

TMEM16F-mediated phospholipid flip-flop on cell surface facilitates syncytialization and placental development.
Abstract
Cell-cell fusion or syncytialization is fundamental to the reproduction, development, and homeostasis of multicellular organisms. In addition to various cell type–specific fusogenic proteins, cell surface externalization of phosphatidylserine (PS), a universal eat-me signal in apoptotic cells, has been observed in different cell fusion events. Nevertheless, the molecular underpinnings of PS externalization and cellular mechanisms of PS-facilitated cell-cell fusion are unclear. Here, we report that TMEM16F, a Ca2+-activated phospholipid scramblase (CaPLSase), plays an essential role in placental trophoblast fusion by translocating PS to cell surface independent of apoptosis. The placentas from the TMEM16F knockout mice exhibit deficiency in trophoblast syncytialization and placental development, which lead to perinatal lethality. We thus identified a new biological function of TMEM16F CaPLSase in trophoblast fusion and placental development. Our findings provide insight into understanding cell-cell fusion mechanism of other cell types and on mitigating pregnancy complications such as miscarriage, intrauterine growth restriction, and preeclampsia.
INTRODUCTION
Cell-cell fusion is a fundamental cellular process essential for sexual reproduction, development, and homeostasis in organisms ranging from fungi to humans (1–3). Sperm-egg fusion determines the success of fertilization. Myoblast fusion into multinucleated myofiber is required for the growth, maintenance, and regeneration of skeletal muscles. Fusion of macrophages into bone-absorbing osteoclasts and giant cells is critical for bone homeostasis and immune response, respectively. In the human and murine placentas, mononucleated cytotrophoblasts continuously fuse with overlying syncytiotrophoblasts to sustain barrier function and transport activities (4). Consistent with its critical physiological roles, malfunction of cell-cell fusion has been implicated in a variety of diseases, including infertility, myopathies, osteoporosis, cancer, and preeclampsia (1, 3, 5, 6).
Despite its importance in biology and pathology, cell-cell fusion remains poorly understood compared to other membrane fusion processes (3). The recent progresses in cell-cell fusion mechanisms have successfully identified a number of fusogenic proteins or fusogens in mammals, such as myomaker (TMEM8C) and myomixer (Gm7325/Minion), that are specifically expressed in vertebrate myoblasts (7, 8), as well as syncytin proteins of retroviral origin that are solely expressed in placental trophoblasts under physiological conditions (9–12). The identifications of the fusogens greatly advanced our understanding of the mechanisms of cell-cell fusion. Cell type–specific expression of the fusogens further implies that cell-cell fusion is a highly diverse and specialized cellular process (1–3).
Phosphatidylserine (PS) is an anionic phospholipid that predominantly resides in the inner leaflet of the plasma membrane of all eukaryotic cells (13). Surface exposure of PS is known to play multifaceted signaling roles, including attracting blood clotting factors to enable blood coagulation and serving as an eat-me signal to recruit phagocytes (13–15). PS surface exposure has also been observed in various cell-cell fusion events such as myoblast fusion, sperm-egg fusion, and trophoblast fusion (3, 13). Compared to cell type–specific fusogens, PS exposure seems to be a more generalized feature for cell-cell fusion of various cell types. The notion of PS exposure as an early hallmark of apoptosis led to the prevailing view of PS exposure during cell-cell fusion: Apoptosis is responsible for PS exposure–mediated cell-cell fusion (16, 17). Nevertheless, syncytialization determines the reproduction and survival of a species. Fusing apoptotic cells to the physiologically important multinucleated cells seems harmful rather than beneficial to the fused cells. It is thus critical to understand the molecular underpinnings of PS exposure during cell fusion and the mechanism on how PS exposure mediates cell-cell fusion.
Here, we report that TMEM16F, a Ca2+-activated phospholipid scramblase (CaPLSase) that translocates PS to platelet surface and facilitates blood coagulation (18, 19), mediates PS exposure during trophoblast fusion independent of apoptosis. Genetic ablation of TMEM16F abolishes fusion among BeWo cells, a human choriocarcinoma trophoblast cell line, while reintroducing TMEM16F to the knockout (KO) cells rescues BeWo cell fusion. Consistent with the in vitro findings, the placentas of TMEM16F-deficient mice exhibit deficiencies in trophoblast fusion and fetal blood vessel development. These placental developmental defects lead to intrauterine growth restriction and partial perinatal lethality. Our findings thus uncover a cell-cell fusion mechanism that requires CaPLSase-mediated PS exposure as an important trophoblast fusion signal. The discovery of TMEM16F CaPLSase as a critical protein in trophoblast fusion and placental development may shine light on understanding placenta-related pregnancy complications such as miscarriage, stillbirth, intrauterine growth restriction, and preeclampsia.
RESULTS
PS surface externalization is required for trophoblast fusion
Forskolin-induced BeWo cell fusion is widely used to study the cellular mechanism of villous trophoblast fusion in vitro (20). Using Di-8-ANEPPS dye to mark the plasma membrane (Fig. 1A), the fusion index of live BeWo cells, which is defined as the percentage of nuclei from cells with three or more nuclei (to exclude potential dividing cells, see Materials and Methods for details), can be reliably quantified (21). Forskolin (30 μM) stimulation significantly boosts the fusion index from 0.03 ± 0.01 to 0.38 ± 0.02 (Fig. 1, B and E), whereas application of annexin V (AnV), a specific PS-binding protein, almost completely suppresses forskolin-induced BeWo cell fusion (Fig. 1, C and E). Our results support that PS surface exposure plays a critical role in trophoblast fusion, which is consistent with the previous reports showing that masking surface-exposed PS with PS-specific monoclonal antibodies inhibits BeWo cell fusion (22, 23).
Fig. 1. Ca2+-activated lipid scrambling is required for trophoblast fusion.
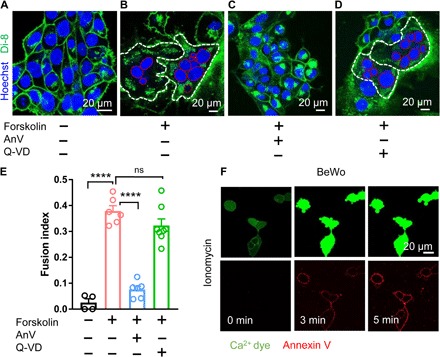
(A) Di-8-ANEPPS (Di-8), a membrane-bound voltage-sensitive dye, delineates BeWo trophoblast boundaries (green). Hoechst labels nuclei (blue). (B) Forskolin (30 μM) induces BeWo trophoblast fusion in 48 hours. White and red dotted lines delineate the plasma membrane and the nuclei of the fused cells, respectively. (C) AnV (0.5 μg/ml), a PS-binding protein, inhibits forskolin-induced BeWo cell fusion. (D) Pan-caspase inhibitor Q-VD-OPh (Q-VD; 10 μM) does not affect forskolin-induced BeWo cell fusion. (E) Summary of forskolin, AnV, and Q-VD-OPh effects on BeWo cell fusion. (F) BeWo cells exhibit robust CaPLSase activities when triggered with 1 μM ionomycin. Ca2+ dye (Calbryte 520) and fluorescently tagged AnV proteins (AnV-CF594) are used to measure the dynamics of intracellular Ca2+ and PS externalization, respectively. Unpaired two-sided Student’s t test for (E). ****P < 0.0001. ns, not significant. Error bars indicate ±SEM. Each dot represents the average of fusion indexes of six random fields from one coverslip (see Materials and Methods for detailed quantification). All fluorescence images are representatives of at least three biological replicates.
Ca2+-activated, but not caspase-activated, phospholipid scrambling is critical for trophoblast fusion
Phospholipid scramblases are passive phospholipid transporters on cell membranes that catalyze PS surface exposure (13). There are two major types of phospholipid scramblases. Caspase-dependent lipid scramblases such as Xkr8 induce PS externalization (24), which serves as an eat-me signal in apoptotic cells to attract phagocytes and to initiate phagocytosis (15). CaPLSases, on the other hand, mediate rapid PS surface exposure in viable cells in response to intracellular Ca2+ elevation (13, 18). To pinpoint the specific type of phospholipid scramblases that are responsible for PS externalization in trophoblast-derived BeWo cells, we first treated the cells with Q-VD-OPh, a pan-caspase inhibitor. The lack of inhibition on the fusion index by Q-VD-OPh (Fig. 1, D and E) indicates that caspase-dependent lipid scrambling is not required for BeWo cell fusion. Our result is consistent with previous findings, suggesting that apoptosis does not make major contributions to trophoblast fusion (25, 26). Instead, the human trophoblasts exhibited robust CaPLSase activity measured by an optimized phospholipid scrambling assay (fig. S1A) (27). We stimulated both BeWo and primary human term placental trophoblasts with ionomycin, a Ca2+ ionophore (Fig. 1F, fig. S1B, and movie S1). Upon stimulation, fluorescently tagged AnV rapidly accumulates on trophoblast surface following intracellular Ca2+ elevation (fig. S1A), indicating that functional CaPLSases are abundantly expressed in human trophoblasts. Consistent with the observed dominant CaPLSase activity in trophoblasts (Fig. 1F) and the importance of PS externalization in trophoblast fusion (Fig. 1E), disruption of Ca2+ homeostasis by reducing extracellular Ca2+ (fig. S1C) hinders BeWo cell fusion. Therefore, our results show that CaPLSase-mediated PS exposure plays an essential role in trophoblast fusion.
TMEM16F Ca2+-activated lipid scramblase is highly expressed in human trophoblasts
All currently known CaPLSases belong to the TMEM16 family (18, 28, 29). To pinpoint the molecular identity of CaPLSase in trophoblasts, we measured the relative mRNA levels of the TMEM16 family in both BeWo (Fig. 2A) and primary human term trophoblast cells (fig. S2A) by quantitative real-time polymerase chain reaction (qRT-PCR). Of these, TMEM16F, a CaPLSase critical for platelet PS externalization during blood coagulation (18, 19), shows the highest expression level in BeWo and human primary trophoblasts, whereas the expression levels of other TMEM16F members are negligible. Immunofluorescence staining confirms that TMEM16F protein is highly expressed in both BeWo and human primary trophoblasts (Fig. 2B). In human first trimester and term placentas, TMEM16F is highly expressed in both syncytiotrophoblasts and cytotrophoblasts in the chorionic villi (Fig. 2C and fig. S2B). Our results thus demonstrate that TMEM16F is highly expressed in human trophoblasts and is the major CaPLSase responsible for PS surface exposure.
Fig. 2. TMEM16F CaPLSase is highly expressed in human placental trophoblasts.
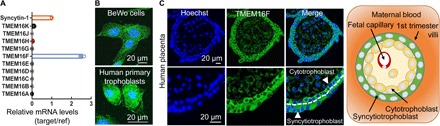
(A) qRT-PCR of TMEM16 family members in BeWo cells. All genes are normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and then normalized to Syncytin-1. (B) Immunofluorescence of TMEM16F (green) in BeWo cells (upper) and the primary human placental trophoblasts from a term placenta (lower). The nuclei are stained with Hoechst (blue). (C) Immunofluorescence of TMEM16F (anti-TMEM16F, green) and nuclei (Hoechst, blue) in a human first trimester placenta villi (upper). Higher magnifications are shown in the lower panels. TMEM16F is highly expressed in both cytotrophoblasts and syncytiotrophoblasts (arrowheads). The white dotted line demarcates the basal membrane of the syncytiotrophoblast. Schematic of the maternal-fetal interface in the first trimester placental villi is shown on the right. Cytotrophoblasts tightly pack against the basal membrane of syncytiotrophoblasts, which form the barrier between maternal blood (orange) and fetal capillaries located in the villous stroma. Images and diagram are shown in cross sections of the villi.
TMEM16F-CaPLSase is indispensable for trophoblast fusion in vitro
To further examine TMEM16F function in trophoblast fusion, we generated a TMEM16F KO BeWo cell line using CRISPR-Cas9 methods. Genetic ablation of TMEM16F completely eliminates the protein expression in BeWo cells as examined by Western blotting and immunostaining (fig. S3, A and B). Distinct from the Cas9 control cells that show strong CaPLSase activity (fig. S3C), the TMEM16F KO BeWo cells lack Ca2+-induced PS exposure when stimulated with ionomycin (Fig. 3A and movie S2). The intracellular Ca2+ elevation in response to ionomycin remain intact (Fig. 3A and movie S2), indicating that the deficiency in phospholipid scrambling in the TMEM16F KO BeWo cells is due to the lack of functional CaPLSases but not impaired Ca2+ response.
Fig. 3. TMEM16F CaPLSase is indispensable for BeWo trophoblast fusion.
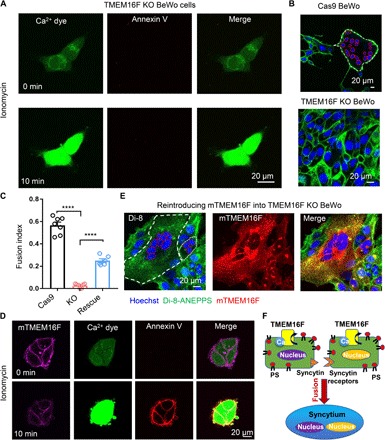
(A) CaPLSase activity is eliminated in TMEM16F KO BeWo cells when triggered with 1 μM ionomycin (see also movie S2). (B) Representative images of Cas9 control cells (upper) and TMEM16F KO BeWo cells (lower) after 48-hour forskolin treatment. (C) Genetic ablation of TMEM16F CaPLSase inhibits forskolin-induced BeWo cell fusion, and reintroducing mTMEM16F partially rescues the fusion deficiency. Each dot represents the average of fusion indexes of six random fields from one coverslip. Unpaired two-sided Student’s t test. ****P < 0.0001. Error bars indicate ±SEM. (D) Overexpression of mTMEM16F in the TMEM16F KO BeWo cells reintroduces CaPLSase activity (see also movie S3). Ionomycin (1 μM) was used to stimulate mTMEM16F. (E) Representative images of the TMEM16F KO BeWo cells overexpressing mTMEM16F after 48-hour forskolin treatment. (F) A cell-cell fusion mechanism requires CaPLSase-induced PS externalization on cell surface. All fluorescence images are representatives of at least three biological replicates. Nuclei and membranes are labeled with Hoechst (blue) and Di-8-ANEPPS (green), respectively, in (B) and (E). White and red dotted lines delineate the plasma membrane and the nuclei of the fused cells, respectively.
Consistent with the critical role of PS externalization in BeWo cell fusion (Fig. 1C), the TMEM16F-deficient BeWo cells lacking CaPLSase activity (Fig. 3A) fail to undergo fusion after forskolin stimulation (Fig. 3, B and C). Another independent TMEM16F KO BeWo cell line, which was generated using a different single-guide RNA (sgRNA), also shows the same deficiencies in CaPLSase activity and cell fusion (fig. S3, D to G), ruling out potential off-target effects of CRISPR-Cas9 genome engineering.
To further validate our finding, we overexpressed murine TMEM16F (mTMEM16F) in the TMEM16F-deficient BeWo cells. Reintroducing mTMEM16F not only restores their CaPLSase activity (Fig. 3D and movie S3) but also rescues cell-cell fusion (Fig. 3, C and E). Together, our TMEM16F ablation and rescue experiments in vitro explicitly demonstrate that TMEM16F CaPLSase plays an indispensable role in BeWo trophoblast fusion. TMEM16F CaPLSase-mediated PS exposure may work in concert with trophoblast-specific fusogenic proteins such as syncytins and their receptors to enable trophoblast fusion (Fig. 3F).
TMEM16F KO mice exhibit deficiency on trophoblast fusion, placental development, and perinatal viability
To understand the function of TMEM16F CaPLSase in trophoblast physiology and placental development in vivo, we examined the pregnant mice from a TMEM16F-deficient line (19). After 10 generations of backcrossing to C57BL/6J genetic background, heterozygous × heterozygous breeding scheme shows a significant reduction of postnatal homozygous TMEM16F−/− (KO) mice (14.7%) from the expected Mendelian inheritance (25%) (Fig. 4A and table S1). Furthermore, the weights of TMEM16F−/− placentas and embryos are significantly reduced compared to wild-type (WT) (TMEM16F+/+) placentas (Fig. 4, B and C), indicating signs of intrauterine growth restriction in the TMEM16F−/− mice.
Fig. 4. TMEM16F KO mice exhibit deficiency in trophoblast fusion, placental development defects, and perinatal lethality.
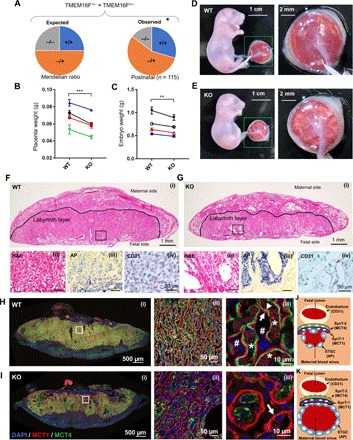
(A) Significant loss of TMEM16F−/− (target deletion) mice from the expected Mendelian inheritance around weaning age. *P < 0.05, χ2 test. (B and C) The TMEM16F−/− mice show markedly decreased placenta weight (B) and embryo weight (C). Note that each data point represents the averages of all the littermates with the same genotype from a pregnant mouse. Each line links the WT and KO fetuses from the same litter. Two-way analysis of variance (ANOVA). ***P < 0.001, **P < 0.01. All data represent means ± SEM. (D and E) Representative embryos and placentas from the WT (D) and TMEM16F KO (E) mice at embryonic day 18.5 (E18.5). The right panels show higher magnifications of the placentas with the fetal side facing up. Note that the opaque puncta appear on the TMEM16F−/− placenta surface, indicating deficiency on fetal vasculature. (F and G) H&E staining (i and ii), immunostaining against CD31 (iii), and AP (iv) of the TMEM16F WT (F) and KO (G) placentas at E18.5. CD31 and AP staining label fetal blood vessels and STGCs that enclose maternal blood sinuses, respectively. The TMEM16F−/− placentas have dramatically enlarged maternal blood sinuses (G, ii and iv) and markedly reduced fetal blood vasculature (G, iii). (H and I) MCT1 and MCT4 immunofluorescence staining of the TMEM16F WT (H) and KO (I) placentas at E18.5. MCT1 specifically expresses in the SynT-1 layer that faces maternal blood sinuses, while MCT4 specifically stains the SynT-2 layer that encloses fetal blood vessels. Panel (i) shows cross sections of the entire placenta, and panels (ii) and (iii) show enlarged views. Arrow and arrowhead indicate SynT-1 ayer nd the SynT-2, respectively. Asterisks and number sign mark fetal blood vessels and maternal blood sinuses, respectively. Note that the SynT-2 layer is missing in a large portion of the TMEM16F−/− labyrinth layer. (J and K) Schematics of the fetomaternal exchange anatomy from TMEM16F WT (J) and KO (K) mouse placentas. The red dotted line in (K) demarcates the missing SynT-2 layer. The TMEM16F−/− placentas have deficiency in forming the SynT-2 layer, which may lead to deficiency in fetomaternal exchange, enlarged maternal blood sinuses, diminished fetal angiogenesis, growth restriction, and partial perinatal lethality.
Detailed histological analysis confirms the placental defects in the TMEM16F−/− mice. The KO placentas appear pale from the fetal surface and exhibit markedly decreased and unevenly distributed vascularization in both placental and extra-embryonic membranes (Fig. 4, D and E). Consistent with these gross morphological changes, hematoxylin and eosin (H&E) staining of KO placentas reveals markedly enlarged cavities in the labyrinth layer, where syncytiotrophoblasts reside [Fig. 4, F (i and ii) and G (i and ii)]. The enlarged cavities are concentrated proximal to the fetal interface. Using alkaline phosphatase (AP) staining (30) to assess for the sinusoidal trophoblast giant cells (STGCs), we confirmed that the enlarged cavities in the KO placentas are maternal blood sinuses, which are enclosed by STGCs [Fig. 4, F (iii) and G (iii)]. In addition, CD31 immunostaining labels the fetal blood vessel in purple [Fig. 4G (iv) and fig. S4A], which is greatly diminished at the fetal interface of the KO labyrinth layer. The reduction of CD31 staining demonstrates that the fetal vessels in the KO placentas fail to develop and branch properly in the labyrinth layer especially the region near the fetal surface. This is consistent with the pale color and deficient vascularization observed from the fetal surface of the KO placentas (Fig. 4E).
As TMEM16F CaPLSase plays an essential role in trophoblast fusion in vitro, we investigated whether TMEM16F KO placentas have defects on syncytiotrophoblast formation. Mouse placentas have two syncytiotrophoblast layers, SynT-1 and SynT-2, to separate fetal blood vessel and maternal blood sinuses (Fig. 4J) (12). To characterize potential syncytialization defects in TMEM16F KO placentas, we stained MCT1 and MCT4, two monocarboxylate transporters that specifically express in the SynT-1 and SynT-2 layers, respectively (31, 32). MCT1 and MCT4 are always colocalized in the TMEM16F WT labyrinth layer, where the fetal blood vessels and maternal blood sinuses are intermingled and uniformly distributed (Fig. 4H). Notably, the MCT4+ SynT-2 layer is essentially absent from a large portion of TMEM16F KO labyrinth layer (Fig. 4I). This defect is more dramatic especially in the regions surrounding the enlarged maternal blood sinuses. As the SynT-2 layer encloses fetal blood vessels in the labyrinth (Fig. 4J), lack of MCT4 staining suggests that fetal blood vessels are not properly developed in these regions, consistent with the diminished anti-CD31 staining [Fig. 4G (iii) and fig. S4A]. AP staining of the STGCs is prominently enhanced around the enlarged maternal blood sinuses in the KO placentas [Fig. 4, F (iii) and G (ii)]. The two layers of syncytiotrophoblasts form the tight exchange barriers between maternal blood and fetal vessels in the mouse labyrinth layer (Fig. 4J) (33). Lack of the SynT-2 layer and missing fetal blood vessels in large portion of the labyrinth and enhanced AP staining surrounding the enlarged maternal blood sinuses collectively suggest that the fetal-maternal exchange in the TMEM16F KO labyrinth layer may be compromised (Fig. 4K). The defect of trophoblast syncytialization in the SynT-2 layer and subsequent maternofetal exchange deficiency might stress the developing KO placentas (Fig. 4I), which eventually causes fetal vascularization defects [Fig. 4G (iv) and fig. S4A], placental insufficiency (Fig. 4, E and G), and perinatal lethality (Fig. 4A and table S1).
DISCUSSION
TMEM16F has been shown to play critical roles in blood coagulation (18, 19), bone mineralization (34), immune response (35), and HIV infection (36). In this study, we have uncovered that TMEM16F also plays an essential role in trophoblast fusion and placental development. When TMEM16F is genetically ablated and CaPLSase-mediated PS exposure is eliminated, BeWo trophoblasts can no longer fuse (Fig. 3C). Reintroducing mouse TMEM16F into TMEM16F KO BeWo cells rescues CaPLSase activity and cell-cell fusion (Fig. 3, D and E). Furthermore, the TMEM16F KO mice show impaired syncytiotrophoblast formation, abnormally enlarged maternal blood sinuses in the labyrinth layer, and malformation of fetal blood vessels, which collectively cause intrauterine growth restriction and perinatal lethality (Fig. 4 and fig. S4).
In stark contrast to the prevailing view that PS exposure during cell-cell fusion is a consequence of apoptosis and caspase activation (16, 17, 37), our experiments demonstrate that apoptosis is dispensable for CaPLSase-mediated cell-cell fusion. Application of Q-VD-OPh, a pan-caspase inhibitor, has no effects on trophoblast fusion (Fig. 1, D and E), suggesting that apoptosis and caspase-activated lipid scramblases play a dispensable role in PS exposure during trophoblast fusion. Considering the facts that (i) multinucleated syncytial cells need to stay healthy to fulfill critical physiological functions, (ii) one of the advantages of cell-cell fusion is to supply the fused cells with additional genetic and metabolic materials, and (iii) multinucleated cell may not have the robust phagocytic machinery as phagocytes to handle apoptotic cells, cell fusion with apoptotic cells would increase the burden of the fused cells rather than benefit their survival and sustained functions. With more in-depth understanding of TMEM16 CaPLSases (18) and caspase-activated lipid scramblases (24), it is apparent that a viable cell with high CaPLSase expression can effectively externalize PS to cell surface (38) without triggering apoptosis (27, 39, 40). Our findings thus support that under physiological conditions, cell-cell fusion preferentially occurs between healthy cells instead of involving apoptotic cells.
Cell-cell fusion is a highly coordinated, tightly regulated, multistep process, which requires synergistic actions of multiple fusion machineries (1–3, 6). The known mammalian fusogens show clear cell type–specific distribution (1–3). In the placenta, syncytins are believed to interact with their receptors to promote trophoblast fusion (9, 10, 41). Considering the broad expression of TMEM16 scramblases (42), CaPLSase-mediated PS exposure may thus serve as a universal prerequisite for cell-cell fusion. CaPLSase-mediated PS exposure might work synergistically with cell type–specific fusogens to facilitate cell-cell fusion (Fig. 3F). The exposed PS on cell surface may prime the cell-cell fusion sites and create an environment to enable fusogens and their partners to form stable interactions, which eventually lead to fusion pore formation. On the other hand, it is also likely that CaPLSase-mediated PS surface exposure in one fusing cell recruits some cell surface PS receptors from another fusing cell. The interaction between PS and PS receptors, similar to the interaction between PS on the apoptotic cell surface and PS receptors in phagocytes during phagocytosis (13, 15, 43), could provide a simple and universal phospholipid-protein interaction that promotes membrane-membrane contact and fusion of various cell types. Therefore, cell type–specific fusogens and their partners might work in concert with CaPLSases to render cell-cell fusion with high efficiency and high specificity. The existence of multiple, highly coordinated yet seemingly redundant cell-cell fusion mechanisms can thus provide evolutionary advantages to ensure the reproduction and survival of an organism. All these hypotheses need to be tested in future studies.
In this study, we used the congenic TMEM16F KO mice that was generated by targeted deletion and have been backcrossed at least 10 generations to C57BL/6J genetic background. In contrast to the significant perinatal lethality observed in these further backcrossed congenic animals (Fig. 4A and table S1), we did not notice obvious lethality during our initial characterization of the TMEM16F KO mice when the animals were backcrossed between two and five generations to C57BL/6J genetic background (19). We suspect that the discrepancy might be derived from the differences in strain background. To further validate our in vivo findings using the TMEM16F congenic KO mice, we characterized a different TMEM16F KO mouse line generated using gene trap (see Materials and Methods for details). The perinatal lethality of the gene trap KO line is even more prominent (table S1), with only a 4.8% postnatal survival rate in homozygotes, which is similar to the lethality reported in the third TMEM16F KO mouse line (7.4% postnatal survival in homozygotes) (34). Consistent with the defects observed in our targeted deletion TMEM16F KO placentas, the gene trap TMEM16F KO placentas also show severe changes in morphology including the enlarged maternal blood sinuses in the labyrinth layer (fig. S5D) and deficiency in fetal blood vessel development (fig. S5B), as well as markedly reduced sizes of the placentas and the fetuses (fig. S5B). On the basis of the characterizations of two independently developed TMEM16F KO mouse lines, we conclude that genetic ablation of TMEM16F causes severe defects in placental and fetal development, which eventually lead to perinatal lethality. Our current findings also shine light on understanding why Scott syndrome, a mild bleeding disorder due to loss-of-function mutations of TMEM16F, is an extremely rare disease (44). It is likely that majority of the homozygous TMEM16F mutant Scott fetuses could not survive during pregnancy due to potential placental defects and perinatal lethality. Future clinical investigations are needed to test this hypothesis.
Two layers of syncytiotrophoblasts, SynT-1 and SynT-2, establish the exchange barrier between the maternal fluid and fetal blood vessels in mouse placentas (11, 12). The SynT-2 layer, which is labeled by MCT4 antibody, is essentially absent in a large portion of the TMEM16F KO labyrinth. Genetic ablation of syncytin-B (SynB) also specifically impairs syncytial formation of the SynT-2 layer without having obvious effect on the SynT-1 layer (12). There are unfused SynT-2 progenitor cells in the SynT-2 layer of the SynB KO placentas. Future histological characterizations including electron microscopy are needed to demonstrate whether the SynT-2 layer of the TMEM16F KO placentas also packs with unfused SynT-2 progenitor cells.
The retrovirus-derived syncytins are the known fusogens for trophoblast fusion (9). In mice, there are two syncytin genes, SynA and SynB (12). Similar to the phenotypes observed in our TMEM16F KO mice, the SynB KO mice also exhibit enlarged maternal sinuses, growth retardation in late gestation, and partial neonatal and postnatal lethality. It is worth noting that both TMEM16F KO placentas and SynB KO placentas have intact SynT-1 layers (12). On the other hand, SynA KO mice show 100% embryonic lethality, growth retardation in early gestation, trophoblast overexpansion, and unfused SynT-1 layer (11). It is unclear what the relationships are between TMEM16F scramblase and the two syncytins in controlling trophoblast fusion and placental development (Fig. 3F). Our current finding that TMEM16F ablation specifically impairs the SynT-2 layer, not the SynT-1 layer, suggests that TMEM16F-mediated lipid scrambling might be required for SynB-controlled trophoblast fusion in the SynT-2 layer. TMEM16F-mediated lipid scrambling might be dispensable for SynA-controlled trophoblast fusion in the SynT-1 layer in mice. Future studies are required to dissect the relationships between TMEM16F and the syncytins in trophoblast differentiation and placental development.
To conclude, in this study, we discovered that TMEM16F CaPLSase plays an essential role in trophoblast fusion and placental development. Our findings will shine new light on understanding the pathophysiology of placenta-related pregnancy complications such as intrauterine growth restriction, miscarriage, preterm birth, and preeclampsia. In addition, TMEM16 CaPLSase–mediated cell-cell fusion mechanism will help to understand other cell-cell fusion events and related diseases, including muscular dystrophy, infertility, viral infection, and cancer.
MATERIALS AND METHODS
Cell lines
The BeWo cell line was a gift from S. Permar at Duke University. The cell line was authenticated by Duke University DNA Analysis Facility. BeWo cells were cultured in Dulbecco’s modified Eagle’s medium–Ham’s F12 (DMEM/F12) medium (Gibco, catalog no. 11320-033) supplemented with 10% fetal bovine serum (Sigma, catalog no. F2442) and 1% penicillin/streptomycin (Gibco, catalog no. 15-140-122). All cells were cultured in a humidified atmosphere with 5% CO2 at 37°C.
Human placenta tissue and primary cultured human trophoblast cells
Placenta tissues were collected under the institutional review board approval (IRB# PRO00014627 and XHEC-C-2018-089) with the waiver of consent to obtain deidentified tissue that was not to be used for clinical purposes. Tissue was transported to the laboratory in DMEM/F12 medium (Gibco, catalog no. 11320-033). For primary trophoblast culture, the placentas were collected from women who underwent planned cesarean delivery at term (39 to 40 gestational weeks), without labor and current or previous pregnancy complications. Placental cytotrophoblast cells were prepared using a modified method of Kliman as described previously (45). In brief, villous tissues were removed randomly from the maternal side of the placenta. The isolated tissues were minced after washing with normal saline and digested with 0.125% trypsin (Sigma) and 0.03% deoxyribonuclease (DNase) I (Sigma, catalog no. 11284932001) in DMEM (Gibco, catalog no. 11995-065) and incubated for 30 min at 37°C with agitation. The supernatant from the first digestion was discarded. Three more rounds of digestions were followed. For each digestion, the dispersed placental cells were filtered through 70-μm cell strainer (BD Falcon, catalog no. 352350) and centrifuged at 1000 rpm for 10 min. The pooled cells were then purified using a 5 to 50% Percoll (Sigma, catalog no. P1644) gradient at stepwise increments of 5%. The cytotrophoblasts between densities of 1.049 and 1.062 g/ml were collected and plated in fibronectin (10 μg/ml)–coated cover glass for culture at 37°C in 5% CO2–95% air in DMEM containing 10% fetal calf serum (Sigma, catalog no. F2442) and 1% antibiotics (Gibco, catalog no. 15-140-122).
Mice
The gene targeting KO line was a gift from L. Jan and has been reported previously (19). The heterozygous (het) mice have been backcrossed 10 generations into a C57BL6 strain. The het × het crosses generated TMEM16F−/− KO mice. The gene trap TMEM16F/Ano6 KO line was purchased from the Jackson Laboratory (C57BL/6-Ano6Gt(EUCJ0166e09)Hmgu, #024798). PCR genotyping was performed using tail DNA extraction. Mouse handling and usage were carried out in a strict compliance with protocols approved by the Institutional Animal Care and Use Committee at Duke University, in accordance with National Institutes of Health guidelines.
TMEM16F KO cell lines
Stable Cas9-expressing cells were generated by transducing BeWo cells with lentiCAS9-blast (Addgene, catalog no. 52962). To generate TMEM16F/ANO6 KO cells, we designed sgRNAs targeting exon 2 using CHOPCHOP (https://chopchop.cbu.uib.no/) and cloned these sequences into lentiguide-puro (Addgene, catalog no. 52962). All lentiviruses were prepared by cotransfecting human embryonic kidney (HEK) 293T cells with the lentivector, psPAX2, and pMD2.g using TransIT-LT1 (Mirus, catalog no. MIR 2304). All transductions were done at a multiplicity of infection (MOI) of <1 in the presence of polybrene (4 μg/ml). Twenty-four hours after infection, cells were selected with blasticidin (10 μg/ml) or puromycin (2 μg/ml) for 48 to 72 hours and then expanded. Genomic DNA was harvested from cells 1 week after transduction with sgRNAs. The TMEM16F exon 2 locus was PCR-amplified from genomic DNA and used in Surveyor assays (Integrated DNA Technologies, catalog no. 706020) to confirm the presence of insertions and deletions.
The BeWo cell populations were selected for single-cell colonies by serial dilution into 96-well plates. The cells were grown for 10 to 14 days and then expanded to larger culture plates. Western blots, immunofluorescent staining, and phospholipid scrambling assay were used to screen the single-cell colonies that have no TMEM16F expression and CaPLSase function. The following sequences were used: sgRNAs used to generate TMEM16F-KO BeWo are AATAGTACTCACAAACTCCG and TTTCCAGTGATCCCAAATCG. TMEM16F forward and reverse primers used in Surveyor assays are TTTTCAGTGGTAGACCTTGCCT and AAGTTCAGCAACCTATTCCCAA, respectively.
Expression of mTMEM16F in TMEM16F-KO BeWo cells
The mTMEM16F with a C-terminal mCherry tag was transduced into TMEM16F-deficient BeWo cells using lentivirus. The lentivirus was produced by cotransfecting the packaging plasmid pCMV-deltaR8.9, envelope plasmid pCMV-VSVg, and ANO6-pLVX-mCherry-C1 vector (Addgene catalog no. 62554) into HEK293T cells using PEI Max 40000 (1 mg/ml; Polysciences, catalog no. 24765-1). After 48 hours of transfection, the lentivirus was concentrated by ultracentrifugating the transfected HEK293T culture supernatant at 19700 rpm for 2 hours at 17°C. The concentrated virus was used to infect TMEM16F-KO BeWo cells at an MOI of 4 in the presence of polybrene (2 μg/ml). TMEM16F expression was detected by the mCherry signal, and the medium was replaced with the fresh medium after 48 hours of infection. The transduced cells were expanded 72 hours after transduction.
Fluorescence imaging of Ca2+ and scramblase-mediated PS exposure
To monitor Ca2+ dynamics, cells were incubated with 1 μM Calbryte 590 AM Ca2+ dye (AAT Bioquest, catalog no. 20701) or Calbryte 520 AM (AAT Bioquest, catalog no. 20650) for 50 min at 37°C. After loading, the cells were washed with Hank’s balanced salt solution (HBSS) solution twice at room temperature to remove excessive Calbryte dyes. The Calbryte 590–loaded cells were then incubated in HBSS buffer containing CF 488–tagged AnV (0.5 μg/ml; Biotium, catalog no. 29011), a fluorescence PS probe. Ionomycin (1 μM) was added to increase intracellular Ca2+. A Zeiss 780 inverted confocal microscope was used to simultaneously image Ca2+ dynamics and CaPLSase-mediated PS exposure at a 5-s interval. ZEN and MATLAB were used for imaging analysis.
Quantitative RT-PCR
Total RNA was extracted using the RNeasy Mini Kit (Qiagen, catalog no. 74104) and quantified using the NanoDrop Spectrophotometer (NanoDrop, Wilmington, DE). Total RNA (1 μg) was used to generate complementary DNA (cDNA) using SuperScript III and Oligo(dT) (Thermo Fisher Scientific, catalog no. 18080051) following the manufacturer’s protocols. cDNA (25 ng) was used for qRT-PCR using 2× IQ SYBR Green supermix cocktail (Bio-Rad, catalog no. 1708880). Primers used for all genes are listed in table S2. Primers used for internal control gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were forward (CATGAGAAGTATGACAACAGCCT) and reverse (AGTCCTTCCACGATACCAAAGT). The iCycler was programmed for an initial denaturation step of 95°C for 2 min, followed by a two-step amplification phase of 35 cycles of 95°C for 30 s and 60°C for 1 min while sampling for fluorescein emission. Samples were run in duplicates, and the mean cycle threshold (Ct) was normalized to the average GAPDH Ct. Fold changes were calculated using the ΔΔCt method after normalization (all gene expressions were compared with Syncytin-1).
Immunofluorescence staining
Cells grown in monolayer cultures were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS), permeabilized with 0.2% Triton X-100, and blocked with 10% goat serum after antibody staining. Specific primary antibodies were added at 1:500 dilution overnight. Fluorescent staining was developed using the Alexa Fluor 488 or Alexa Fluor 594 fluorescence system (Molecular Probes, catalog no. 35552). Coverslips were mounted onto slides with Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories Inc., catalog no. H-1200). Fluorescent images were collected by using a Zeiss 780 inverted confocal microscope. ZEN and ImageJ were used for imaging analysis.
Immunoblotting
After the cells reached 80 to 90% confluence in 96-mm culture dish, trypsinization was used to collect the cells, which were then pelleted by centrifugating at 900 rpm, 4°C. After washing twice with cold PBS, the cell pellet was lysed with radioimmunoprecipitation assay (RIPA) lysate buffer (Thermo Fisher Scientific, catalog no. 89900) in the presence of 5 mM EDTA (Thermo Fisher Scientific, catalog no. 1861274) and 1× protease inhibitor cocktail (Thermo Fisher Scientific, catalog no. 1862209). After incubating on ice for 30 min, the lysate was centrifuged at 11,000 rpm for 20 min at 4°C. The supernatant was collected and incubated with 1× Laemmli (Bio-Rad, catalog no. 161-0747) supplemented with 112 mM dithiothreitol (Bio-Rad, catalog no. 161-0611) for 30 min at room temperature and then separated on SDS–polyacrylamide gel electrophoresis (PAGE) gel. After that, Trans-Blot Turbo Transfer system (Bio-Rad) was used to transfer the proteins from the SDS-PAGE gel to polyvinylidene difluoride membrane. The membrane was incubated in Ponceau S stain for 15 min. After three-time washing with 5% acetic acids, the Ponceau S–stained membrane was imaged using ChemiDoc XRS+ System (Bio-Rad). Nonfat milk (5%)/Tween 20 (0.1%) (Sigma, catalog no. P9416) was then used to block the membrane for 1 hour at room temperature. The membrane was incubated with anti-TMEM16F antibody (Millipore-Sigma, catalog no. HPA038958) with gentle shaking overnight at 4°C. Following three-time PBST (PBS supplemented with 0.1% Tween 20) washes, the membrane was then incubated with corresponding secondary antibody for 1 hour at room temperature. The Clarity Western ECL Substrate Kit (Bio-Rad, catalog no. 170-5060) was used to activate chemiluminescent signals from protein bands. The chemiluminescent signals were detected and analyzed by ChemiDoc XRS+ System (Bio-Rad) and Bio-Rad Imaging software. The TMEM16F band intensity from the chemiluminescence was normalized to the total protein loading detected by Ponceau S.
Cell-cell fusion and fusion index quantification
Syncytialization of BeWo cells was induced with forskolin, and fusion index was measured using a simple live cell imaging method (21). Briefly, BeWo cells were seeded on poly-l-lysine (Sigma, catalog no. P2636)–coated no. 0 cover glass for 24 hours at 5% CO2 at 37°C. After 24 hours, BeWo cells were treated with 30 μM forskolin (Cell Signaling Technology, catalog no. 3828s) for 48 hours to induce cell fusion. The culture medium containing 30 μM forskolin was replaced every 24 hours. After treatments, the cultured cells were incubated with Hoechst (Invitrogen, catalog no. H3570, 1:1500) and Di-8-ANEPPS (Invitrogen, catalog no. D3167, 2 μM) at 5% CO2 at 37°C for 15 to 20 min and subsequently washed in dye-free medium for twice at room temperature. Fluorescent images of six fields were randomly captured with a Zeiss 780 inverted confocal microscope using a 63×/1.4 NA (numerical aperture) Oil Plan-Apochromat DIC objective. ZEN and MATLAB were used for imaging analysis. Cells with more than two nuclei are defined as fused cells. To quantify cell fusion, fusion index (FI) of each sample was calculated as
where fi is the fusion index of each individual random field, nf is the number of nuclei from a fused cell, and N is the total number of nuclei for each field.
To interfere with BeWo cell fusion, AnV (0.5 μg/ml; Biotium) or 10 μM Q-VD-OPh (Sigma, catalog no. SML0063) was added to the culture medium 12 hours before forskolin treatment and remained throughout the experiment.
Mouse placenta histological analysis
Mice were deeply anesthetized using isoflurane. Placentas and embryos were freshly collected and fixed in 4% paraformaldehyde (Electron Microscopy Sciences, catalog no. 15710) for 2 days at 4°C and then transferred to 70% alcohol for 3 to 5 days before being processed by using 4-hour tissue processing setting with Leica ASP6025 (Leica Biosystems). Placentas were embedded in paraffin (HistoCore Arcadia H and Arcadia C, Leica Biosystems) right after the processing and then sectioned at 5 μm by using Leica RM2255 (Leica Biosystems, Buffalo Grove, IL) for histological staining.
H&E staining was performed through deparaffinization at 60°C followed by xylene twice (Fisher Chemical, catalog no. X5S-4) and a graded alcohol series (100%, 95%, 70%). Tissue was rinsed in MilliQ water for 5 min and then incubated in Hematoxylin 560 MX (Leica Biosystems, catalog no. 3801575) for 2 min and in running water for 1 min. Tissue was dipped seven times in 0.3% ammonium hydroxide (Fisher Chemical, catalog no. A669S-500) and then rinsed in running water for 1 min. Tissue was incubated in 70% ethanol for 1 min followed by 10 dips in Alcoholic Eosin Y 515 (Leica Biosystems, catalog no. 3801615). Dehydration was performed through an alcohol gradient of 95%, 100%, and xylene twice, 1 min each. All slides were mounted using DPX Mounting Medium (Electron Microscopy Sciences, catalog no. 13512).
CD31 immunohistochemistry (IHC) staining was performed by using rabbit polyclonal to CD31 (Abcam, catalog no. ab28364) with the Avidin/Biotin Blocking Kit (Vector Laboratories Inc., catalog no. SP-2001), VECTASTAIN Elite ABC HRP Kit (Peroxidase, Standard) (Vector Laboratories Inc., catalog no. PK-6100), and VECTOR VIP Peroxidase (HRP) Substrate Kit (Vector Laboratories Inc., catalog no. SK-4600). E-cadherin (Cdh1) IHC staining was done by using mouse monoclonal (HECD-2) to E-cadherin (Abcam, catalog no. ab1416) with the M.O.M. Kit (Vector Laboratories Inc., catalog no. BMK2202) incorporated with the kits used for CD31 staining. Both E-cadherin and CD31 staining have nuclear counterstaining with VECTOR Methyl Green (Vector Laboratories Inc., catalog no. H-3402-500). Microscopic imaging of histology samples was taken at 20× by using an Olympus microscope (BX63l, Olympus, Center Valley, PA) with cellSens Dimension software.
MCT1 is the specific marker of SynT-1, and MCT4 stains the basal membrane of SynT-2. Immunofluorescence staining against MCT1 and MCT4 is performed on TMEM16F WT and KO placenta by using MCT1 antibody (Millipore-Sigma, AB1286-I), which stains the fused SynT-1 layer facing the maternal blood sinuses, and MCT4 antibody (Santa Cruz Biotechnology Inc., sc-376140), which stains the fused SynT-2 layer facing the fetal blood vessels.
AP staining was used to assess for differentiation of STGCs. Briefly, sections were deparaffinized and rinsed in PBS and incubated in wash buffer (NTMT: 0.1 M NaCl; 0.1 M tris, pH 9.5; 0.05 M MgCl2; and 0.1% Tween 20, prepared fresh), and AP activity was detected by using BCIP-NBT (bromochloroindolyl phosphate–nitro blue tetrazolium) substrate to form a blue precipitate as previously described (46). The reaction was stopped in PBS, and cells were counterstained in Nuclear Fast Red (Vector Laboratories, catalog no. H-3403) for 1 min, followed by dehydration through a graded ethanol series. Images were collected using a Leica DMR light microscope (fluorescence and bright-field) and an EVOS XL Core microscope (bright-field; Invitrogen, USA).
Statistical analysis
The analyses of placenta and embryo weight were based on two-way ANOVA. The χ2 test was applied to test whether the genotype distribution of the mice from the het × het breeding scheme obeyed the Mendelian ratios. Other statistical analyses were based on a Student’s two-tailed unpaired t test and were performed using GraphPad Prism (GraphPad Software). Unless otherwise described, the data are representative of mean ± SEM. P values less than 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
We are grateful to L. Y. Jan for providing the TMEM16F-deficient mice, S.-Y. Kim for the assistance on generating the CRISPR BeWo TMEM16F KO cell lines, and M. Flamand and K. Meyer for helping with lentivirus generation. We appreciate M. Parast and J. Everitt for their help with placenta histology, S. Le for molecular biology, and C. Lockwood, V. Bennet, M. Boyce, Z. Zhang, P. Dong, and S. Le for their constructive comments on the manuscript. Funding: This work was supported by the NIH (NIH-DP2GM126898 to H.Y.) and the USF Nexus Initiative (H.P.). Author contributions: H.Y. conceived and supervised the project. Y.Z. and H.Y. designed the research and performed cell fusion experiments. Y.Z. characterized the KO mice. T.L. selected and characterized TMEM16F KO cells and worked on all molecular biology. T.L., Y.Z., and H.Y. developed the lipid scrambling assay. Z.M. and L.F. provided human placenta tissue and performed PCR. Y.Z., R.G., H.P., H.K., and D.R.N. performed histology. H.Y. and Y.Z. wrote the manuscript with input from L.F., D.R.N., and H.P. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data generated or analyzed during this study are included in this published article (and its supplementary information files). Raw data and code used are available from the corresponding author upon reasonable request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/19/eaba0310/DC1
REFERENCES AND NOTES
- 1.Chen E. H., Olson E. N., Unveiling the mechanisms of cell-cell fusion. Science 308, 369–373 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Hernández J. M., Podbilewicz B., The hallmarks of cell-cell fusion. Development 144, 4481–4495 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Brukman N. G., Uygur B., Podbilewicz B., Chernomordik L. V., How cells fuse. J. Cell Biol. 218, 1436–1451 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Potgens A. J., Schmitz U., Bose P., Versmold A., Kaufmann P., Frank H. G., Mechanisms of syncytial fusion: A review. Placenta 23( suppl. A), S107–S113 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Duelli D., Lazebnik Y., Cell-to-cell fusion as a link between viruses and cancer. Nat. Rev. Cancer 7, 968–976 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Ogle B. M., Cascalho M., Platt J. L., Biological implications of cell fusion. Nat. Rev. Mol. Cell Biol. 6, 567–575 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Bi P., Ramirez-Martinez A., Li H., Cannavino J., Anally J. R., Shelton J. M., Sánchez-Ortiz E., Bassel-Duby R., Olson E. N., Control of muscle formation by the fusogenic micropeptide myomixer. Science 356, 323–327 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Millay D. P., O'Rourke J. R., Sutherland L. B., Bezprozvannaya S., Shelton J. M., Bassel-Duby R., Olson E. N., Myomaker is a membrane activator of myoblast fusion and muscle formation. Nature 499, 301–305 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mi S., Lee X., Li X., Veldman G. M., Finnerty H., Racie L., LaVallie E., Tang X. Y., Edouard P., Howes S., Keith J. C. Jr., McCoy J. M., Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 403, 785–789 (2000). [DOI] [PubMed] [Google Scholar]
- 10.Blond J. L., Lavillette D., Cheynet V., Bouton O., Oriol G., Chapel-Fernandes S., Mandrand B., Mallet F., Cosset F. L., An envelope glycoprotein of the human endogenous retrovirus HERV-W is expressed in the human placenta and fuses cells expressing the type D mammalian retrovirus receptor. J. Virol. 74, 3321–3329 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dupressoir A., Vernochet C., Bawa O., Harper F., Pierron G., Opolon P., Heidmann T., Syncytin-A knockout mice demonstrate the critical role in placentation of a fusogenic, endogenous retrovirus-derived, envelope gene. Proc. Natl. Acad. Sci. U.S.A. 106, 12127–12132 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dupressoir A., Vernochet C., Harper F., Guégan J., Dessen P., Pierron G., Heidmann T., A pair of co-opted retroviral envelope syncytin genes is required for formation of the two-layered murine placental syncytiotrophoblast. Proc. Natl. Acad. Sci. U.S.A. 108, E1164–E1173 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bevers E. M., Williamson P. L., Getting to the outer leaflet: Physiology of phosphatidylserine exposure at the plasma membrane. Physiol. Rev. 96, 605–645 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Penberthy K. K., Ravichandran K. S., Apoptotic cell recognition receptors and scavenger receptors. Immunol. Rev. 269, 44–59 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Segawa K., Nagata S., An apoptotic 'Eat Me' signal: Phosphatidylserine exposure. Trends Cell Biol. 25, 639–650 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Hochreiter-Hufford A. E., Lee C. S., Kinchen J. M., Sokolowski J. D., Arandjelovic S., Call J. A., Klibanov A. L., Yan Z., Mandell J. W., Ravichandran K. S., Phosphatidylserine receptor BAI1 and apoptotic cells as new promoters of myoblast fusion. Nature 497, 263–267 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park S. Y., Yun Y., Lim J. S., Kim M. J., Kim S. Y., Kim J. E., Kim I. S., Stabilin-2 modulates the efficiency of myoblast fusion during myogenic differentiation and muscle regeneration. Nat. Commun. 7, 10871 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suzuki J., Umeda M., Sims P. J., Nagata S., Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468, 834–838 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Yang H., Kim A., David T., Palmer D., Jin T., Tien J., Huang F., Cheng T., Coughlin S. R., Jan Y. N., Jan L. Y., TMEM16F forms a Ca2+−activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell 151, 111–122 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wice B., Menton D., Geuze H., Schwartz A. L., Modulators of cyclic AMP metabolism induce syncytiotrophoblast formation in vitro. Exp. Cell Res. 186, 306–316 (1990). [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y., Yang H., A simple and robust fluorescent labeling method to quantify trophoblast fusion. Placenta 77, 16–18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katsuragawa H., Kanzaki H., Inoue T., Hirano T., Mori T., Rote N. S., Monoclonal antibody against phosphatidylserine inhibits in vitro human trophoblastic hormone production and invasion. Biol. Reprod. 56, 50–58 (1997). [DOI] [PubMed] [Google Scholar]
- 23.Lyden T. W., Ng A. K., Rote N. S., Modulation of phosphatidylserine epitope expression by BeWo cells during forskolin treatment. Placenta 14, 177–186 (1993). [DOI] [PubMed] [Google Scholar]
- 24.Suzuki J., Denning D. P., Imanishi E., Horvitz H. R., Nagata S., Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 341, 403–406 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Das M., Xu B., Lin L., Chakrabarti S., Shivaswamy V., Rote N. S., Phosphatidylserine efflux and intercellular fusion in a BeWo model of human villous cytotrophoblast. Placenta 25, 396–407 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Guilbert L. J., Riddell M., Winkler-Lowen B., Caspase activation is not required for villous cytotrophoblast fusion into syncytiotrophoblasts. Placenta 31, 982–988 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Le T., Le S. C., Yang H., Drosophila Subdued is a moonlighting transmembrane protein 16 (TMEM16) that transports ions and phospholipids. J. Biol. Chem. 294, 4529–4537 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki J., Fujii T., Imao T., Ishihara K., Kuba H., Nagata S., Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J. Biol. Chem. 288, 13305–13316 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griffin D. A., Johnson R. W., Whitlock J. M., Pozsgai E. R., Heller K. N., Grose W. E., Arnold W. D., Sahenk Z., Hartzell H. C., Rodino-Klapac L. R., Defective membrane fusion and repair in Anoctamin5-deficient muscular dystrophy. Hum. Mol. Genet. 25, 1900–1911 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simmons D. G., Fortier A. L., Cross J. C., Diverse subtypes and developmental origins of trophoblast giant cells in the mouse placenta. Dev. Biol. 304, 567–578 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Perez-Garcia V., Fineberg E., Wilson R., Murray A., Mazzeo C. I., Tudor C., Sienerth A., White J. K., Tuck E., Ryder E. J., Gleeson D., Siragher E., Wardle-Jones H., Staudt N., Wali N., Collins J., Geyer S., Busch-Nentwich E. M., Galli A., Smith J. C., Robertson E., Adams D. J., Weninger W. J., Mohun T., Hemberger M., Placentation defects are highly prevalent in embryonic lethal mouse mutants. Nature 555, 463–468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagai A., Takebe K., Nio-Kobayashi J., Takahashi-Iwanaga H., Iwanaga T., Cellular expression of the monocarboxylate transporter (MCT) family in the placenta of mice. Placenta 31, 126–133 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Rossant J., Cross J. C., Placental development: Lessons from mouse mutants. Nat. Rev. Genet. 2, 538–548 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Ehlen H. W., Chinenkova M., Moser M., Munter H. M., Krause Y., Gross S., Brachvogel B., Wuelling M., Kornak U., Vortkamp A., Inactivation of anoctamin-6/Tmem16f, a regulator of phosphatidylserine scrambling in osteoblasts, leads to decreased mineral deposition in skeletal tissues. J. Bone Miner. Res. 28, 246–259 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Ousingsawat J., Wanitchakool P., Kmit A., Romao A. M., Jantarajit W., Schreiber R., Kunzelmann K., Anoctamin 6 mediates effects essential for innate immunity downstream of P2X7 receptors in macrophages. Nat. Commun. 6, 6245 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Zaitseva E., Zaitsev E., Melikov K., Arakelyan A., Marin M., Villasmil R., Margolis L. B., Melikyan G. B., Chernomordik L. V., Fusion stage of HIV-1 entry depends on virus-induced cell surface exposure of phosphatidylserine. Cell Host Microbe 22, 99–110.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noubissi F. K., Ogle B. M., Cancer cell fusion: Mechanisms slowly unravel. Int. J. Mol. Sci. 17, E1587 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watanabe R., Sakuragi T., Noji H., Nagata S., Single-molecule analysis of phospholipid scrambling by TMEM16F. Proc. Natl. Acad. Sci. U.S.A. 115, 3066–3071 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Le T., Jia Z., Zhang Y., Le S. C., Chen J., Yang H., An inner activation gate controls TMEM16F phospholipids scrambling. Biophys. J. 116, 25a–26a (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Segawa K., Suzuki J., Nagata S., Constitutive exposure of phosphatidylserine on viable cells. Proc. Natl. Acad. Sci. U.S.A. 108, 19246–19251 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Potgens A. J., Drewlo S., Kokozidou M., Kaufmann P., Syncytin: The major regulator of trophoblast fusion? Recent developments and hypotheses on its action. Hum. Reprod. Update 10, 487–496 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Pedemonte N., Galietta L. J., Structure and function of TMEM16 proteins (anoctamins). Physiol. Rev. 94, 419–459 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Lemke G., How macrophages deal with death. Nat. Rev. Immunol. 19, 539–549 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zwaal R. F., Comfurius P., Bevers E. M., Scott syndrome, a bleeding disorder caused by defective scrambling of membrane phospholipids. Biochim. Biophys. Acta 1636, 119–128 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Sun K., Yang K., Challis J. R., Differential regulation of 11 beta-hydroxysteroid dehydrogenase type 1 and 2 by nitric oxide in cultured human placental trophoblast and chorionic cell preparation. Endocrinology 138, 4912–4920 (1997). [DOI] [PubMed] [Google Scholar]
- 46.Natale D. R., Starovic M., Cross J. C., Phenotypic analysis of the mouse placenta. Methods Mol. Med. 121, 275–293 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/19/eaba0310/DC1