

Lifestyle and germline factors determine specific gastric carcinogenesis of East Asians.
Abstract
Germline and environmental effects on the development of gastric cancers (GC) and their ethnic differences have been poorly understood. Here, we performed genomic-scale trans-ethnic analysis of 531 GCs (319 Asian and 212 non-Asians). There was one distinct GC subclass with clear alcohol-associated mutation signature and strong Asian specificity, almost all of which were attributable to alcohol intake behavior, smoking habit, and Asian-specific defective ALDH2 allele. Alcohol-related GCs have low mutation burden and characteristic immunological profiles. In addition, we found frequent (7.4%) germline CDH1 variants among Japanese GCs, most of which were attributed to a few recurrent single-nucleotide variants shared by Japanese and Koreans, suggesting the existence of common ancestral events among East Asians. Specifically, approximately one-fifth of diffuse-type GCs were attributable to the combination of alcohol intake and defective ALDH2 allele or to CDH1 variants. These results revealed uncharacterized impacts of germline variants and lifestyles in the high incidence areas.
INTRODUCTION
Gastric cancer (GC) is the third leading cause of cancer mortality worldwide (1). The GC incidence shows strong geographic variations, with the highest incidence in East Asia, especially Japan and Korea (2). Although there are several reports that suggest hereditary and lifestyle factors, including strains of Helicobacter pylori (H. pylori), as contributors to these geographic variations (3, 4), no reports so far have analyzed these factors with genomic resolution in a trans-ethnic manner. Although recent whole-genome/exome sequencing has contributed to the global characterization of somatic genetics and driver genes of GCs (5–8), the precise interplays among lifestyles, germline variations, and somatic mutations, especially in the contexts of ethnic variations, are not fully clarified to date. To elucidate this point, we conducted whole-exome sequencing for a large cohort of GCs with detailed etiological information among Japanese, a population with one of the highest incidences of GCs in the world. These results were integrated with The Cancer Genome Atlas (TCGA) dataset to conduct trans-ethnic analysis of somatic and germline genetics of GCs.
RESULTS
Trans-ethnic mutational signature of GC
The overall profile of the whole-exome sequencing for 531 GC cases (288-case TCGA dataset and our 243-case Japanese dataset) exhibited substantial diversity in the somatic mutation profiles among GCs (Fig. 1A). Comparing the distributions of GC subtypes from the perspective of the TCGA subtypes (9), Epstein-Barr virus (EBV)–associated GC and microsatellite instability (MSI)–GC were more frequent in the TCGA dataset, while genomically stable (GS)–GC was more frequent in the Japanese cohort (table S1). Cancer genomes are known to have acquired various, but specific, somatic mutational patterns during carcinogenesis depending on the carcinogenic etiologies they experienced, recently called as mutational “Signatures” (10). We used a pan-cancer catalog of 30 Signatures from the COSMIC (Catalogue Of Somatic Mutations In Cancer) database (11) for this analysis (see Materials and Methods). According to the hierarchical clustering based on the mutational signature contributions in each case (see Materials and Methods), GCs were subdivided into six groups (marked by color bars at the top of the heat map), each of which exhibited dominant contribution of Signatures 1, 3, 6, 15, 16, 17, and others (Fig. 1A). Single-nucleotide variants (SNVs) attributable to Signatures 1, 3, 6, and 15 are known to be associated with the aging of the patients, BRCA deficiency, and mismatch repair (MMR) deficiency, respectively (10). To the contrary, theoretical etiologies of Signature 17 have not been determined to date.
Fig. 1. Trans-ethnic mutational signature analysis of GC.
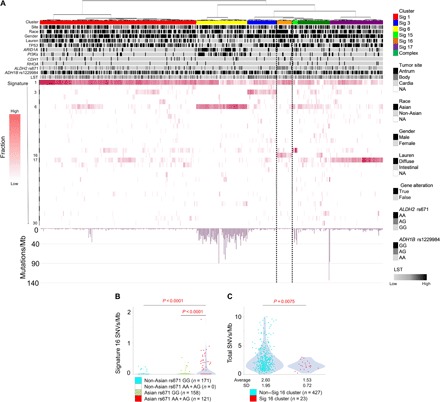
(A) The overall genetic profiles of 531 GCs, including Japanese GC cohort and TCGA GC datasets, are shown, on the basis of the hierarchical clustering of mutational signatures. GCs were genetically subdivided into six subgroups, each of which is characterized by Signatures 1 (red), 6 (yellow), 3 (blue), 16 (orange, surrounded by the dotted line), 15 (green), and 17 (purple). Clinicopathological information, representative somatic driver gene alterations, and germline variations of ALDH2 rs671/ADH1B rs1229984 are indicated as black/white columns at the top. White-to-red colored columns in the hierarchical clustering map represent the contribution rates (red bar on the left side) of the mutational signatures in each case. Mutation frequencies per megabase (Mb) are indicated at the bottom as bar graphs. PI3Ks: PIK3CA and PTEN mutations. (B) The numbers of the Signature 16 SNVs/Mb in GC cases are shown in relation to the races and ALDH2 genotypes of the patients. P values were calculated using unpaired Wilcoxon rank sum test. (C) The numbers of total SNVs/Mb in cases of Signature 16 cluster and those of other clusters are shown. Cases with hypermutator signature (yellow and green bars) were excluded. P values were calculated using the unpaired Wilcoxon rank sum test. NA, not applicable.
To identify any unknown interplays between SNV Signatures and other factors, we examined possible clinicopathological factors in each of the clusters, including patient races and well-known germline variants. We then found that a subgroup (Fig. 1A, orange bar) was strongly contributed by Signature 16, whose contributions consisted of approximately 28% of somatic SNVs, and almost all the patients in this cluster were of Asian ethnic background (90.5%), and a high proportion of these patients (16 of 23 = 69.6%) harbored a well-known inactive ALDH2 allele (rs671 AA or AG) (table S2). We further confirmed this observation of the correlation between Signature 16 and the ALDH2 allele by analyzing the Signature 16 contributions and ALDH2 genotypes in all the 531 cases, finding their positive correlation in an Asian-specific manner (Fig. 1B; P < 0.0001, unpaired Wilcoxon rank sum test). As is reported (12), inactive ALDH2 rs671-A allele is specific to Asian populations; thus, the correlation of the ALDH2 genotype with Signature 16 contribution is considered attributable specifically in the Asian populations.
Although the GC cluster with Signature 16 had no characteristic patterns of major driver gene mutations, this cluster was relatively enriched for diffuse-type histology (12 of 23) (Fig. 1A and table S2). The overall mutation burdens in this cluster were significantly smaller than those of the other GCs (P = 0.0075; Fig. 1, A and C), while the age at onset and Signature 1 mutations of the GC cases in the Signature 16 cluster were comparable to the other GCs (fig. S1). In view of chromosomal aberrations, we compared large-scale state transition (LST) scores (13) between all the Signature clusters (fig. S2), revealing that the LST scores of Signature 16 cluster were among the lowest, except for hypermutators (Signature 6/15 clusters) that are already known to harbor fewer chromosomal abnormalities (5, 9) (fig. S2). Together, the Signature 16 cluster is suggested not only as an Asian-specific subclass but also as a distinct biological entity.
Etiological factors of Signature 16 GC
With these findings and recent reports showing that Signature 16 is suggestive of an alcohol-associated signature in liver, esophageal, and several other cancers (14–16), we then performed an in-depth analysis of our Japanese cohort with more information of their lifestyles and etiologies to investigate the relationship among Signature 16, patient ALDH2 genotypes, and alcohol intake. We also analyzed how strongly the combination of germline genetics and lifestyles affects the high-GC incidences in the Japanese population. The existence of the Signature 16 GC cluster was clearly reconfirmed (Fig. 2A), and the cluster contained 6.6% (16 of 243) of Japanese GCs. A high portion of patients (11 of 16, 68.8%) in the Signature 16 GC cluster were characterized as alcohol consumers with inactive ALDH2 allele (table S3). Therefore, the Signature 16 mutations in the GC context were most probably attributable to the combined effects of alcohol intake and loss-of-function allele of ALDH2 (table S3). To quantitatively confirm this phenomenon, we investigated the correlation between the number of Signature 16 SNVs and the combination of the alcohol intake habit and ALDH2 allele among all the Japanese GCs (Fig. 2B). While the effects of either alcohol intake or the ALDH2 allele alone were minimal on the number of Signature 16 mutations, the combination of both the alcohol intake behavior and inactive ALDH2 allele resulted in an 11.1-fold increase of the Signature 16 burdens (P < 0.0001, unpaired Wilcoxon rank sum test; Fig. 2B). ALDH2 is an enzyme that metabolizes acetaldehyde to acetic acid, the former of which exhibits significant genotoxic effects (17). As reported, the gastric mucosa expresses ALDH2, and the acetaldehyde levels in the gastric contents of ALDH2-inactive individuals were 5.6 times higher than those in the ALDH2-active individuals, after intragastric alcohol infusion (18, 19). Therefore, the accumulated acetaldehyde due to the low ALDH2 activity most likely induced the Signature 16 mutation patterns in gastric epithelial cells. The Signature 16 SNV occurred at the transcribed strands significantly more frequently than at the untranscribed strands (fig. S3; P < 0.0001, Fisher’s exact test), suggesting its association with transcription-coupled nucleotide excision repair (20). In summary, we conclude that the mutational Signature 16 significantly contributes to GCs in Asian populations and is attributable to the combined effects of the germline factor and patients’ lifestyle, which are alcohol intake behavior and loss-of-function allele of ALDH2. It was revealed that patients with GC with high numbers of Signature 16 SNVs did not always consume large amounts of alcohol; therefore, it can be concluded that a large amount of alcohol is not necessarily required to induce Signature 16 SNVs in ALDH2-inactive individuals. Mutational Signature 16 is also observed in esophageal squamous cell carcinoma and is related to alcohol intake with inactive ADH1B genotypes (rs1229984-GG) (16). In our Japanese GC cohort, however, such a correlation was not statistically confirmed (fig. S4, A and B), partly because of the limited statistical power due to the low minor allele frequency (MAF) of the ADH1B rs1229984 locus.
Fig. 2. Mutational signature analysis of Japanese GCs with lifestyle and germline information.

(A) The overall genetic profiles of 243 Japanese GCs are shown as a hierarchical clustering heat map as in Fig. 1A. Clinicopathological information, germline variations of ALDH2 rs671 and ADH1B rs1229984, and alcohol and smoking habits are indicated as black/white columns at the top. The contribution rate of the mutational signatures in each case is shown as in Fig. 1A. The germline and somatic variations of the CDH1 and BRCA pathway genes are indicated at the bottom of the figure. Red and blue columns indicate truncation and missense variants, respectively, with the proviso that somatic mutations in hypermutators (GCs with Signature 6 contribution, yellow column) and BRCA pathway variations without BRCA signatures (both somatic and germline) are shown as transparent columns. (B) The numbers of Signature 16 SNVs are plotted according to the patient subgroups defined by ALDH2 genotype and alcohol consumption habit. P values were calculated using unpaired Wilcoxon rank sum test. (C) The numbers of Signature 16 SNVs are plotted according to the patient subgroups defined by the combinations of ALDH2 genotype, alcohol consumption, and smoking habit. P values were calculated using the unpaired Wilcoxon rank sum test.
It is well known that alcohol consumption together with smoking epidemiologically increases the risk of esophageal squamous cell carcinoma (21). Conversely, it is not fully evident whether or not smoking synergizes with alcohol consumption regarding the GC risk. Here, detailed etiological analysis using Japanese GC cohort revealed that smoking habit has synergistic effects on Signature 16 mutations, specifically among GCs of alcohol consumers with ALDH2 defective allele (rs671 AA/AG) (P = 0.0339; Fig. 2C). This synergic effect was not observed among patients with ALDH2-proficient genotypes (rs671 GG) nor was there an increase in the numbers of Signature 16 mutations among smokers without drinking habit. Therefore, it can be considered that combination of alcohol consumption and smoking habit has synergic effects on the generation of Signature 16 SNVs, specifically among people with ALDH2 defective allele. To the contrary, the so-called smoking signature (Signature 4) was not apparently dominant among patients in the Signature 16 cluster (Figs. 1A and 2A), and it was also revealed that Signature 4 SNVs were not synergized by alcohol consumption (fig. S4C), indicating a possible one-side effect of smoking habit on the generation of alcohol-related signatures during GC carcinogenesis.
Immunological features of alcohol-related GC
Because of the distinct nature of the Signature 16 GCs, especially their smaller mutation burdens, we were then motivated to investigate whether this GC subgroup has characteristic biology including immunological features (22). Bioinformatics approach using gene expression profiles of 184 (of 243 cases of exome-sequenced) Japanese GCs pointed out that Signature 16 GCs exhibited characteristic immunological microenvironments (Fig. 3A), where we estimated compositions of 22 types of tumor-infiltrating immune cells in each GC using CIBERSORT (cell-type identification by estimating relative subsets of RNA transcripts) algorism (see Materials and Methods) (23). It was revealed that Signature 16 GCs showed skewed distributions in principal components analysis in view of the compositions of infiltrating immune cells (Fig. 3A), and higher B cell infiltration was the most prominent feature of the Signature 16 GCs (Fig. 3B). This trend of B cell infiltration was commonly observed among Signature 16 GCs regardless of the tumor subtypes, either diffuse or intestinal types. It was also noted that expression of CXCL13, a well-established B cell–recruiting chemokine, was significantly higher among Signature 16 GCs compared to the other groups of GCs (Fig. 3C). To confirm these observations, we histopathologically evaluated the B cell infiltration and CXCL13 expression in Signature 16 GCs. It was revealed that cancer cells themselves, in addition to well-characterized follicular dendritic cells, expressed CXCL13, and in total, 68.8% (11 of 16) of Japanese Signature 16 GCs showed immunohistochemically recognizable positivity for CXCL13 in cancer cells, either in a focal or wide-spread manner (Fig. 3D). In these cases, cancer cell nests with CXCL13 positivity were occasionally accompanied with B cell infiltrations around the nests. Representatively, one case showed massive infiltration of B cells within the cancer area as well as high-level expression of cancer cell–intrinsic CXCL13 (Fig. 3D). Pathway enrichment analysis using RNA sequencing (RNA-seq) data (see Materials and Methods) revealed significant enrichment of B cell receptor signaling, cytokine/chemokine signal, and Fc g receptor signaling pathways in the Signature 16 GCs compared to others (table S4), which further supports the characteristic B cell infiltrations in Signature 16 GCs. In summary, it is suggested that Signature 16 GCs frequently express CXCL13 in a cancer cell–intrinsic manner and thus recruit the infiltration of B cells within cancer area.
Fig. 3. Cancer-recruited B cell infiltration in alcohol-related GCs.
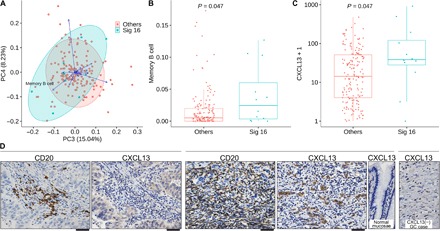
(A) Principal components analysis of 184 Japanese GC cases based on their profiles of the proportions of tumor-infiltrating immune cells defined by CIBERSORT algorism (see Materials and Methods). PC3 and PC4 components are shown. Green and red circles indicate Signature 16 GC and other GCs, respectively. Green and red circular areas indicate 95% confidence intervals of Signature 16 GC and other GCs, respectively. Arrows represent the correlations between the principal components (PC) and the variables (compositions of immune cells). (B) The proportion of B cells infiltrating in the GC tissues determined by the CIBERSORT algorism (see Materials and Methods) is shown. Green and red dots indicate Signature 16 GCs and other GCs, respectively. P value was calculated by unpaired Wilcoxon rank sum test. (C) Gene expression levels of B cell–attracting chemokine CXCL13 determined by RNA sequencing of bulk GC tissues (see Materials and Methods) are shown. Y axes are shown as log scale. Green and red dots indicate Signature 16 GCs and other GCs, respectively. P value was calculated by unpaired Wilcoxon rank sum test. (D) Representative cases of Signature 16 GCs with immunohistochemical staining of CD20 and CXCL13 are shown. In a GC case on the left side, as in other Signature 16 GCs, cancer cell–intrinsic CXCL13 expression and B cell infiltration around the tumor nest are observed. The GC case shown in the middle panels represents the prominent expression of tumor cell–intrinsic CXCL13 expression and massive infiltrations of B cells, followed by a picture at the right side, indicating negative CXCL13 staining in the normal gastric mucosae of the same specimen, as a control. A GC case at the rightest side indicates negative staining of CXCL13. Black bars under pictures, 50 μm.
Impact of germline variations in Asian GC
Germline variations of E-cadherin (CDH1) are well known to be responsible for hereditary diffuse-type GC (HDGC) (4), and recent exome sequencing identified germline mutations of the BRCA1/2 pathway genes as causative factors for GCs especially with familial histories (24). However, most of these data were obtained in non-Asian cases. Meanwhile, in Asian countries, the global burdens of GC-causing germline variants and their effects on familial aggregations have remained unclear to date. We summarized possible pathogenic germline variants of 624 cancer-related genes (25) in our Japanese GC cohort after filtering out the common and/or presumably nonpathogenic variants in the Japanese population (see Materials and Methods). While several truncating variants were observed in the BRCA pathway genes, we unexpectedly found that the CDH1 gene has the highest variant frequency and density (Table 1, bold), suggesting that the germline variants of the CDH1 gene significantly contribute to the genetic backgrounds of GCs in the Japanese population. Among Genome-Wide Association Study (GWAS)–reported GC-susceptive genes (26–30), we found that PLCE1 variants were mildly enriched among Japanese GC patients compared to the general Japanese population (table S5; 2.0-fold, P = 0.039, Fisher’s exact test).
Table 1. Summary of germline variants that are presumably pathogenic.
Genes with presumably pathogenic germline variants are shown in the order of their densities of nonsilent variants (left, only genes with 5% or higher variant frequency among our cohort are shown) and numbers of truncation variants (right). AA, number of amino acid residues.
| Nonsilent | Truncation | ||||||
| Gene | N | Amino acid (AA) size |
Density (N/AA) |
Gene | N | Gene | N |
| CDH1 | 16 | 882 | 0.018 | ZFHX3 | 11 | POLD1 | 1 |
| PRDM1 | 13 | 789 | 0.016 | GJB2 | 8 | POLE | 1 |
| ARID1B | 27 | 2231 | 0.012 | CRIPAK | 7 | POLQ | 1 |
| BLM | 16 | 1417 | 0.011 | SLC25A13 | 7 | RAD51D | 1 |
| ABCC4 | 13 | 1325 | 0.010 | BRCA2 | 2 | RUNX1T1 | 1 |
| PIK3C2G | 14 | 1445 | 0.010 | PIK3C2G | 2 | CYP2D6 | 1 |
| RICTOR | 16 | 1708 | 0.009 | ATM | 1 | ERBB2 | 1 |
| SETD2 | 18 | 2061 | 0.009 | B4GALT3 | 1 | ERCC4 | 1 |
| EPPK1 | 18 | 2420 | 0.007 | BRIP1 | 1 | EXO1 | 1 |
| ROS1 | 17 | 2347 | 0.007 | CUX1 | 1 | FANCA | 1 |
| SLX4 | 13 | 1834 | 0.007 | FGFR3 | 1 | SBDS | 1 |
| ZFHX3 | 26 | 3703 | 0.007 | GSK3B | 1 | SF1 | 1 |
| DOCK8 | 13 | 2031 | 0.006 | HLA-G | 1 | SMARCB1 | 1 |
| POLE | 14 | 2286 | 0.006 | ITGAV | 1 | TLR4 | 1 |
| FAT1 | 25 | 4588 | 0.005 | PALB2 | 1 | ||
| MGA | 16 | 3114 | 0.005 | ||||
| COL7A1 | 15 | 2944 | 0.005 | ||||
| APC | 14 | 2843 | 0.005 | ||||
| TRRAP | 14 | 3830 | 0.004 | ||||
In our detailed analysis of the BRCA pathway genes, we found 20 (in 22 of 243 cases, 9.1%) germline variants among cases with BRCA-related mutational signature (Signature 3) [Figs. 2A and 4A (blue bars) and table S6]. This frequency was not statistically different from that in the TCGA non-Asian GC population (19 of 212 cases, 9.0%; P = 1.0000, Fisher’s exact test). According to reported studies (31, 32), it has also been concluded that frequencies of BRCA1/2 pathogenic variants among general Japanese and West European populations are almost comparable. Several cancers with the BRCA signature are reported to be good responders to platinum chemotherapeutics and poly(adenosine diphosphate–ribose) polymerase inhibitors (33). The Signature 3 cluster (blue bars in Figs. 1A and 2A) consists of a roughly equal fraction of GCs in both Japanese and TCGA non-Asians (25 of 243, 10.3% and 19 of 212, 9.4%, respectively; P = 0.8735, Fisher’s exact test), which should be appropriate targets of such therapeutics.
Fig. 4. The landscape of germline variations in East Asian and non-Asian GCs.
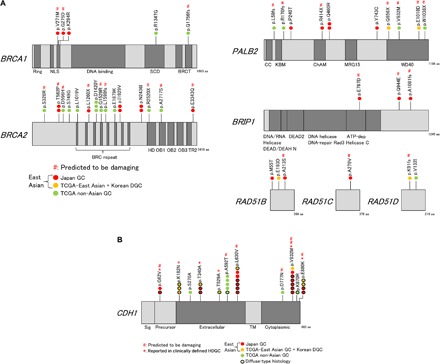
(A) Distributions of germline variations of BRCA pathway genes found in combined datasets [Japanese GCs, TCGA GCs (9), and Korean DGCs (6)]. Only variants found in cases linked to the BRCA signature are shown. Red, Japanese; green, TCGA non-Asian. None of the patients were TCGA East Asians/Koreans (orange) who met this criterion. #, predicted to be damaging in silico; NLS, nuclear localization signal; SCD, serine cluster domain; HD, helical domain; OB, oligonucleotide binding; TR2, second RAD51-binding domain; aa, amino acid; ATP, adenosine triphosphate. (B) Distributions of germline variations of CDH1 in the same datasets as in (A). *, reported in clinically defined HDGC (strong familial aggregation or extremely early-onset case). Bold circle, diffuse-type histology. SIG, signal; TM, transmembrane domain.
As for the CDH1 germline variants, after filtering out common variations (see Materials and Methods), we summarized all nonsilent germline variants in our 243-case Japanese GC cohort (Fig. 4B, Table 2, and table S7), discovering 18 variants in total. This observed frequency (18 of 243, 7.4%) was higher than the reported consensus in the Japanese population (34–36). In contrast to the variants found in TCGA non-Asians (Fig. 4B, green colored circle), most (14 of 18 cases) of the variants in the Japanese population were linked to diffuse-type histology (Fig. 4B, black outlined red circle), which provides a strong biological support to conclude their pathogenic roles (P = 0.0060, Fisher’s exact test) (37). The variant frequency of CDH1 in the Japanese DGC patients (14 of 105, 13.3%) was approximately 4.1-fold higher than that in the TCGA non-Asian GC populations (2 of 62, 3.2%; P = 0.0535, Fisher’s exact test; Table 2). It is intriguing that distributions of the CDH1 germline variants in the Japanese population were distinct from those of the TCGA non-Asians and specifically fell into five SNVs (p.G62V, p.T340A, p.L630V, p.V832M, and p.E880K). Among these five SNVs, four were predicted to be damaging in silico, four were reported in cases that met the HDGC criteria [strong familial aggregation (34, 35, 38) or extremely early onset (6)], and at least two were molecularly confirmed to be causative of malignancies due to the deficits in cell-cell adhesion and/or in the inhibition of invasion (Table 2) (39, 40). When germline variants found in a large Korean sporadic DGC dataset (6) were combined, it was revealed that these five recurrent SNVs were shared by both Japanese and Korean populations at measurable frequencies (Fig. 4B). This trend shows a clear contrast to the germline BRCA pathway variants (Fig. 4A), where no shared variants were found between Japanese and Korean GCs. The overall variant frequency of the 11 SNV loci found in this study was significantly higher among Japanese DGC patients than the general Japanese populations (4.0-fold; P < 0.0001, Fisher’s exact test) and also higher among the general East Asian population than the general European population (6.0-fold; P = 0.00124, Fisher’s exact test) (Table 2), supporting the pathogenic characteristics of these variants and confirming their enrichments in East Asian populations.
Table 2. Summary of germline CDH1 variants in East Asia (Japan and Korea) and TCGA datasets.
Germline CDH1 variants in East Asia (Japan and Korea) and TCGA datasets are shown. ToMMo, Tohoku Medical Megabank Organization.
| SNVs |
Polyphen2 (HumDiv) |
Reported in HDGC* |
Reported in Korea† |
Japanese GC |
TCGA non-Asian GC |
Japanese DGC |
TCGA non-Asian DGC |
Japanese DGC versus non-Asian DGC |
Japan ToMMo |
Japanese DGC versus Japan ToMMo |
1000 Genomes EAS |
1000 Genomes EUR |
1000 Genomes EAS versus EUR |
Note | |
| Case | 243 | 212 | 105 | 62 | 1070 | 504 | 503 | ||||||||
| 1 | p.G62V | Probably damaging |
Yes | 2 | 1 | 3 | (35) | ||||||||
| 2 | p.K182N | Benign | Yes | Yes | 1 | 1 | (6) | ||||||||
| 3 | p.S270A | Benign | 1 | ||||||||||||
| 4 | p.T340A | Benign | Yes | Yes | 2 | 2 | 6 | 1 | (6, 38, 40) | ||||||
| 5 | p.T529A | Benign | Yes | Yes | (6) | ||||||||||
| 6 | p.A592T | Possibly damaging |
5 | 1 | 2 | (48) | |||||||||
| 7 | p.L630V | Probably damaging |
Yes | 6 | 4 | 9 | 6 | (6) | |||||||
| 8 | p.D777N | Probably damaging |
1 | ||||||||||||
| 9 | p.V832M | Probably damaging |
Yes | Yes | 6 | 5 | 10 | 2 | (34, 40, 49) | ||||||
| 10 | p.K870R | Benign | 1 | 1 | |||||||||||
| 11 | p.E880K | Possibly damaging |
Yes | Yes | 2 | 2 | 7 | 2 | (6) | ||||||
| Sum | 7.4% (18) | 3.8% (8) | 13.3% (14) | 3.2% (2) |
4.1-fold (P = 0.0535) |
3.4% (36) |
4.0-fold (P < 0.0001) |
2.4% (12) | 0.4% (2) |
6.0-fold (P = 0.0124) |
*Reported in cases that met the clinical criteria of HDGC.
†Reported in TCGA Korean and in (6).
Familial history of germline CDH1 variant carriers
Out of 18 Japanese GC patients with germline CDH1 variants, 11 had mild familial cancer histories (table S5), including one case with lobular carcinoma of the breast, a well-known trace of germline CDH1 variants. However, although the information available was limited, only one case (DGC at 39 years old) met the clinical criteria of the HDGC (4). On the other hand, seven individuals had no familial histories of cancers and were clinically considered sporadic cases. Therefore, it should be noted that substantial portions of GCs that are clinically assumed as sporadic incidences could be attributable to germline variations of the patients. In view of molecular carcinogenesis pathways, it is hypothesized that the clinical phenotypes of CDH1 germline variations are significantly modified by other environmental factors such as H. pylori infection and food contents. We investigated H. pylori copy numbers at noncancerous mucosa of our GC patient cohort, finding that GC cases with germline variants of CDH1 exhibited statistically higher H. pylori copy numbers compared to others (fig. S5). This finding is consistent with a previous report showing that ongoing infections of H. pylori are more frequent in DGCs (41), since almost all the CDH1 germline variant patients harbor DGCs (table S7). H. pylori is known to disrupt proper function of CDH1 (42), which might suggest that H. pylori infection works as an additional second hit for the CDH1 germline variant population in the GC development. This observation and our speculations, however, are not scientifically conclusive because the copy number of H. pylori is influenced by the existence of mucosal metaplasia and the longevity of chronic infection (43). Even if the familial histories or clinical phenotypes did not straightforwardly resemble the definitions of familial GCs clinically, at least a portion of GC cases can be attributable to CDH1 germline variants. The p.V832M (34) and p.G62V (35) variants, for instance, were reported in families with strong familial aggregations of GCs (7 of 12 and 7 of 13 family members examined, respectively); however, only four of eight cases with these variants in our Japanese GCs showed, at most, slight familial histories, and none of them fulfilled the clinical criteria of HDGC, although the family information was available only for the first-degree relatives.
DISCUSSION
Large-scale whole-exome sequencing for East Asian GCs in combination with the public dataset has clarified important lifestyle and germline predispositions of GC development among high-incident East Asian populations. Alcohol intake behavior with genetically inactive ALDH2 background predisposes individuals to GCs with Signature 16. Besides, synergetic effects of alcohol intake and smoking habit were observed among the ALDH2-inactive individuals. Alcohol intake and the incidence of various types of cancers have extensively been studied so far (14–16, 21). Although conflicting reports have indicated the correlations between alcohol intake and GC risk based on indirect epidemiological data (44, 45), no reports have shown direct biological evidence of the effects of alcohol on GC carcinogenesis. Global genetic classifications combined with clear quantification of alcohol contributions defined by mutational signatures, as taken by this study, could succeed in unraveling the ethnicity-specific intercorrelations of etiologies in GC development in the high-incidence areas. Alcohol-related GC is a biologically distinct subgroup with low mutation burdens and characteristic immunological profiles of increased B cell infiltration due to cancer cell–derived CXCL13 chemokine. While there are several reports of alcohol-induced dysregulation of adaptive immunity including B cells (46, 47), its biological mechanism and its relation to alcohol exposure are unclear and remain to be investigated further. Our results show that Signature 16 GCs have distinct immunological features, which attracts further attention in relation to the responsiveness of these GCs to current immune therapies as well as their applicability for alternative immunotherapies.
In addition to the BRCA pathways, this study also clarified that germline variations of CDH1 exist at a higher-than-expected frequency among East Asian GCs. The shared distributions of the recurrent CDH1 variants between Japanese and Koreans strongly suggest the existence of common ancestral events and widespread distributions of these CDH1 pathogenic variants in the East Asian areas. Thus, these variants affect the prevalence of GCs in these areas, although their precise geographic distributions are yet to be determined. It is important that many of these cases are clinically recognized as sporadic cases and have been overlooked so far without special attention to the preventive cares for family members, such as periodic endoscopies and/or prophylactic surgeries (4). The incomplete penetrance and environmental dependency of CDH1 variants are likely to have led to the controversy about their pathogenicity so far. Several groups have tried to experimentally show the evidence of pathogenicity of the variants, but their results were often conflicting (6, 40, 48, 49), partially because of the differences in the assays used and the difficulties in proving the biological function of the variants with mild effects. Our large-scale analysis of genomics and clinical data in this study clearly showed multiple evidences of the pathogenic nature of these CDH1 variants.
Approximately one-fifth (22 of 105, 21.0%) of the DGCs in our study were attributable either to the alcohol consumption coupled with defective ALDH2 allele or to the germline loss-of-function variants of CDH1. Our findings clarified previously unrecognized impacts of the defined germline and lifestyle factors on the high incidences of GCs in East Asian areas and provided us strong motivations for clinical intervention in lifestyles and familial cares coupled with precision germline genotyping in these areas.
MATERIALS AND METHODS
Informed consent and sample preparation
Fresh-frozen GC and paired normal stomach tissues were obtained from essentially consecutive patients who underwent gastrectomy at the University of Tokyo Hospital (n = 169) and Yokohama City University Hospital (n = 126). Informed consent was obtained from each individual, and this study was approved by the institutional review boards at the University of Tokyo, Yokohama City University, and Tokyo Medical and Dental University. QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) and RNeasy Mini Kit (Qiagen) were used to extract 295 paired DNA and RNA samples from these tissues, respectively, as per the manufacturer’s instructions.
Clinicopathological information
Patient clinicopathological information [gender, age at diagnosis, alcoholic drinking habits, smoking habits, Lauren classification, tumor site, stage (UICC TNM classification), and histopathological features] were collected with informed consents. Patients were classified as “consuming alcohol” if they reported socially associated or more severe alcohol usage or otherwise as “not consuming alcohol.” Patients who smoked at least one cigarette/day on average for at least 1 year during their lifetime were defined as “smokers” or otherwise defined as “nonsmokers.” Tumor TNM staging including an assessment of the tumor size and lymph node and distant metastases were reviewed by at least two pathologists and defined according to the American Joint Committee on Cancer (eighth edition). Detailed data for the 243 patients are shown in table S9. Mix-type GCs (i.e., coexistence of diffuse and intestinal components) were regarded as diffuse-type GCs for this study, as is suggested by guidelines (50).
H. pylori infection status
H. pylori infection status of the patients at the time of surgeries was determined by quantitative polymerase chain reaction (qPCR), using the extracted DNA from noncancerous gastric tissues of patients with GC. The ratio of qPCR quantities of H. pylori per human genomic DNA was considered the H. pylori copy number in the subjected specimens. Human genomic DNA was quantified by the TaqMan method targeting ribonuclease P gene locus (Thermo Fisher Scientific, MA, USA), and the amount of H. pylori was measured by SYBR Green method targeting ureC gene, which was reported to be the most sensitive and specific marker for H. pylori (51). The ureC gene primers were 5′-GCATGCAATTGAATAAAGCC-3′ (forward) and 5′-GCCGCTATAACGGATCAAAT-3′ (reverse) as reported (51). TaqMan PCR was performed as per the manufacturer’s protocol, and SYBR Green qPCR was performed 50 cycles for denature, 95°C for 15 s, and extension, 60°C for 1 min. In total, we obtained informative data of H. pylori copy numbers for 170 of 243 Japanese GC patients. Because of the substantial paucities of genomic DNA, it was not possible to perform qPCRs for the remaining 73 cases.
Exome capture, library construction, and whole-exome sequencing
Whole-exome sequencing was performed for 295 GCs and paired normal gastric tissues, as previously described (8). Each DNA sample (1.1 mg) was sheared using a Covaris SS Ultrasonicator (Covaris, MA, USA) according to the manufacturer’s instructions. A Sciclone next-generation sequencing workstation (Caliper Life Sciences, MA, USA) was used to automatically construct the DNA sequence libraries, according to the manufacturer’s instruction. Exome capture was performed using the Agilent SureSelect Human All Exon Kit v4 and v5+ LincRNA (Agilent Technologies, CA, USA). Each sample was sequenced using an Illumina HiSeq 2000 platform (Illumina, CA, USA) and the provided protocol for producing 2 × 100–base pair (bp) pair-end reads. Image analyses and base calling were performed using an Illumina pipeline with default settings. The median depth of tumor/normal coding sequence is 170/84 in Japan and 165/91 in TCGA.
Mutation calling
We obtained FASTQ files of the TCGA dataset [published in 2014 (9)] from the TCGA website (https://tcga-data.nci.nih.gov). Then, we performed mutation calling of our dataset and TCGA dataset using exactly the same pipeline from the beginning of sequence mapping procedure as follows. Paired-end reads were aligned to the human reference genome (GRCh37) using NovoAlign (http://novocraft.com/products/novoalign/) for both tumor and nontumor samples. Probable PCR duplications, for which paired-end reads aligned to the same genomic position, were removed, and pileup files were generated using SAMtools (52) and some in-house programs. To find somatic point mutations (SNVs and short indels), the following cutoff values were used for base selection: (i) a mapping quality score of at least 20 and (ii) a base quality score of at least 10. Somatic mutations were selected using the following filtering conditions: (iii) The numbers of reads supporting a mutation for tumor were at least 4 and 8, with at least one base with a quality score greater than 30, when tumor variant allele frequency (TVAF) ≥ 0.15 and 0.15 > TVAF ≥ 0.05, respectively; (iv) variant allele frequency (VAF) of matched nontumor sample was less than 0.03 with a read depth of at least 8. Since sequence errors are considered to occur in a sequence-specific manner, the sequence read information of all nontumor samples was combined together to accurately discriminate true positives from false positives. Then, the following filter was applied: (v) VAF of grouped nontumor sample (NVAF) was less than 0.03 and 0.01, when TVAF ≥ 0.15 and 0.15 > TVAF ≥ 0.05, respectively; (vi) the ratio (TVAF/NVAF) of VAF of tumor (TVAF) and grouped nontumor samples (NVAF) must be more than 20. After mutation calling, (vii) mutations with a strand bias (between forward and reverse reads) greater than 95% were removed.
TCGA dataset
TCGA GC dataset [published in 2014 (9)] was obtained from the TCGA website (https://tcga-data.nci.nih.gov). It includes 288 (212 non-Asians and 76 Asians) GC cases with information regarding their somatic mutations, list of reads per kilobase per million (RPKM) mapped reads (for the 262 GCs and 29 normal stomach tissues), and clinical data. No ethnicity information was provided for 41 GC cases (obtained from 38 German and 3 Canadian patients); therefore, we considered these to be non-Asian cases. BAM files were obtained from the TCGA website archives and used to analyze germline variations for the TCGA dataset.
Classifications of Japanese GC cases by the TCGA subtypes
We classified our Japanese GC cases into four subgroups: EBV-associated GC, MSI-GC, GS-GC, and chromosomal instability (CIN)–GC (9). First, we defined EBV-GC by detecting viral sequences in the RNA-seq data using PathSeq (53). If the sequence reads unique to the EBV were more than 1 × 10−5 of the reads unique to human genome per individual, these GCs were classified as EBV-GC. This cutoff was determined in accordance with the data of Epstein-Barr virus-encoded small RNA (EBER) in situ hybridization. Second, we defined MSI-GC by mutational signature analysis. Signatures 6 and 15 are well-known resultants of the MMR deficiencies; therefore, we identified MSI-GC as those in Signature 6 or 15 clusters. Last, we divided the rest of GCs into GS- or CIN-GCs. These two subtypes were distinguished by the presence or absence of extensive somatic copy number aberrations based on the hierarchical clustering. To analyze copy number aberrations, the read depth was compared between normal and tumor for each capture target region. The tumor/normal depth ratios were calculated, and the values were smoothed using a moving average. Using these values, clustering was done in R based on Euclidean distance using Ward’s method as was performed in the TCGA (9).
Mutational signature analysis
We obtained somatic SNVs for 531 GC cases (288 TCGA cases and 243 Japanese cases). We used the pan-cancer catalog of 30 Signatures referenced in the COSMIC database (11) (http://cancer.sanger.ac.uk/cosmic), and the contribution of each mutational Signature was calculated by deconstructSigs software (table S10) (54). Unsupervised hierarchical clustering was performed on the Signature contribution scores for each GC case, by using pheatmap v1.0.2 software in R to define the GC subgroups (Fig. 1A). The ward.D was used as the clustering method, and Euclidean distances (Japan and TCGA datasets; Fig. 1A) and correlation distances (Japanese GCs; Fig. 2A), respectively, were used to assemble the GC clusters. The incidence of Signature 4– or Signature 16–associated SNVs/Mb was calculated on the basis of the contribution scores of Signature 16 in each case, as defined above. We excluded hypermutator GCs from the analyses of the frequencies of SNVs between subgroups, since large numbers of SNVs in hypermutators could extremely affect the statistical analyses. We defined the hypermutator GCs as those with mutation burdens over 10/Mb (55).
RNA sequencing
For 184 Japanese GC cases, total RNA was extracted from frozen sections (10 sections × 10 μm thickness) of GC specimens by RNeasy Mini Kit (Qiagen). Quantification and qualification of extracted RNAs were determined by Agilent Bioanalyzer (Agilent Technologies, CA, USA). RNA-seq libraries were constructed by the TruSeq Stranded mRNA Sample Prep Kit (Illumina, CA, USA) according to the manufacturer’s protocol. Each RNA-seq library was sequenced on an Illumina HiSeq platform (Illumina) by 2 × 100-bp paired-end sequencing as per the manufacturer’s protocol. FASTQ files were processed using Kallisto version 0.43.1 with Ensemble transcriptome (release 79) to create gene expression profiles.
Estimation of LSTs
We estimated the numbers of LSTs by the following three steps: (i) copy number ratio calculation, (ii) detection of large copy number changes, and (iii) LST detection. First, initial copy number ratios were obtained by comparing read depth information for tumor and normal samples, and the copy number estimates were adjusted by tumor purity as described previously (8). Next, copy number ratios within 100-kbp window were averaged and further smoothed by moving average filters with a window of five points. Then, circular binary segmentation implemented in python (https://github.com/jeremy9959/cbs) was applied with 10,000 random permutations. To detect only highly confident copy number variation (CNV) regions, the cutoff value was set to P = 0. The detected segments were merged when the difference of mean log ratios between the regions ≤ 0.3. Last, LSTs were detected on the basis of the definition of LST as previously described (13). After filtering and smoothing of all variations less than 3 Mb, a chromosomal break between adjacent regions of at least 10 Mb was defined as LST. The number of LSTs in the tumor genome was estimated for each chromosome arm independently.
Detection of differentially expressed genes among Signature 16 GCs
To detect any differentially expressed gene sets among Signature 16 GCs, we compared the fragments per kilobase of exon per million reads mapped (FPKM) between Signature 16 GCs (n = 12) and the other GCs (n = 172). Initially, we calculated q values, but there were no genes with statistical significances, probably because of the few numbers of Signature 16 GCs; therefore, we used P values to define differentially expressed genes. We selected genes with P values of <0.05 and, at the same time, the average FPKM of the genes among Signature 16 GCs > the other GCs. Last, we identified 1303 genes. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was performed using The Database for Annotation, Visualization and Integrated Discovery (DAVID) platform.
CIBERSORT
CIBERSORT (23) was applied to 184 Japanese GC tissues to enumerate the proportions of 22 types of immune cells [B cells naïve, B cells memory, plasma cells, T cells CD8, T cells CD4 naïve, T cells CD4 memory resting, T cells CD4 memory activated, T cells follicular helper, T cells gamma delta, T cells regulatory, natural killer (NK) cells resting, NK cells activated, monocytes, macrophages M0, macrophages M1, macrophages M2, dendritic cells resting, dendritic cells activated, mast cells resting, mast cells activated, eosinophils, and neutrophils] using the LM22 dataset. CIBERSORT was run in “absolute mode,” and “disable quantile normalization” option was selected as recommended for RNA-seq data.
Immunohistochemistry
Immunohistochemistry staining was performed onto formalin-fixed and paraffin-embedded (FFPE) specimens of GC cases. FFPE tissues were deparaffinized by xylene (Wako Pure Chemical Industries, Japan), and antigens were retrieved by autoclave treatments in citrate buffer (pH 6.0) (Abcam, Cambridge, UK). Endogenous peroxidases were removed by 3% H2O2 (Sigma-Aldrich, MO, USA). Then, 2% bovine serum albumin (Sigma-Aldrich) in phosphate-buffered saline was used for a blocking solution. Antibodies α-CD20 #ab78237 (Abcam) and α-CXCL13 #20927-1-AP (Proteintech Group, IL, USA) were used as primary antibodies. Histostar (MBL, Japan) and Histostar diaminobenzidine (DAB) substrate solution (MBL) were used for detecting signals.
Signature 16 transcriptional strand bias
Transcriptional strand biases of the Signature 16 were determined as follows. All SNVs called by Karkinos were divided into transcribed or untranscribed SNVs. The contribution of each mutational Signature was calculated by deconstructSigs software (54) on transcribed or untranscribed SNVs.
Germline variant analysis
We analyzed germline variants by using GATK software (https://software.broadinstitute.org/gatk/) and identified 85,006 germline nonsilent (missense and nonframeshift indels) and 7000 germline truncation variants (nonsense, nonstop, and frameshift indels) using the following parameters: sequence depth ≧30 and <1% of population-level MAF, according to the Tohoku Medical Megabank Organization (ToMMo) (56), 1000 Genomes (57), and Exome Aggregation Consortium (ExAC) (58) databases. We retained 66,438 nonsilent variants and 3952 truncation variants after the removal of variants with VAF <0.2 and/or GATK quality value score < 500. We further screened the 262 gastric tumors and 29 normal stomach tissues sourced from TCGA according to whether they achieved a C score (59) ≧10 and a RPKM value of >1 and resultantly generated a list of 34,622 nonsilent and 2424 truncation variants. Last, we focused on 624 cancer-associated genes selected using a previously described pan-cancer germline variant analysis method (25) and obtained 1777 nonsilent variants (in 390 genes) and 60 truncation variants (in 28 genes) (fig. S6). Table 1 shows genes with presumed pathogenic germline variants identified in the 243 Japanese GC cases. For the focused analysis of CDH1, we included the TCGA GC dataset whose germline variants were called using GATK by a filtering depth of ≧30, <1% of population-level MAF, VAF ≧0.2, and a quality score of ≧500 (as described above) (9) and the germline variants called in Korean DGC dataset (6). We identified 11 SNVs in total (Table 2).
The truncation germline variants identified in the 243 Japanese GC cases are also summarized in Table 1. According to a previous paper of comprehensive breast cancer study (60), BRCA pathway genes include BRCA1, BRCA2, PALB2, RAD51, BARD1, and BRIP1; thus, we selected GC cases that exhibited nonsilent and/or truncation variants in these BRCA genes and were associated with Signature 3 contributions (the BRCA signature) using a filtering depth of ≧30, <1% population-level MAF, and VAF ≧0.2, and a quality score of ≧500 (table S3) from the Japanese cohort and TCGA dataset (9).
Supplementary Material
Acknowledgments
We thank K. Shiina, K. Nakano, S. Kawanabe, S. Yuba, and A. Yamamoto for technical assistances. Funding: This study was supported by AMED Project for Cancer Research and Therapeutic Evolution (P-CREATE) (JP19cm0106502) (to H.Abu.), JSPS KAKENHI Grant-in-Aid for Scientific Research (S) 24221011 (to H.Abu.), JSPS KAKENHI Grant-in-Aid for Scientific Research(A) 16H02481 (to S.I.), AMED P-CREATE (JP19cm0106551) (to S.I.), and AMED Practical Research for Innovative Cancer Control (JP16ck0106265) (to T.S.). The supercomputing resource was provided by the Human Genome Center (University of Tokyo). Author contributions: S.I. and H.Abu. designed the study. A.S. processed samples and organized exome sequencing and data analyses. M.K., H.Abe, T.Us., T.M., E.S., T.O., S.N., Y.S., Y.R., and M.F. coordinated acquisitions of surgical samples. H.K., D.K., A.T., H.Abe, T.Us., M.F., and S.I. carried out pathological reviews and analyses. S.F., T.Um., H.U., K.T., Y.T., and S.Y. performed computational analyses. A.S., H.K., D.K., and S.I. wrote the manuscript. G.N., T.S., A.N., M.F., S.I., and H.Abu. involved in supervisions of experiments and analyses, critical reviews, and discussion. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in this paper are present in this paper, the Supplementary Materials, or available at the following repository: [JGA (Japanese Genotype-phenotype Archive) database; https://www.ddbj.nig.ac.jp/jga/index-e.htm] with the accession number JGAS00000000228 and JGAS00000000229. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/19/eaav9778/DC1
REFERENCES AND NOTES
- 1.Ferlay J., Soerjomataram I., Dikshit R., Eser S., Mathers C., Rebelo M., Parkin D. M., Forman D., Bray F., Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 136, E359–E386 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Torre L. A., Bray F., Siegel R. L., Ferlay J., Lortet-Tieulent J., Jemal A., Global cancer statistics, 2012. CA Cancer J. Clin. 65, 87–108 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Tsugane S., Sasazuki S., Diet and the risk of gastric cancer: Review of epidemiological evidence. Gastric Cancer 10, 75–83 (2007). [DOI] [PubMed] [Google Scholar]
- 4.van der Post R. S., Vogelaar I. P., Carneiro F., Guilford P., Huntsman D., Hoogerbrugge N., Caldas C., Schreiber K. E. C., Hardwick R. H., Ausems M. G. E. M., Bardram L., Benusiglio P. R., Bisseling T. M., Blair V., Bleiker E., Boussioutas A., Cats A., Coit D., DeGregorio L., Figueiredo J., Ford J. M., Heijkoop E., Hermens R., Humar B., Kaurah P., Keller G., Lai J., Ligtenberg M. J. L., O’Donovan M., Oliveira C., Pinheiro H., Ragunath K., Rasenberg E., Richardson S., Roviello F., Schackert H., Seruca R., Taylor A., ter Huurne A., Tischkowitz M., Joe S. T. A., van Dijck B., van Grieken N. C. T., van Hillegersberg R., van Sandick J. W., Vehof R., van Krieken J. H., Fitzgerald R. C., Hereditary diffuse gastric cancer: Updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J. Med. Genet. 52, 361–374 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Y., Sethi N. S., Hinoue T., Schneider B. G., Cherniack A. D., Sanchez-Vega F., Seoane J. A., Farshidfar F., Bowlby R., Islam M., Kim J., Chatila W., Akbani R., Kanchi R. S., Rabkin C. S., Willis J. E., Wang K. K., McCall S. J., Mishra L., Ojesina A. I., Bullman S., Pedamallu C. S., Lazar A. J., Sakai R.; Cancer Genome Atlas Research Network, Thorsson V., Bass A. J., Laird P. W., Comparative molecular analysis of gastrointestinal adenocarcinomas. Cancer Cell 33, 721–735.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cho S. Y., Park J. W., Liu Y., Park Y. S., Kim J. H., Yang H., Um H., Ko W. R., Lee B. I., Kwon S. Y., Ryu S. W., Kwon C. H., Park D. Y., Lee J.-H., Lee S. I., Song K. S., Hur H., Han S.-U., Chang H., Kim S.-J., Kim B.-S., Yook J.-H., Yoo M.-W., Kim B.-S., Lee I.-S., Kook M.-C., Thiessen N., He A., Stewart C., Dunford A., Kim J., Shih J., Saksena G., Cherniack A. D., Schumacher S., Weiner A.-T., Rosenberg M., Getz G., Yang E. G., Ryu M.-H., Bass A. J., Kim H. K., Sporadic early-onset diffuse gastric cancers have high frequency of somatic CDH1 alterations, but low frequency of somatic RHOA mutations compared with late-onset cancers. Gastroenterology 153, 536–549.e26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang K., Yuen S. T., Xu J., Lee S. P., Yan H. H. N., Shi S. T., Siu H. C., Deng S., Chu K. M., Law S., Chan K. H., Chan A. S. Y., Tsui W. Y., Ho S. L., Chan A. K. W., Man J. L. K., Foglizzo V., Ng M. K., Chan A. S., Ching Y. P., Cheng G. H. W., Xie T., Fernandez J., Li V. S. W., Clevers H., Rejto P. A., Mao M., Leung S. Y., Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 46, 573–582 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Kakiuchi M., Nishizawa T., Ueda H., Gotoh K., Tanaka A., Hayashi A., Yamamoto S., Tatsuno K., Katoh H., Watanabe Y., Ichimura T., Ushiku T., Funahashi S., Tateishi K., Wada I., Shimizu N., Nomura S., Koike K., Seto Y., Fukayama M., Aburatani H., Ishikawa S., Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat. Genet. 46, 583–587 (2014). [DOI] [PubMed] [Google Scholar]
- 9.The Cancer Genome Atlas Research Network , Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alexandrov L. B., Nik-Zainal S., Wedge D. C., Aparicio S. A. J. R., Behjati S., Biankin A. V., Bignell G. R., Bolli N., Borg A., Børresen-Dale A.-L., Boyault S., Burkhardt B., Butler A. P., Caldas C., Davies H. R., Desmedt C., Eils R., Eyfjörd J. E., Foekens J. A., Greaves M., Hosoda F., Hutter B., Ilicic T., Imbeaud S., Imielinski M., Jäger N., Jones D. T. W., Jones D., Knappskog S., Kool M., Lakhani S. R., López-Otín C., Martin S., Munshi N. C., Nakamura H., Northcott P. A., Pajic M., Papaemmanuil E., Paradiso A., Pearson J. V., Puente X. S., Raine K., Ramakrishna M., Richardson A. L., Richter J., Rosenstiel P., Schlesner M., Schumacher T. N., Span P. N., Teague J. W., Totoki Y., Tutt A. N. J., Valdés-Mas R., van Buuren M. M., van’t Veer L., Vincent-Salomon A., Waddell N., Yates L. R.; Australian Pancreatic Cancer Genome Initiative; ICGC Breast Cancer Consortium; ICGC MMML-Seq Consortium; ICGC PedBrain, Zucman-Rossi J., Futreal P. A., McDermott U., Lichter P., Meyerson M., Grimmond S. M., Siebert R., Campo E., Shibata T., Pfister S. M., Campbell P. J., Stratton M. R., Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Forbes S. A., Bindal N., Bamford S., Cole C., Kok C. Y., Beare D., Jia M., Shepherd R., Leung K., Menzies A., Teague J. W., Campbell P. J., Stratton M. R., Futreal P. A., COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 39, D945–D950 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H., Borinskaya S., Yoshimura K., Kal’ina N., Marusin A., Stepanov V. A., Qin Z., Khaliq S., Lee M.-Y., Yang Y., Mohyuddin A., Gurwitz D., Mehdi S. Q., Rogaev E., Jin L., Yankovsky N. K., Kidd J. R., Kidd K. K., Refined geographic distribution of the oriental ALDH2*504Lys (nee 487Lys) variant. Ann. Hum. Genet. 73, 335–345 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Popova T., Manié E., Rieunier G., Caux-Moncoutier V., Tirapo C., Dubois T., Delattre O., Sigal-Zafrani B., Bollet M., Longy M., Houdayer C., Sastre-Garau X., Vincent-Salomon A., Stoppa-Lyonnet D., Stern M.-H., Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 72, 5454–5462 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Garaycoechea J. I., Crossan G. P., Langevin F., Mulderrig L., Louzada S., Yang F., Guilbaud G., Park N., Roerink S., Nik-Zainal S., Stratton M. R., Patel K. J., Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 553, 171–177 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Letouzé E., Shinde J., Renault V., Couchy G., Blanc J.-F., Tubacher E., Bayard Q., Bacq D., Meyer V., Semhoun J., Bioulac-Sage P., Prévôt S., Azoulay D., Paradis V., Imbeaud S., Deleuze J.-F., Zucman-Rossi J., Mutational signatures reveal the dynamic interplay of risk factors and cellular processes during liver tumorigenesis. Nat. Commun. 8, 1315 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang J., Tan W., Ling Z., Xi R., Shao M., Chen M., Luo Y., Zhao Y., Liu Y., Huang X., Xia Y., Hu J., Parker J. S., Marron D., Cui Q., Peng L., Chu J., Li H., Du Z., Han Y., Tan W., Liu Z., Zhan Q., Li Y., Mao W., Wu C., Lin D., Genomic analysis of oesophageal squamous-cell carcinoma identifies alcohol drinking-related mutation signature and genomic alterations. Nat. Commun. 8, 15290 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang M., McIntee E. J., Cheng G., Shi Y., Villalta P. W., Hecht S. S., Identification of DNA adducts of acetaldehyde. Chem. Res. Toxicol. 13, 1149–1157 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Yin S. J., Liao C. S., Wu C. W., Li T. T., Chen L. L., Lai C. L., Tsao T. Y., Human stomach alcohol and aldehyde dehydrogenases: Comparison of expression pattern and activities in alimentary tract. Gastroenterology 112, 766–775 (1997). [DOI] [PubMed] [Google Scholar]
- 19.Maejima R., Iijima K., Kaihovaara P., Hatta W., Koike T., Imatani A., Shimosegawa T., Salaspuro M., Effects of ALDH2 genotype, PPI treatment and L-cysteine on carcinogenic acetaldehyde in gastric juice and saliva after intragastric alcohol administration. PLOS ONE 10, e0120397 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fousteri M., Mullenders L. H., Transcription-coupled nucleotide excision repair in mammalian cells: Molecular mechanisms and biological effects. Cell Res. 18, 73–84 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Ishiguro S., Sasazuki S., Inoue M., Kurahashi N., Iwasaki M., Tsugane S.; JPHC Study Group , Effect of alcohol consumption, cigarette smoking and flushing response on esophageal cancer risk: A population-based cohort study (JPHC study). Cancer Lett. 275, 240–246 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Katoh H., Komura D., Konishi H., Suzuki R., Yamamoto A., Kakiuchi M., Sato R., Ushiku T., Yamamoto S., Tatsuno K., Oshima T., Nomura S., Seto Y., Fukayama M., Aburatani H., Ishikawa S., Immunogenetic profiling for gastric cancers identifies sulfated glycosaminoglycans as major and functional B cell antigens in human malignancies. Cell Rep. 20, 1073–1087 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Newman A. M., Liu C. L., Green M. R., Gentles A. J., Feng W., Xu Y., Hoang C. D., Diehn M., Alizadeh A. A., Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sahasrabudhe R., Lott P., Bohorquez M., Toal T., Estrada A. P., Suarez J. J., Brea-Fernández A., Cameselle-Teijeiro J., Pinto C., Ramos I., Mantilla A., Prieto R., Corvalan A., Norero E., Alvarez C., Tapia T., Carvallo P., Gonzalez L. M., Cock-Rada A., Solano A., Neffa F., Della Valle A., Yau C., Soares G., Borowsky A., Hu N., He L.-J., Han X.-Y.; Latin American Gastric Cancer Genetics Collaborative Group, Taylor P. R., Goldstein A. M., Torres J., Echeverry M., Ruiz-Ponte C., Teixeira M. R., Carvajal-Carmona L. G., Echeverry M., Bohorquez M., Prieto R., Suarez J., Mateus G., Bravo M. M., Bolaños F., Vélez A., Corvalan A., Carvallo P., Torres J., Carvajal-Carmona L., Germline mutations in PALB2, BRCA1, and RAD51C, which regulate DNA recombination repair, in patients with gastric cancer. Gastroenterology 152, 983–986.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu C., Xie M., Wendl M. C., Wang J., McLellan M. D., Leiserson M. D. M., Huang K.-l., Wyczalkowski M. A., Jayasinghe R., Banerjee T., Ning J., Tripathi P., Zhang Q., Niu B., Ye K., Schmidt H. K., Fulton R. S., McMichael J. F., Batra P., Kandoth C., Bharadwaj M., Koboldt D. C., Miller C. A., Kanchi K. L., Eldred J. M., Larson D. E., Welch J. S., You M., Ozenberger B. A., Govindan R., Walter M. J., Ellis M. J., Mardis E. R., Graubert T. A., Dipersio J. F., Ley T. J., Wilson R. K., Goodfellow P. J., Raphael B. J., Chen F., Johnson K. J., Parvin J. D., Ding L., Patterns and functional implications of rare germline variants across 12 cancer types. Nat. Commun. 6, 10086 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu N., Wang Z., Song X., Wei L., Kim B. S., Freedman N. D., Baek J., Burdette L., Chang J., Chung C., Dawsey S. M., Ding T., Gao Y.-T., Giffen C., Han Y., Hong M., Huang J., Kim H. S., Koh W.-P., Liao L. M., Mao Y. M., Qiao Y.-L., Shu X.-O., Tan W., Wang C., Wu C., Wu M.-J., Xiang Y.-B., Yeager M., Yook J. H., Yuan J.-M., Zhang P., Zhao X.-K., Zheng W., Song K., Wang L.-D., Lin D., Chanock S. J., Goldstein A. M., Taylor P. R., Abnet C. C., Genome-wide association study of gastric adenocarcinoma in Asia: a comparison of associations between cardia and non-cardia tumours. Gut 65, 1611–1618 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abnet C. C., Freedman N. D., Hu N., Wang Z., Yu K., Shu X.-O., Yuan J.-M., Zheng W., Dawsey S. M., Dong L. M., Lee M. P., Ding T., Qiao Y.-L., Gao Y.-T., Koh W.-P., Xiang Y.-B., Tang Z.-Z., Fan J.-H., Wang C., Wheeler W., Gail M. H., Yeager M., Yuenger J., Hutchinson A., Jacobs K. B., Giffen C. A., Burdett L., Fraumeni J. F., Tucker M. A., Chow W.-H., Goldstein A. M., Chanock S. J., Taylor P. R., A shared susceptibility locus in PLCE1 at 10q23 for gastric adenocarcinoma and esophageal squamous cell carcinoma. Nat. Genet. 42, 764–767 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi Y., Hu Z., Wu C., Dai J., Li H., Dong J., Wang M., Miao X., Zhou Y., Lu F., Zhang H., Hu L., Jiang Y., Li Z., Chu M., Ma H., Chen J., Jin G., Tan W., Wu T., Zhang Z., Lin D., Shen H., A genome-wide association study identifies new susceptibility loci for non-cardia gastric cancer at 3q13.31 and 5p13.1. Nat. Genet. 43, 1215–1218 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Wang Z., Dai J., Hu N., Miao X., Abnet C. C., Yang M., Freedman N. D., Chen J., Burdette L., Zhu X., Chung C. C., Ren C., Dawsey S. M., Wang M., Ding T., Du J., Gao Y.-T., Zhong R., Giffen C., Pan W., Koh W.-P., Dai N., Liao L. M., Yan C., Qiao Y.-L., Jiang Y., Shu X.-O., Chen J., Wang C., Ma H., Su H., Zhang Z., Wang L., Wu C., Xiang Y.-B., Hu Z., Yuan J.-M., Xie L., Zheng W., Lin D., Chanock S. J., Shi Y., Goldstein A. M., Jin G., Taylor P. R., Shen H., Identification of new susceptibility loci for gastric non-cardia adenocarcinoma: pooled results from two Chinese genome-wide association studies. Gut 66, 581–587 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Study Group of Millennium Genome Project for Cancer, Genetic variation in PSCA is associated with susceptibility to diffuse-type gastric cancer. Nat. Genet. 40, 730–740 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Hall M. J., Reid J. E., Burbidge L. A., Pruss D., Deffenbaugh A. M., Frye C., Wenstrup R. J., Ward B. E., Scholl T. A., Noll W. W., BRCA1 and BRCA2 mutations in women of different ethnicities undergoing testing for hereditary breast-ovarian cancer. Cancer 115, 2222–2233 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Momozawa Y., Iwasaki Y., Parsons M. T., Kamatani Y., Takahashi A., Tamura C., Katagiri T., Yoshida T., Nakamura S., Sugano K., Miki Y., Hirata M., Matsuda K., Spurdle A. B., Kubo M., Germline pathogenic variants of 11 breast cancer genes in 7,051 Japanese patients and 11,241 controls. Nat. Commun. 9, 4083 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farmer H., McCabe N., Lord C. J., Tutt A. N. J., Johnson D. A., Richardson T. B., Santarosa M., Dillon K. J., Hickson I., Knights C., Martin N. M. B., Jackson S. P., Smith G. C. M., Ashworth A., Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Yabuta T., Shinmura K., Tani M., Yamaguchi S., Yoshimura K., Katai H., Nakajima T., Mochiki E., Tsujinaka T., Takami M., Hirose K., Yamaguchi A., Takenoshita S., Yokota J., E-cadherin gene variants in gastric cancer families whose probands are diagnosed with diffuse gastric cancer. Int. J. Cancer 101, 434–441 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Shinmura K., Kohno T., Takahashi M., Sasaki A., Ochiai A., Guilford P., Hunter A., Reeve A. E., Sugimura H., Yamaguchi N., Yokota J., Familial gastric cancer: Clinicopathological characteristics, RER phenotype and germline p53 and E-cadherin mutations. Carcinogenesis 20, 1127–1131 (1999). [DOI] [PubMed] [Google Scholar]
- 36.Iida S., Akiyama Y., Ichikawa W., Yamashita T., Nomizu T., Nihei Z., Sugihara K., Yuasa Y., Infrequent germ-line mutation of the E-cadherin gene in Japanese familial gastric cancer kindreds. Clin. Cancer Res. 5, 1445–1447 (1999). [PubMed] [Google Scholar]
- 37.Takeichi M., Cadherin cell adhesion receptors as a morphogenetic regulator. Science 251, 1451–1455 (1991). [DOI] [PubMed] [Google Scholar]
- 38.Moreira-Nunes C. A., Barros M. B. L., do Nascimento Borges B., Montenegro R. C., Lamarão L. M., Ribeiro H. F., Bona A. B., Assumpção P. P., Rey J. A., Pinto G. R., Burbano R. R., Genetic screening analysis of patients with hereditary diffuse gastric cancer from northern and northeastern Brazil. Hered. Cancer Clin. Pract. 12, 18 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mateus A. R., Simões-Correia J., Figueiredo J., Heindl S., Alves C. C., Suriano G., Luber B., Seruca R., E-cadherin mutations and cell motility: A genotype–phenotype correlation. Exp. Cell Res. 315, 1393–1402 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Suriano G., Mulholland D., de Wever O., Ferreira P., Mateus A. R., Bruyneel E., Nelson C. C., Mareel M. M., Yokota J., Huntsman D., Seruca R., The intracellular E-cadherin germline mutation V832 M lacks the ability to mediate cell–cell adhesion and to suppress invasion. Oncogene 22, 5716–5719 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Kwak H.-W., Choi I. J., Cho S.-J., Lee J. Y., Kim C. G., Kook M.-C., Ryu K. W., Kim Y.-W., Characteristics of gastric cancer according to Helicobacter pylori infection status. J. Gastroenterol. Hepatol. 29, 1671–1677 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Murata-Kamiya N., Kurashima Y., Teishikata Y., Yamahashi Y., Saito Y., Higashi H., Aburatani H., Akiyama T., Peek R. M., Azuma T., Hatakeyama M., Helicobacter pylori CagA interacts with E-cadherin and deregulates the β-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene 26, 4617–4626 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Genta R. M., Graham D. Y., Comparison of biopsy sites for the histopathologic diagnosis of Helicobacter pylori: A topographic study of H. pylori density and distribution. Gastrointest. Endosc. 40, 342–345 (1994). [DOI] [PubMed] [Google Scholar]
- 44.Duell E. J., Sala N., Travier N., Muñoz X., Boutron-Ruault M. C., Clavel-Chapelon F., Barricarte A., Arriola L., Navarro C., Sánchez-Cantalejo E., Quirós J. R., Krogh V., Vineis P., Mattiello A., Tumino R., Khaw K.-T., Wareham N., Allen N. E., Peeters P. H., Numans M. E., Bueno-de-Mesquita H. B., van Oijen M. G. H., Bamia C., Benetou V., Trichopoulos D., Canzian F., Kaaks R., Boeing H., Bergmann M. M., Lund E., Ehrnström R., Johansen D., Hallmans G., Stenling R., Tjønneland A., Overvad K., Ostergaard J. N., Ferrari P., Fedirko V., Jenab M., Nesi G., Riboli E., González C. A., Genetic variation in alcohol dehydrogenase (ADH1A, ADH1B, ADH1C, ADH7) and aldehyde dehydrogenase (ALDH2), alcohol consumption and gastric cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC) cohort. Carcinogenesis 33, 361–367 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Shimazu T., Tsuji I., Inoue M., Wakai K., Nagata C., Mizoue T., Tanaka K., Tsugane S.; Research Group for the Development and Evaluation of Cancer Prevention Strategies in Japan , Alcohol drinking and gastric cancer risk: An evaluation based on a systematic review of epidemiologic evidence among the japanese population. Jpn. J. Clin. Oncol. 38, 8–25 (2008). [DOI] [PubMed] [Google Scholar]
- 46.Drew P. A., Clifton P. M., LaBrooy J. T., Shearman D. J., Polyclonal B cell activation in alcoholic patients with no evidence of liver dysfunction. Clin. Exp. Immunol. 57, 479–486 (1984). [PMC free article] [PubMed] [Google Scholar]
- 47.Pasala S., Barr T., Messaoudi I., Impact of alcohol abuse on the adaptive immune system. Alcohol Res. 37, 185–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keller G., Vogelsang H., Becker I., Plaschke S., Ott K., Suriano G., Mateus A. R., Seruca R., Biedermann K., Huntsman D., Döring C., Holinski-Feder E., Neutzling A., Siewert J. R., Höfler H., Germline mutations of the E-cadherin(CDH1) and TP53 genes, rather than of RUNX3 and HPP1, contribute to genetic predisposition in German gastric cancer patients. J. Med. Genet. 41, e89 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Curtis M. W., Ly Q. P., Wheelock M. J., Johnson K. R., Evidence that the V832M E-cadherin germ-line missense mutation does not influence the affinity of α -catenin for the cadherin/catenin complex. Cell Commun. Adhes. 14, 45–55 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Chen Y.-C., Fang W.-L., Wang R.-F., Liu C.-A., Yang M.-H., Lo S.-S., Wu C.-W., Li A. F.-Y., Shyr Y.-M., Huang K.-H., Clinicopathological Variation of Lauren Classification in Gastric Cancer. Pathol. Oncol. Res. 22, 197–202 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Shukla S. K., Prasad K. N., Tripathi A., Ghoshal U. C., Krishnani N., Nuzhat H., Quantitation of Helicobacter pylori ureC gene and its comparison with different diagnostic techniques and gastric histopathology. J. Microbiol. Methods 86, 231–237 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup , The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kostic A. D., Ojesina A. I., Pedamallu C. S., Jung J., Verhaak R. G. W., Getz G., Meyerson M., PathSeq: Software to identify or discover microbes by deep sequencing of human tissue. Nat. Biotechnol. 29, 393–396 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rosenthal R., McGranahan N., Herrero J., Taylor B. S., Swanton C., deconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 17, 31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Campbell B. B., Light N., Fabrizio D., Zatzman M., Fuligni F., de Borja R., Davidson S., Edwards M., Elvin J. A., Hodel K. P., Zahurancik W. J., Suo Z., Lipman T., Wimmer K., Kratz C. P., Bowers D. C., Laetsch T. W., Dunn G. P., Johanns T. M., Grimmer M. R., Smirnov I. V., Larouche V., Samuel D., Bronsema A., Osborn M., Stearns D., Raman P., Cole K. A., Storm P. B., Yalon M., Opocher E., Mason G., Thomas G. A., Sabel M., George B., Ziegler D. S., Lindhorst S., Issai V. M., Constantini S., Toledano H., Elhasid R., Farah R., Dvir R., Dirks P., Huang A., Galati M. A., Chung J., Ramaswamy V., Irwin M. S., Aronson M., Durno C., Taylor M. D., Rechavi G., Maris J. M., Bouffet E., Hawkins C., Costello J. F., Meyn M. S., Pursell Z. F., Malkin D., Tabori U., Shlien A., Comprehensive analysis of hypermutation in human cancer. Cell 171, 1042–1056.e10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nagasaki M., Yasuda J., Katsuoka F., Nariai N., Kojima K., Kawai Y., Yamaguchi-Kabata Y., Yokozawa J., Danjoh I., Saito S., Sato Y., Mimori T., Tsuda K., Saito R., Pan X., Nishikawa S., Ito S., Kuroki Y., Tanabe O., Fuse N., Kuriyama S., Kiyomoto H., Hozawa A., Minegishi N., Douglas Engel J., Kinoshita K., Kure S., Yaegashi N.; ToMMo Japanese Reference Panel Project, Yamamoto M., Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. Nat. Commun. 6, 8018 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.1000 Genomes Project Consortium , A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lek M., Karczewski K. J., Minikel E. V., Samocha K. E., Banks E., Fennell T., O’Donnell-Luria A. H., Ware J. S., Hill A. J., Cummings B. B., Tukiainen T., Birnbaum D. P., Kosmicki J. A., Duncan L. E., Estrada K., Zhao F., Zou J., Pierce-Hoffman E., Berghout J., Cooper D. N., Deflaux N., DePristo M., Do R., Flannick J., Fromer M., Gauthier L., Goldstein J., Gupta N., Howrigan D., Kiezun A., Kurki M. I., Moonshine A. L., Natarajan P., Orozco L., Peloso G. M., Poplin R., Rivas M. A., Ruano-Rubio V., Rose S. A., Ruderfer D. M., Shakir K., Stenson P. D., Stevens C., Thomas B. P., Tiao G., Tusie-Luna M. T., Weisburd B., Won H.-H., Yu D., Altshuler D. M., Ardissino D., Boehnke M., Danesh J., Donnelly S., Elosua R., Florez J. C., Gabriel S. B., Getz G., Glatt S. J., Hultman C. M., Kathiresan S., Laakso M., McCarroll S., McCarthy M. I., McGovern D., McPherson R., Neale B. M., Palotie A., Purcell S. M., Saleheen D., Scharf J. M., Sklar P., Sullivan P. F., Tuomilehto J., Tsuang M. T., Watkins H. C., Wilson J. G., Daly M. J., MacArthur D. G.; Exome Aggregation Consortium , Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kircher M., Witten D. M., Jain P., O’Roak B. J., Cooper G. M., Shendure J., A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Polak P., Kim J., Braunstein L. Z., Karlic R., Haradhavala N. J., Tiao G., Rosebrock D., Livitz D., Kübler K., Mouw K. W., Kamburov A., Maruvka Y. E., Leshchiner I., Lander E. S., Golub T. R., Zick A., Orthwein A., Lawrence M. S., Batra R. N., Caldas C., Haber D. A., Laird P. W., Shen H., Ellisen L. W., D’Andrea A. D., Chanock S. J., Foulkes W. D., Getz G., A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 49, 1476–1486 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/19/eaav9778/DC1