars showing means ± SD (**P ≤ 0.01 and ***P ≤ 0.001).
We fixed samples and immunostained against pan-neuronal marker TUJ1 to visualize growth of brain-like tissue in our model (Fig. 5B). After 1 week of HSV-1 infection, regions of neuronal loss were visible even at low magnification (arrows). We wanted to first confirm the penetration of the virus by immunostaining (Fig. 5C). We also wanted to understand whether observations made in monolayer cultures would also be observed in our 3D brain tissue model. Robust Aβ+ PLFs were observed only in the HSV-1–infected samples. We also performed SEM on the 3D human brain–like tissue constructs and found morphological changes in HSV-1–infected samples, namely, the presence of both small and large PLF structures (Fig. 5D), that were reminiscent of AD plaques. Upon closer examination of PLFs by confocal microscopy, these structures also stained positive for ThT and Tau1 (fig. S3).
We went on to assay the expression of previously characterized AD mediators—such as APP, BACE1, PSEN1, and PSEN2 (Fig. 5, F to H)—and found that these mediators were similarly regulated as in monolayer cultures, with significant decreases in both APP and BACE1, and a corresponding increase in PSEN1/2. We further wanted to discern whether reactive gliosis and inflammation were occurring in these 3D cultures as in our previous experiments. In accordance with previous observations, GFAP expression was significantly increased as indicated by both immunostaining (Fig. 5I) and qPCR (Fig. 5J). Similarly, TNFα expression was also significantly up-regulated in HSV-1–infected hiNSC donuts (Fig. 5K) when compared to uninfected controls. To further understand this AD-like pathology, we performed a qPCR array of known AD mediators (Qiagen RT Profiler Array for AD) to compare expression levels between mock- and HSV-1–infected 3D human brain–like tissue constructs (fig. S4 and table S3). Forty genes in this AD-specific array were significantly up-regulated in HSV-1–infected samples.
HSV-1–treated 3D human brain–like tissue cultures show significantly less electrophysiological activity reminiscent of impaired functionality in patients with AD
Last, the gold standard of any bioengineered tissue model is functionality. To be considered “brain-like tissue,” the tissue construct must generate and maintain electrical activity. Furthermore, we hypothesized that our herpes-induced AD brain model would also recapitulate patient symptoms with regard to diminished brain activity. High levels of Aβ oligomers have been shown to cause synapse malfunction (41) and reduce long-term potentiation, which correlates with memory loss (42).
LFP is an electrophysiological recording generated by the total electric current flowing by resident neurons present in a small area of nervous tissue. Voltage is produced across this region by synaptic activity, which can be measured and quantified. Briefly, a microelectrode is placed on the tissue, which reads the voltage change in that region over time. These relative voltage changes or “spikes” are indicative of actively firing neurons present in the tissue (Fig. 5, L and M). LFP recordings were taken from both mock- and HSV-1–infected donut cultures maintained for 5 weeks. Representative traces (Fig. 5N) and the averages of spikes per minute for n = 6 donuts per treatment (Fig. 5O) demonstrated that HSV-1–treated donuts showed significantly less activity compared to controls, which is reminiscent of the impaired brain functionality seen in patients with AD.
DISCUSSION
Generating in vitro human models of sporadic AD poses a considerable challenge because of the complex, undefined etiology and slow disease progression that naturally occurs in patients. Here, we describe a multidisciplinary approach to address this critical need by generating robust and physiologically relevant 3D human tissues for studying sporadic AD through the use of hiNSC technology and HSV-1 in combination with a bioengineered brain tissue model.
A number of in vitro human models of AD have been developed. Human-induced pluripotent stem cell (hiPSC) technology has provided a useful platform with which to study these types of neurodegenerative diseases; however, it is not without limitations. One benefit to the hiPSC methodology is the ability to convert specific patient cells to a pluripotent state. In this way, cells can be harvested from patients with known disorders and/or genetic mutations, reprogrammed, and then differentiated to essentially create a “disease in a dish.” A number of protocols have been developed to differentiate iPSCs into various neuronal phenotypes; however, these protocols are often very time consuming, require multiple complicated intermediate steps such as the formation of nonadherent embryoid bodies and neurospheres, and result in large variability in achieving neuronal differentiation (43). A number of studies have been conducted using cells derived from patients with familial EOAD, which is associated with mutations in APP, PSEN1, or PSEN2 (2). As these genetic mutations provide the stimulus for initiating AD in vitro, these studies do not address the issue of disease onset or progression in most AD cases, which occur sporadically. Furthermore, because the process by which somatic cells are converted into iPSCs is thought to epigenetically “reset” the cells into an embryonic-like state, it has been suggested that these cell lines lack an aging profile upon reprogramming. Studies using iPSC-derived neurons demonstrate an immature phenotype and a lack of aging markers (44), which poses an issue when attempting to model a neurodegenerative disease in which the primary risk factor is age. It has been proposed that direct reprogramming or transdifferentiation, methods that bypass the pluripotent state, more efficiently maintains an aging signature in resulting neurons (45).
Other studies have taken alternative approaches to generate these types of human disease models by synthetically creating an AD-like microenvironment. While some studies have shown certain characteristics of AD, such as neuronal loss (46) or Tau phosphorylation (47), most of these models are often unable to fully recapitulate the more complex features of AD, such as plaque formation, reactive gliosis, and loss of neural tissue functionality. In addition, to generate some of these neurodegenerative models, many of these systems require months of culture before establishing any discernible phenotype (33). Perhaps, most, if not all, studies to date specifically require the incorporation of known AD mediators to initiate an AD-like state. For example, multiple studies induce AD in their respective model systems through the use of viruses overexpressing AD mediators such as PSEN1 (33), pro-inflammatory cytokines (48), and/or incorporation of detrimental Aβ peptides (33, 35, 48). The requirement of the use of exogenous mediators of AD to artificially induce plaque formation is not reflective of the in vivo condition, especially considering that the actual cause of AD is so elusive.
There is increasing evidence to suggest a clinical correlation between pathogens such as herpes and the development of sporadic AD (20, 21), including the findings that HSV-1 DNA was detected in the brain tissue of patients with AD (24–26) and recent epidemiological studies even reporting HSV-1 infection as a significant risk factor of later AD onset (27). While there is no published correlative evidence of active herpes infections in patients with AD, these clinical findings suggest that active infection may not be a requirement for AD, but rather that prior history of herpes infection is sufficient to initiate AD pathogenesis. Ours is not the first to describe an experimental link between HSV-1 and AD. Several reports have described in vitro monolayer cell culture models in which HSV-1 has led to neuronal death (32, 49), Tau phosphorylation (50), and intracellular expression of various isoforms of APP cleavage products (51–53). While the finding that certain proteins known to be involved in the amyloidogenic pathway are also up-regulated in response to HSV-1 infection is exciting, few studies have reported the formation of multicellular plaque-like structures in neuron cultures. These PLFs are composed of multiple merged infected hiNSCs, averaging ~19 nuclei per individual PLF (data not shown). A recent study suggested an intracellular basis for the ultimate formation of these extracellular Aβ plaques (54). Briefly, small Aβ oligomers form and get transported to vesicular bodies, after which a phagocytic event occurs to generate multicellular plaques that apoptose releasing contents into the extracellular space. It is plausible that similar mechanisms occur in our HSV-mediated in vitro system, as the PLFs formed in these cultures are multicellular and apoptotic.
Furthermore, most of these HSV-AD studies have relied on the use of primary cells derived from rodent models (49, 52) and/or human cancer cell lines (50–53) and often require the use of exogenous AD mediators (51, 53). Recent work by Eimer et al. (34) reported that Aβ proteins sequestered herpes viruses in 3D human neural cell cultures using an immortalized cell line. However, this study required the use of lentiviruses overexpressing human b-APP or APP and PSEN1, containing familial AD mutations, which is not truly physiologically reflective of sporadic AD pathogenesis. In addition, this model did not demonstrate multiple aspects of AD physiology including multicellular plaque formation, reactive gliosis, or loss of brain tissue functionality.
While our study demonstrates multiple phenotypic characteristics of patients with AD, it was somewhat unexpected that BACE1 expression was decreased in response to HSV-1 infection, a finding that was consistent across all experiments. We demonstrated a significant decrease in APP expression, which aligns with results from previous research (51). Two enzymes, β-secretase and γ-secretase, sequentially cleave transmembrane protein APP to generate Aβ peptides. As the initial BACE1-mediated cleavage event is believed to initiate Aβ production, inhibition of this enzyme has emerged as a target for therapeutic treatment of AD. Because HSV-1 causes the production of Aβ1–42 in hiNSCs, given its role in the amyloidogenic processing of the APP protein, the hypothesis is that HSV-1 would also increase BACE1 expression (2) as it has previously in HSV-1–infected Vero cell cultures (31). BACE1 functions in other capacities outside of APP regulation. BACE1 KO (BACE1−/−) mice have been reported to exhibit smaller size, decreased survival, spontaneous seizures (55), and memory deficits (56). BACE1 is highly expressed in the developing nervous system, suggestive of its critical role in neuronal proliferation, differentiation, and maturation. In addition, conditional KO of BACE1 in adult animals resulted in disorganization of the hippocampus, suggesting that BACE1 inhibitor drugs developed for AD treatment might have off-target detrimental effects on learning and memory (57), such as neuronal differentiation. Because BACE1 is so intricately involved in many of these developmental processes, we postulate that, in our neural stem cell system, BACE1 functions in other capacities outside of APP regulation and that the HSV-1–induced decrease in BACE1 expression is the consequence of impaired differentiation of hiNSCs.
To identify potential factors that may compensate for BACE1 in APP regulation, we performed a qPCR array of known AD mediators (Qiagen RT Profiler Array for AD) to compare expression levels between mock- and HSV-1–infected 3D brain-like tissue constructs (fig. S4 and table S3). Forty genes in this AD-specific array were significantly up-regulated in HSV-1–infected 3D brain-like constructs. Of particular interest were cathepsin G (CTSG) (86.18-fold up-regulation) and BACE2 (19.03-fold up-regulation) (table S3). These two AD-associated factors, which are both highly up-regulated by HSV-1 in 3D human brain–like constructs, have both been suggested to play similar roles to BACE1 in AD pathogenesis. CTSG was shown to cleave APP, and its expression has been localized to the temporal cortex of both AD and aged human brain tissue, often associated with Aβ plaques (58). In addition, while the role of BACE2, a β-secretase that has 45% homology with BACE1, has often been debated, several recent reports have directly implicated BACE2 with AD pathogenesis. Unexpectedly high levels of BACE2 expression were identified in AD brain tissue, demonstrating higher enzymatic activity in samples taken from patients at preclinical stages of AD with expression colocalizing with reactive astrocytes (59). Furthermore, BACE2 was also recently shown to function as a conditional β-secretase as a novel mechanism of AD pathogenesis (60). Together, we suggest that BACE1 is down-regulated in our system, likely because of its prominent role in differentiation of neural stem cells, and that CTSG and/or BACE2 are likely candidates that compensate for this lack of BACE1 expression by functioning in the primary cleavage event of APP that initiates Aβ plaque generation and AD pathogenesis.
While previous reports have suggested a potential link between HSV-1 and up-regulation of certain AD mediators (31), to date, no one has shown direct causality of AD by herpes virus in a 3D human brain model. Furthermore, ours is the first to report a human model of AD that displays so many physiologically relevant features of human disease in one system, including the generation of large, multicellular, dense Aβ+ fibrillar PLFs, neuronal loss, reactive gliosis, neuroinflammation, and diminished neural network functionality. We also demonstrate efficacy in reducing herpes-induced AD phenotypes through the use of clinically relevant antiviral medication, suggesting that this type of pharmaceutical intervention might be useful for preventing and/or treating AD in patients. Importantly, our 3D human brain–like tissue model requires a shorter time period to achieve an AD-like phenotype relative to other 3D systems of AD (33) and uses unmodified stem cells completely in the absence of any established mediators known to induce AD specifically (33–35). Together, we describe a rapid and reproducible 3D human brain–like tissue model of herpes-induced sporadic AD, which yields a physiologically relevant phenotype that closely mimics pathological findings in human patients. This robust bioengineered system will provide the opportunity to further elucidate mechanisms of sporadic AD pathogenesis and to ultimately develop more efficacious strategies for treating this complex and devastating disease.
Supplementary Material
Acknowledgments
We thank R. Itzhaki of The University of Manchester for incredibly helpful discussion. Funding: This research was supported by the Allen Discovery Center program through the Paul G. Allen Frontiers Group (12171) and the NIH (R01NS092847, P41EB027062, and U19AI13115). Competing interests: The authors declare that they have no competing interests. Author contributions: D.M.C. conceived the idea, performed the experiments and data analysis, and wrote the manuscript. N.R., R.N.P., and K.G.W. performed the experiments and data analysis. L.G. and D.L.K. supervised the project. All authors commented on the manuscript. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/19/eaay8828/DC1
REFERENCES AND NOTES
- 1.Alzheimer's Association , 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 12, 459–509 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Naj A. C., Schellenberg G. D.; Alzheimer’s Disease Genetics Consortium (ADGC) , Genomic variants, genes, and pathways of Alzheimer’s disease: An overview. Am. J. Med. Genet. B Neuropsychiatr. Genet. 174, 5–26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weyer S. W., Klevanski M., Delekate A., Voikar V., Aydin D., Hick M., Filippov M., Drost N., Schaller K. L., Saar M., Vogt M. A., Gass P., Samanta A., Jäschke A., Korte M., Wolfer D. P., Caldwell J. H., Müller U. C., APP and APLP2 are essential at PNS and CNS synapses for transmission, spatial learning and LTP. EMBO J. 30, 2266–2280 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelleher R. J. III, Shen J., Presenilin-1 mutations and Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 114, 629–631 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Strooper B., Vassar R., Golde T., The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 6, 99–107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takahashi R. H., Nagao T., Gouras G. K., Plaque formation and the intraneuronal accumulation of β-amyloid in Alzheimer’s disease. Pathol. Int. 67, 185–193 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Perez-Nievas B. G., Serrano-Pozo A., Deciphering the astrocyte reaction in Alzheimer’s disease. Front. Aging Neurosci. 10, 114 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ho G. J., Drego R., Hakimian E., Masliah E., Mechanisms of cell signaling and inflammation in Alzheimer’s disease. Curr. Drug Targets Inflamm. Allergy 4, 247–256 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Reddy P. H., Mani G., Park B. S., Jacques J., Murdoch G., Whetsell W. Jr., Kaye J., Manczak M., Differential loss of synaptic proteins in Alzheimer’s disease: Implications for synaptic dysfunction. J. Alzheimers Dis. 7, 103–117 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Arber C., Lovejoy C., Wray S., Stem cell models of Alzheimer’s disease: Progress and challenges. Alzheimers Res. Ther. 9, 42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balin B. J., Little C. S., Hammond C. J., Appelt D. M., Whittum-Hudson J. A., Gérard H. C., Hudson A. P., Chlamydophila pneumoniae and the etiology of late-onset Alzheimer’s disease. J. Alzheimers Dis. 13, 371–380 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Miklossy J., Emerging roles of pathogens in Alzheimer disease. Expert Rev. Mol. Med. 13, e30 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Itzhaki R. F., Lathe R., Balin B. J., Ball M. J., Bearer E. L., Braak H., Bullido M. J., Carter C., Clerici M., Cosby S. L., del Tredici K., Field H., Fulop T., Grassi C., Griffin W. S. T., Haas J., Hudson A. P., Kamer A. R., Kell D. B., Licastro F., Letenneur L., Lövheim H., Mancuso R., Miklossy J., Otth C., Palamara A. T., Perry G., Preston C., Pretorius E., Strandberg T., Tabet N., Taylor-Robinson S. D., Whittum-Hudson J. A., Microbes and Alzheimer’s disease. J. Alzheimers Dis. 51, 979–984 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harris S. A., Harris E. A., Herpes simplex virus type 1 and other pathogens are key causative factors in sporadic alzheimer’s disease. J. Alzheimers Dis. 48, 319–353 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Readhead B., Haure-Mirande J. V., Funk C. C., Richards M. A., Shannon P., Haroutunian V., Sano M., Liang W. S., Beckmann N. D., Price N. D., Reiman E. M., Schadt E. E., Ehrlich M. E., Gandy S., Dudley J. T., Multiscale analysis of independent alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 99, 64–82.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giuffrida M. L., Caraci F., Pignataro B., Cataldo S., de Bona P., Bruno V., Molinaro G., Pappalardo G., Messina A., Palmigiano A., Garozzo D., Nicoletti F., Rizzarelli E., Copani A., β-amyloid monomers are neuroprotective. J. Neurosci. 29, 10582–10587 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miklossy J., Alzheimer’s disease - a neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J. Neuroinflammation 8, 90 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pisa D., Alonso R., Juarranz A., Rábano A., Carrasco L., Direct visualization of fungal infection in brains from patients with Alzheimer’s disease. J. Alzheimers Dis. 43, 613–624 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Bates K. A., Fonte J., Robertson T. A., Martins R. N., Harvey A. R., Chronic gliosis triggers Alzheimer’s disease-like processing of amyloid precursor protein. Neuroscience 113, 785–796 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Itzhaki R. F., Corroboration of a major role for herpes simplex virus type 1 in Alzheimer’s disease. Front. Aging Neurosci. 10, 324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Itzhaki R. F., Herpes simplex virus type 1 and Alzheimer’s disease: Possible mechanisms and signposts. FASEB J. 31, 3216–3226 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Liu H., Qiu K., He Q., Lei Q., Lu W., Mechanisms of blood-brain barrier disruption in herpes simplex encephalitis. J. Neuroimmune Pharmacol. 14, 157–172 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Ball M. J., “Limbic predilection in Alzheimer dementia: Is reactivated herpesvirus involved?”. Can. J. Neurol. Sci. 9, 303–306 (1982). [DOI] [PubMed] [Google Scholar]
- 24.Jamieson G. A., Maitland N. J., Wilcock G. K., Craske J., Itzhaki R. F., Latent herpes simplex virus type 1 in normal and Alzheimer’s disease brains. J. Med. Virol. 33, 224–227 (1991). [DOI] [PubMed] [Google Scholar]
- 25.Jamieson G. A., Maitland N. J., Wilcock G. K., Yates C. M., Itzhaki R. F., Herpes simplex virus type 1 DNA is present in specific regions of brain from aged people with and without senile dementia of the Alzheimer type. J. Pathol. 167, 365–368 (1992). [DOI] [PubMed] [Google Scholar]
- 26.Wozniak M. A., Mee A. P., Itzhaki R. F., Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J. Pathol. 217, 131–138 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Itzhaki R. F., Lathe R., Herpes viruses and senile dementia: First population evidence for a causal link. J. Alzheimers Dis. 64, 363–366 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Cairns D. M., Chwalek K., Moore Y. E., Kelley M. R., Abbott R. D., Moss S., Kaplan D. L., Expandable and rapidly differentiating human induced neural stem cell lines for multiple tissue engineering applications. Stem Cell Reports 7, 557–570 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cairns D. M., Boorgu D. S. S. K., Levin M., Kaplan D. L., Niclosamide rescues microcephaly in a humanized in vivo model of Zika infection using human induced neural stem cells. Biol. Open 7, bio031807 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chwalek K., Tang-Schomer M. D., Omenetto F. G., Kaplan D. L., In vitro bioengineered model of cortical brain tissue. Nat. Protoc. 10, 1362–1373 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wozniak M. A., Frost A. L., Preston C. M., Itzhaki R. F., Antivirals reduce the formation of key Alzheimer’s disease molecules in cell cultures acutely infected with herpes simplex virus type 1. PLOS One 6, e25152 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bourgade K., Le Page A., Bocti C., Witkowski J. M., Dupuis G., Frost E. H., Fülöp T. Jr., Protective effect of Amyloid-β peptides against herpes simplex virus-1 infection in a neuronal cell culture model. J. Alzheimers Dis. 50, 1227–1241 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Choi S. H., Kim Y. H., Hebisch M., Sliwinski C., Lee S., D’Avanzo C., Chen H., Hooli B., Asselin C., Muffat J., Klee J. B., Zhang C., Wainger B. J., Peitz M., Kovacs D. M., Woolf C. J., Wagner S. L., Tanzi R. E., Kim D. Y., A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515, 274–278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eimer W. A., Vijaya Kumar D. K., Navalpur Shanmugam N. K., Rodriguez A. S., Mitchell T., Washicosky K. J., György B., Breakefield X. O., Tanzi R. E., Moir R. D., Alzheimer’s disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron 100, 1527–1532 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Papadimitriou C., Celikkaya H., Cosacak M. I., Mashkaryan V., Bray L., Bhattarai P., Brandt K., Hollak H., Chen X., He S., Antos C. L., Lin W., Thomas A. K., Dahl A., Kurth T., Friedrichs J., Zhang Y., Freudenberg U., Werner C., Kizil C., 3D culture method for alzheimer’s disease modeling reveals interleukin-4 rescues Aβ42-induced loss of human neural stem cell plasticity. Dev. Cell 46, 85–101.e8 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Bondareff W., Age-related changes in brain extracellular space affect processing of amyloid-β peptides in Alzheimer’s disease. J. Alzheimers Dis. 35, 1–6 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Carmichael J. C., Yokota H., Craven R. C., Schmitt A., Wills J. W., The HSV-1 mechanisms of cell-to-cell spread and fusion are critically dependent on host PTP1B. PLOS Pathog. 14, e1007054 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Biancalana M., Koide S., Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta 1804, 1405–1412 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simpson J. E., Ince P. G., Lace G., Forster G., Shaw P. J., Matthews F., Savva G., Brayne C., Wharton S. B.; MRC Cognitive Function and Ageing Neuropathology Study Group , Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol. Aging 31, 578–590 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Decourt B., Lahiri D. K., Sabbagh M. N., Targeting tumor necrosis factor alpha for Alzheimer’s disease. Curr. Alzheimer Res. 14, 412–425 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shankar G. M., Bloodgood B. L., Townsend M., Walsh D. M., Selkoe D. J., Sabatini B. L., Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walsh D. M., Klyubin I., Fadeeva J. V., Cullen W. K., Anwyl R., Wolfe M. S., Rowan M. J., Selkoe D. J., Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Hu B. Y., Weick J. P., Yu J., Ma L. X., Zhang X. Q., Thomson J. A., Zhang S. C., Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc. Natl. Acad. Sci. U.S.A. 107, 4335–4340 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patani R., Lewis P. A., Trabzuni D., Puddifoot C. A., Wyllie D. J. A., Walker R., Smith C., Hardingham G. E., Weale M., Hardy J., Chandran S., Ryten M., Investigating the utility of human embryonic stem cell-derived neurons to model ageing and neurodegenerative disease using whole-genome gene expression and splicing analysis. J. Neurochem. 122, 738–751 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mertens J., Reid D., Lau S., Kim Y., Gage F. H., Aging in a dish: iPSC-derived and directly induced neurons for studying brain aging and age-related neurodegenerative diseases. Annu. Rev. Genet. 52, 271–293 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.LeBlanc A., Liu H., Goodyer C., Bergeron C., Hammond J., Caspase-6 role in apoptosis of human neurons, amyloidogenesis, and Alzheimer’s disease. J. Biol. Chem. 274, 23426–23436 (1999). [DOI] [PubMed] [Google Scholar]
- 47.Lund E. T., McKenna R., Evans D. B., Sharma S. K., Mathews W. R., Characterization of the in vitro phosphorylation of human tau by tau protein kinase II (cdk5/p20) using mass spectrometry. J. Neurochem. 76, 1221–1232 (2001). [DOI] [PubMed] [Google Scholar]
- 48.Park J., Wetzel I., Marriott I., Dréau D., D’Avanzo C., Kim D. Y., Tanzi R. E., Cho H., A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 21, 941–951 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zambrano A., Solis L., Salvadores N., Cortés M., Lerchundi R., Otth C., Neuronal cytoskeletal dynamic modification and neurodegeneration induced by infection with herpes simplex virus type 1. J. Alzheimers Dis. 14, 259–269 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Wozniak M. A., Frost A. L., Itzhaki R. F., Alzheimer’s disease-specific tau phosphorylation is induced by herpes simplex virus type 1. J. Alzheimers Dis. 16, 341–350 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Wozniak M. A., Itzhaki R. F., Shipley S. J., Dobson C. B., Herpes simplex virus infection causes cellular β-amyloid accumulation and secretase upregulation. Neurosci. Lett. 429, 95–100 (2007). [DOI] [PubMed] [Google Scholar]
- 52.De Chiara G., Marcocci M. E., Civitelli L., Argnani R., Piacentini R., Ripoli C., Manservigi R., Grassi C., Garaci E., Palamara A. T., APP processing induced by herpes simplex virus type 1 (HSV-1) yields several APP fragments in human and rat neuronal cells. PLOS One 5, e13989 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Santana S., Recuero M., Bullido M. J., Valdivieso F., Aldudo J., Herpes simplex virus type I induces the accumulation of intracellular β-amyloid in autophagic compartments and the inhibition of the non-amyloidogenic pathway in human neuroblastoma cells. Neurobiol. Aging 33, 430.e19–430.e33 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Friedrich R. P., Tepper K., Rönicke R., Soom M., Westermann M., Reymann K., Kaether C., Fändrich M., Mechanism of amyloid plaque formation suggests an intracellular basis of Aβ pathogenicity. Proc. Natl. Acad. Sci. U.S.A. 107, 1942–1947 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hu X., Zhou X., He W., Yang J., Xiong W., Wong P., Wilson C. G., Yan R., BACE1 deficiency causes altered neuronal activity and neurodegeneration. J. Neurosci. 30, 8819–8829 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Laird F. M., Cai H., Savonenko A. V., Farah M. H., He K., Melnikova T., Wen H., Chiang H. C., Xu G., Koliatsos V. E., Borchelt D. R., Price D. L., Lee H. K., Wong P. C., BACE1, a major determinant of selective vulnerability of the brain to amyloid-β amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J. Neurosci. 25, 11693–11709 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ou-Yang M. H., Kurz J. E., Nomura T., Popovic J., Rajapaksha T. W., Dong H., Contractor A., Chetkovich D. M., Tourtellotte W. G., Vassar R., Axonal organization defects in the hippocampus of adult conditional BACE1 knockout mice. Sci. Transl. Med. 10, eaao5620 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Savage M. J., Iqbal M., Loh T., Trusko S. P., Scott R., Siman R., Cathepsin G: Localization in human cerebral cortex and generation of amyloidogenic fragments from the β-amyloid precursor protein. Neuroscience 60, 607–619 (1994). [DOI] [PubMed] [Google Scholar]
- 59.Holler C. J., Webb R. L., Laux A. L., Beckett T. L., Niedowicz D. M., Ahmed R. R., Liu Y., Simmons C. R., Dowling A. L. S., Spinelli A., Khurgel M., Estus S., Head E., Hersh L. B., Murphy M. P., BACE2 expression increases in human neurodegenerative disease. Am. J. Pathol. 180, 337–350 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Z., Xu Q., Cai F., Liu X., Wu Y., Song W., BACE2, a conditional β-secretase, contributes to Alzheimer’s disease pathogenesis. JCI Insight 4, 123431 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/19/eaay8828/DC1