

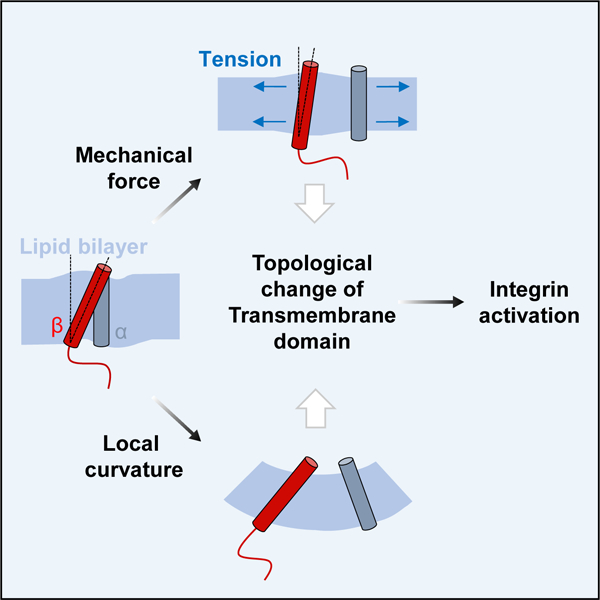
SUMMARY
Cells can sense and respond to various mechanical stimuli from their surrounding environment. One of the explanations for mechanosensitivity, a lipid bilayer model, suggests that a stretch of the membrane induced by mechanical force alters the physical state of lipid bilayer, driving mechanosensors to assume conformations better matched to the altered membrane. However, mechanosensors of this class are restricted to ion channels. Here, we reveal that integrin αIIbβ3, a prototypic adhesion receptor, can be activated by various mechanical stimuli including stretch, shear stress, and osmotic pressure. The force-induced integrin activation was not dependent on its known intracellular activation signaling events and even observed in reconstituted cell-free liposomes. Instead, these mechanical stimuli were found to alter the lipid embedding of integrin β3 transmembrane domain (TMD) and subsequently weaken the αIIb-β3 TMD interaction, which results in activation of the receptor. Moreover, artificial modulation of the membrane curvature near integrin αIIbβ3 can induce its activation in cells as well as in lipid nanodiscs, suggesting that physical deformation of the lipid bilayer, either by mechanical force or curvature, can induce integrin activation. Thus, our results establish the adhesion receptor as a bona fide mechanosensor that directly senses and responds to the force-modulated lipid environment. Furthermore, this study expands the lipid bilayer model by suggesting that the force-induced topological change of TMDs and subsequent alteration in TMD interactome is a molecular basis of sensing mechanical force transmitted via the lipid bilayer.
Keywords: membrane protein, transmembrane domain, lipid bilayer, mechanical force, mechanosensor
Graphical Abstract
eTOC Blurb
Kim et al. propose an integral membrane protein, integrin αIIbβ3, as a mechanosensor and clarifies its force-dependent activity control mechanism. Force-sensitive topological change of transmembrane domains within the lipid bilayer is suggested as a key feature of mechanosensitivity.
INTRODUCTION
Direct sensing of mechanical force by cells has been explained by two alternative hypotheses, a tethered model and a lipid bilayer model [1]. The tethered model suggests that mechanosensors are tethered to a relatively stationary position, such as cell-extracellular matrix (ECM) contact through the cytoskeleton. Mechanical force applied to the cells is transmitted via the cytoskeleton to induce tension in the mechanosensor and cause its structural change, which exposes a cryptic protein binding site or changes its enzymatic activity [2]. The lipid bilayer model suggests the mechanical force acting on the cell membrane causes tension in the lipid bilayer, which induces changes in the structure and activity of mechanosensors within the lipid bilayer [3, 4]. One important aspect of mechanosensors in this model is that their mechanical force-dependent activity changes in a cell-free lipid bilayer system [5]. So far, some ion channels, such as TRAAK, TREK-1 and Piezo1 have been proven to meet this criterion [6, 7]. Because transmembrane domains (TMDs) are the only regions that directly interact with lipids, their structural or topological changes for adaptation to a mechanically altered lipid environment could be the basis of mechanosensitivity of those channels. If this is the case, it can be expected that not only ion channels but also other types of transmembrane proteins, of which activities can be regulated by topological changes of their TMDs, could work as mechanosensors.
Integrins are transmembrane adhesion receptors consisting of α and β subunits [8]. Their affinity states toward ligands are controlled by α-β TMD interaction [9, 10] that depends on the precise tilt angle of the β TMD relative to the normal axis of the membrane [11]. Indeed, altering the topology of the β3 TMD disrupts the TMD interaction and also induces the high-affinity state of integrin αIIbβ3 [12]. Moreover, previous work indicated that integrins can be activated in lipid rafts [13, 14], where the relatively ordered and thick membrane may influence the topology of their TMDs. Considering the tilt angle-dependent TMD interaction and its importance in their signaling properties, integrins might be activated by mechanical force-dependent topological changes of their TMDs, thus being mechanosensors of the lipid bilayer. Here, we provide evidences in support of this hypothesis and establish that the prototypic platelet integrin αIIbβ3 is a bona fide mechanosensor.
RESULTS
Stretching cells induces integrin activation.
To test whether mechanical force can cause integrin activation, we established Chinese hamster ovary (CHO) cells expressing integrin αIIbβ3 (CHO/αIIbβ3) (Figure 1A), cultured them on collagen-coated stretch chambers (Figure 1B), and directly stretched the cells in the presence of FITC-fibrinogen. In this analysis, fibrinogen binding to integrin αIIbβ3 was increased by stretching (Figure 1C). Contributions from another fibrinogen-binding integrin αVβ3 seemed minimal because its level was at most 8% of that of integrin αIIbβ3 (Figure S1A). Moreover, the stretch-induced fibrinogen binding was blocked by an integrin αIIbβ3-specific antagonist (eptifibatide) to the level of CHO cells (Figure 1C), showing that fibrinogen binding is mainly mediated by integrin αIIbβ3. Because talin binding to the cytoplasmic tail of integrin β3 subunit [15] has been proposed as a final step of the integrin activation signaling [16], we generated CHO/αIIbβ3(Y747A) cells (Figure 1A) in which integrin β3 was replaced with the talin-binding defective mutant β3(Y747A) [17] and tested the requirement of talin binding in this process. Surprisingly, stretch caused similar increases in fibrinogen binding to CHO/αIIbβ3(Y747A) (Figure 1C), while overexpression of talin head domain (THD), mimicking an activated talin [15], induced integrin activation only in CHO/αIIbβ3 cells (Figure 1D).
Figure 1. Effects of stretching on integrin activation and its TMD interaction.

(A) The surface expression of integrin αIIbβ3 on CHO/αIIbβ3 and CHO/αIIbβ3(Y747A). (B) Schematic diagram of the stretching chamber. (C) CHO, CHO/αIIbβ3 (WT), or CHO/αIIbβ3(Y747A) cells (YA) were stretched in the presence of in FITC-fibrinogen. Fluorescence intensity from bound fibrinogen was normalized to protein concentration, and shown as a bar graph. (D) The degrees of fibrinogen binding in talin head domain (THD)-transfected cells. Error bars in (C) and (D) represent s.d. (n = 3) (E) Diagram of 3xFLAG-αIIb(TMD-tail)-TAP and Tac-β3(TMD-tail). TAP; tandem affinity purification. (F) CHO cells transfected with a and b TMD-tail constructs were stretched and maintained for 0, 1, or 5 min before their lysis. The TMD interaction was measured by detecting Tac-β3(TMD-tail) in precipitates of αIIb(TMD-tail)-TAP. Relative TMD interaction compared to upstretched control is shown. *P < 0.05 (one-way ANOVA followed by Tukey multiple comparisons post hoc test). (G, H) Effect of Y747A mutation or cytochalasin D on stretch-induced disruption of αIIb-β3 TMD interaction. Error bars in (F), (G), and (H) represent s.e. (n = 3). *P < 0.05, **P < 0.001, ***P < 0.0001. See also Figure S1.
Disruption of αIIb-β3 TMD interaction is essential for integrin activation [18, 19]. To provide a mechanistic basis, we next examined the effect of cell stretching on the TMD interaction using integrin αIIb and β3 TMD constructs (Figure 1E, [12]). The αIIb-β3 TMD interaction observed in non-stretch condition was immediately abolished when cells were stretched (Figure 1F). The stretch-induced TMD separation was partially recovered after prolonged incubation under stretched condition (Figure 1F, lanes 3–4), implying that the cells may resolve stretch-induced tension, e.g., by vesicle fusion to the membrane or by osmotic swelling. The stretch-induced disruption of the αIIb-β3 TMD interaction was independent of talin binding (Figure 1G) or the intact actin cytoskeleton (Figure 1H, Figure S1B). Taken together, our results showed that integrin αIIbβ3 can be activated by mechanical forces and that the mechanical force-induced integrin activation does not depend on the final integrin activation signaling event but rather involves the direct disruption of αIIb-β3 TMD interactions.
Shear stress can activate integrin αIIbβ3 in a signaling-independent manner.
To further confirm integrin activation by mechanical force, we adapted fluid-induced shear stress, another form of mechanical force. Cells on a collagen-coated flow chamber were subjected to laminar flow of a buffer containing fluorescein isothiocyanate-labeled fibrinogen (FITC-fibrinogen), and fibrinogen binding to the cells was monitored by fluorescence microscopy (Figure 2A). When the flow was applied for 5 min to induce a shear stress at 40 dyne/cm2, fibrinogen binding to both wild type αIIbβ3 (Figure 2B) and its Y747A mutant (Figure 2C) was significantly increased. The shear stress-induced fibrinogen binding was completely blocked by eptifibatide, indicating that fibrinogen binding is again mediated by integrin αIIbβ3 (Figure 2B–D). The bound FITC-fibrinogen co-localized with integrin αIIbβ3 (Figure S1C). Collagen-coating (Figure 2E, Figure S1D) or intact actin cytoskeleton (Figure 2F, Figure S1E) was not essential for the shear stress response. In addition, the shear stress-induced fibrinogen binding was observed even in CHO/αIIbβ3(Y747A/∆750) cells (Figure 2G, Figure S1F–G) where the C-terminal region after Ala750 of integrin β3 tail was deleted in the Y474A background, thereby removing the binding site of another integrin activator, kindlin-2 [20]. Shear stress-induced fibrinogen binding appears to require a threshold shear stress between 20 and 30 dyne/cm2 (Figure S1H).
Figure 2. Talin-binding independent activation of integrin αIIbβ3 under shear stress.

(A) Fibrinogen binding assay in the flow chamber. (B, C) Binding of fibrinogen to CHO/αIIbβ3 (B) or CHO/αIIbβ3(Y747A) cells (C) in static or flow condition was analyzed. Representative fluorescence images and the merged differential interference contrast (DIC) images are shown. Scale bar, 50 µm. (D) Average intensity of FITC-fibrinogen bound to cells in each condition was measured and normalized against that of CHO/αIIbβ3 cells under flow condition (n = 60, 66, 34, 61). (E) Shear stress-dependent fibrinogen binding to CHO/αIIbβ3 cells grown on collagen- or poly-L-lysine-coated flow chamber (n = 41, 45, 44, 43). (F) The effect of cytochalasin D on the flow response of CHO/αIIbβ3 (n = 50, 41, 40). (G) Fibrinogen binding to CHO/αIIbβ3 and CHO/αIIbβ3(Y747A∆750) under shear stress (n = 36, 65, 44, 58). Representative images for (D), (F), and (G) are shown in Figures S1D, S1E, and S1G, respectively. In all panels, error bars indicate s.d., ***P < 0.0001. See also Figure S1.
Next, we tested the involvement of phosphoinositide 3-kinase (PI3K) signaling previously shown to be important in mechanical force-induced integrin αVβ3 activation [21]. To this end, we applied flow with biotinylated fibrinogen-containing media, and measured the fibrinogen binding by western blotting. In this experimental setting, shear stress also activated integrin αIIbβ3 (Figure 3A–B). However, pretreatment of PI3K inhibitor did not impair fibrinogen binding to CHO/αIIbβ3 cells (Figure 3A–B). Therefore, though there could be a context-dependent mechanotransduction pathway for integrin activation, a large portion of shear stress-induced integrin αIIbβ3 activation appears to be independent of previously described integrin activation signaling. In agreement with this, addition of fibrinogen to flow-experienced cells right after flow was stopped did not cause fibrinogen binding (Figure 3C–D), implying that the shear stress-induced integrin activation might be controlled by a rapid on-and-off mechanism without prolonged activation of a series of signaling molecules. However, we do not exclude the possibility that the force-mediated immediate and transient active state of the integrin can be replaced by the more sustainable talin-mediated active state if talin is activated by a mechanotransduction pathway involving PI3K [22] or other signaling pathways. Indeed, binding of the activated version of talin, THD, to integrin αIIbβ3 was significantly increased in stretched cells (Figure S2), suggesting that the mechanosensitive transient activation mechanism may facilitate talin binding presumably by providing the separated α-β tail for better recruitment of activated talin.
Figure 3. Signaling-independent activation of integrin αIIbβ3 under shear stress.

(A) CHO/αIIbβ3 cells on flow chambers were pretreated with 100 nM wortmannin (Wort) or not, and applied with biotinylated fibrinogen-containing HBSS under static or flow condition (~40 dyne/cm2) in the presence of 2 mM EDTA or 10 mg/ml LIBS6 (integrin β3 activating antibody). Bound fibrinogen was measured by western blotting, with integrin b3 as an internal loading control. Phosphorylation level of Akt, a downstream effector of PI3K, was determined. (B) The degrees of fibrinogen binding in each condition normalized against EDTA-treated and LIBS6-treated samples (n=3). ***P < 0.0001. (C) CHO/αIIbβ3 cells were incubated under flow (40 dyne/cm2) for 5 min in FITC-fibrinogen-containing media (F5), or incubated first without fibrinogen but under flow for 5 min, and then further incubated with fibrinogen in static condition for 5 min (F5S5). Scale bar, 50 µm. (D) Average intensity of FITC-fibrinogen bound to cells in each condition (n = 41, 43). ***P < 0.0001 (Unpaired t test). In all panel, error bars indicate s.d. See also Figure S2.
Shear stress promotes soluble fibrinogen binding to integrin αIIbβ3 on immobilized platelets.
Once platelets adhere to an injured vessel wall during the hemostatic process, they experience shear stress caused by the velocity gradient of blood flow from the inner surface to the middle of a blood vessel. Thus, to verify our finding in a more physiological setting, we sought to determine whether shear stress can activate integrin αIIbβ3 on the surface of vessel-attached platelets. To minimize interference from other blood components, we prepared Walsh buffer containing only FITC-fibrinogen. Shear stress significantly induced fibrinogen binding to integrin αIIbβ3 on platelets (Figure 4A–B) with approximately 2,655 fibrinogen molecules bound per platelet (Figure S3A). At static or low shear condition (≤ 20 dyne/cm2), fibrinogen binding was rarely detected (Figure 4C, Figure S3B), while treatment with thrombin, a platelet agonist, induced fibrinogen binding under static condition (Figure S3C). Remarkably, the shear stress-induced fibrinogen binding was not inhibited by the phospholipase C inhibitor U73122 (Figure 4D–E), which can block agonist-induced fibrinogen binding to platelets (Figure 4F). In addition, the shear stress-induced increase in fibrinogen binding did not arise from platelet activation, as evident by annexin V staining [23] (Figure 4G–H). As in CHO cells, collagen coating was not essential either for shear stress-induced fibrinogen binding (Figure 4I, Figure S3D). Importantly, the increase in fibrinogen binding under flow was similarly observed in platelets from the talin-binding impaired β3 L746A mice [24] (Figure S3E), suggesting again that the fibrinogen binding is not talin binding-dependent. Finally, under flow condition, human platelets also showed an increase in binding to PAC1, the activation-specific human integrin αIIbβ3 antibody (Figure S3F).
Figure 4. Shear stress-induced fibrinogen-integrin αIIbβ3 interaction on immobilized platelets.

(A, B) FITC-fibrinogen binding to immobilized mouse platelets was tested under static, flow (40 dyne/cm2), or flow plus 10 µM eptifibatide condition. Average intensities of FITC fluorescence were measured, and relative fibrinogen binding compared to that under flow condition was shown as bar graph (n = 68, 70, 56). (C) The degree of fibrinogen binding to platelets under different flow rates (n = 74, 76, 71, 72, 54). Representative images are shown in Figure S3B. (D, E) Effect of 10 mM U73122 on the shear stress-induced fibrinogen binding to mouse platelets. Relative fibrinogen binding was shown as in (B) (n = 120, 106, 123). (F) Mouse platelets suspended in Walsh buffer containing FITC-fibrinogen were stimulated with 10 mM adenosine diphosphate (ADP) in the presence or absence of 10 mM U73122. Fibrinogen binding was analyzed under flow cytometry. (G) Platelets were incubated for 5 min under static (with or without thrombin) or flow (40 dyne/cm2), and then stained with FITC-conjugated annexin V. (H) Average intensities of annexin V-FITC fluorescence are shown (n = 49, 37, 43). (I) The shear stress responses of platelets on flow chambers coated with either collagen or poly-L-lysine were compared (n = 62, 70, 58, 49). Representative images are shown in Figure S3D. In all panels, error bars indicate s.d. ***P < 0.0001. Scale bar, 50 µm. See also Figures S3 and S4.
Osmotic force activates integrins in liposomes.
The observation of force-dependent but signal- and talin binding-independent integrin activation led us to hypothesize that integrin αIIbβ3 may constitute a bona fide mechanosensor, which, at least in part, can be directly activated by its own force-sensing machinery. Indeed, integrins are important connections between the ECM and the actin cytoskeleton, thereby serving as candidate mechanosensors in the tethered model [25–27]. Because stretch-induced disruption of the αIIb-β3 TMD interaction and shear-induced integrin activation were observed here even without an intact actin cytoskeleton, the tethered model cannot explain all aspects of force-dependent integrin activation. As an additional explanation, we hypothesized integrin αIIbβ3 may act as a mechanosensor according to the lipid bilayer model. To test this possibility, we created a cell-free liposome system wherein only purified lipid bilayer-embedded integrin αIIbβ3 is present, applied osmotic force to the liposomes by putting them in a hypotonic solution (Figure 5A), and measured their binding to fibrinogen-coated surface. The degree of liposome binding to non-coated surfaces was negligible, whereas addition of Mn2+, which activates the integrin possibly by stabilizing the active conformation outside, showed similar degrees of binding to fibrinogen-coated surfaces under all conditions (Figure S4A), confirming that the assay is functional. Importantly, the higher the applied osmotic force, the higher was the integrin liposome binding to the fibrinogen-coated surface (Figure 5B). In sharp contrast, fibrinogen binding of integrin αIIbβ3 embedded in nanodiscs (Figure 5C), a disc-shaped planar lipid bilayer [28] in which osmotic force cannot be exerted, remained unchanged (Figure 5D). Thus, we conclude that osmotic pressure applied to the lipid bilayer can shift the equilibrium in favor of the active conformation of the receptor, which makes integrin αIIbβ3 to serve as a genuine mechanosensor of tension applied to the lipid bilayer without involvement of any other cellular machinery.
Figure 5. Integrin αIIbβ3 activation in a cell-free lipid bilayer system.

(A) Integrin liposomes in isotonic or hypotonic solution. (B) The integrin liposomes generated in the presence of 100 mM glucose were added to a solution with final 100, 60, or 40 mM glucose, and incubated for 10 min on fibrinogen-coated surfaces. The integrin liposome binding to fibrinogen-coated surface was analyzed by measuring the amount of phosphatidylcholine left on the surface. Degree of binding in the presence of eptifibatide was subtracted to calculate specific binding in each condition, which was shown as a bar graph. *P = 0.0292, ***P < 0.0001. (C) Integrin αIIbb3 embedded in nanodiscs. (D) The effects of different glucose concentrations on the fibrinogen binding of the integrin nanodiscs were tested as in (B). (E) The amino acid sequence of integrin β3 TMD-tail with TMD region boxed. (F) Liposomes containing L694C-, I721C-, or S752C-bimane peptides were added into hypotonic or isotonic solution, right before emission spectra from these liposomes were monitored every 2 min for 10 min. Fluorescence intensities were normalized to the maximum fluorescence intensity at 0 min. In all panels, error bars indicate s.d. (n=3). See also Figures S4 and S5.
Mechanical force alters the lipid embedding of the β3 TMD.
The αIIb-β3 TMD interaction depends on the proper TMD orientation relative to the lipid bilayer [29], and, thus, changes of this orientation disrupts αIIb-β3 TMD interaction leading to integrin αIIbβ3 activation [30, 31]. Because the orientation of TMDs is maintained by lipid-protein interactions [4, 32], we hypothesized that the tension in the lipid bilayer may change the β3 TMD-lipid interaction and alter the lipid embedding of integrin β3. To test this, we purified β3 TMD-tail proteins with an engineered cysteine at the N-terminal (L694C) or C-terminal end (I721C) of the TMD, or in the middle of the cytoplasmic tail (S752), and labeled them with the environment-sensitive dye, bimane (Figure 5E). The bimane-conjugated peptides were then inserted into liposomes containing 100 mM glucose inside and diluted with hypotonic solutions. We confirmed that glucose, unlike other solutes such as sucrose, did not interfere with the emission spectrum of bimane (Figure S4B–C). Immediately after the dilution, their fluorescence emission was measured during osmotic swelling (Figure S4D). The osmotic force caused a gradual decrease in the bimane fluorescence intensity from both the N- and C-terminal ends of the β3 TMD peptides, while the fluorescence of bimane in the middle of the tail was not changed (Figure 5F). In contrast, no fluorescence changes were observed in the liposomes in isotonic solution (Figure 5F) or in nanodiscs (Figure S4E). These results show that the mechanical stimuli acting on the lipid bilayer changes the lipid embedding of β3 TMD. Therefore, alterations in β3 TMD embedding by mechanical force applied to the lipid bilayer can provide a basis of mechanical force-induced integrin activation. To confirm this idea, we perturbed the embedding by generating integrin liposomes with different acyl chain length and tested their relative binding to fibrinogen (Figure S4F). In the absence of force, the binding efficiencies of integrin liposomes with short-chain (C12:0) and long-chain lipids (C24:1) were significantly higher than those with normal length (C14:0 and C16:0). The higher degree of fibrinogen binding for the long-chain lipids may relate to differences in lipid packing due to the long acyl chain of the lipids and/or the unsaturated bond introduced to lower the melting temperature. In any case, these results confirm that lipid bilayer perturbation, regardless of thinning or thickening, can activate integrin. In short or long lipids, the degree of integrin activation was not changed further by osmotic pressure (Figure S4F), suggesting that the TMD topology in thin or thick lipid bilayer remains perturbed upon mechanical forces.
When the tilt angle of β3 TMD is decreased by mechanical force, some of its hydrophobic residues would be unfavorably exposed to the aqueous solution. Because one way for integral membrane proteins to relieve such hydrophobic mismatch is to cluster together via their TMDs [32–34], we tested whether the reported tendency of integrin β3 to form homomeric TMD oligomer in the abnormal lipid environment, e.g. in globular micelles [35], can be induced in mechanically perturbed membrane. Due to weak and possibly transient β3-β3 TMD interaction in cell membrane, cross linking was required to observe such interaction (Figure S5A–B, lane 3). Under stretch, we could detect a complex with a molecular weight corresponding to the β3 TMD-tail dimer (lane 7). We also detected α-β TMD interaction which was decreased under stretch (Figure S5A–B), as shown in the TMD binding assay without cross linking (Figure 1F). Thus, once mechanical force is applied to the cells, the αIIb-β3 interaction is disrupted in relation to an altered embedding of β3 TMD, and the thermodynamically unstable β3 TMD can be stabilized, at least transiently, by the homomeric TMD interaction.
Membrane curvature induces integrin αIIbβ3 activation.
The observation of integrin activation caused by changes in membrane embedding in the force-modulated lipid bilayer made us to assume that any other physical deformation of the lipid bilayer would potentially cause integrin activation. To verify this idea, we examined the effect of membrane curvature which is known to activate mechanosensitive channels [36] presumably by affecting lipid packing and membrane tension asymmetrically in the inner and outer leaflet of the membrane [37]. We employed the curvature-inducing motif of the epsin N-terminal homology (ENTH) domain (Figure 6A) which converts liposomes into tubules with ~15 nm radius [38], and fused it to the F3 domain of talin containing an integrin binding site for its targeting near integrins and to green fluorescence protein (GFP) for detection. The activating effect of talin F3, due to lack of additional element in the THD, is substantially reduced compared to the THD (Figure S6A), even though its binding to integrin αIIbβ3 was readily detected (Figure S6B). Nonetheless, the ENTH fusion to talin F3 induced a marked increase in PAC1 binding (Figure 6B–C). The membrane-binding ability of ENTH was essential for the activation, because introduction of K76A and L6E, known lipid-binding defective mutations of ENTH [38, 39], significantly reduced PAC1 binding (Figure 6D) although they can still interact with the integrin (Figure 6E). Deletion of the curvature-forming amphipathic helix [40] also largely inhibited the PAC1 binding (Figure S7A). In addition, two mutations, L6Q and L6W, known to possess different curvature-forming activities but similar lipid binding capacities [38], showed contrasting results. The normal curvature-forming L6W mutant markedly induced integrin activation while the curvature-forming defective L6Q mutant did not induce any increase (Figure 6F). The binding of these mutants to the integrin was similar (Figure 6G). To rule out the contribution of talin F3, we utilized an F3 mutant, F3(L325R), that retains integrin binding activity but does not trigger integrin activation [41]. The activating effect of GFP-F3(L325R) was negligible, but ENTH fusion to the mutant significantly induced integrin activation (Figure S7B). Together, these results suggest that increased integrin activation by the ENTH-F3 is due to its ability to induce curvature localized near the integrin.
Figure 6. Regulation of integrin activation by membrane curvature.

(A) The crystal structure of ENTH (PBD: 1H0A). The N-terminal amphipathic helix responsible for inducing membrane curvature is indicated with red color. (B) CHO/αIIbβ3 cells transfected with various constructs were tested for their ability to activate integrin αIIbβ3. Representative dot plots are shown. Means of PAC1 binding in cells expressing certain levels of GFP are indicated with red dots. (C) Using the means of PAC1 binding and the levels of GFP expression in (B), the dot plots were converted into a line graph. (D, E, F, G) Effects of ENTH mutants, K76A, L6E, L6W and L6Q, on ENTH-F3-mediated integrin activation and their binding to integrin αIIbβ3 were analyzed. In all panels, error bars indicate s.d. (n=3). See also Figures S6 and S7.
While the activation effect of ENTH only was minimal (Figure 6B–C), fluorescence microscopy revealed that both ENTH and ENTH-F3 similarly localized to the membrane (Figure S7C). Because ENTH is perceived to induce local curvature with a radius of curvature of about 15 nm [38], we assumed that the affected integrin should localize within this radius of curvature. However, even though ENTH is targeted to the membrane, this condition may not be met in the cell system. To verify this assumption, we utilized nanodiscs with a diameter of ~10 nm where limited membrane surface ensures that ENTH will be proximal to integrin. In this system, the ENTH domain without talin F3 fusion should induce membrane curvature near integrins, which was confirmed by staining the nanodisc with a fluorescent dye, 5-hexadecanoylaminofluorescein, that senses membrane curvature [42] (Figure S7D). When integrin nanodiscs (Figure S7E–F) were incubated with the ENTH domain, we observed that ENTH domain alone significantly increased fibrinogen binding to integrin nanodiscs (Figure 7A–B, Figure S7G). In contrast, the effects of ENTH mutant lacking the amphipathic helix (ENTH∆AH) on integrin nanodiscs (Figure 7B) were negligible. In addition, when various ENTH domains were added to nanodiscs containing the bimane-conjugated β3 TMD peptide, the bimane fluorescence intensities were changed in a dose-dependent manner only in the normal curvature-forming ENTH proteins, wild type and ENTH(L6W) (Figure 7C), suggesting that the ENTH domain-mediated curvature can induce topological change of the β3 TMD. Altogether, these results further show that membrane curvature can influence integrin activation.
Figure 7. Curvature-induced integrin activation in a cell-free lipid bilayer system.

(A) Integrin nanodisc binding assay. (B) Integrin αIIbβ3 reconstituted in nanodiscs were incubated with purified ENTH or ENTH AH, and their binding to fibrinogen-coated surface was analyzed. Error bars indicates s.d. (n=3), ***P < 0.0001. (C) Nanodiscs containing β3 TMD-tail peptide labelled with bimane at L694C was incubated with different amount of each ENTH proteins (wild type, ∆AH, L6E, K76A, L6Q, L6W) for 15 min and emission spectra from the mixtures were measured. All samples were solubilized in 3% SDS and their emission spectra were measured again to verify that the difference in fluorescence depends on topology but not concentration of the β3 TMD-tail peptide. See also Figure S7.
DISCUSSION
Our study suggests that alterations in the physical state of the lipid bilayer caused by either mechanical force or membrane curvature can induce changes in the membrane embedding of β3 TMD, disruption of the αIIb-β3 TMD interaction, and subsequent integrin activation. Thus, we propose that physical events that change the membrane structure may constitute a physiological integrin activation mechanism.
Many studies have revealed the importance of intracellular activators for integrin activation. For example, platelets from a patient with integrin β3 tail truncated at Arg724 or mice bearing the talin binding-impaired integrin β3(Y747A) fail to aggregate in response to physiological agonists [24, 43]. Although those platelets also show defects in agonist-induced spreading on a fibrinogen matrix, however, they still adhere to the matrix [24, 44]. In contrast, blockade of αIIbβ3 function with eptifibatide can fully inhibit platelet adhesion [45]. The seemingly normal adhesion achieved by these mutant integrins suggests there might be ways to induce fibrinogen binding independently of the intracellular activators. Biophysical studies have suggested that, because of long repelling glycoproteins crowding the plasma membrane, integrins with relatively short projections need to be “pushed” to reach their ligands [46]. Considering that pushing the membrane near integrins will likely induce membrane tension and curvature, we suggest that the proposed integrin activation involving changes in the lipid bilayer properties can be one of such mechanisms.
Nonetheless, this signaling-independent mechanism may not, by itself, replace talin-induced integrin activation. Instead, this mechanism may facilitate the integrin-talin interaction (Figure S2). During hemostasis, for example, the shear stress-induced activation of integrin αIIbβ3 may enhance transient attachment of fibrinogen to adherent platelets before agonist-induced signaling further stabilizes adhesion by recruiting talin. Likewise, during platelet spreading on fibrinogen matrix, growth of intracellular actin filaments may push membrane, changing membrane tension and curvature, and prime integrins in areas of high curvature such as the tips of filopodia named as “sticky fingers” [47] before their full activation by the intracellular activation machinery. We further emphasize that the proposed mechanism may not be solely responsible for the integrin activation under mechanical force. Rather, the immediate integrin activation, induced by changes in membrane structure, could shift the equilibrium in favor of integrin activation that can be perpetuated by slower but sustainable force-induced mechanotransduction pathways involving intracellular integrin activating machinery. As mentioned above, previous studies have well established that stretch or flow can activate integrin αVβ3 in endothelial cells via activation of VEGFR2 and PI3 kinase [22], implying possible cooperative roles of these two distinct mechanical responses on integrin activation depending on cellular context.
The alteration of β3 TMD topology upon mechanical force may be explained in part by the fact that the β3 TMD exist as tilted form inducing distortion of the surrounding lipid bilayer. Flattening of the lipid bilayer caused by membrane tension seems to energetically favor the less tilted topology of the TMD, similar to the mechanosensitive TRAAK channel’s preference for a cylinder shape over a wedge shapes in the presence of membrane tension [7]. Alternatively, mechanical force may change the physical properties of the lipid bilayer, such as the thickness [48] or the degree of lipid packing/fluidity [49], which can in turn alter the β3 TMD topology. The ~15% increase in size of liposomes measured during osmotic swelling (Figure S4D) is expected to result in an approximately 24% reduction in lipid bilayer thickness when assuming the product of the surface area and thickness of liposomes is constant during osmotic stretching [50]. Because the precise topology of β3 TMD is essential for the α-β TMD interaction [12], any alteration in embedding of β3 TMD in lipid bilayer caused by changes in membrane thickness, regardless of less or more lipid embedding, can potentially disrupt the TMD interaction and activate integrin αIIbβ3 as shown in Figure S4F. Indeed, integrins are thought to be active in lipid rafts [14] where relatively long lipids may modulate their embedding. Therefore, a physical deformation of the lipid bilayer induced by mechanical force and adaptation of integrin TMDs to the force-modulated lipid environment can be a molecular basis of force-sensing mechanism of integrin αIIbβ3.
In conclusion, this study establishes integrin αIIbβ3 as a genuine mechanoreceptor that can be activated by mechanical force and by membrane curvature. More generally, our study suggests that force-induced changes in TMD topology and subsequent alteration in TMD interactome can serve as a general principal of sensing mechanical force transmitted via the lipid bilayer.
STAR METHODS
Lead contact and materials availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Chungho Kim (chungho@korea.ac.kr). All plasmids generated in this study are available from the Lead Contact without restriction.
Experimental model and subject details
Chinese hamster ovary (CHO) cells and established stable cell lines used in this study are listed in key resource table. Cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) fetal bovine serum, 2 mM L-glutamine, non-essential amino acids and penicillin-streptomycin. Cells were maintained at 37 °C with 5% CO 2 in an incubator. For platelet experiments, blood was collected from 8-week-old male C57Bl/6 mice. Animal studies were approved by the Institutional Animal Care and Use Committee of Korea University. Human platelets were isolated from healthy donors. The use of human platelets was approved by institutional review board of Korea University Anam Hospital (IRB No. 2018AN0389).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-IL2Rα | Santa Cruz Biotechnology | sc-665 RRID: AB_631114 |
| Rabbit polyclonal anti-integrin β3 (Rb8053) | [18] | N/A |
| Mouse monoclonal anti-Akt | Cell Signaling Technology | 2920 RRID: AB_1147620 |
| Rabbit monoclonal anti-pAkt | Cell Signaling Technology | 9271 RRID: AB_329825 |
| Mouse monoclonal anti-GFP | Santa Cruz Biotechnology | sc-9996 RRID: AB_627695 |
| Mouse monoclonal anti-Strep-tag | IBA Lifesciences | 2–1507-001 RRID: AB_513133 |
| Mouse monoclonal anti-HA | Wako | 014–21881 |
| Mouse monoclonal anti-integrin β3 | Santa Cruz Biotechnology | sc-365679 RRID: AB_10844835 |
| Anti-rabbit IgG-APC | Thermo Fisher | A10931 RRID: AB_141369 |
| Anti-mouse IgG-Alexa Fluor 488 | Thermo Fisher | A11001 RRID: AB_2534069 |
| Anti-mouse IgG-Alexa Fluor 350 | Thermo Fisher | A11068 RRID:AB_2534112 |
| Anti-mouse IgG-TRITC | Jackson Laboratory | 715–025-151 RRID: AB_2340767 |
| Anti-mouse IgG-APC | Thermo Fisher | 17–4010-82 RRID: AB_2573203 |
| Anti-mouse IgM-APC | Thermo Fisher | 17–5790-82 RRID: AB_469458 |
| Mouse monoclonal anti-integrin αIIbβ3 (PAC1) | BD Biosciences | 340535 RRID: AB_400048 |
| Mouse monoclonal anti-integrin αIIbβ3 (D57) | [12] | N/A |
| Mouse monoclonal anti-integrin αIIbβ3 (LIBS6) | [12] | N/A |
| Bacterial and Virus Strains | ||
| Escherichia coli strain DH5α | Enzynomics | CP011 |
| Escherichia coli strain Stbl3 | New England Biolabs | C3040I |
| Escherichia coli strain BL21(DE3) | Enzynomics | CP111 |
| Lentivirus | This study | N/A |
| Biological Samples | ||
| Human: Platelet | Healthy donors | Korea University Anam Hospital (IRB No. 2018AN0389) |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dulbecco’s Modified Eagle Medium (DMEM) | Thermo Fisher | SH30243.01 |
| Fetal Bovine Serum (FBS) | Thermo Fisher | SH30919.03 |
| Penicillin-streptomycin | Thermo Fisher | SV30010 |
| L-glutamine | Welgene | LS 002–01 |
| MEM Non-Essential Amino Acids Solution | Thermo Fisher | 11140050 |
| Trypsin-EDTA | Thermo Fisher | SH30042.01 |
| UltraCULTURE™ Serum-free Medium | Lonza | 12–725F |
| PolyJet™ In Vitro DNA Transfection Reagent | SignaGen | SL100688 |
| Eptifibatide acetate (Integrilin) | Santa Cruz Biotechnology | sc-205675A |
| Fibrinogen from human plasma | Sigma-Aldrich | F4883–500MG |
| Fluorescein isothiocyanate isomer I (FITC) | Sigma-Aldrich | F7250 |
| EZ-Link™ NHS-Biotin | Thermo Fisher | 20217 |
| Streptavidin APC Conjugate | Thermo Fisher | 17–4317-82 |
| Streptavidin-Peroxidase Polymer | Merk | S2438–250UG |
| Cytochalasin D | Sigma-Aldrich | C8273–1MG |
| Hank’s Balanced Salt Solution (HBSS) | Welgene | LB 203–04 |
| Type I Collagen Solution | Advanced BioMatrix | #5005 |
| Poly-L-Lysine Solution | Sigma-Aldrich | P4707–50ML |
| Wortmannin | Sigma-Aldrich | W1928 |
| Adenosine 5′-diphosphate sodium salt | Sigma-Aldrich | A2754–100MG |
| U-73122 | Sigma-Aldrich | U6756 |
| Thrombin | Sigma-Aldrich | T4648–1KU |
| Annexin V-FITC | Merk | APOAF-50TST |
| D-(+)-Glucose | Sigma-Aldrich | G7021 |
| Sucrose | Samchun Chemicals | DMS-S0012 |
| Triton X-100 | Sigma-Aldrich | T9284–100ML |
| Sodium deoxycholate | Sigma-Aldrich | 30970–25G |
| Tris | Affymetrix | J75825 |
| Manganese dichloride | Sigma-Aldrich | 221279–100G |
| Sodium chloride | Duchefa Biochemie | S0520.5000 |
| Calcium chloride | Sigma-Aldrich | C1016–500G |
| Magnesium chloride | Sigma-Aldrich | M8266–100G |
| Bromobimane | Santa Cruz Biotechnology | sc-214629 |
| 12:0 PG | Avanti Polar Lipids | 840435P-25MG |
| 12:0 PC | Avanti Polar Lipids | 850335P-25MG |
| 14:0 PG | Avanti Polar Lipids | 850445P-25MG |
| 14:0 PC | Avanti Polar Lipids | 850345P-25MG |
| 16:0 PG | Avanti Polar Lipids | 840455P-25MG |
| 16:0 | Avanti Polar Lipids | 850355P-25MG |
| 24:1 PC | Avanti Polar Lipids | 850399P-25MG |
| Phosphatidylinositol 4,5-bisphosphate diC16 (PI(4,5)P2 diC16) | Echelon Biosciences | P-4516 |
| 5-hexadecanoylaminofluorescein | Thermo Fisher | H110 |
| Strep-Tactin Superflow | IBA Lifesciences | 2–1208-010 |
| Calmodulin-Sepharose 4B | GE Healthcare | 17–0529-01 |
| HisPur™ Ni-NTA Resin | Thermo Fisher | 88221 |
| Bio-Beads SM-2 Adsorbents | Bio-Rad | 152–3920 |
| DSS (disuccinimidyl suberate) | Thermo Fisher | 21555 |
| Zeba™ Spin Desalting Column | Thermo Fisher | 89892 |
| HiPrep™ 16/60 Sephacryl® S-200 HR | GE Healthcare Life Sciences | 17–1166-01 |
| HiTrap Q FF anion exchange chromatography column | GE Healthcare Life Sciences | 17515601 |
| Rhodamine Phalloidin | Thermo Fisher | R415 |
| Antifade Mounting Medium with DAPI | Vector Laboratories | H-1200 |
| Chloroform | Sigma-Aldrich | 154733–1L |
| Methanol | Sigma-Aldrich | 154903–1L |
| Critical Commercial Assays | ||
| GeneArt Seamless Cloning and Assembly Kit | Thermo Fisher | A13288 |
| Phosphatidylcholine Assay Kit | Cell Biolabs | STA-600 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Hamster: CHO | Korean Collection for Type Cultures (KCTC) | RRID: CVCL_0214 |
| Hamster: CHO/αIIbβ3 | This study | N/A |
| Hamster: CHO/αIIbβ3(Y747A) | This study | N/A |
| Hamster: CHO/αIIbβ3(Y747A/∆750) | This study | N/A |
| Hamster: CHO/αIIbTST-β3 | This study | N/A |
| Human: Lenti-X 293T | Clontech Laboratories | 632180 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | OrientBio | N/A |
| Mouse: C57BL/6 ITGB3 (WT/WT) | [24] | N/A |
| Mouse: C57BL/6 ITGB3 (L746A/L746A) | [24] | N/A |
| Oligonucleotides | ||
| See Table S1 (Primer sequences for PCR) | This study | |
| Recombinant DNA | ||
| pcDNA3.1 | [18] | N/A |
| pcDNA3.1-THD (Talin head domain)-GFP | [18] | N/A |
| pcDNA3.1-F3 (Talin F3 domain)-GFP | This study | N/A |
| pcDNA3.1-GFP | This study | N/A |
| pEGFPC1-EPSIN1- | Pietro De Camilli | Addgene Plasmid #21339 |
| pcDNA3.1-ENTH-F3-GFP | This study | N/A |
| pcDNA3.1-ENTH-F3(L325R)-GFP | This study | N/A |
| pcDNA3.1-ENTH(L6E)-F3-GFP | This study | N/A |
| pcDNA3.1-ENTH(K76A)-F3-GFP | This study | N/A |
| pcDNA3.1-ENTH(L6Q)-F3-GFP | This study | N/A |
| pcDNA3.1-ENTH(L6W)-F3-GFP | This study | N/A |
| pcDNA3.1-ENTH(ΔAH)-F3-GFP | This study | N/A |
| pFUGW-ITA2B | This study | N/A |
| pFUGW-ITGB3 | This study | N/A |
| pFUGW-ITGB3(Y747A) | This study | N/A |
| Software and Algorithms | ||
| ImageJ | [51] |
https://imagej.nih.gov/ij/ RRID: SCR_003070 |
| GraphPad Prism 5.0 | GraphPad Software |
https://www.graphpad.com/ RRID: SCR_002798 |
| MATLAB R2014a | The MathWorks Inc. |
https://www.mathworks.com/products/matlab.html RRID: SCR_001622 |
| FlowJo 10.6.1 | Becton, Dickinson and Company |
https://www.flowjo.com/ RRID: SCR_008520 |
| WinMDI 2.8 | The Scripps Institute, Flow Cytometry Core Facility | RRID: SCR_013745 |
| NIS-Elements | Nikon | RRID: SCR_014329 |
| Other | ||
| Stretch chamber | Strex Cell | STB-CH-10 |
| Manual stretching machine | Strex Cell | STB-100–10 |
| µ-Slide I Luer (Flow chamber) | ibidi GmbH | 80166 |
| Eclipse Ti (Fluorescence microscope) | Nikon | Nikon Eclipse Ti-E |
| Bio imaging analyzer | GE Healthcare Life Sciences | LAS 4000 mini |
| Flow cytometer | BD Biosciences | FACS Calibur |
| Flow cytometer | BD Biosciences | FACS Accuri C6 |
| Chromatography system | GE Healthcare Life Sciences | ÄKTA™ start |
| Fluorescence Spectrometers | HORIBA Scientific | FluoroMAx-4 |
| Cell culture dish | SPL Life Sciences | 20100 |
| 96 Well Black Polystyrene Microplate | Merk | CLS3603 |
Method details
Plasmids and cloning:
pcDNA3.1/3xFLAG-αIIb(TMD-tail)-TAP, pcDNA3.1/Tac-β3(TMD-tail), and pcDNA3.1/2xHA-β3(TMD-tail) were generated as previously described [18]. To generate pcDNA3.1/F3-GFP, the talin F3 region and enhanced GFP (EGFP) were amplified by polymerase chain reaction (PCR) and inserted into pcDNA3.1 vector using GeneArt™ Seamless Cloning and Assembly Enzyme Mix. EcoRI and NotI recognition sites were then inserted in the upstream region of F3. pcDNA3.1/GFP was generated by excising the F3 region from pcDNA3.1/F3-GFP. The ENTH domain (Met1-Ala163) was amplified by PCR using primers that introduced the EcoRI and NotI sites on the ends of the amplicon. The EcoRI/NotI digested PCR fragment was then inserted into pcDNA3.1/F3-GFP or pcDNA3.1/GFP. Insertion of the EcoRI/NotI sites, deletion of F3 region, mutagenesis of ENTH domain (∆AH, L6E, K76A, L6Q, and L6W) were performed by PCR-mediated site-directed mutagenesis. pEGFPC1-EPSIN 1 was a gift from Pietro De Camilli (Addgene plasmid # 22228). Primers used in this study are listed in Table S1.
Fluorescence labeling:
Fibrinogen from human plasma (Sigma-Aldrich, F-4883) was labeled with FITC (Sigma-Aldrich, F7250) according to the manufacturer’s recommendation. In brief, 20 mg of fibrinogen was incubated with 20-fold molar excess of FITC for 2 h at 4 °C with rotation. To remove free FITC, 20 mM Tris-Cl was added to the reaction and the mixture was desalted in DPBS [phosphate buffered saline (PBS) supplemented with 1 mM CaCl2 and 1 mM MgCl2; Hyclone] using Zeba™ Spin Desalting Columns (Thermo Fisher Scientific, 89892). Further purification was accomplished by using anion exchange chromatography column (HiTrap Q FF; GE Healthcare, 17–5053-01) to remove heavily labeled non-functional fibrinogen. The resulting fraction containing functional FITC-fibrinogen was then diluted with Hank’s Balanced Salt Solution (HBSS; WELGENE) containing 1.3 mM CaCl2 and 1 mM MgSO5. Due to the aggregation-prone nature of the labeled fibrinogen, labeling was performed immediately prior to each experiment.
Isolation of platelets:
After mice were anesthetized by isoflurane, the blood was drawn from mice by cardiac puncture using a 25G syringe containing 150 µL ACD solution (85 mM sodium citrate, 65 mM citric acid, and 104 mM D-glucose, pH 4.4). Blood was added to the solution mixed with 175 µL ACD solution and 875 µL Modified Tyrode’s solution (140 mM NaCl, 2.7 mM KCl, 0.4 mM NaH2PO4, 10 mM NaHCO3, 5 mM dextrose, and 10 mM HEPES, pH 7.4). The mixture was treated with 1 U/µl apyrase (Sigma-Aldrich) and centrifuged at 200 g for 5 min at room temperature without braking to obtain platelet-rich plasma (PRP). PRP was treated with 1 U/µl apyrase, incubated at room temperature for 10 min, and then centrifugated at 700 g for 5 min at room temperature without breaking. The platelet pellet was resuspended in Walsh buffer (137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 3.3 mM NaH2PO4, and 20 mM HEPES, pH 7.4) and incubated at room temperature for 30 min before performing the experiment. Mice bearing β3(L746A) mutation was previously described [24].
Fibrinogen binding assay:
Isolated mouse platelets in Walsh buffer were allowed to attach to the collagen-coated flow chamber (µ– Slide I Luer; ibidi GmbH, 80166) for 15 min. After the unbound platelets were removed, Walsh buffer containing 20 µg/mL FITC-fibrinogen was applied to the chamber at the rate of 10 mL/min for 5 min using a syringe pump (Revodix Inc.). The flow chamber containing platelets was gently washed twice, and bound fibrinogen was observed under a fluorescence microscope (Ti-E inverted microscope; Nikon). Fluorescence images were captured using a charge-coupled device camera (DS-Qi2; Nikon) and processed using NIS-Elements AR (Nikon). To measure fibrinogen binding to CHO/αIIbβ3 or CHO/αIIbβ3(Y747A) cells, these cells were seeded on type I collagen-coated flow chamber and incubated in 37 °C in a CO2 incubator for 1 day. HBSS containing 20 µg/mL FITC-fibrinogen was applied to the chamber and fibrinogen binding was measured by the same procedure used for platelets. To calculate the relative fibrinogen binding (the fold increase of flow-induced fibrinogen binding against basal binding), background-subtracted FITC fluorescence intensities were first measured in each platelet/cell in all conditions and then the mean of FITC fluorescence in each condition was divided by that in flow condition.
Cell stretch experiment:
Cells were seeded on collagen-coated stretch chamber (Strex Cell, STB-CH-10) and incubated at 37 °C in a CO2 incubator until they reached 70~80% confluency. The chamber was washed three times with HBSS and then stretched uniaxially to 130% of its original length for 40 sec in the presence of 50 µg/mL FITC-fibrinogen. After washing, cells were lysed with RIPA extraction buffer (Thermo Fisher Scientific). FITC fluorescence and total protein concentration in the clarified lysates were measured using FluoroMax-4 spectrofluorometer (HORIBA Scientific) and BCA Colorimetric Assay kit (Thermo Fisher Scientific), respectively, and fluorescence intensities normalized to protein concentrations were calculated. Non-specific fibrinogen binding was measured by performing the stretch experiment in the presence of 10 µM eptifibatide. To measure αIIbβ3 TMD interaction under stretch condition, CHO cells were cultured and transfected with pcDNA3.1/3xFLAG-αIIb(TMD-tail)-TAP, pcDNA3.1/Tac-β3(TMD-tail), or their mutant constructs. After washing with DPBS, cells on the chamber were stretched as described above, lysed immediately after removal of DPBS, and used for pull-down experiment as described previously [18].
Proteoliposome and nanodisc preparation:
Inactive human integrin αIIbβ3 and β3(TMD-tail) peptides were prepared as described previously [30]. The homogeneous 1:1 lipid mixture of 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC; Avanti Polar Lipids) and 1,2-dimyristoyl-sn-glycero-3-phospho-(1ʹ-rac-glycerol) (DMPG; Avanti Polar Lipids) was solubilized with buffer A (20 mM Tris-Cl, 100 mM NaCl, and 0.1% Triton X-100, pH 7.4) containing 100 mM glucose. Purified integrin αIIbβ3 or β3(TMD-tail) peptide was added to the lipid solution. To promote proteoliposome formation, the detergent in the solution was removed by stepwise addition of SM-2 Biobeads (Bio-Rad, 152–3920). The solution was then extruded through a 200 nm pore diameter membrane filter using an extruder, to generate integrin liposomes. Formation of liposomes was confirmed by electron microscopy. Integrin nanodiscs were prepared as previously described [30]. Briefly, 1:1 mixture of DMPC and DMPG were solubilized with buffer A in most except in the ENTH experiments. For the ENTH experiments (Fig. 7B, 7C and Supplementary Fig. 5D), phosphatidylinositol 4,5-bisphosphate C16 (Echelon) was added to 1:1 mixture of its C16 counterparts, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC; Avanti Polar Lipids) and 1,2-dipalmitoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (DPPG; Avanti Polar Lipids), at final 1% for better ENTH recruitment. After the purified integrin αIIbβ3 or β3(TMD-tail) peptide and membrane scaffold protein 1D1 were then added to the lipid mixture, 4 volumes of SM-2 Biobeads were added for detergent removal. The resulting nanodiscs were further purified using size exclusion columns and/or concentrated using Amicon Ultra centrifugal filter units (Merck Millipore), as needed.
Proteoliposome binding assay:
Clear bottom black plates (96-well) were coated with 10 µg/mL fibrinogen (Sigma-Aldrich) followed by blocking with 2% (w/v) bovine serum albumin. Integrin liposomes containing 100 mM glucose were diluted with binding buffer (20 mM Tris-Cl, 100 mM NaCl, 0.5 mM CaCl2, and 0.5 mM MgCl2, pH 7.4) containing 100 mM (303 mOsm/L), 60 mM (263 mOsm/L), or 40 mM (243 mOsm/L) glucose. The diluted liposomes were then added on fibrinogen-coated plate and incubated for 10 min at room temperature in the presence or absence of 10 µM eptifibatide or 1 mM MnCl2. After washing, the amount of fibrinogen-bound liposome was quantified using the Phosphatidylcholine Assay kit (Cell Biolabs, STA-600).
Strep-β3 pull-down assay:
CHO/αIIbTSTβ3 cells were transfected with various GFP-tagged constructs. One day after transfection, cells were lysed with lysis buffer (30 mM HEPES, 150 mM NaCl, 0.4% Triton X-100, pH 7.4, and protease inhibitor cocktail) and the lysate clarified by centrifugation. The clarified lysates were then incubated with Strep-Tactin Sepharose (IBA Lifesciences, 2–1201-010) for 6 h at 4 °C. Eluted proteins were analyzed by western blotting using anti-strep antibody (IBA Lifesciences, 2–1507-001) or anti-GFP antibody (Santa Cruz Biotechnology, sc-9996).
Flow cytometry:
To test the effect of ENTH on integrin αIIbβ3 activation, CHO/αIIbβ3 cells were transiently transfected with various ENTH-F3-GFP constructs, F3-GFP, or GFP. At 24 h after transfection, cells were detached and incubated with 4 mg/ml PAC1 (BD Biosciences) on ice. After washing, those cells were further stained with allophycocyanin-conjugated anti-mouse IgM antibody (Thermo Fisher Scientific), and then analyzed by flow cytometry as described previously [18]. To identify surface integrin αIIbβ3 expression of established cell lines, cells were stained with D57 and further stained with allophycocyanin-conjugated anti-mouse IgG antibody (Thermo Fisher Scientific).
Quantification and statistical analysis
Unless otherwise stated in the figure legend, statistical analyses were done by One way ANOVA followed by Bonferroni’s post-hoc comparisons tests using GraphPad Prism 5.0, GraphPad Software, La Jolla California USA. Standard error (s.e.) or standard deviation (s.d.) are indicated in figure legends.
Supplementary Material
Highlights.
Mechanical stretch of the membrane changes the topology of β3 transmembrane domain.
Mechanical force disrupts integrin αIIb-β3 transmembrane domain interaction.
Physical deformation of the membrane can directly activate integrin αIIbβ3.
Integrin αIIbβ3 is a mechanoreceptor sensing mechanical forces in the lipid bilayer.
ACKNOWLEDGMENTS
This work was supported by the Basic Science Research Program NRF-2019R1A2C2008067 (C.K.), the Bio & Medical Technology Development Program NRF-2018M3A9A8017949 (S.J.H.), National Institutes of Health NHLBI grant HL117061 (B.G.P.), American Heart Association Grant #18TPA34170481 (T.S.U.), and a Korea University Grant (C.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
Data and code availability
Source data that support the findings of this study are available from the corresponding author upon request.
REFERENCES
- 1.Hamill OP, and Martinac B. (2001). Molecular basis of mechanotransduction in living cells. Physiol Rev 81, 685–740. [DOI] [PubMed] [Google Scholar]
- 2.Hu X, Margadant FM, Yao M, and Sheetz MP. (2017). Molecular stretching modulates mechanosensing pathways. Protein Sci 26, 1337–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haswell ES, Phillips R, and Rees DC. (2011). Mechanosensitive channels: what can they do and how do they do it? Structure 19, 1356–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anishkin A, Loukin SH, Teng J, and Kung C. (2014). Feeling the hidden mechanical forces in lipid bilayer is an original sense. Proc Natl Acad Sci U S A 111, 7898–7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arnadottir J, and Chalfie M. (2010). Eukaryotic mechanosensitive channels. Annu Rev Biophys 39, 111–137. [DOI] [PubMed] [Google Scholar]
- 6.Brohawn SG, Su Z, and MacKinnon R. (2014). Mechanosensitivity is mediated directly by the lipid membrane in TRAAK and TREK1 K+ channels. Proc Natl Acad Sci U S A 111, 3614–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brohawn SG, Campbell EB, and MacKinnon R. (2014). Physical mechanism for gating and mechanosensitivity of the human TRAAK K+ channel. Nature 516, 126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hynes RO. (2002). Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687. [DOI] [PubMed] [Google Scholar]
- 9.Kim M, Carman CV, and Springer TA. (2003). Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science 301, 1720–1725. [DOI] [PubMed] [Google Scholar]
- 10.Luo BH, Springer TA, and Takagi J. (2004). A specific interface between integrin transmembrane helices and affinity for ligand. PLoS Biol 2, e153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lau TL, Kim C, Ginsberg MH, and Ulmer TS. (2009). The structure of the integrin alphaIIbbeta3 transmembrane complex explains integrin transmembrane signalling. EMBO J 28, 1351–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim C, Schmidt T, Cho EG, Ye F, Ulmer TS, and Ginsberg MH. (2011). Basic amino-acid side chains regulate transmembrane integrin signalling. Nature 481, 209–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun X, Fu Y, Gu M, Zhang L, Li D, Li H, Chien S, Shyy JY, and Zhu Y. (2016). Activation of integrin alpha5 mediated by flow requires its translocation to membrane lipid rafts in vascular endothelial cells. Proc Natl Acad Sci U S A 113, 769–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leitinger B, and Hogg N. (2002). The involvement of lipid rafts in the regulation of integrin function. J Cell Sci 115, 963–972. [DOI] [PubMed] [Google Scholar]
- 15.Calderwood DA, Zent R, Grant R, Rees DJ, Hynes RO, and Ginsberg MH. (1999). The Talin head domain binds to integrin beta subunit cytoplasmic tails and regulates integrin activation. J Biol Chem 274, 28071–28074. [DOI] [PubMed] [Google Scholar]
- 16.Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, de Pereda JM, Ginsberg MH, and Calderwood DA. (2003). Talin binding to integrin beta tails: a final common step in integrin activation. Science 302, 103–106. [DOI] [PubMed] [Google Scholar]
- 17.Garcia-Alvarez B, de Pereda JM, Calderwood DA, Ulmer TS, Critchley D, Campbell ID, Ginsberg MH, and Liddington RC. (2003). Structural determinants of integrin recognition by talin. Mol Cell 11, 49–58. [DOI] [PubMed] [Google Scholar]
- 18.Kim C, Lau TL, Ulmer TS, and Ginsberg MH. (2009). Interactions of platelet integrin alphaIIb and beta3 transmembrane domains in mammalian cell membranes and their role in integrin activation. Blood 113, 4747–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu P, and Luo BH. (2015). Integrin alphaIIbbeta3 transmembrane domain separation mediates bi-directional signaling across the plasma membrane. PLoS One 10, e0116208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma YQ, Qin J, Wu C, and Plow EF. (2008). Kindlin-2 (Mig-2): a co-activator of beta3 integrins. J Cell Biol 181, 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katsumi A, Naoe T, Matsushita T, Kaibuchi K, and Schwartz MA. (2005). Integrin activation and matrix binding mediate cellular responses to mechanical stretch. J Biol Chem 280, 16546–16549. [DOI] [PubMed] [Google Scholar]
- 22.Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, and Schwartz MA. (2005). A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437, 426–431. [DOI] [PubMed] [Google Scholar]
- 23.Ramstrom S, O’Neill S, Dunne E, and Kenny D. (2010). Annexin V binding to platelets is agonist, time and temperature dependent. Platelets 21, 289–296. [DOI] [PubMed] [Google Scholar]
- 24.Petrich BG, Fogelstrand P, Partridge AW, Yousefi N, Ablooglu AJ, Shattil SJ, and Ginsberg MH. (2007). The antithrombotic potential of selective blockade of talin-dependent integrin alpha IIb beta 3 (platelet GPIIb-IIIa) activation. J Clin Invest 117, 2250–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alon R, and Dustin ML. (2007). Force as a facilitator of integrin conformational changes during leukocyte arrest on blood vessels and antigen-presenting cells. Immunity 26, 17–27. [DOI] [PubMed] [Google Scholar]
- 26.Chen W, Lou J, Evans EA, and Zhu C. (2012). Observing force-regulated conformational changes and ligand dissociation from a single integrin on cells. J Cell Biol 199, 497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng S, Lu X, Resendiz JC, and Kroll MH. (2006). Pathological shear stress directly regulates platelet alphaIIbbeta3 signaling. Am J Physiol Cell Physiol 291, C1346–1354. [DOI] [PubMed] [Google Scholar]
- 28.Bayburt TH, and Sligar SG. (2003). Self-assembly of single integral membrane proteins into soluble nanoscale phospholipid bilayers. Protein Sci 12, 2476–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Situ AJ, Kang SM, Frey BB, An W, Kim C, and Ulmer TS. (2018). Membrane Anchoring of alpha-Helical Proteins: Role of Tryptophan. J Phys Chem B 122, 1185–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim C, Ye F, Hu X, and Ginsberg MH. (2012). Talin activates integrins by altering the topology of the beta transmembrane domain. J Cell Biol 197, 605–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ye F, Yang C, Kim J, MacNevin CJ, Hahn KM, Park D, Ginsberg MH, and Kim C. (2017). Epigallocatechin gallate has pleiotropic effects on transmembrane signaling by altering the embedding of transmembrane domains. J Biol Chem 292, 9858–9864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nyholm TK, Ozdirekcan S, and Killian JA. (2007). How protein transmembrane segments sense the lipid environment. Biochemistry 46, 1457–1465. [DOI] [PubMed] [Google Scholar]
- 33.Milovanovic D, Honigmann A, Koike S, Gottfert F, Pahler G, Junius M, Mullar S, Diederichsen U, Janshoff A, Grubmuller H, et al. (2015). Hydrophobic mismatch sorts SNARE proteins into distinct membrane domains. Nat Commun 6, 5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harayama T, and Riezman H. (2018). Understanding the diversity of membrane lipid composition. Nat Rev Mol Cell Biol 19, 281–296. [DOI] [PubMed] [Google Scholar]
- 35.Li R, Babu CR, Lear JD, Wand AJ, Bennett JS, and DeGrado WF. (2001). Oligomerization of the integrin alphaIIbbeta3: roles of the transmembrane and cytoplasmic domains. Proc Natl Acad Sci U S A 98, 12462–12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cox CD, Bavi N, and Martinac B. (2019). Biophysical Principles of Ion-Channel-Mediated Mechanosensory Transduction. Cell Rep 29, 1–12. [DOI] [PubMed] [Google Scholar]
- 37.Yesylevskyy SO, Rivel T, and Ramseyer C. (2017). The influence of curvature on the properties of the plasma membrane. Insights from atomistic molecular dynamics simulations. Sci Rep 7, 16078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ford MG, Mills IG, Peter BJ, Vallis Y, Praefcke GJ, Evans PR, and McMahon HT. (2002). Curvature of clathrin-coated pits driven by epsin. Nature 419, 361–366. [DOI] [PubMed] [Google Scholar]
- 39.Hussain NK, Yamabhai M, Bhakar AL, Metzler M, Ferguson SS, Hayden MR, McPherson PS, and Kay BK. (2003). A role for epsin N-terminal homology/AP180 N-terminal homology (ENTH/ANTH) domains in tubulin binding. J Biol Chem 278, 28823–28830. [DOI] [PubMed] [Google Scholar]
- 40.Zeno WF, Baul U, Snead WT, DeGroot ACM, Wang L, Lafer EM, Thirumalai D, and Stachowiak JC. (2018). Synergy between intrinsically disordered domains and structured proteins amplifies membrane curvature sensing. Nat Commun 9, 4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wegener KL, Partridge AW, Han J, Pickford AR, Liddington RC, Ginsberg MH, and Campbell ID. (2007). Structural basis of integrin activation by talin. Cell 128, 171–182. [DOI] [PubMed] [Google Scholar]
- 42.Hatzakis NS, Bhatia VK, Larsen J, Madsen KL, Bolinger PY, Kunding AH, Castillo J, Gether U, Hedegard P, and Stamou D. (2009). How curved membranes recruit amphipathic helices and protein anchoring motifs. Nat Chem Biol 5, 835–841. [DOI] [PubMed] [Google Scholar]
- 43.Wang R, Shattil SJ, Ambruso DR, and Newman PJ. (1997). Truncation of the cytoplasmic domain of beta3 in a variant form of Glanzmann thrombasthenia abrogates signaling through the integrin alpha(IIb)beta3 complex. J Clin Invest 100, 2393–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hodivala-Dilke KM, McHugh KP, Tsakiris DA, Rayburn H, Crowley D, Ullman-Cullere M, Ross FP, Coller BS, Teitelbaum S, and Hynes RO. (1999). Beta3-integrin-deficient mice are a model for Glanzmann thrombasthenia showing placental defects and reduced survival. J Clin Invest 103, 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bye AP, Unsworth AJ, and Gibbins JM. (2018). Screening and High-Throughput Platelet Assays. Methods Mol Biol 1812, 81–94. [DOI] [PubMed] [Google Scholar]
- 46.Atilgan E, and Ovryn B. (2009). Nucleation and growth of integrin adhesions. Biophys J 96, 3555–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Galbraith CG, Yamada KM, and Galbraith JA. (2007). Polymerizing actin fibers position integrins primed to probe for adhesion sites. Science 315, 992–995. [DOI] [PubMed] [Google Scholar]
- 48.Cohen BE. (2018). Membrane Thickness as a Key Factor Contributing to the Activation of Osmosensors and Essential Ras Signaling Pathways. Front Cell Dev Biol 6, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamamoto K, and Ando J. (2013). Endothelial cell and model membranes respond to shear stress by rapidly decreasing the order of their lipid phases. J Cell Sci 126, 1227–1234. [DOI] [PubMed] [Google Scholar]
- 50.Lin CM, Wu DT, Tsao HK, and Sheng YJ. (2012). Membrane properties of swollen vesicles: growth, rupture, and fusion. Soft Matter 8, 6139–6150. [Google Scholar]
- 51.Schneider CA, Rasband WS, and Eliceiri KW. (2012). NIH Image to ImageJ: 25 years of image analysis. Nature methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.