

Abstract
Cellular gatekeepers are essential to maintain order within a cell and anticipate signals of stress to promote survival. BAX inhibitor-1 (BI-1), also named transmembrane BAX inhibitor motif containing-6 (TMBIM6), is a highly conserved endoplasmic reticulum (ER) transmembrane protein. Originally identified as an inhibitor of BAX-induced apoptosis, its pro-survival properties have been expanded to include functions targeted against ER stress, calcium imbalance, reactive oxygen species accumulation and metabolic dysregulation. Nevertheless, the structural biology and biochemical mechanism of action of BI-1 are still under debate. BI-1 has been implicated in several diseases, including chronic liver disease, diabetes, ischemia/reperfusion injury, neurodegeneration and cancer. While most studies have demonstrated a beneficial role for BI-1 in the ubiquitous maintenance of cellular homeostasis, its expression in cancer cells seems most often to contribute to tumorigenesis and metastasis. Here we summarize what is known about BI-1 and encourage future studies on BI-1’s contribution to cellular life and death decisions to advocate its potential as a target for drug development and other therapeutic strategies.
Keywords: TMBIM6, ER stress, Cell death, Calcium, Disease
Graphical abstract
Disturbances in normal cell function activate evolutionarily conserved stress responses that aim to compensate for damage but can eventually trigger cell death. Bax Inhibitor-1 (BI-1) functions as a molecular rheostat, interacting with a broad range of partners to promote cell survival. Here we review the proposed models and mechanisms of BI-1 and its role in various diseases.

Introduction
A large number of regulatory proteins are involved in the signaling pathways that determine cell fate. BAX Inhibitor-1 (BI-1) is the founding member of the transmembrane BAX Inhibitor-1-containing motif (TMBIM) family, which includes ancestral regulators of cell death due to their high degree of genetic and functional conservation[1, 2]. These hydrophobic TMBIM proteins share a UPF0005 consensus motif in the C-terminus that codifies for six or seven transmembrane-spanning regions and is conserved in several species[2]. The cytoprotective function of the TMBIM family was found to regulate evolutionarily conserved mechanisms of stress resistance in animals, plants and yeast[3, 4]. While all six TMBIM family members seem to protect against apoptosis, as discussed in extenso [2, 5, 6], this review will concentrate on the function of TMBIM6, the original BI-1, in the animal kingdom.
Initially, a functional screen for BAX suppressors in yeast identified BI-1 to have a protective functional effect over BAX-induced cell death[7]. While it seemed reasonable to name the protein after its observed function, the cytoprotective function of BI-1 was surprisingly not due to a direct physical interaction with BAX. Rather, BI-1 co-immunoprecipitated with the anti-apoptotic BCL2 family proteins BCL2 and BCLXL, but not the pro-apoptotic proteins BAX or BAK due the requirement of a BH4 domain for BI-1 interaction[7]. In mammalian cell lines, BI-1 overexpression protected against, while its antisense promoted, apoptosis[7].
A recent neuroscience review has provided a brief overview of BI-1-like proteins and focused on the functions of BI-1 to threshold ER stress-induced oxidative damage and inflammation[8]. The present review rather compiles the most recent findings supporting all cytoprotective functions of BI-1 (ER stress and IRE1α signaling, calcium flux, as well as ROS regulation), highlights the implication of BI-1 signaling in disease (i.e. liver, neurological, immune disorders) and provides a synthesis of the evidence linking BI-1 to cancer.
I. Structure and distribution of BI-1
BI-1 is a highly hydrophobic protein containing 237 amino acids (∼26kDa) located almost exclusively on the ER, as illustrated by immunofluorescence microscopy and fractionation studies[7, 9]. For the most part, BI-1 is characterized to have six fully transmembrane domains, with a cytosolic orientation of both N and C termini, and a C-terminal re-entrant loop[7, 10, 11] (Figure 1). A number of interacting partners bind to increase the anti-apoptotic activity of BI-1, including BCL2 and BCLxL at the N-terminal, and IRE1α, inositol 1,4,5-trisphosphate receptor (IP3R), NADPH-P450 reductase (NPR) and cytochrome P450 species 2E1 (CYP2E1) at the C-terminal[12]. Indeed, deletion of a C-terminal domain of BI-1 abrogates its cytoprotective activity to suppress BAX-induced cell death in yeast[3]. The first BLAST searches revealed that the human and rat genes of BI-1 were essentially identical to the testis enhanced gene transcript due to its presence in the testis[13, 14]. BI-1 is also abundantly expressed in the liver and kidney, and is detected in the heart, skeletal muscle, spleen, brain, placenta, lung and pancreas[7, 9]. Although BI-1-deficient mice are viable and develop normally, they exhibit increased vulnerability to tissue damage when challenged[15].
Figure 1: Structure of BAX Inhibitor-1 (BI-1).

BAX Inhibitor-1 (BI-1) is an endoplasmic reticulum (ER)-resident protein with six fully transmembrane domains and a C-terminal re-entrant loop. Listed are select interacting partners that bind either to the N- or C-terminal domain of BI-1 (BCL2 apoptosis regulator (BCL2); BCL2 like 1 (BCLxL); inositol-requiring enzyme 1 alpha (IRE1α); inositol 1,4,5-trisphosphate receptor (IP3R); NADPH-P450 reductase (NPR); cytochrome P450 species 2E1 (CYP2E1). The membrane topology model was generated using Protter [82] with the BI1_Human protein sequence from the UniProt protein knowledgebase.
II. The various cytoprotective roles of BI-1
BI-1 interacts with a broad range of partners to protect against many facets of cell death (Figure 2). Nonetheless, apoptosis is initially a defense mechanism. Both physiological and pathological stimuli can trigger apoptosis through caspase activation classified into two main pathways: 1) intrinsic apoptosis engaged by intracellular stress conditions (DNA damage or accumulation of unfolded proteins, cytosolic Ca2+ or reactive oxygen species); and 2) extrinsic apoptosis that transduces signals from extracellular ligands through death receptors. The activation of either pathway results in mitochondrial outer membrane permeabilization (MOMP) and the release of apoptogenic factors, such as cytochrome c, that activate the caspase cascade of apoptosis. Interestingly, fibroblasts, hepatocytes and neurons isolated from BI-1-deficient mice are selectively hypersensitive to apoptosis induced by pharmacological ER stress activators such as thapsigargin and tunicamycin, compared to other intrinsic or extrinsic apoptotic stimuli[15].
Figure 2: Overview of the cytoprotective functions of BI-1.

BI-1 confers cellular protection as an anti-apoptotic protein by limiting multiple stress-inducing pathways surrounding the ER and mitochondria. Nevertheless, BI-1 can be ubiquitinated by the ER-associated RING-type E3 ligase bifunctional apoptosis regulator (BAR) for degradation by the proteasome, thus reducing BI-1 levels in the cell. BI-1 inhibits the activities of the ER stress sensor IRE1α directly and by blocking BAX/BAK binding. BI-1 can increase its anti-apoptotic function through the association with BCL2, which can inhibit BAX/BAK translocation to the mitochondria. BI-1 can regulate Ca2+ balance in the ER and mitochondria, notably by sensitizing IP3R function into controlled Ca2+ release into the cytosol. BI-1 can impact mitochondrial bioenergetics mediated by reduced [Ca2+], reduced reactive oxygen species (ROS) release and oxidative stress. BI-1 can interact with the microsomal monooxygenase system, composed of NADPH-P450 reductase (NPR) and cytochrome P450 member CYP2E1, to limit the harmful conversion of endogenous or exogenous compounds to ROS, which promote cytosolic acidification and oxidative stress. BI-1 can promote an antioxidant response via NRF2 and heme oxygenase-1 (HO-1) that counteracts ROS. Finally, BI-1 expression can reduce pH in lysosomes, consequently increasing lysosome activity and autophagy. (apoptosis signal regulating kinase 1 (ASK1); BCL2 associated X, apoptosis regulator/BCL2 antagonist/killer 1 (BAX/BAK); BCL2 apoptosis regulator (BCL2); DNA damage inducible transcript 3 (CHOP); inositol 1,4,5-trisphosphate receptor (IP3R); JUN N-terminal kinase (JNK); NLR family pyrin domain containing 3 (NLRP3); TNF receptor associated factor 2 (TRAF2); thioredoxin interacting protein (TXNIP); unfolded protein response (UPR); X-box binding protein 1 (XBP1)).
A. Implication of BI-1 in ER stress-induced cell death and the IRE1α pathway
The ER functions as a platform, central to several stress signaling pathways. In overwhelming conditions that cause unfolded or misfolded proteins to accumulate in the ER lumen, the ER activates the unfolded protein response (UPR) signaling cascade via the transmembrane sensors inositol-requiring enzyme 1 (IRE1), PKR-like ER kinase (PERK) and activating transcription factor (ATF6). The UPR first aims to alleviate cellular stress and re-establish proteostasis, but may activate cell death in response to irreversible cell damage.
ER stress-induced apoptosis was shown to require the pro-apoptotic members BAX and BAK of the BCL2 family to initiate mitochondrial dysfunction and cell death[16]. Afterwards, a study reported for the first time that BAX and BAK can colocalize to the IRE1α sensor on the ER membrane, uncovering a direct link between an essential gateway for cell death and the most conserved pathway of the UPR. BAX and BAK physically form a protein complex with the cytosolic C-terminal domain of IRE1α that appears essential for IRE1α activation and amplified apoptosis[17]. Interestingly, BI-1 binds to this same cytosolic region, directly inhibiting IRE1α signaling and downstream XBP1 mRNA splicing[18, 19]. The presence of BI-1 drastically reduces the binding of BAX to IRE1α, suggesting that BI-1 and BAX/BAK compete for their binding site on IRE1α[18], with the winner dictating IRE1α activity and ultimately deciding cell fate. However, BI-1 knockdown in BAX/BAK double-knockout cells did not restore the normal levels of XBP1 mRNA splicing, suggesting that BI-1 operates upstream of BAX and BAK in the control of the IRE1α-XBP1 pathway[18]. An added foe of BI-1 in IRE1α regulation is the bifunctional apoptosis regulator (BAR), an ER-associated RING-type E3 ligase that ubiquitinates BI-1 to tag it for subsequent proteosomal-mediated degradation[20]. The dynamic post-translational regulation of BI-1 by BAR at the ER membrane thus impedes the inhibitory influence of BI-1 on IRE1α signaling. Hence, prolonged activation of IRE1α during ER stress may promote cell death through the kinase-dependent pathway TRAF2-JNK or the RNase-dependent pathways involving XBP1 splicing and RIDD of mRNAs encoding essential cell-survival proteins or miRs[21]. In parallel, CHOP may also direct the subsequent activation of the pro-apoptotic BCL2 family member pathway by the transcriptional induction of BIM[22], inducing cell death[21].
B. Proposed mechanisms of BI-1-regulated calcium flux
The ER is the primary cellular store for Ca2+. The ER mainly orchestrates Ca2+ efflux through IP3R channels that passively release Ca2+ from the ER and Ca2+ influx through sarcoplasmic/ER calcium ATPase (SERCA) channels that actively pump Ca2+ into the ER. Due to its location on the ER membrane, BI-1 is involved in [Ca2+] regulation in the ER lumen. BI-1-overexpressing cells have lower resting ER Ca2+ levels and higher ER Ca2+ leakage[23]. An advantage of lower [Ca2+] in the ER due to BI-1 overexpression is the controlled discharge of Ca2+ from the ER to the cytosol, leading to reduced Ca2+ uptake by mitochondria[23], thus protecting the cell from Ca2+ overload that triggers apoptosis. To better study its function, potential membrane topologies of BI-1 have been illustrated but remain controversial. While algorithms seem consistent in predicting the first six transmembrane domains of BI-1 with a cytoplasmic N-terminal, they differ in the structural characterization of the last ∼20 residues of the protein that argue for or against the presence of a seventh transmembrane domain. As the HA-tagged C-terminal of BI-1 mapped to the cytosol, this region was suggested to bear a membranous re-entrant loop or a short cytosolic helix tail[10]. While still under debate, several mechanisms have been proposed to explain how BI-1 might mediate Ca2+ efflux from the ER as a/an: 1) Ca2+/H+ antiporter; 2) Ca2+ leak channel; or 3) IP3R sensitizer (Figure 3). It remains important to note that although these mechanisms have been studied independently, they may not be mutually exclusive.
Figure 3: Proposed models of BI-1 Ca2+ regulation.
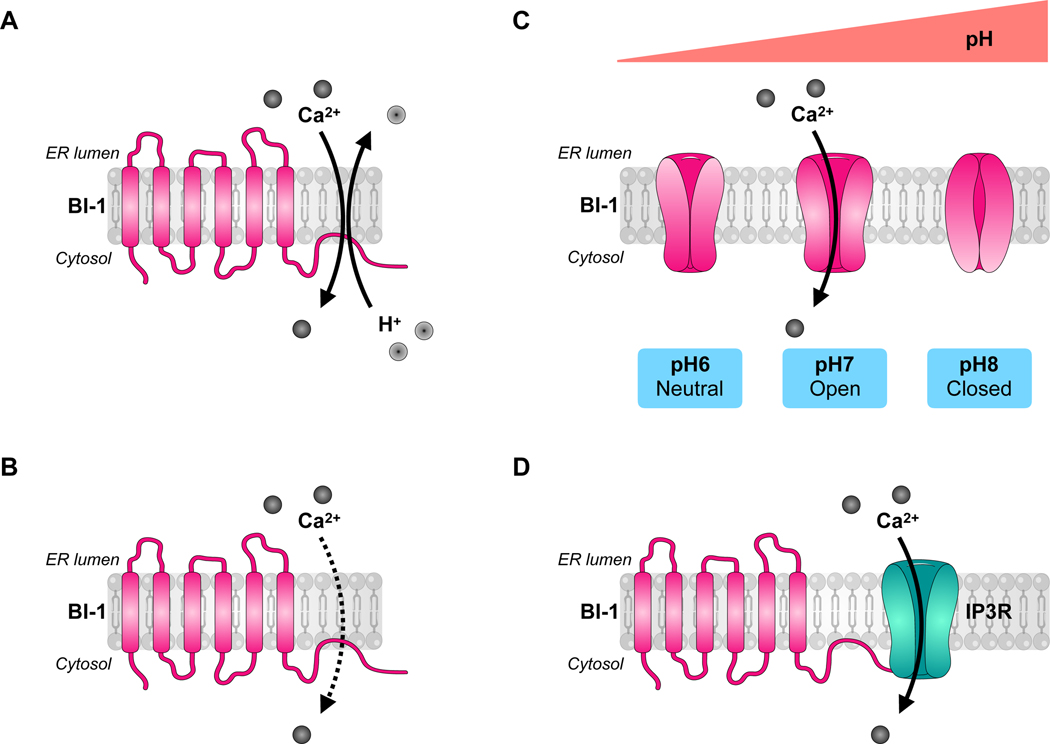
The C-terminal of BI-1 may have Ca2+channel or sensitizing properties and be regulated by changes in intracellular pH. A) BI-1 could function as a Ca2+/H+ antiporter. B) BI-1 could passively release Ca2+ into the cytosol as a Ca2+ leak channel. C) A simplified view of the status of the Ca2+-permeable pore of BI-1 in terms of intracellular pH, endorsing the model in B). D) BI-1 could function as an inositol 1,4,5-trisphosphate receptor (IP3R) sensitizer, directing ER Ca2+ flux control.
i. The first hypothesis -
states that BI-1 functions as a Ca2+/H+ antiporter, allowing Ca2+ release into the cytosol[24, 25] (Figure 3A). To regulate ER Ca2+ levels, the lysine-rich C-terminal cytosolic domain (EKDKKKEKK) of BI-1 was suggested to mediate Ca2+ channel gating in a pH-dependent manner, as BI-1-overexpressing cells showed increased release of Ca2+ in response to acidic conditions[26]. Although most evidence supports a cytoprotective function for BI-1 in fine-tuning the Ca2+ leak from the ER, a cell death-promoting phenotype for BI-1 was observed under low pH conditions (5.0 – 6.4), since acidification in BI-1-overexpressing cultures dangerously increased Ca2+ release from the ER and dose-dependently reduced cell viability[26].
While resting cells have largely monomeric forms of BI-1, cytosolic acidification in stressed cells induces the oligomerization and antiporter activity of BI-1, whose initial function is to exchange ER-stored Ca2+ with H+, reduce cytosolic acidification and promote cell survival[24]. The Ca2+/H+ antiporter activities of BI-1 may be stimulated by interaction with BH4 domains (present in BCL2 and BCLxL) through enhanced protein oligomerization[25]. Nevertheless, no direct evidence has been shown for the association of BI-1 with proton ions and Bultynck et al. commented that such a Ca2+/H+ antiporter function appears unlikely[27]. They claim the C-terminal of BI-1 does not have the biochemical properties to sense acidic pH changes (< 6.8), as its lysine-rich regions might only be adapted to sense more basic pH changes. They further question whether persistent H+ gradients even occur in physiological conditions, given the high permeability of ER membranes for protons that renders the exchange of Ca2+ with H+ futile[27]. In that respect, a pH-sensitive Ca2+ leak channel function seems more probable.
ii. The second hypothesis -
states that the presumed C-terminal ER-membrane dipping domain of BI-1 located after its six transmembrane domains forms a Ca2+-channel pore responsible for a Ca2+ leak from the ER to the cytosol[11] (Figure 3B). To clarify the effect of pH on BI-1 function, Kiviluoto et al. optimized pH assays specifically in intact ER of permeabilized cells, as opposed to using artificial microscomal preparations or proteoliposomes. While it was corroborated that the Ca2+ leak property of BI-1 was pH-dependent, the Ca2+ flux peaks at a physiological pH and decreases under increasingly acidic conditions[28]. Specifically, BI-1 is less efficient to provoke Ca2+-release at pH values < 6.8, with BI-1-mediated Ca2+ leakage completely blocked at pH 6.0[28]. The examination of the crystal structure of the bacterial homolog of BI-1 from Bacillus subtilis (BsYetJ) led to its characterization as a bona fide functional bacterial BI-1 homolog of human BI-1 (but with seven transmembrane domains and a luminal N-terminal) with Ca2+ leak activity dependent on pH-driven conformational changes[29]. This group recently established, with functional and computational data, that the modulation of BsYetJ activity was ion-dependent, with the protonation of its aspartate residues (Asp171–195 dyad) controlling pore opening and pH-dependent Ca2+ binding[5, 30]. Thus, BI-1-regulated Ca2+ flux is rather pH dependent in bell-shaped fashion: at pH 6, critical aspartate residues forming the Ca2+ pore in BI-1 become protonated, resulting in an open structure but loss of negative charges critical for Ca2+ binding; pH 7 allows for formation of a negatively charged pore for Ca2+ passage; and at pH 8, BI-1 is closed due to deprotonation of aspartate residues and formation of hydrogen bonds that lock the structure[5, 30]. A simplified illustration of BI-1’s pH-dependent pore function features an neutral pore impearmeable to Ca2+ at pH 6, an open electronegative pore permeable to Ca2+ at pH 7, and a closed pore at pH 8[27] (Figure 3C).
iii. The third hypothesis -
states that BI-1 can sensitize IP3R independently of its Ca2+ channel activity, contributing to reduce steady-state Ca2+ concentration in the ER[31, 32] (Figure 3D). Purified and ectopically-expressed BI-1 from transfected HeLa cells co-immunoprecipitated with IP3R, further identified to bind at the C-terminal of BI-1[31]. This suggests that BI-1 and IP3R can directly influence each other’s channel activity by forming protein complexes. By increasing controlled ER Ca2+ leakage into the cytosol, BI-1 sensitization of IP3R reduces otherwise-destined Ca2+ transfer from the ER into the mitochondria at MAMs[32]. By regulating steady-state levels of ER Ca2+ via IP3R, BI-1 influences mitochondrial bioenergetics and stimulates autophagy to promote cellular recovery when faced with metabolic stress[32]. Similar to BI-1-overexpressing cells, BAX/BAK double-knockout cells have reduced steady-state ER Ca2+ levels due to increased ER Ca2+ leakage driven by IP3R[33]. On the contrary, BCL2 appears to prevent IP3R-mediated Ca2+ leakage through the inhibitory binding of its BH4 domain to the regulatory and coupling domain of IP3R[34, 35]. Knowing that BI-1 interacts directly with BCL2 as well as IP3R adds yet another degree of complexity to understanding cellular [Ca2+] regulation and anti-apoptotic function (as extensively reviewed in [36, 37]). While small leakage of Ca2+ is necessary to avoid overload in the ER, too much ER Ca2+ depletion disturbs protein production and the Ca2+-dependent activities of many intraluminal chaperones, suggesting that many actors are certainly at play to fine-tune [Ca2+] in fundamental organelles and the cell itself. In certain circumstances, BI-1 may even play both the role of calcium Ca2+ leak channel itself as well as IP3R sensitizer. Whether BI-1 is able to use both properties at once or chooses to alternate between either mechanism has yet to be determined.
C. Potential for BI-1 to limit ROS-induced cell death
If produced in excess, reactive oxygen species (ROS), a natural by-product of metabolic pathways, leads to oxidative stress that can damage cellular architecture and initiate apoptosis. Due to its protein folding function, the ER is closely associated with ROS production. The microsomal monooxygenase (MMO) system of the ER, also called microsomal electron transfer chain, leads to the release of ROS such as O-2 and H2O2[38]. BI-1 may protect against ROS accumulation by interacting with the MMO members NPR and CYP2E1 in conditions of ER stress[39–41]. BI-1 overexpression limited the production of H2O2 in a dose-dependent and exponential decaying manner, seemingly due to the direct interaction of BI-1’s C-terminal with NPR and CYP2E1 to block electron transfer and ROS production[39]. Likewise, BI-1 overexpression in ER stress-exposed cells led to reduced CYP2E1 and membrane lipid peroxidation. Enhanced lysosomal activity in BI-1-overexpressing cells appears linked to CYP2E1 degradation[41] and BI-1 was reported to maintain high lysosomal activity under ER stress, leading to lysosomal acidification and increased autophagy[42]. Forced expression of BI-1 in HEK293 cells and MEFs suppressed mitochondria-mediated ROS production and associated apoptosis due to activated ERK signaling[43]. Another possible way for BI-1 to limit ROS production is by stimulating the antioxidant response. Cells overexpressing BI-1 showed increased levels of the redox-sensitive transcription factor NRF2 and its antioxidant enzyme target heme oxygenase-1 (HO-1), expressed to counteract ROS accumulation[44]. Conversely, siRNA-mediated knockdown of BI-1 led to reduced HO-1 levels and increased sensitivity to cell death[44]. However, BI-1-deficient MEFs were later shown to exhibit no differences in HO-1 transcription levels[18]. Therefore, more work must be done to clarify this connection.
III. Implication of BI-1 in disease
Increasing reports investigate the physiopathological role of BI-1 in various contexts, from metabolic diseases affecting the liver[19, 45, 46] or kidneys[47] to lung disease[48], infection [49], neurological disorders[50, 51] and cancer. Most of the literature associates the anti-apoptotic function of BI-1 with cancer development (Table 1). Nevertheless, the expression of BI-1 has yet to be shown to correlate with cancer prognosis. Although seemingly ubiquitous, the endogenous expression of BI-1 may differ among tissues or cell types and at different stages of development, leading to various cell-type and time-specific roles. While non-exhaustive, protein profiling by two-dimensional electrophoresis and mass spectrometry from liver, brain, heart, lung and kidney tissue samples identified altered differential protein expressions of GRP75, peroxiredoxin 6, fumarylacetoacetate hydrolase, selenium-binding protein 2, phosphatidylethanolamine-binding protein 1 and ferritin light chain 1 in tissues from BI-1−/− compared to BI-1+/+ mice, leaving clues about BI-1 function in those organs[52]. Despite this interest, few researchers have addressed the implication of BI-1 in disease.
Table 1:
Overview of BI-1 expression in diseases referenced.
| Disease | BI-1 levels | Observations | Reference |
|---|---|---|---|
| Hepatic disorders | |||
| Insulin resistance | down | BI-1 overexpression improves hepatic insulin signaling. | [19] [40] |
| Non-Alcoholic Fatty Liver Disease | down | BI-1 KO mice are vulnerable to acute ER stress- and HFD-induced liver injury. | [54] [46] |
| Hypoxia/ischemia-reperfusion | up | After IR, BI-1 KO mice exhibit ER stress and cell death while BI-1 is upregulated in recovering WT mice. | [55] |
| CCl4 | down | BI-1 levels increase after mesenchymal stem cell transplantation therapy. | [58] |
| Partial hepatectomy | down | BI-1 KO mice have accelerated liver regeneration. | [45] |
| Immune system regulation and infection | |||
| Autoimmune encephalomyelitis | ? | BI-1 KO mice display later onset of clinical symptoms with attenuated T- and B-cell function. | [59] |
| Influenza | ? | BI-1 overexpression suppresses virus-induced cell death. | [49] |
| Neurological disorders | |||
| Stroke | ? | BI-1 KO mice are vulnerable to cerebral artery occlusion injury. BI-1 transgenic mice have reduced brain injury and better recovery. |
[15] [50] |
| Depression | ? | BI-1 transgenic mice are protected against learned helplessness and anhedonia. BI-1 mice are vulnerable to stress-induced depression-like behavior. |
[51] [62] |
| Alzheimer’s disease | ? | BI-1 is a stable presenilin 1 interacting partner. | [63] |
| Other disorders | |||
| Cardiac ischemia-reperfusion | down | BI1 transgenic mice are protected from microvascular injury and mitochondrial dysfunction. | [56] |
| Neonatal hypoxic-ischemic injury | up | BI-1 overexpression increases the production of antioxidant enzymes and attenuates inflammation after injury. | [57] |
| Nephrotoxicity | down | BI-1 KO mice are vulnerable to cyclosporine A-induced kidney injury. | [47] |
| Pulmonary fibrosis | ? | BI-1 KO mice exhibit high sensitivity and low survival in response to bleomycin-induced lung injury. | [48] |
| Cancer | |||
| Liver cancer | down | Human BI-1 expression is downregulated as liver damage progresses. | [67] |
| Breast cancer | up | BI-1 is upregulated in human breast cancer, though to varying degrees in common breast cancer cell lines. High-dose estrogen can induce Klf10 to suppress BI-1 transcription and induce apoptosis. |
[66] [68] |
| Prostate cancer | up | BI-1 downregulation induces prostate carcinoma cell death. | [69] |
| Testicular cancer | up | BI-1 may serve a protective role against cisplatin-associated reproductive toxicity. | [70] |
| Nasopharyngeal carcinoma | up | BI-1 downregulation induces cell death, while BI-1 and Bcl-2 family interactions may contribute to apoptosis-resistance. BI-1 downregulation inhibits tumor growth and increases apoptosis. |
[71] [72] |
| Lung cancer | (up) | BI-1 is upregulated in a subtype of non-small cell lung cancers. BI-1 levels vary according to metastatic potential, and patients with BI-1- positive adenocarcinoma have relatively favorable prognoses. |
[73] [74] |
A. Hepatic disorders
The fact that BI-1 is prominently expressed in the liver has incited scientists to investigate the contribution of BI-1 in hepatic disorders. Studies characterize BI-1 as a hepatoprotective factor in metabolic disorders associated with obesity, ischemia-reperfusion injury and liver regeneration.
i. Insulin resistance and Non-Alcoholic Fatty Liver Disease
BI-1 was identified as a critical regulator of ER stress responses in the development of obesity-associated insulin resistance[19]. BI-1 mRNA expression was downregulated by at least 30% in livers from ob/ob, db/db and 12-week high-fat diet (HFD)-fed mice compared to lean controls. Adenoviral transfer to restore hepatic BI-1 expression in these mice severely blunted the IRE1α-dependent splicing of XBP1 mRNA and improved insulin sensitivity and glucose tolerance due to dramatically reduced expression of G6Pase and PEPCK responsible for gluconeogenesis[19]. Hepatic BI-1 was further demonstrated to protect against insulin resistance by limiting ER stress and CYP2E1-dependent ROS accumulation in livers of mice fed a HFD for 8 weeks[40]. In BI-1-deficient hepatocytes, the knockdown of CYP2E1 significantly reduced fatty acid-induced ROS production and improved insulin signaling as represented by increased phosphorylated AKT, FOXO-1 and GSK3 levels[40].
Hepatic insulin resistance is strongly associated with hepatic lipid accumulation in a pathology called Non-Alcoholic Fatty Liver Disease, in which patients also exhibit increased expression of ER stress markers[53]. During chronic HFD feeding, BI-1 was proposed to mediate fatty acid synthesis and ApoB lipoprotein secretion associated with the ER oxidative folding environment in the liver[54]. Livers of BI-1-deficient mice fed a HFD revealed excessive post-translational oxidation of protein disulfide isomerase and ER stress associated with lipid accumulation[54]. Our results on liver disease progression highlight the important role of BI-1 in limiting IRE1α activity from driving the transition from steatosis (fatty liver) to Non-Alcoholic Steatohepatitis characterized by inflammation, hepatocyte apoptosis and often fibrosis. In livers of patients with Non-Alcoholic Fatty Liver Disease, we found BI-1 mRNA downregulation to be accompanied by greater IRE1α RNase signaling[46]. Our current data also reveal that BI-1 confers protection against NLRP3 inflammasome activation and caspase-1 or caspase-11-dependent pyroptosis[46].
ii. Liver injury inflicted by ischemia-reperfusion or CCl4 injection
Hypoxia and ischemia-reperfusion insults activate ER stress-associated cell death. Livers from BI-1-deficient mice subjected to ischemia-reperfusion presented greater hepatocellular injury and higher expression of CHOP, spliced XBP1, cleaved ATF6 and phosphorylated JNK compared to control mice[55]. Interestingly after ischemia-reperfusion, livers of wild-type mice contained a significant 2-fold upregulation in BI-1 mRNA levels, possibly in an effort to limit tissue injury[55]. Similarly, BI-1 protects against microvascular injury and mitochondrial dysfunction due to cardiac ischemia-reperfusion injury[56] and inflammation, ER and oxidative stress in the context of neonatal hypoxic-ischemic injury[57]. The cytoprotective role of BI-1 was also shown to have a detoxifying effect in mice with CCl4-induced liver injury and ROS accumulation[58]. The transplantation of mesenchymal stem cells as a therapeutic intervention promoted an antioxidant response and rescued BI-1 mRNA levels during the recovery from CCl4-induced liver injury[58].
iii. Liver regeneration
In response to massive hepatocellular injury, the liver demonstrates significant regenerative capacity to restore liver mass, thus providing a context for studying in vivo mechanisms of cell growth regulation. Regenerating hepatocytes from BI-1-deficient mice subjected to a two-third liver resection entered the cell cycle faster than control mice[45]. This was demonstrated by increased incorporation of BrdUrd, accelerated expression of cyclin family proteins (cyclin D1, cyclin D3, Cdc2, Cdk2 and Cdk4) with hyperphosphorylation of retinoblastoma protein and faster degradation of p27 in regenerating BI-1-deficient hepatocytes[45]. Faster regeneration in livers from BI-1-deficient mice also correlated with increased dephosphorylation and nuclear translocation of NFAT1, a Ca2+-dependent event linked to calcineurin activation that promotes cytokine gene expression and facilitates hepatocyte entry into the cell cycle[45]. This suggests that the effects of BI-1 deficiency on ER Ca2+ homeostasis may account for the accelerated proliferation of hepatocytes.
B. Immune system regulation and infection
Lymphocytic BI-1 was suggested to play a key role in the adaptive immune response by regulating intracellular Ca2+ homeostasis. Spleens of BI-1-deficient mice presented significant alterations in B-cell maturation due increased ER and cytosolic Ca2+ levels that activated NFkB signaling[59]. Purified B cells from BI-1-deficienct mice were further associated with attenuated function and increased spontaneaous cell death[59]. Recently, BI-1 was suggested to protect against influenza virus by inhibiting ROS-mediated cell death[49].
C. Neurological disorders
BI-1 has been reported to possess a neuroprotective role, even at the earliest of developmental stages to promote neurogenesis. Mouse embryonic stem cells with ectopic BI-1 expression were protected from apoptosis, through suppression of p38 activation, and primed for neuronal differentiation, mediated by the MEK/ERK pathway, towards a neuroectodermal lineage[60]. In mature neurons in vitro, BI-1 limited neuronal cell death induced by oxygen-glucose-deprivation or pharmacological ER stress-induced toxicity with thapsigargin[61]. In forms of acute brain injury inflicted by middle cerebral artery occlusion, BI-1 deficiency in mice increased sensitivity to stroke[15], while enforced neuronal BI-1 expression reduced CHOP expression and protected against stroke injury[50]. In response to traumatic brain injury delivered by cortical impact, neuronal BI-1 overexpression protected against impaired motor function, lesion volume and percentage of CHOP-positive and TUNEL-positive cells[50].
BI-1 was also found to promote resilience in animal models of depression. Mice with enforced neuronal expression of BI-1 exhibited spontaneous recovery from learned helplessness and were protected from developing a depressive-like phenotype induced by serotonin or catecholamine depletion[51]. A possible explanation for improved behavior was altered Ca2+ dynamics in primary cortical neuron cultures from BI-1 transgenic mice. Interestingly, neurons with enhanced BI-1 expression had lower basal cytosolic Ca2+ levels and a limited increase in intracellular [Ca2+] from thapsigargin-induced SERCA inhibition than neurons from wild-type mice[51]. The contribution of BI-1 in Ca2+-related psychiatric conditions was further investigated in BI-1−/− mice shown to be more vulnerable to chronic mild stress-induced depressive-like behavior than wild-type mice[62]. After 6 weeks of exposure to a variety of unpredictable environmental, social and physical stressors, the brains of BI-1-deficient mice exhibited ROS accumulation and slight ER stress with a slight but significant decrease in the levels of the monoamines serotonin, norepinephrine and dopamine compared to wild-type mice[62]. This was proposed to be mediated by BI-1-dependent Ca2+ regulation and associated monoamine oxidase-A activity responsible for regulating serotonin, norepinephrine and dopamine levels[62].
Results have seldom been shown for BI-1 in the context of neurodegenerative diseases. Alzheimer’s disease is characterized by hundreds of mutations in presenilin, the catalytic subunit of γ-secretase responsible for the generation of β-amyloid peptides from the amyloid precursor protein[63], and BI-1 was recently reported to be a binding protein of free presenilin 1, independent of intact γ-secretase[63]. While this finding is a first step towards opening investigations on BI-1 implication in neurodegenerative disease, it’s a wonder the cytoprotective role for BI-1 has not been explored with greater therapeutic interest in such ER stress-associated diseases characterized by the accumulation of misfolded proteins, protein aggregates and neuronal cell death.
D. Cancer development and progression
Cancer results from an imbalance between apoptosis and cell proliferation, fueled by the formation of a microenvironment with hypoxic, nutrient-deprived and acidic conditions. As BI-1 is characterized as an anti-apoptotic protein, it is not surprising that BI-1 is found to be upregulated in numerous different tumor samples (as reviewed in [2, 6, 64, 65]. Besides the frequent upregulation of BI-1 expression, BI-1 downregulation was observed human patients with stomach, colon, kidney, lung and rectal cancers[66]. With the expression of BI-1 in cancer cells depending on tumor type, the relevance of BI-1 expression in clinical outcomes remains to be stated clearly for therapeutic intervention.
i. Liver cancer
As mentioned above, the noteworthy ability of the liver to regenerate rapidly may predispose this organ to carcinogenesis. As regenerating hepatocytes in BI-1-deficient mice duplicate and proliferate faster[45], one would assume BI-1 to protect against hepatocellular carcinoma development. The examination of liver biopsies from patients with hepatocellular carcinoma confirmed significantly downregulated BI-1 mRNA expression in tumoral and surrounding cirrhotic tissues[67].
ii. Breast cancer
BI-1 expression was observed in several human breast cancer cell lines, with highest levels of expression in estrogen-independent MDA-MB-453 compared to HCC70, T-47D, MCF-7, ZR-75–1 and MDA-MB-231 breast carcinoma cells (listed from highest to lowest expression)[66]. A cancer profiling array from different human tumors and corresponding normal tissues from individual female patients revealed upregulated BI-1 expression in breast cancer, but also in ovarian and cervical cancer[66]. While the expression of BI-1 in breast cancer was initially observed to be hormone independent, an estrogen-induced Krüppel-like zinc finger transcription factor (Klf10) directly suppressed BI-1 transcription, leading to increased cytosolic [Ca2+] concentration and apoptosis in breast cancer cells[68]. Treatment with estradiol-17β revealed a dose-dependent decrease in BI-1 protein levels in estrogen-dependent MCF-7 breast cancer cells, and the knockdown of Klf10 reversed the effects of estradiol-17β-induced BI-1 downregulation[68]. Klf10 overexpression in MCF-7 cells led to lower BI-1 and higher apoptotic marker protein expression[68], contradicting the previous report of unaffected apoptosis in MCF-7 cells after BI-1 knockdown[66].
iii. Prostate and testicular cancer
Upregulated gene expression of BI-1 was singled out of a cDNA array analysis comparing a set of capsule-invasive prostate tumor and matched normal prostate tissue and confirmed by quantitative RT-PCR in epithelial tissue samples from prostate cancer patients[69]. Downregulation of BI-1 in human PC-3, LNCaP, and DU-145 prostate carcinoma cells lowered cell viability attributable to increased apoptosis and possibly necrosis, visible by higher active caspase-3-positive staining and in situ end labeling that recognizes DNA strand breaks[69]. Additionally, a study explored the protective function of BI-1 in the testis against cisplatin, a gold-standard chemotherapy drug for testicular cancer, which can cause reproductive toxicity[70]. Cisplatin-treated BI-1-deficient mice presented greater testicular damage and reduced testosterone-synthesis-associated proteins compared to control mice[70]. BI-1 protection was attributed to the regulation of ROS metabolism by the antioxidant AKT-Nrf2-HO-1 axis and improved ER folding capacitance[70]. While lentivirus-mediated testicular expression of BI-1 ultimately rescued BI-1-deficient mice from testicular injury and reduced testosterone production, experimentation using models of testicular cancer will be needed to further characterize these toxicity mechanisms associated with anticancer therapy.
iv. Nasopharyngeal carcinoma
BI-1 expression was explored in CNE-2Z and CNE-1 human nasopharyngeal carcinoma cell lines[71]. The downregulation of BI-1 in these cells led to apoptosis triggered by a decrease in the anti-apoptotic/pro-apoptotic ratios of BCL-XL/BAX and BCL2/BAX and subsequent increase in active caspase-3[71]. Subsequently, BI-1 downregulation as a target for human gene therapy was tested by lentivirus-mediated RNA interference[72]. Administration of lenti-shBI-1 in a nasopharyngeal carcinoma xenograft model established by inoculating nude mice with was CNE-1 cells led to significantly inhibited tumor growth[72], demonstrating the therapeutic value of BI-1 inhibition in nasopharyngeal carcinoma treatment.
v. Lung cancer
The characterization of BI-1 expression in lung cancer remains ambiguous. One study found that BI-1 was positively expressed in all pulmonary adenocarcinomas with a bronchioloalveolar carcinoma (17 of 32 tumors examined), while absolutely no expression was detected in the other histological subtypes classified as papillary, acinar and/or solid tumors[73]. Clinically, bronchioloalveolar carcinoma (now referred to as “lepidic predominant adenocarcinoma”, a subtype of non-small cell lung cancers) is characterized by well-differentiated but noninvasive tumors and a favorable prognosis. Hence, although positive BI-1 expression was present in this type of lung cancer, it is not associated with adenocarcinomas having more aggressive behaviors. More recently, BI-1 expression was detected in primary lesions and metastasis lymph nodes of clinical non-small cell lung cancer samples, although no information on the histological cancer subtype of the samples was provided[74]. Using human lung adenocarcinoma cell lines, BI-1 was less expressed in the highly metastatic Anip973 than in its parental line AGZY83-a associated with low metastatic capacity[74]. Thus, the differential expression of BI-1 in cells with varying metastatic potential and classified into separate subtypes of lung adenocarcinoma requires further investigation.
vi. Metastasis
During metastasis, cancer cells may break away from one of the primary cancers aforementioned, travel through the blood or lymph system and form new tumors in other parts of the body. BI-1 may mediate actin polymerization through its dynamic Ca2+ regulation and actin binding site, promoting cell adhesion[75]. In a vicious cycle, actin polymerization can enhance ER Ca2+ release[76], while high [Ca2+] can drive actin polymerization in a mechanism recently named calcium-mediated actin reset (CaAR)[77]. Varying Ca2+ levels under cellular stress is hence relevant in conditions ranging from wound healing to cancer progression as it may be responsible for the reorganization of the actin cytoskeleton, cell shape and movement. BI-1 was further found to promote metastasis by altering glucose metabolism through increased glycolysis and activating the Na+/H+ exchanger (NHE)[78]. Glycolytic metabolism imposes an increased acid load on tumor cells. The pH of BI-1 overexpressing cells was lower as cells excreted more H+ and increased NHE activity to compensate, leading to enhanced cell invasiveness and mobility as well as acid-sensitive matrix metalloproteinase-2/9 activity[78]. The viability of BI-1 overexpressing cells was found to be promoted by the intracellular pH regulation through NHE[78]. Furthermore, the interconnection between BI-1, NHE and cancer metastasis was joined by the monoamine carboxylate transporter (MCT) system in the regulation of acidic extracellular pH[79]. Ultimately, blocking NHE or MCT activity inhibited BI-1-induced cellular acidity and cancer metastasis, leading to cell death[79]. Targeting ion exchangers or transporters may provide a therapeutic strategy aimed at limiting BI-1 activity and cancer metastasis.
Understanding the regulatory mechanisms of the BI-1 family in the tumor microenvironment will most surely help to develop and optimize cancer therapy. BI-1 was previously shown to be a pharmacological target of the chemotherapeutic drugs doxorubicin and daunorubicin that lowered cell viability in ER-stressed cells by inhibiting the Ca2+/H+ antiporter-like activity of BI-1 recreated in proteolisosomes in vitro[80]. An immunotherapeutic approach was also suggested for acute myeloid leukemia, since the HLA-A24-binding peptide derived from BI-1 was identified as a novel immunogenic tumor-associated antigen capable of generating leukemia-specific cytotoxic T lymphocytes[81]. BI-1 may thus be a potential tool for cancer vaccine development. Although the functions of BI-1 in cancer cells should be further investigated, these studies collectively suggest that BI-1 activities are associated with cancer progression and propagation. Importantly, regardless of the type tumor analyzed, it remains unknown how a BI-1 signature would impact a patient’s outcome. In addition, no correlations have been reported between the level of BI-1 expression and the clinical response of cancer patients to chemotherapeutic drugs. Future work must be done to define BI-1 as a potentially relevant cancer therapeutic target, and to design specific BI-1-targeted novel strategies.
Conclusion
BI-1 is the best-characterized member of the TMBIM family and a highly conserved protein. Multiple hypotheses have emerged on the true function of BI-1 as a transmembrane protein but most agree that it acts as a guardian of ER homeostasis, notably bridging UPR functions and [Ca2+] regulation. As an anti-apoptotic protein, BI-1 controls ER Ca2+ as well as ROS production, both potent inducers of cell death if released in excess. That being said, the fact that the structural biochemistry of BI-1 remains to be confirmed may challenge the conclusions made on its alternative functions. The highly hydrophobic nature of BI-1 also makes it difficult to use appropriate antibodies. Mechanistic studies must still be conducted to answer fundamental questions that will pave the way for therapeutic manipulation.
This review has contributed to combine recent progress made to understand the role of BI-1 in cell death and disease. Nevertheless, many questions remain: “What are the other molecular triggers on the fine line between ER stress-induced adaptation to apoptosis and how do they cooperate with BI-1? Besides IRE1α, can BI-1 directly affect the two other transmembrane sensors of the UPR and their downstream signaling? How can BI-1 lower [Ca2+] in the ER as well in the cytosol if it functions as an ER Ca2+ leak channel? When does an anti-apoptotic protein like BI-1 protect cancer cells at the expense of normal cells, and is BI-1 able to recognize one from the other? How does BI-1 expression correlate with the various phases of cancer? The relatively ubiquitous expression of BI-1 has made it an interesting player in multiple diseases. Future studies and a consolidation of BI-1 function in cellular life and death situations will undeniably lead to novel therapeutic strategies.
Acknowledgements
We thank Dr. Randal J. Kaufman for helpful discussions. This work was supported by grants from INSERM (France), the University of Nice and the French Government (National Research Agency, ANR-18-CE14–0022-01, IRE1inNASH) and through the ‘Investments for the Future’ LABEX SIGNALIFE: program reference #ANR-11-LABX-0028–01, and charities (Societe Francophone du Diabete (MSD, Allocation Exceptionnelle) and Association pour la Recherche sur le Cancer (ARC; #20171206287), La Ligue Contre le Cancer to D.V.). M.B is supported by the French Government National. C.L. is a member of the UCSD Diabetes Research Center and is supported by the NIH grant T32DK007494.
Abbreviations
- ATF6
activating transcription factor
- BI-1
BAX Inhibitor-1
- BAR
bifunctional apoptosis regulator
- CYP2E1
cytochrome P450 species 2E1
- HO-1
heme oxygenase-1
- HFD
high-fat diet
- IP3R
inositol 1,4,5-trisphosphate receptor
- IRE1
inositol-requiring enzyme 1
- Klf10
Krüppel-like zinc finger transcription factor
- MOMP
mitochondrial outer membrane permeabilization
- MCT
monoamine carboxylate transporter
- NHE
Na+/H+ exchanger
- NPR
NADPH-P450 reductase
- PERK
PKR-like ER kinase
- ROS
reactive oxygen species
- SERCA
sarcoplasmic/ER calcium ATPase
- TMBIM
transmembrane BAX Inhibitor-1-containing motif
- UPR
unfolded protein response
Footnotes
Conflicts of Interest
None to declare.
References
- 1.Henke N, Lisak DA, Schneider L, Habicht J, Pergande M & Methner A (2011) The ancient cell death suppressor BAX inhibitor-1, Cell Calcium. 50, 251–60. [DOI] [PubMed] [Google Scholar]
- 2.Rojas-Rivera D & Hetz C. (2015) TMBIM protein family: ancestral regulators of cell death, Oncogene. 34, 269–80. [DOI] [PubMed] [Google Scholar]
- 3.Chae H-J, Ke N, Kim H-R, Chen S, Godzik A, Dickman M & Reed JC (2003) Evolutionarily conserved cytoprotection provided by Bax Inhibitor-1 homologs from animals, plants, and yeast, Gene. 323, 101–113. [DOI] [PubMed] [Google Scholar]
- 4.Ishikawa T, Watanabe N, Nagano M, Kawai-Yamada M & Lam E (2011) Bax inhibitor-1: a highly conserved endoplasmic reticulum-resident cell death suppressor, Cell Death Differ. 18, 1271–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Q (2017) TMBIM-mediated Ca(2+) homeostasis and cell death, Biochim Biophys Acta Mol Cell Res. 1864, 850–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carrara G, Parsons M, Saraiva N & Smith GL (2017) Golgi anti-apoptotic protein: a tale of camels, calcium, channels and cancer, Open Biol. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu Q & Reed JC (1998) Bax Inhibitor-1, a Mammalian Apoptosis Suppressor Identified by Functional Screening in Yeast, Molecular Cell. 1, 337–346. [DOI] [PubMed] [Google Scholar]
- 8.Doycheva D, Kaur H, Tang J & Zhang JH (2019) The characteristics of the ancient cell death suppressor, TMBIM6, and its related signaling pathways after endoplasmic reticulum stress, J Neurosci Res. [DOI] [PubMed] [Google Scholar]
- 9.Lisak DA, Schacht T, Enders V, Habicht J, Kiviluoto S, Schneider J, Henke N, Bultynck G & Methner A (2015) The transmembrane Bax inhibitor motif (TMBIM) containing protein family: Tissue expression, intracellular localization and effects on the ER CA(2)(+)-filling state, Biochim Biophys Acta. 1853, 2104–14. [DOI] [PubMed] [Google Scholar]
- 10.Carrara G, Saraiva N, Gubser C, Johnson BF & Smith GL (2012) Six-transmembrane topology for Golgi anti-apoptotic protein (GAAP) and Bax inhibitor 1 (BI-1) provides model for the transmembrane Bax inhibitor-containing motif (TMBIM) family, J Biol Chem. 287, 15896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bultynck G, Kiviluoto S, Henke N, Ivanova H, Schneider L, Rybalchenko V, Luyten T, Nuyts K, De Borggraeve W, Bezprozvanny I, Parys JB, De Smedt H, Missiaen L & Methner A (2012) The C terminus of Bax inhibitor-1 forms a Ca2+-permeable channel pore, J Biol Chem. 287, 2544–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robinson KS, Clements A, Williams AC, Berger CN & Frankel G (2011) Bax inhibitor 1 in apoptosis and disease, Oncogene. 30, 2391–400. [DOI] [PubMed] [Google Scholar]
- 13.Walter L, Dirks B, Rothermel E, Heyens M, Szpirer C, Levan G & Günther E (1994) A novel, conserved gene of the rat that is developmentally regulated in the testis, Mammalian Genome. 5, 216–221. [DOI] [PubMed] [Google Scholar]
- 14.Walter L, Marynen P, Szpirer J, Levan G & Günther E (1995) Identification of a Novel Conserved Human Gene, TEGT, Genomics. 28, 301–304. [DOI] [PubMed] [Google Scholar]
- 15.Chae HJ, Kim HR, Xu C, Bailly-Maitre B, Krajewska M, Krajewski S, Banares S, Cui J, Digicaylioglu M, Ke N, Kitada S, Monosov E, Thomas M, Kress CL, Babendure JR, Tsien RY, Lipton SA & Reed JC (2004) BI-1 regulates an apoptosis pathway linked to endoplasmic reticulum stress, Mol Cell. 15, 355–66. [DOI] [PubMed] [Google Scholar]
- 16.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB & Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death, Science. 292, 727–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antonsson B, Brandt GS, Iwakoshi NN, Schinzel A, Glimcher LH & Korsmeyer SJ (2006) Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha, Science. 312, 572–6. [DOI] [PubMed] [Google Scholar]
- 18.Lisbona F, Rojas-Rivera D, Thielen P, Zamorano S, Todd D, Martinon F, Glavic A, Kress C, Lin JH, Walter P, Reed JC, Glimcher LH & Hetz C (2009) BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1alpha, Mol Cell. 33, 679–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bailly-Maitre B, Belgardt BF, Jordan SD, Coornaert B, von Freyend MJ, Kleinridders A, Mauer J, Cuddy M, Kress CL, Willmes D, Essig M, Hampel B, Protzer U, Reed JC & Bruning JC (2010) Hepatic Bax inhibitor-1 inhibits IRE1alpha and protects from obesity-associated insulin resistance and glucose intolerance, J Biol Chem. 285, 6198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rong J, Chen L, Toth JI, Tcherpakov M, Petroski MD & Reed JC (2011) Bifunctional apoptosis regulator (BAR), an endoplasmic reticulum (ER)-associated E3 ubiquitin ligase, modulates BI-1 protein stability and function in ER Stress, J Biol Chem. 286, 1453–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tabas I & Ron D (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress, Nat Cell Biol. 13, 184–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Puthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, Gotoh T, Akira S, Bouillet P & Strasser A (2007) ER stress triggers apoptosis by activating BH3-only protein Bim, Cell. 129, 1337–49. [DOI] [PubMed] [Google Scholar]
- 23.Xu C, Xu W, Palmer AE & Reed JC (2008) BI-1 regulates endoplasmic reticulum Ca2+ homeostasis downstream of Bcl-2 family proteins, J Biol Chem. 283, 11477–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahn T, Yun CH, Chae HZ, Kim HR & Chae HJ (2009) Ca2+/H+ antiporter-like activity of human recombinant Bax inhibitor-1 reconstituted into liposomes, FEBS J. 276, 2285–91. [DOI] [PubMed] [Google Scholar]
- 25.Ahn T, Yun CH, Kim HR & Chae HJ (2010) Cardiolipin, phosphatidylserine, and BH4 domain of Bcl-2 family regulate Ca2+/H+ antiporter activity of human Bax inhibitor-1, Cell Calcium. 47, 387–96. [DOI] [PubMed] [Google Scholar]
- 26.Kim HR, Lee GH, Ha KC, Ahn T, Moon JY, Lee BJ, Cho SG, Kim S, Seo YR, Shin YJ, Chae SW, Reed JC & Chae HJ (2008) Bax Inhibitor-1 Is a pH-dependent regulator of Ca2+ channel activity in the endoplasmic reticulum, J Biol Chem. 283, 15946–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bultynck G, Kiviluoto S & Methner A (2014) Bax Inhibitor-1 Is Likely a pH-Sensitive Calcium Leak Channel, Not a H+/Ca2+ Exchanger, Science Signaling. 7, pe22. [DOI] [PubMed] [Google Scholar]
- 28.Kiviluoto S, Luyten T, Schneider L, Lisak D, Rojas-Rivera D, Welkenhuyzen K, Missaen L, De Smedt H, Parys JB, Hetz C, Methner A & Bultynck G (2013) Bax Inhibitor-1-mediated Ca2+ leak is decreased by cytosolic acidosis, Cell Calcium. 54, 186–92. [DOI] [PubMed] [Google Scholar]
- 29.Chang Y, Bruni R, Kloss B, Assur Z, Kloppmann E, Rost B, Hendrickson WA & Liu Q (2014) Structural basis for a pH-sensitive calcium leak across membranes, Science. 344, 1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo G, Xu M, Chang Y, Luyten T, Seitaj B, Liu W, Zhu P, Bultynck G, Shi L, Quick M & Liu Q (2019) Ion and pH Sensitivity of a TMBIM Ca(2+) Channel, Structure. 27, 1013–1021 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kiviluoto S, Schneider L, Luyten T, Vervliet T, Missiaen L, De Smedt H, Parys JB, Methner A & Bultynck G (2012) Bax inhibitor-1 is a novel IP(3) receptor-interacting and -sensitizing protein, Cell death & disease. 3, e367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sano R, Hou YC, Hedvat M, Correa RG, Shu CW, Krajewska M, Diaz PW, Tamble CM, Quarato G, Gottlieb RA, Yamaguchi M, Nizet V, Dahl R, Thomas DD, Tait SW, Green DR, Fisher PB, Matsuzawa S & Reed JC (2012) Endoplasmic reticulum protein BI-1 regulates Ca(2)(+)-mediated bioenergetics to promote autophagy, Genes Dev. 26, 1041–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T & Korsmeyer SJ (2005) Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum, Proceedings of the National Academy of Sciences of the United States of America. 102, 105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rong Y-P, Bultynck G, Aromolaran AS, Zhong F, Parys JB, De Smedt H, Mignery GA, Roderick HL, Bootman MD & Distelhorst CW (2009) The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor, Proceedings of the National Academy of Sciences. 106, 14397–14402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Monaco G, Decrock E, Akl H, Ponsaerts R, Vervliet T, Luyten T, De Maeyer M, Missiaen L, Distelhorst CW, De Smedt H, Parys JB, Leybaert L & Bultynck G (2012) Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl, Cell Death Differ. 19, 295–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ivanova H, Vervliet T, Monaco G, Terry LE, Rosa N, Baker MR, Parys JB, Serysheva II, Yule DI & Bultynck G (2019) Bcl-2-Protein Family as Modulators of IP3 Receptors and Other Organellar Ca(2+) Channels, Cold Spring Harb Perspect Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vervliet T, Parys JB & Bultynck G (2016) Bcl-2 proteins and calcium signaling: complexity beneath the surface, Oncogene. 35, 5079–92. [DOI] [PubMed] [Google Scholar]
- 38.Davydov DR (2011) Microsomal monooxygenase as a multienzyme system: the role of P450-P450 interactions, Expert Opinion on Drug Metabolism & Toxicology. 7, 543–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim HR, Lee GH, Cho EY, Chae SW, Ahn T & Chae HJ (2009) Bax inhibitor 1 regulates ER-stress-induced ROS accumulation through the regulation of cytochrome P450 2E1, J Cell Sci. 122, 1126–33. [DOI] [PubMed] [Google Scholar]
- 40.Lee GH, Oh KJ, Kim HR, Han HS, Lee HY, Park KG, Nam KH, Koo SH & Chae HJ (2016) Effect of BI-1 on insulin resistance through regulation of CYP2E1, Sci Rep. 6, 32229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee GH, Kim HR & Chae HJ (2012) Bax inhibitor-1 regulates the expression of P450 2E1 through enhanced lysosome activity, Int J Biochem Cell Biol. 44, 600–11. [DOI] [PubMed] [Google Scholar]
- 42.Lee GH, Kim DS, Kim HT, Lee JW, Chung CH, Ahn T, Lim JM, Kim IK, Chae HJ & Kim HR (2011) Enhanced lysosomal activity is involved in Bax inhibitor-1-induced regulation of the endoplasmic reticulum (ER) stress response and cell death against ER stress: involvement of vacuolar H+-ATPase (V-ATPase), J Biol Chem. 286, 24743–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim JH, Lee ER, Jeon K, Choi HY, Lim H, Kim SJ, Chae HJ, Park SH, Kim S, Seo YR, Kim JH & Cho SG (2012) Role of BI-1 (TEGT)-mediated ERK1/2 activation in mitochondria-mediated apoptosis and splenomegaly in BI-1 transgenic mice, Biochim Biophys Acta. 1823, 876–88. [DOI] [PubMed] [Google Scholar]
- 44.Lee GH, Kim HK, Chae SW, Kim DS, Ha KC, Cuddy M, Kress C, Reed JC, Kim HR & Chae HJ (2007) Bax inhibitor-1 regulates endoplasmic reticulum stress-associated reactive oxygen species and heme oxygenase-1 expression, J Biol Chem. 282, 21618–28. [DOI] [PubMed] [Google Scholar]
- 45.Bailly-Maitre B, Bard-Chapeau E, Luciano F, Droin N, Bruey JM, Faustin B, Kress C, Zapata JM & Reed JC (2007) Mice lacking bi-1 gene show accelerated liver regeneration, Cancer Res. 67, 1442–50. [DOI] [PubMed] [Google Scholar]
- 46.Lebeaupin C, Vallee D, Rousseau D, Patouraux S, Bonnafous S, Adam G, Luciano F, Luci C, Anty R, Iannelli A, Marchetti S, Kroemer G, Lacas-Gervais S, Tran A, Gual P & Bailly-Maitre B (2018) Bax inhibitor-1 protects from nonalcoholic steatohepatitis by limiting inositol-requiring enzyme 1 alpha signaling in mice, Hepatology. 68, 515–532. [DOI] [PubMed] [Google Scholar]
- 47.Yadav RK, Lee GH, Lee HY, Li B, Jung HE, Rashid HO, Choi MK, Yadav BK, Kim WH, Kim KW, Park BH, Kim W, Lee YC, Kim HR & Chae HJ (2015) TMBIM6 (transmembrane BAX inhibitor motif containing 6) enhances autophagy and reduces renal dysfunction in a cyclosporine A-induced nephrotoxicity model, Autophagy. 11, 1760–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee MR, Lee GH, Lee HY, Kim DS, Chung MJ, Lee YC, Kim HR & Chae HJ (2014) BAX inhibitor-1-associated V-ATPase glycosylation enhances collagen degradation in pulmonary fibrosis, Cell death & disease. 5, e1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hossain MK, Saha SK, Abdal Dayem A, Kim JH, Kim K, Yang GM, Choi HY & Cho SG (2018) Bax Inhibitor-1 Acts as an Anti-Influenza Factor by Inhibiting ROS Mediated Cell Death and Augmenting Heme-Oxygenase 1 Expression in Influenza Virus Infected Cells, Int J Mol Sci. 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krajewska M, Xu L, Xu W, Krajewski S, Kress CL, Cui J, Yang L, Irie F, Yamaguchi Y, Lipton SA & Reed JC (2011) Endoplasmic reticulum protein BI-1 modulates unfolded protein response signaling and protects against stroke and traumatic brain injury, Brain Res. 1370, 227–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hunsberger JG, Machado-Vieira R, Austin DR, Zarate C, Chuang DM, Chen G, Reed JC & Manji HK (2011) Bax inhibitor 1, a modulator of calcium homeostasis, confers affective resilience, Brain Res. 1403, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li B, Reed JC, Kim HR & Chae HJ (2012) Proteomic profiling of differentially expressed proteins from Bax inhibitor-1 knockout and wild type mice, Mol Cells. 34, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lebeaupin C, Proics E, de Bieville CH, Rousseau D, Bonnafous S, Patouraux S, Adam G, Lavallard VJ, Rovere C, Le Thuc O, Saint-Paul MC, Anty R, Schneck AS, Iannelli A, Gugenheim J, Tran A, Gual P & Bailly-Maitre B (2015) ER stress induces NLRP3 inflammasome activation and hepatocyte death, Cell death & disease. 6, e1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee HY, Lee GH, Bhattarai KR, Park BH, Koo SH, Kim HR & Chae HJ (2016) Bax Inhibitor-1 regulates hepatic lipid accumulation via ApoB secretion, Sci Rep. 6, 27799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bailly-Maitre B, Fondevila C, Kaldas F, Droin N, Luciano F, Ricci J-E, Croxton R, Krajewska M, Zapata JM, Kupiec-Weglinski JW, Farmer D & Reed JC (2006) Cytoprotective gene bi-1 is required for intrinsic protection from endoplasmic reticulum stress and ischemia-reperfusion injury, Proceedings of the National Academy of Sciences of the United States of America. 103, 2809–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou H, Shi C, Hu S, Zhu H, Ren J & Chen Y (2018) BI1 is associated with microvascular protection in cardiac ischemia reperfusion injury via repressing Syk-Nox2-Drp1-mitochondrial fission pathways, Angiogenesis. 21, 599–615. [DOI] [PubMed] [Google Scholar]
- 57.Doycheva D, Xu N, Tang J & Zhang J (2019) Viral-mediated gene delivery of TMBIM6 protects the neonatal brain via disruption of NPR-CYP complex coupled with upregulation of Nrf-2 post-HI, J Neuroinflammation. 16, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cho K-A, Woo S-Y, Seoh J-Y, Han H-S & Ryu K-H (2012) Mesenchymal stem cells restore CCl4-induced liver injury by an antioxidative process, Cell Biology International. 36, 1267–1274. [DOI] [PubMed] [Google Scholar]
- 59.Lisak D, Schacht T, Gawlitza A, Albrecht P, Aktas O, Koop B, Gliem M, Hofstetter HH, Zanger K, Bultynck G, Parys JB, De Smedt H, Kindler T, Adams-Quack P, Hahn M, Waisman A, Reed JC, Hovelmeyer N & Methner A (2016) BAX inhibitor-1 is a Ca(2+) channel critically important for immune cell function and survival, Cell Death Differ. 23, 358–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jeon K, Lim H, Kim J-H, Han D, Lee E-R, Yang G-M, Song M-K, Kim J-H & Cho S-G (2012) Bax inhibitor-1 enhances survival and neuronal differentiation of embryonic stem cells via differential regulation of mitogen-activated protein kinases activities, Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1823, 2190–2200. [DOI] [PubMed] [Google Scholar]
- 61.Dohm CP, Siedenberg S, Liman J, Esposito A, Wouters FS, Reed JC, Bähr M & Kermer P (2006) Bax inhibitor-1 protects neurons from oxygen-glucose deprivation, Journal of Molecular Neuroscience. 29, 1–8. [DOI] [PubMed] [Google Scholar]
- 62.Lee HY, Lee GH, Marahatta A, Lin SM, Lee MR, Jang KY, Kim KM, Lee HJ, Lee JW, Bagalkot TR, Chung YC, Lee YC, Kim HR & Chae HJ (2013) The protective role of Bax inhibitor-1 against chronic mild stress through the inhibition of monoamine oxidase A, Sci Rep. 3, 3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu S, Song W, Wong CCL & Shi Y (2019) Bax inhibitor 1 is a gamma-secretase-independent presenilin-binding protein, Proc Natl Acad Sci U S A. 116, 141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kerstin R, Claudia YUC, Vesna B & Peter MV (2008) The Bax Inhibitor-1 (BI-1) Family in Apoptosis and Tumorigenesis, Current Molecular Medicine. 8, 148–156. [DOI] [PubMed] [Google Scholar]
- 65.Li B, Yadav RK, Jeong GS, Kim HR & Chae HJ (2014) The Characteristics of Bax Inhibitor-1 and its Related Diseases, Current Molecular Medicine. 14, 603–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grzmil M, Kaulfuss S, Thelen P, Hemmerlein B, Schweyer S, Obenauer S, Kang TW & Burfeind P (2006) Expression and functional analysis of Bax inhibitor-1 in human breast cancer cells, J Pathol. 208, 340–9. [DOI] [PubMed] [Google Scholar]
- 67.Kotsafti A, Farinati F, Cardin R, Burra P & Bortolami M (2010) Bax Inhibitor-1 down-regulation in the progression of chronic liver diseases, BMC Gastroenterology. 10, 35–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hsu CF, Sui CL, Wu WC, Wang JJ, Yang DH, Chen YC, Yu WC & Chang HS (2011) Klf10 induces cell apoptosis through modulation of BI-1 expression and Ca2+ homeostasis in estrogen-responding adenocarcinoma cells, Int J Biochem Cell Biol. 43, 666–73. [DOI] [PubMed] [Google Scholar]
- 69.Grzmil M, Thelen P, Hemmerlein B, Schweyer S, Voigt S, Mury D & Burfeind P (2003) Bax Inhibitor-1 Is Overexpressed in Prostate Cancer and Its Specific Down-Regulation by RNA Interference Leads to Cell Death in Human Prostate Carcinoma Cells, The American Journal of Pathology. 163, 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim HK, Yadav RK, Bhattarai KR, Jung HW, Kim HR & Chae HJ (2018) Transmembrane BAX Inhibitor Motif-6 (TMBIM6) protects against cisplatin-induced testicular toxicity, Hum Reprod. 33, 378–389. [DOI] [PubMed] [Google Scholar]
- 71.Zhang M, Li X, Zhang Y & Zhou K (2010) Bax inhibitor-1 mediates apoptosis-resistance in human nasopharyngeal carcinoma cells, Mol Cell Biochem. 333, 1–7. [DOI] [PubMed] [Google Scholar]
- 72.Li XY, Lai YK, Zhang JF, Luo HQ, Zhang MH, Zhou KY & Kung HF (2011) Lentivirus-mediated RNA interference targeting Bax inhibitor-1 suppresses ex vivo cell proliferation and in vivo tumor growth of nasopharyngeal carcinoma, Hum Gene Ther. 22, 1201–8. [DOI] [PubMed] [Google Scholar]
- 73.Tanaka R, Ishiyama T, Uchihara T, Inadome Y, Iijima T, Morishita Y, Kano J, Goya T & Noguchi M (2006) Expression of the Bax inhibitor-1 gene in pulmonary adenocarcinoma, Cancer. 106, 648–53. [DOI] [PubMed] [Google Scholar]
- 74.Lu B, Li Y, Li H, Zhang Y, Xu J, Ren L, Fu S & Zhou Y (2015) Bax inhibitor-1 is overexpressed in non-small cell lung cancer and promotes its progression and metastasis, International Journal of Clinical and Experimental Pathology. 8, 1411–1418. [PMC free article] [PubMed] [Google Scholar]
- 75.Lee GH, Ahn T, Kim DS, Park SJ, Lee YC, Yoo WH, Jung SJ, Yang JS, Kim S, Muhlrad A, Seo YR, Chae SW, Kim HR & Chae HJ (2010) Bax inhibitor 1 increases cell adhesion through actin polymerization: involvement of calcium and actin binding, Mol Cell Biol. 30, 1800–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang Y, Mattson MP & Furukawa K (2002) Endoplasmic reticulum calcium release is modulated by actin polymerization, Journal of Neurochemistry. 82, 945–952. [DOI] [PubMed] [Google Scholar]
- 77.Wales P, Schuberth CE, Aufschnaiter R, Fels J, Garcia-Aguilar I, Janning A, Dlugos CP, Schafer-Herte M, Klingner C, Walte M, Kuhlmann J, Menis E, Hockaday Kang L, Maier KC, Hou W, Russo A, Higgs HN, Pavenstadt H, Vogl T, Roth J, Qualmann B, Kessels MM, Martin DE, Mulder B & Wedlich-Soldner R (2016) Calcium-mediated actin reset (CaAR) mediates acute cell adaptations, Elife. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee GH, Yan C, Shin SJ, Hong SC, Ahn T, Moon A, Park SJ, Lee YC, Yoo WH, Kim HT, Kim DS, Chae SW, Kim HR & Chae HJ (2010) BAX inhibitor-1 enhances cancer metastasis by altering glucose metabolism and activating the sodium-hydrogen exchanger: the alteration of mitochondrial function, Oncogene. 29, 2130–41. [DOI] [PubMed] [Google Scholar]
- 79.Lee GH, Chae HJ & Kim HR (2011) Monoamine carboxylate transporters are involved in BI-1-associated cancer metastasis in HT1080 colon fibrosarcoma cells, Int J Oncol. 39, 209–16. [DOI] [PubMed] [Google Scholar]
- 80.Yun CH, Chae HJ, Kim HR & Ahn T (2012) Doxorubicin- and daunorubicin-induced regulation of Ca2+ and H+ fluxes through human bax inhibitor-1 reconstituted into membranes, J Pharm Sci. 101, 1314–26. [DOI] [PubMed] [Google Scholar]
- 81.Schmidt SM, Konig T, Bringmann A, Held S, von Schwarzenberg K, Heine A, Holderried TA, Stevanovic S, Grunebach F & Brossart P (2009) Characterization of BAX inhibitor-1 as a novel leukemia-associated antigen, Leukemia. 23, 1818–24. [DOI] [PubMed] [Google Scholar]
- 82.Omasits U, Ahrens CH, Muller S & Wollscheid B (2014) Protter: interactive protein feature visualization and integration with experimental proteomic data, Bioinformatics. 30, 884–6. [DOI] [PubMed] [Google Scholar]