

Abstract
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) assays were developed to measure arylsulfatase A (ARSA) activity in leukocytes and dried blood spot (DBS) using deuterated natural sulfatide substrate. These new assays were highly specific and sensitive. Patients with metachromatic leukodystrophy (MLD) and multiple sulfatase deficiency (MSD) displayed clear deficit in the enzymatic activity, and could be completely distinguished from normal controls. The leukocyte assay reported here will be important for diagnosing MLD and MSD patient and for monitoring the efficacy of therapeutic treatments. On the other hand, ARSA activity was measured in DBS for the first time without an antibody. Currently, this new ARSA DBS assay serves as a second-tier test following the sulfatide measurement in DBS in our large-scale de-identified pilot study for the screening of MLD. This leads to an elimination of most of the false positives identified by the sulfatide assay. We are also evaluating its potential utility as the first-tier test for the newborn screening of MLD.
Keywords: metachromatic leukodystrophy, multiple sulfatase deficiency, arylsulfatase A, enzyme deficiency, leukocyte, dried blood spots, liquid chromatography-tandem mass spectrometry, diagnostics, newborn screening
Graphical Abstract
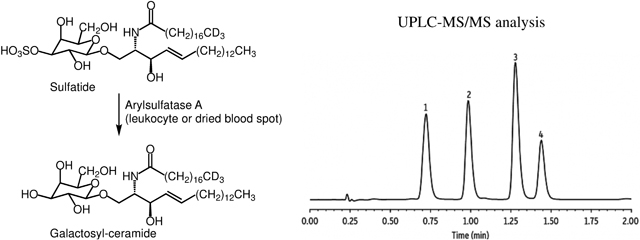
Metachromatic leukodystrophy (MLD) is caused by deficiency of the lysosomal enzyme arylsulfatase A (ARSA) which removes sulfate from 3-sulfo-galactosylceramides (sulfatides). With the development of potential treatments, including gene therapy and hematopoietic stem cell transplantation, and the demonstration that clinical outcomes are optimized if treatments are initiated prior to full onset of symptoms1, 2, newborn screening (NBS) for MLD may be warranted in the near future. Elevated sulfatides in urine and blood are typically seen in MLD patients but not in those carrying pseudodeficiency varaints3. The diagnosis of MLD also relies on a nominally low ARSA enzymatic activity in leukocyte or fibroblast lysate. However, there have been multiple reports detailing the pitfalls of the current ARSA assays using generic sulfatase substrates4, 5. Thus, a sensitive and specific enzymatic assay for ARSA would be an improvement to the current diagnostic test.
We are currently carrying out a large-scale de-identified pilot NBS study for MLD by measuring sulfatides abundance in dried blood spots (DBS)3. After screening approximately 50,000 random newborns, it became apparent that a second-tier test was required to reduce the number of false positives. Therefore, we also sought to develop an assay to measure ARSA enzymatic activity in DBS, so that the test can be performed onsite as part of the screening process without the need to contact families to obtain a different sample specimen like whole blood or urine.
Herein, we developed an in vitro assay to measure ARSA activity in leukocytes lysate that utilized its natural substrate for specificity. The new ARSA leukocyte assay had an exceptionally high sensitivity and precision such that trace amounts of residual enzymatic activity could be detected with statistical significance. An ARSA enzymatic activity assay in DBS was also developed in this study, which was conducted for the first time without an anti-ARSA antiserum. These new ARSA assays will also be useful for diagnosing patients with multiple sulfatase deficiency (MSD), where defect in the formylglycine generating enzyme prohibits the crucial modification on the active site of all sulfatases6.
EXPERIMENTAL SECTIONS
Materials.
Whole blood from a healthy adult donor and DBS from 10 healthy adults were collected with consent and were used as positive controls. Whole blood from MLD and MSD patients were obtained through the Myelin Disorders Biorepository Project at the Children’s Hospital of Philadelphia, the MLD Foundation, Duke University, the Children’s Hospital of Pittsburgh and the San Raffaele Telethon Institute for Gene Therapy (Milan, Italy) with IRB approvals. DBS was prepared by spotting the whole blood onto 903 protein saver cards (GE Healthcare Life Sciences). Additional DBS from MLD patients were obtained through Meyer Children’s Hospital (Florence, Italy). All patients were diagnosed based on clinical, biochemical and genetic evidence of the disease. De-identified newborn DBS, which were previously stored at room temperature for 30–60 days, were acquired through the Washington State Department of Health with the approval from the Washington State Institutional Review Board. Quality Control DBS for lysosomal storage disorders was acquired from the Centers for Disease Control and Prevention (CDC).
d3-C18:0-Sulfatide was purchased from Matreya, LLC (Cat. 1536). d7-C18:0-Galactosyl-ceramide was synthesized in-house as described in the Supplemental Material and was quantified by quantitative 1H-NMR. Stock solutions of sulfatide and galactosyl-ceramide were prepared in 2/1 chloroform/methanol (v/v) and were stored in glass vials with Teflon-septum screw caps at −20 °C. ARSA-containing lymphoblasts (GM14603) and ARSA-lacking lymphoblasts (GM23097) were obtained from the Coriell Institute Cell Repository and were cultured as per the vendor’s instructions.
Preparation of leukocytes and BCA protein assay.
Venous blood (at least 0.5 mL) was collected into a K2EDTA tube and mixed by inversion to distribute the anti-coagulant. The blood was stored at 4 °C prior to overnight shipment in insulated boxes with frozen gel packs. Leukocytes were prepared within 24 hours of blood collection as described before7. Leukocyte processed from whole blood collected in heparin tubes was also suitable for the assay, but required additional validations. Supernatant and leukocyte suspension were then stored at −80 °C. Phosphate-buffered saline should be avoided as inorganic phosphate strongly inhibits ARSA.
Cells were stored at −80 °C for at least 16 hours prior to lysis by thawing on ice. The lysate was centrifuged at 10,000 g for 5 min at room temperature, and the supernatant was transferred to a new tube. The concentration of protein was determined by the Microplate BCA kit (ThermoFisher, Cat. 23252), using bovine serum albumin as the standard. Leukocyte lysates were typically assayed for protein concentration and ARSA activity right after thawing. However, multiple freeze-thaw cycle had minimal effect on the ARSA activity (Figure S6b).
ARSA leukocyte assay.
Assay cocktail consisted of 150 μM d3-C18:0-sulfatide as substrate and 2 μM d7-C18:0-galactosyl-ceramide as internal standard in ARSA assay buffer (80 mM sodium acetate (J.T. Baker, Cat. 3460–01), 2.0 g/L sodium taurodeoxycholate (Carbosyth, Cat. FS45995), pH 4.5 ± 0.02). It was prepared by mixing proper amounts of substrate and internal standard stock solution, then removing the organic solvent with a centrifugal concentrator or a jet of oil-free air at room temperature, and reconstituting the residual with assay buffer with a vortex mixer. Assay cocktail was typically prepared fresh, but it could also be stored frozen at −20 °C.
Leukocyte protein concentration was adjusted to 0.2 μg/μL using 0.9% NaCl in water as diluent. The assay was set up using 10 μL of leukocyte lysate containing 2 μg protein and 30 μL of assay cocktail in a 96-well, polypropylene deep-well plate sealed with a silicone matt. The plate was centrifuged for 1 min at 3000 g to ensure that all liquid was at the bottom of each well. The plate was placed on an orbital shaker (400 rpm on a 3 mm shaking radius) at 37 °C for 16 hours. Reactions were quenched by addition of 300 μL of methanol, and the plate was centrifuged for 5 min at 3000 g. Supernatant (150 μL) was transferred to the autosampler plate followed by addition of 50 μL water per well, and the plate was placed in the cooled (8 °C) autosampler chamber of the LC-MS/MS instrument.
ARSA DBS assay after purification by immuno-precipitation.
Polyclonal anti-ARSA antiserum (R&D System, Cat. AF2485) was immobilized on a high binding plate (PerkinElmer, Cat. 1244–550) at 1 μg anti-ARSA per well as follows. Anti-ARSA (100 μL of 10 μg/mL) in 0.2 M sodium phosphate, pH 6.8 was added in each well, and the plate was sealed with plastic film and incubated overnight on an orbital shaker at room temperature with gentle shaking. The solution was aspirated off, and 300 μL 0.9% NaCl in water was added per well, followed by aspiration. Blocking buffer (250 μL of 0.05 M Tris-HCl, pH 7.8, 0.9% NaCl, 0.05% NaN3, 6.0% D-sorbitol, 1.0% bovine serum albumin (Sigma, Cat. A6003), 1.0 mM CaCl2) was added, and the plate was sealed and incubated overnight on an orbital shaker at room temperature with gentle shaking. After aspiration, to each well was added 300 μL 0.9% NaCl, followed by aspiration. Plates were sealed with plastic film and stored in a sealed container with water for humidification. The coated plates can be stored at 4 °C for at least 6 months.
To each 3 mm DBS punch in a separate 96-well plate, 120 μL extraction buffer (0.1 M sodium acetate, 0.1% (w/v) bovine serum albumin, pH 5.0) was added. The plate was sealed and shaken on an orbital shaker at 37 °C for 1 hour. DBS eluent (100 μL) was transferred to an anti-ARSA coated well, and the plate was sealed and shaken on an orbital shaker at room temperature for 1 hour. The solution was removed by aspiration, and 300 μL of 20 mM sodium acetate, pH 5.0 was added to wash the well, followed by aspiration. To each well, 50 μL of assay cocktail (150 μM d3-C18:0-sulfatide as substrate and 0.2 μM d7-C18:0-galactosyl-ceramide as internal standard in assay buffer) was added. The plate was centrifuged for 1 min at 3000 g to ensure the bottom of each well was covered with liquid. The plate was sealed and placed on an orbital shaker (400 rpm on a 3 mm shaking radius) at 37 °C for 16 hours. The assay workup was the same as for the leukocyte assay.
ARSA DBS after purification by size-exclusion chromatography.
To each 3 mm DBS punch in a 96-well plate, 50 μL extraction buffer (0.8% NH4OH (Millipore, Cat. AX1303) in assay buffer) was added. The plate was sealed and shaken on an orbital shaker at room temperature for 4 hours. Sephadex G-25 resin (GE Healthcare Life Sciences, Cat. 17003201) was swollen in Milli-Q water (10 mL per g of dry resin) for 3 hours prior to use. In a 96-well fritted plate (ThermoFisher, Cat. 278011), 600 μL resin slurry (equivalent to 60 mg dry resin) was added per well using a pipette. Water was removed by centrifugation of the plate at 800 g for 1 min, and the resin was washed 3 times with 600 μL assay buffer, each time the assay buffer was removed by centrifugation at 800 g for 1 min. After the third wash, a clean 96-well plate was placed under the fritted plate as a receiver plate. DBS eluent (30 μL) was loaded on to the resin, followed by 15 μL of assay buffer. These liquids were added with the pipette tip against the side wall of the fritted well so that the resin pellet was not disturbed. The plate was centrifuged at 800 g for 1 min, and the eluate was collected. To the purified DBS eluate in the receiver plate, 10 μL assay cocktail (450 μM d3-C18:0-sulfatide as substrate and 1 μM d7-C18:0-galactosyl-ceramide as internal standard in assay buffer) was added. The plate was centrifuged for 1 min at 3000 g to ensure that all liquid was at the bottom of each well. The plate was sealed and placed on an orbital shaker (400 rpm on a 3 mm shaking radius) at 37 °C for 16 hours. The assay workup was the same as for the leukocyte assay.
UPLC-MS/MS analysis.
UPLC-MS/MS analysis was carried out on a Waters Xevo TQ-S micro mass spectrometer coupled to a Waters AQUITY UPLC I-Class system using multiple reaction monitoring (MRM) in ESI positive mode. MRM settings and ESI source conditions are listed in Supplemental Material.
Separation of the enzymatic substrate and product was achieved at a column temperature of 30 °C using an ACQUITY UPLC CSH Fluoro Phenyl column (1.7 μm, 2.1 mm × 50 mm, Waters Corp., Cat. 186005351) connected to an ACQUITY UPLC CSH Fluoro Phenyl VanGuard Pre-column (1.7 μm, 2.1 mm × 5 mm, Waters Corp., Cat. 186005358) at a flow rate of 0.7 mL/min. Mobile phase A was 50/50 (v/v) water/acetonitrile with 0.1 % formic acid; mobile phase B was 50/50 (v/v) acetonitrile/isopropanol with 0.1% formic acid. The weak needle wash and the strong needle wash were 25/25/50 (v/v/v) methanol/isopropanol/water and 47.5/47.5/5 (v/v/v) methanol/isopropanol/water, respectively. The linear gradient was as followed: 0–0.5 min, 0% solvent B; 0.5–1.0 min, 0 to 90% solvent B; 1.0–1.5 min, 90 to 100% solvent B, 1.5–1.95 min, 100% solvent B, 1.95–2.0 min, 100 to 0% solvent B. All solvents were Optima Grade (Fisher Scientific).
Enzymatic activity calculation.
ARSA activity in leukocyte (nmol/h/mg protein) was calculated by multiplying the ion ratio of ARSA product to ARSA internal standard (blank subtracted) by the nanomoles of internal standard added to the assay, then dividing by the incubation time (h) and the amount of leukocyte protein used (mg). ARSA activity in DBS (μM/h) was calculated by multiplying the ion ratio of ARSA product to ARSA internal standard (blank subtracted) by the μmoles of internal standard added to the assay, then dividing by the incubation time (h) and the volume of the blood (L), assuming each 3 mm DBS punch contained 3.2 μL blood. The product-to-internal standard MRM response ratio was assumed to be unity.
RESULTS
Development of a new ARSA leukocyte assay.
Recently, Han et al. reported the detection of ARSA enzymatic activity where a natural substrate, C18:0-sulfatide, was incubated with leukocyte lysate in buffer containing sodium taurodeoxycholate as detergent8. In this study we used the deuterium labeled sulfatide d3-C18:0-sulfatide as substrate so that the enzymatic product (d3-C18:0-galactosyl-ceramide) also carried the three deuterium labels, and could be selectively quantified without being interfered by the endogenous galactosyl-ceramide. Using this approach, we were able to detect ARSA enzymatic activity in lysates of mixed leukocytes (mainly lymphocytes) by LC-MS/MS. As shown in Figure 1, the de-sulfated product peak eluted at 1.07 min, and was fully separated from the peak of the sulfated substrate eluting at 1.5 min. If leukocyte lysate was omitted, no discernable product peak was observed above the baseline noise (data not shown). The ARSA activity showed a sigmoidal dependence on the detergent concentration (Figure S1), and a concentration of 2.0 g/L sodium taurodeoxycholate gave maximal activity and was used in all of the following studies. Since detergent was present in the assay buffer, MLD patients that are due to dysfunctional Saposin B protein will not be detected with this assay, but these variants are very rare among MLD patients (less than 5%)9. In the previous report, MnCl2 was added to the buffer at a concentration of 33 mM8. This was based on an earlier paper suggesting that this metal ion lowers the critical micellar concentration of the inhibitory ionic form of the bile salt and allows the formation of mixed micelles of taurodeoxycholate and sulfatide10. However, as shown in Figure S2a, we found out that ARSA activity decreased with increasing amount of MnCl2 in assay buffer, thus MnCl2was omitted from the final assay condition. Barium or cerium sulfates are often used to assay sulfatases in complex matrices to sequester inorganic sulfate and phosphate ions, which are potent inhibitors of sulfatases11. However, ARSA activity was found to decrease as the concentration of Ce(Acetate)3 was increased from 0 to 20 mM (Figure S2b), therefore this salt was omitted from the assay. The ARSA activity was pH dependent with an optimal pH at 4.5 (Figure S3). The enzymatic rate displayed hyperbolic kinetics as the substrate concentration varied, giving a KM of 83 μM (Figure S4). The pH optimum of ARSA and the KM value found in our study agreed with the previous reports8, 12. The reaction progress curve shows a falloff from linearity over 20 hours (Figure S5). Even though the progress curve is not linear, the amount of enzymatic product formed after 16 hours of incubation increased linearly with the amount of leukocyte protein used (Figure 2).
Figure 1.
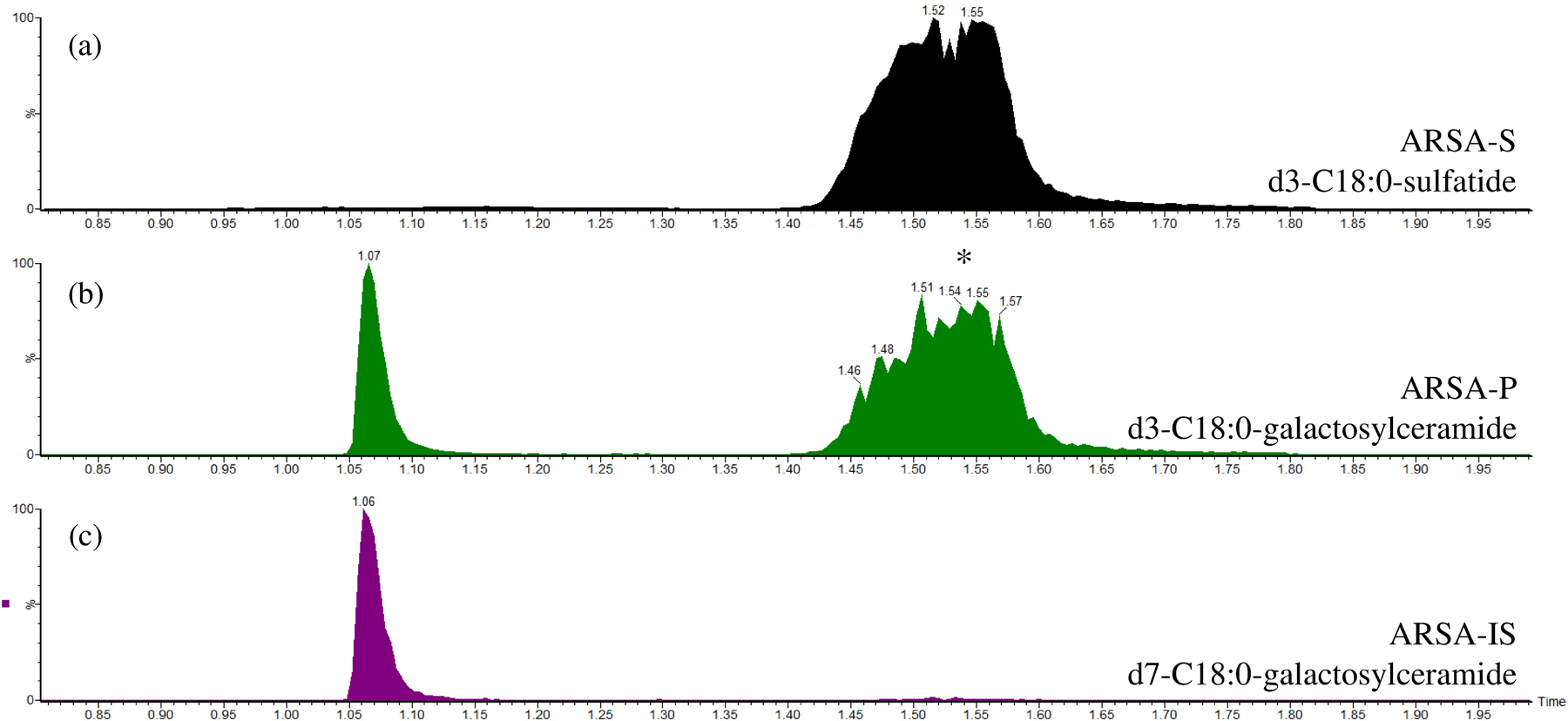
LC-MS/MS chromatography of the (a) ARSA substrate; (b) ARSA enzymatic product; and (c) ARSA internal standard channel. The asterisk in the ARSA enzymatic product channel was from substrate breakdown in the heated ESI source. The x-axis is time (min) and the y-axis is ion counts in the MRM channel after being normalized to the maximum signal (100%).
Figure 2.
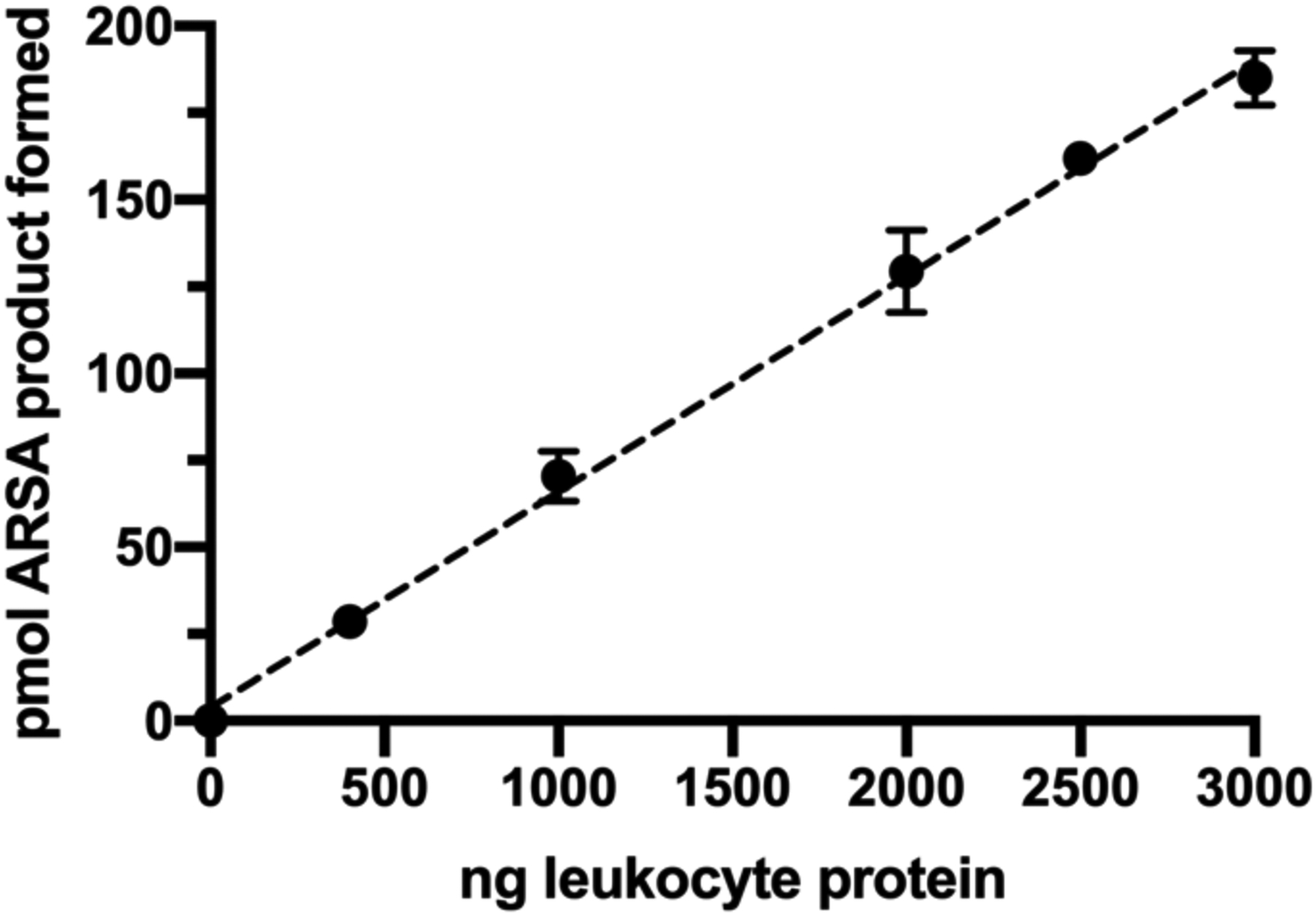
The amount of ARSA enzymatic product formed as a function of the amount of protein in the leukocyte lysate used. Error bars are standard deviations based on triplicate measurement.
We studied the stability of ARSA in whole blood under optimized assay conditions. K2EDTA-treated venous blood was kept at room temperature or 4 °C for up to 4 days prior to the preparation of leukocytes. As shown in Figure S6a, about 14% and 32% of the ARSA activity was lost over the first 24 hours when the whole blood was stored at 4 °C and room temperature, respectively. The ARSA activity continued to decrease with blood storage time under both conditions, with approximately 45% left after 4 days. We thus recommend that whole blood be shipped overnight at 4 °C followed by immediate leukocyte preparation and freezing of the cell pellet. ARSA activity decreased only slightly with the number of freeze/thaw cycles of the mixed leukocytes, with about 20% activity lost after 5 cycles (Figure S6b).
To study the minimal residual ARSA activity that can be detected by this LC-MS/MS assay, various amount of lysate from ARSA-containing lymphoblasts (Coriell Institute Repository, GM14603) was mixed with lysate from ARSA-deficient lymphoblasts (GM23097) (2 μg protein total). With 100% ARSA-deficient cells lysate, no ARSA activity was observed (Figure 3). ARSA activity increased proportionally with the percentage of ARSA-containing cells lysate in the mixture. From the inset of Figure 3, it was clear that as little as 0.1% residual ARSA activity could be detected above baseline with statistical significance.
Figure 3.
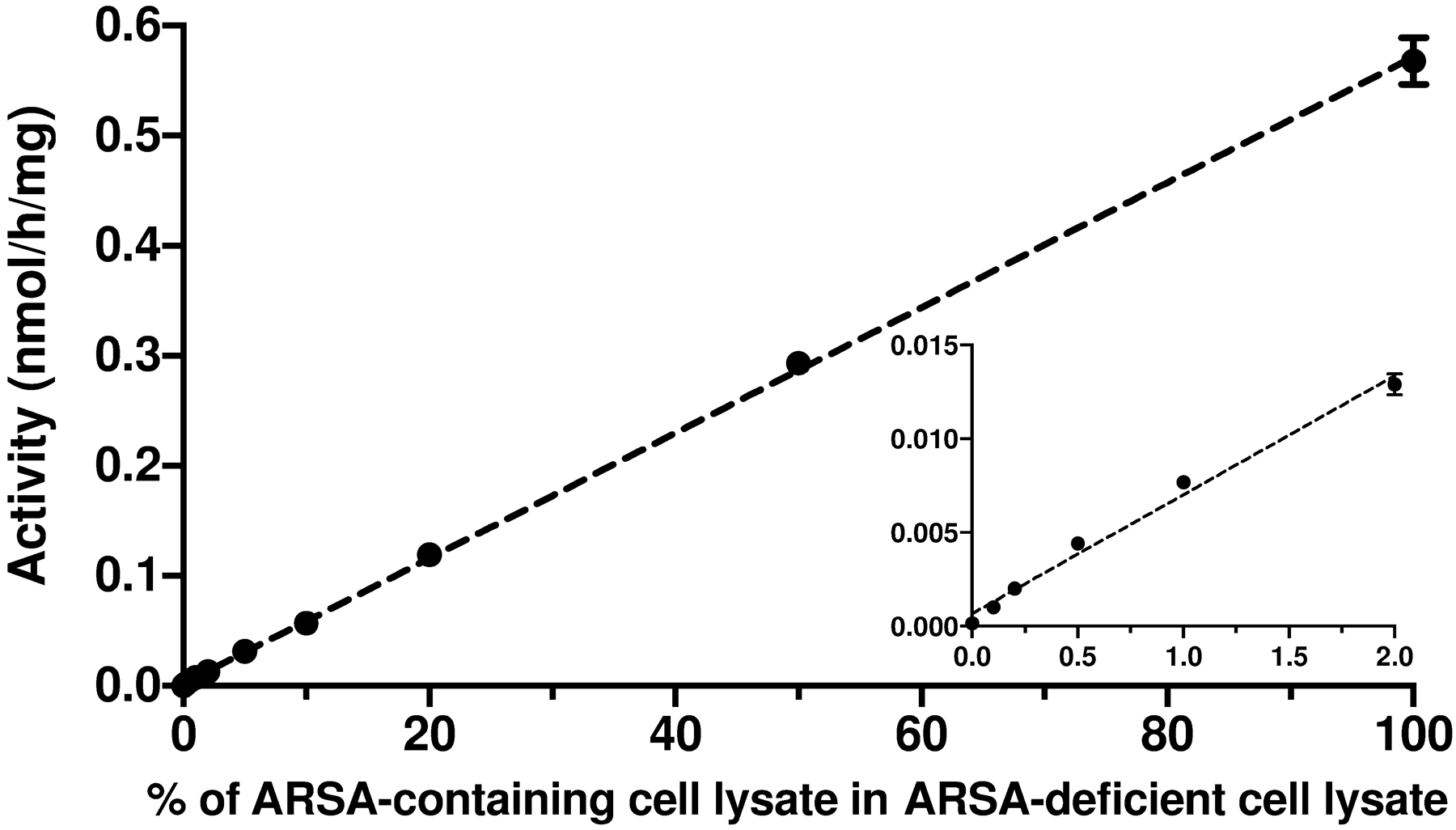
ARSA activity as a function of the fraction of ARSA-containing lymphoblast lysate (GM14603) added to ARSA-deficient lymphoblast lysate (GM23097). A total of 2 μg protein was used per assay. Error bars are standard deviations based on the triplicate measurements. The insert is an expansion of the plot at the lower end.
ARSA activity in leukocyte lysates from MLD and MSD patients.
Table 1 shows the ARSA activity in leukocyte lysates from 22 MLD patients and 1 MSD patient. Genotype information and age of onset (categorized by gross motor or cognitive symptoms), if available, are also provided. ARSA activity in leukocyte lysate was expressed as nmol/h/mg protein or as a percentage of the activity measured in the leukocyte lysate from a single healthy adult donor. MLD patient 21 and 22 were identified through their affected siblings, and were asymptomatic at the time of sampling, therefore their ages of disease onset were unavailable. MLD patient 5 also had Type I Gaucher disease, therefore the MLD variants were not the only contributing factor to the age of onset. The most severe MLD patients (late-infantile onset) in our cohort displayed symptoms between 12–35 months, and had residual ARSA activity in the range of 0.03–5.8% compared to the healthy adult. MLD patient 13 and 14, two late infantile patients, displayed higher residual ARSA activity (4.0% and 5.8%) than the rest of the late-infantile patient cohort. Four MLD patients with juvenile onset (MLD patient 15–19) had residual ARSA activity below 0.4% when compared to the healthy adult. One MLD patient (MLD patient 20) showed initial symptoms at 100 months and displayed 0.18% residual ARSA activity. Therefore, with this limited dataset, there appeared to be no correlation between residual ARSA activity in leukocyte lysate and the age of disease onset. The patient genotypes were not predictive of the age of disease onset as well. For example, MLD patient 7 and 15 had the same genotype (c.459+1G>A//c.1277C>T) and similar residual leukocyte ARSA activity (0.20% and 0.17% of the healthy adult), yet their age of onset was 45 months apart.
Table 1.
Summary of ARSA activity in leukocyte lysates from 22 MLD patients and 1 MSD patient1.
| Genotype | age of onset | ARSA activity (nmol/h/mg protein) | % to adult control | Note | |
|---|---|---|---|---|---|
| MLD patient 1 | late infantile | 0.0011 ± 0.0005 | 0.03 | ||
| MLD patient 2 | c.370G>A+c*96A>G//c.685–1G>A+c.1055A>G | late infantile | 0.0071 ± 0.0002 | 0.18 | |
| MLD patient 3 | c.465+1G>A//c.1108–3C>G | late infantile | 0.0055 ± 0.0007 | 0.14 | |
| MLD patient 4 | c.449C>T//c.474C>A | 12 month | 0.0067 ± 0.0002 | 0.17 | |
| MLD patient 5 | c.178C>T//c.178C>T | 13 month | 0.0081 ± 0.0007 | 0.21 | patient also has Gaucher |
| MLD patient 6 | 14 month | 0.0055 ± 0.0031 | 0.14 | ||
| MLD patient 7 | c.459+1G>A//c.1277C>T | 15 month | 0.0064 ± 0.0002 | 0.17 | |
| MLD patient 8 | 18 month | 0.0198 ± 0.0018 | 0.52 | ||
| MLD patient 9 | 18 month | 0.0056 ± 0.0011 | 0.15 | ||
| MLD patient 10 | 18 month | 0.0052 ± 0.0022 | 0.14 | ||
| MLD patient 11 | p.E198A199del//c.848+1G>A | 25 month | 0.0078 ± 0.0018 | 0.20 | |
| MLD patient 12 | c.1175G>A//c.442G>A | 29 month | 0.0128 ± 0.0003 | 0.33 | |
| MLD patient 13 | c.136T>C//c.115G>A | 30 month | 0.1533 ± 0.0108 | 3.99 | |
| MLD patient 14 | c.1144G>A//c.459 + 1G>A | 35 month | 0.2212 ± 0.0142 | 5.75 | |
| MLD patient 15 | c.459+1G>A//c.1277C>T | 60 month | 0.0078 ± 0.0011 | 0.20 | |
| MLD patient 16 | c.465+1G>A//c.869G>A | 60 month | 0.0139 ± 0.0005 | 0.36 | |
| MLD patient 17 | c.931G>A//c.931G>A | early juvenile | 0.0040 ± 0.0005 | 0.10 | |
| MLD patient 18 | c.465+1 G>A//c.869 G>A | early juvenile | 0.0057 ± 0.0006 | 0.15 | |
| MLD patient 19 | c.1136C>T//c.1136C>T | early juvenile | 0.0063 ± 0.0013 | 0.16 | |
| MLD patient 20 | 100 month | 0.0068 ± 0.0018 | 0.18 | ||
| MLD patient 21 | c.459+1G>A//c.1277C>T | 0.008 ± 0.0004 | 0.21 | ||
| MLD patient 22 | c.459+1G>A//c.1277C>T | 0.0067 ± 0.0002 | 0.17 | ||
| MSD patient 1 | 18 month | 0.0046 ± 0.0015 | 0.12 | ||
| Adult control | 3.8438 ± 0.0971 |
ARSA activity was measured in triplicate on three aliquots from the same batch of leukocyte lysate.
An additional multiplexed assay was used to measure the activity of iduronate-2-sulfatase (I2S), N-acetylgalactosamine-6-sulfatase (GALNS), N-acetylgalactosamine-4-sulfatase (ARSB), α-N-acetylglucosaminidase (NAGLU), and lysosomal β-glucuronidase (GUSB) in these leukocyte lysates containing 2 μg protein using a previously published protocol13. All MLD patients had activities above 50% when compared to the healthy adult donor, whereas the MSD patient had sulfatase (I2S, GALNS and ARSB) activities below 2%, consistent with the diagnosis of MSD (data not shown).
Application of ARSA assay to DBS extracts purified by immune-precipitation.
Initial attempts to detect ARSA enzymatic activity in DBS were unsuccessful. When the leukocyte lysate was substituted with a 3 mm DBS punch in the ARSA assay, no ARSA activity could be detected. Moreover, no ARSA activity was observed when leukocyte lysate was co-incubated with a DBS punch, whereas activity was seen when the leukocyte was incubated with a filter paper punch lacking blood matrix (data not shown). This indicated the presence of some ARSA inhibitor(s) in whole blood. Addition of 5 mM Ce(Acetate)3 or Pd(Acetate)2 to the assay buffer did not rescue ARSA activity in DBS (data not shown), suggesting that the inhibitor(s) in whole blood were not inorganic sulfate or phosphate ions alone.
In an earlier study, Tan et al. were able to detect ARSA enzymatic activity in DBS after immuno-precipitating the ARSA protein in the DBS extract and the activity was then measured by the generic fluorogenic substrate for sulfatases, 4-methylumbelliferyl sulfate14. Inspired by their work, we purified the blood extract from DBS by immuno-precipitation, and the ARSA activity was readily detected by LC-MS/MS using sulfatide as substrate (Figure 4). This is consistent with the previous report and indicated that the ARSA inhibitor(s) from whole blood were removed during the immuno-precipitation. However, we tested six other commercially available anti-ARSA serums, including one polyclonal and five monoclonal, but no ARSA activity could be recovered after immuno-precipitation (Figure S7).
Figure 4.
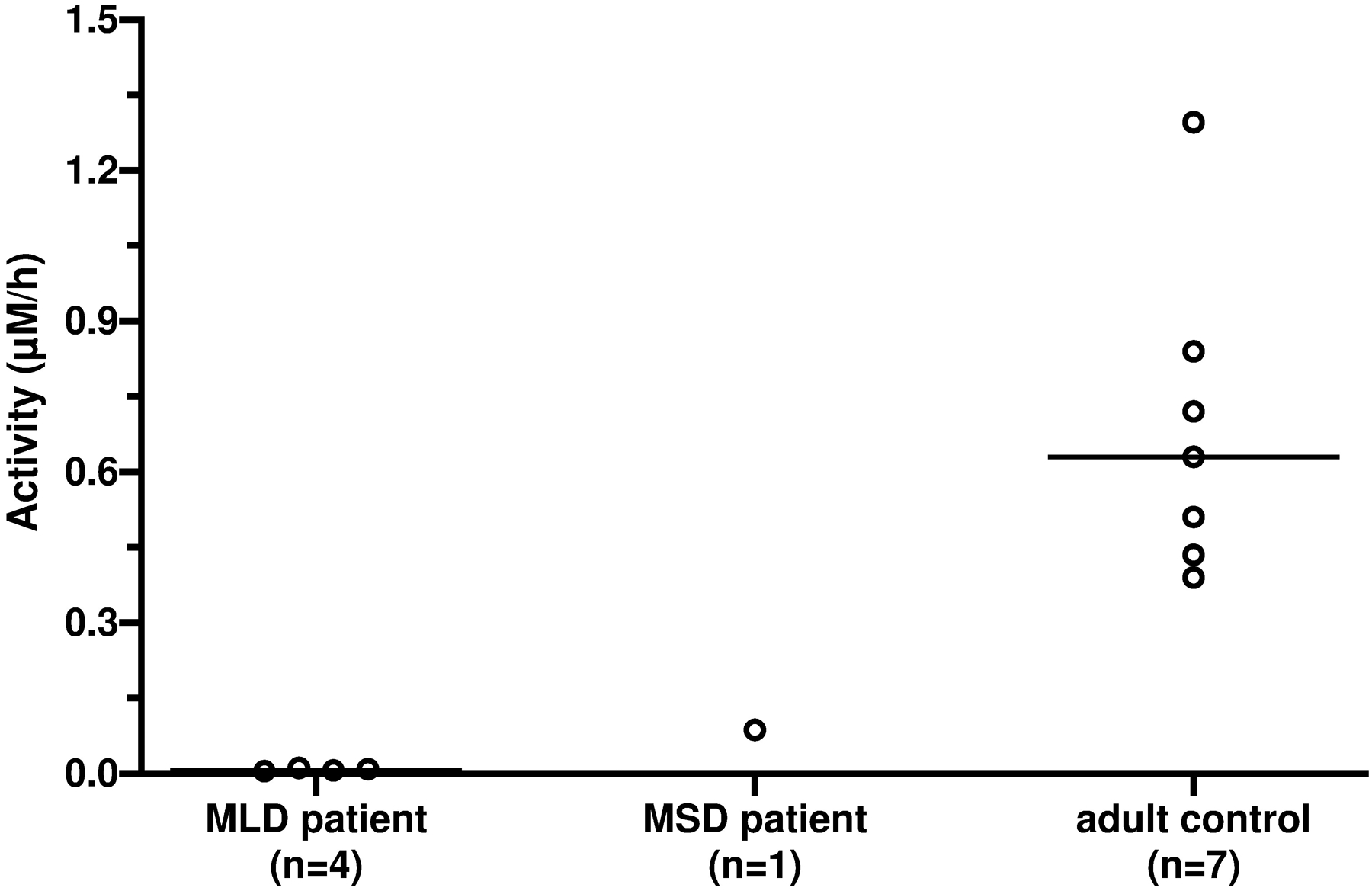
ARSA activity in DBS from 4 MLD patients (median: 0.007 μM/h, range: 0.005–0.011 μM/h), 1 MSD patient (0.087 μM/h), and 7 healthy adults (0.63 μM/h, range: 0.39–1.30 μM/h), after immuno-precipitation purification. The horizontal bar indicates the median of each group.
Figure 4 shows the ARSA enzymatic activity in DBS from 4 MLD patients (median: 0.007 μM/h, range: 0.005–0.011 μM/h), 1 MSD patient (0.087 μM/h), and 7 healthy adults (0.63 μM/h, range: 0.39–1.30 μM/h) after the DBS extract was purified by immune-precipitation. All MLD patients had barely detectable ARSA activity, and the MSD patient displayed 14% ARSA activity when compared to the mean activity of the healthy adults, indicating these patients had essentially no residual ARSA activity.
We then used the ARSA DBS assay with immuno-precipitation to investigate the stability of this enzyme in DBS (Figure S8). When stored at room temperature with desiccants, ARSA activity dropped to 95% after 7 days and to 60% after 14 days. Interestingly, additional activity loss was minimal over the next 100 days. ARSA activity remained stable in DBS when stored at 4 °C or −20 °C with desiccants for over 3 months (Figure S8). These results suggested that DBS should not be kept at room temperature over an extended period.
Application of ARSA assay to DBS extracts purified by size-exclusion chromatography.
Given that the ARSA activity in DBS could only be measured after immuno-precipitation using a commercially available polyclonal antibody, we sought alternative purification methods that do not rely on a reagent in limited quantity. Initial attempts were based on ion exchange chromatography as it was previously reported for the purification of recombinantly expressed ARSA15. However, no ARSA activity was recovered in the DBS extract purified by either cation or anion exchange chromatography (data not shown), probably because NaCl at high concentration is a strong inhibitor of ARSA16. We then pursued size-exclusion chromatography to purify ARSA from the DBS matrix. As shown in Figure S9, ARSA activity was readily detected after DBS extract was purified with resins of various MW cutoff. The low-cost Sephadex G-25 resin with a MW cutoff of 5 k Da gave optimal result, and was used in all the following studies.
Next, we optimized the extraction protocol of ARSA from the DBS punch. It was found that the color of the extract from poorly stored DBS punch was pale compared to extract from DBS stored under optimal conditions, and the ARSA activity measured in such pale extract was substantially lower, suggesting that not all of the protein was extracted from the punch. To improve the protein recovery, different extraction buffers were tested, including ARSA buffer, ammonium hydroxide in water and ammonium hydroxide in ARSA buffer, with a 50% increase in ARSA recovery when 0.5% ammonium hydroxide was in the ARSA buffer (Figure S10). The use of ammonium hydroxide was based on an earlier study showing improved extraction of proteins from DBS using this additive17. Further optimization of the amount of ammonium hydroxide in the ARSA buffer gave the optimal DBS extraction buffer (0.8 % ammonium hydroxide in ARSA buffer) (Figure S11a). The extraction time was optimized, and 4 hours at room temperature was chosen for optimal extraction (Figure S11b). We also varied the amount of resin used per assay for the size-exclusion chromatography, and found that 60 mg dry resin per assay gave better consistency compared to 40 mg per assay, although at a cost of 20% loss in activity (Figure S12).
With the final ARSA DBS assay in hand, we validated the variation in ARSA activity across a series of Quality Control DBS for lysosomal storage disorders from the CDC (base pool (0.023 ± 0.003 μM/h), low (0.048 ± 0.007 μM/h), medium (0.21 ± 0.02 μM/h), and high (0.37 ± 0.02 μM/h) standards) (Figure 5). These QC samples were prepared by mixing various amounts of unprocessed cord blood with base pool blood (prepared from leukocyte-reduced blood and heat-inactivated, charcoal-stripped serum)18. Figure 5 demonstrated the assay had good linear response (R2 > 0.99) and good reproducibility (< 15% CV) with 20 replicates at each point. It should be noted that there was finite ARSA activity in the base pool sample (non-zero intercept of Figure 5) after blank subtraction, showing that not all of the ARSA enzyme was depleted in the base pool DBS.
Figure 5.
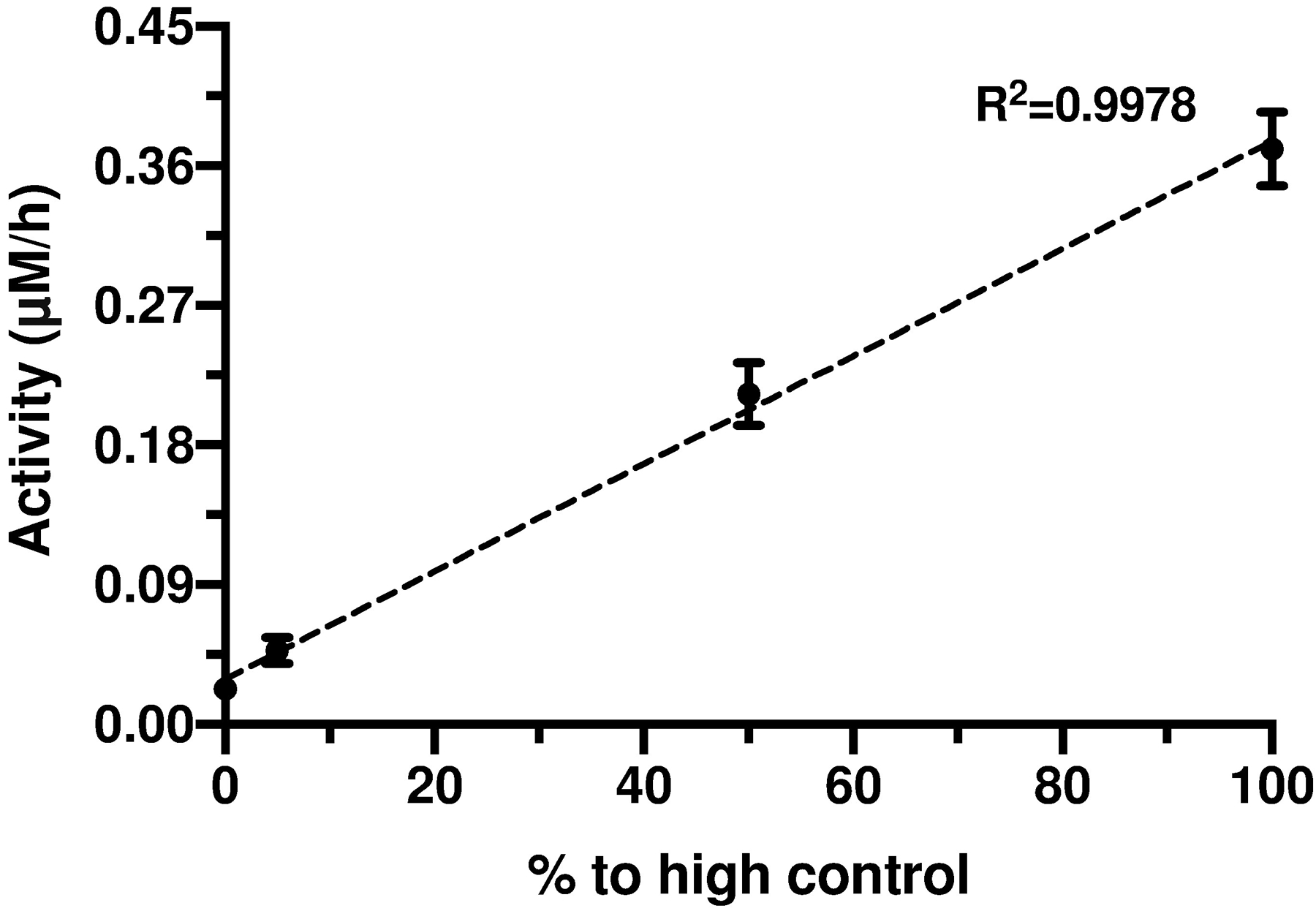
ARSA activity after size-exclusion chromatography purification in CDC Quality Control DBS samples, including base pool (0.023 ± 0.003 μM/h), low (0.048 ± 0.007 μM/h), medium (0.21 ± 0.02 μM/h), and high (0.37 ± 0.02 μM/h) controls, each representing 0, 5%, 50% and 100% to high control, respectively. Each point was measured in 20 replicates. Error bars were standard deviations based on the 20 measurements.
Having fully optimized and validated the ARSA DBS assay, we measured ARSA activity in DBS from 34 MLD patients (median: 0.0015 μM/h, range: 0–0.18 μM/h), 3 MSD patients (median: 0.032 μM/h, range: 0.028–0.076 μM/h), 10 healthy adults (median: 0.80 μM/h, range: 0.45–1.3 μM/h), and 294 presumed random newborns (median: 0.27 μM/h, range: 0.082–0.65 μM/h) (Figure 6a). The two MLD patients that had the highest ARSA DBS activity (0.11 and 0.18 μM/h) were MLD patient 13 and 14 (Table 1), respectively. It should be noted that the random newborn DBS were stored for 1–2 months at room temperature. Presumably about 40% of the ARSA activity had been lost in these samples (based on DBS stability data, Figure S8), thus the range of activities in random newborns showed in Figure 6a was lower than what would be expected in fresh newborn DBS. The patients and normal adult DBS were stored at −20 °C shortly after DBS collection with presumably minimal loss of ARSA activity. Since it was extremely hard to obtain large amount of fresh newborn DBS, an aging experiment was carried out as a compromise. When fresh DBS from 18 MLD (including DBS from MLD patient 13 and 14) and 2 MSD patients were aged at room temperature for 1 month, over 50% of the residual ARSA activity in DBS from MLD patient 13 and 14 was lost. The result demonstrated that patients can be completely distinguished from the normal (random newborns) based on the ARSA DBS activity if they had similar storage condition (Figure 6b).
Figure 6.
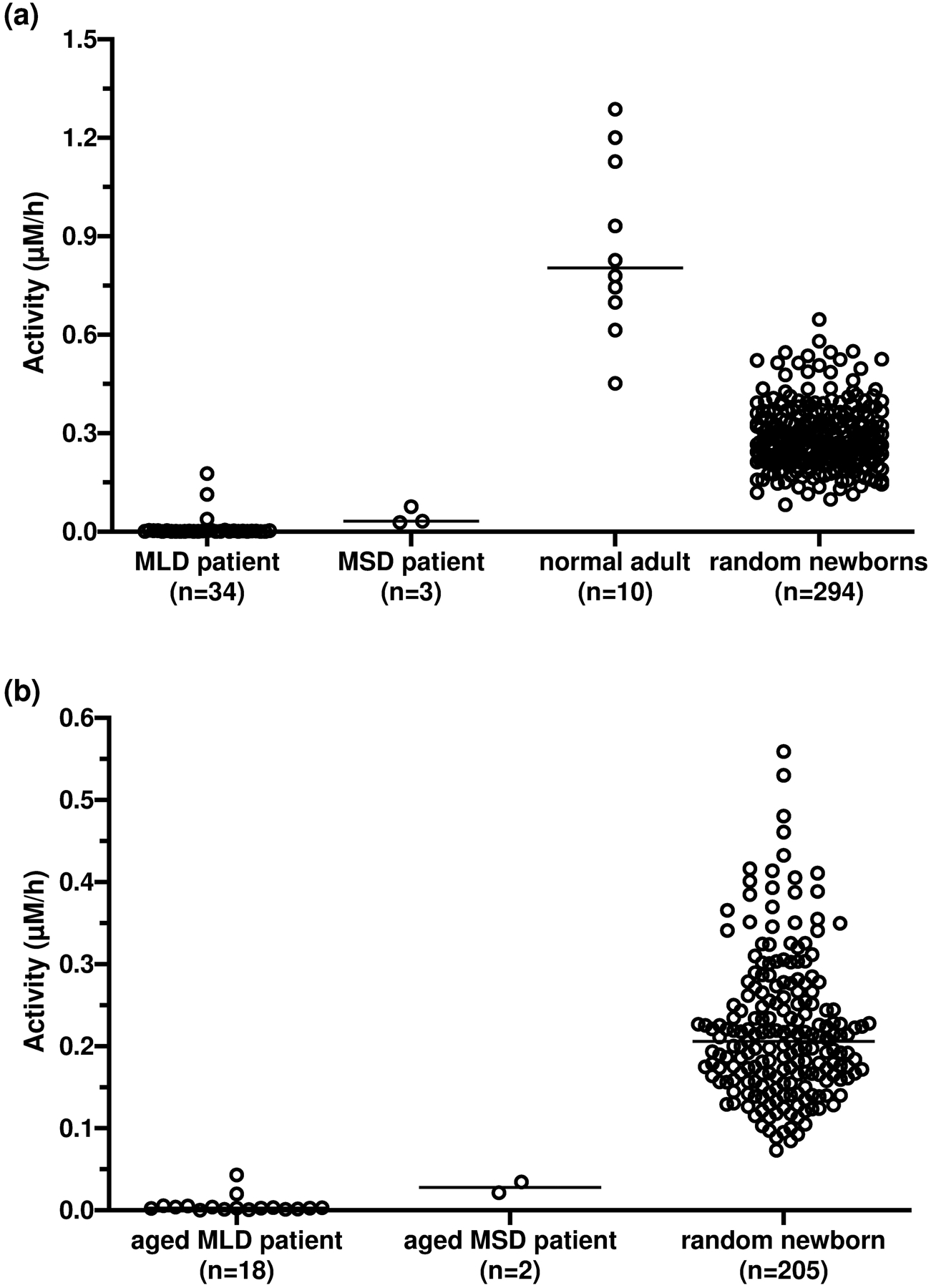
(a) ARSA activity after size-exclusion chromatography purification in DBS from 34 MLD patients (median: 0.0015 μM/h, range: 0–0.18 μM/h), 3 MSD patients (median: 0.032 μM/h, range: 0.028–0.076 μM/h), 10 healthy adults (median: 0.80 μM/h, range: 0.45–1.3 μM/h) and 294 random newborns (median: 0.27 μM/h, range: 0.082–0.65 μM/h). Fresh DBS from patients and healthy adults were used. (b)ARSA activity in aged DBS (stored at room temperature for 1 month before processing) from 18 MLD patients (median: 0.003 μM/h, 0–0.043 μM/h), 2 MSD patients (0.021 and 0.035 μM/h), and 205 random newborns (median: 0.21 μM/h, 0.073–0.56 μM/h. The horizontal bar indicates the median of each group.
DISCUSSION
Recently, Han et al. reported an ARSA leukocytes assay similar to the one reported in the current study8. They used non-deuterated C18:0-sulfatide as substrate and commercially available d35-C18:0-galactosyl-ceramide as internal standard8. In this study, we used deuterium labeled sulfatide as the enzymatic substrate so that the deuterated product can be differentiated from the endogenous galactosyl-ceramide. Moreover, d35-C18:0-galactosyl-ceramide does not co-elute with the enzymatic product due to the 35 deuterium labels, therefore in this study we used the in-house synthesized d7-C18:0-galactosyl-ceramide that co-eluted with the enzymatic product as the internal standard. Co-elution of the analyte and its internal standard is important for LC-MS/MS analysis as there could be different matrix effects on analytes eluting at different retention time. Surprisingly, the ARSA activity in leukocyte lysate from healthy adults reported by Han et al. were approximately 100-fold higher than those measured in the current study. We speculated that the enzymatic product measured by Han et al. was a combination of enzymatic breakdown of sulfatide and the galactosyl-ceramide present in the sample. We also note that the activities reported by Han et al. were in general 30 to 100-fold higher than the activities of other lysosomal enzymes measured in leukocytes7, 19, 20.
The LC-MS/MS ARSA assay developed here is expected to be more accurate and precise than the traditional colorimetric and fluorometric ARSA assays, where generic artificial sulfatase substrates were used21, 22. Since these generic substrates are not specific to ARSA, these assays either required two assays performed in parallel, one with an ARSA specific inhibitor and one without, or required the removal of isoenzymes by ion-exchange chromatography21, 22. Inhibition of ARSA is only partial in these earlier methods as the inhibitors used are neither highly potent, nor ARSA-specific, which compromises the reliability of the assay, especially at the lower end. Usage of assay buffer that favors ARSA activity has been reported, but it is not clear if the residual activity is due to ARSA, or other off-target sulfatases, or both. This is a serious concern as proper evaluation of trace amounts of residual ARSA activity is crucial when diagnosing potential patients. Recently, a new spectrophotometric ARSA assay was developed using sulfatide as substrate12. However, this assay quantified the enzymatic activity by measuring the decrease of substrate instead of the formation of product, therefore it will not be accurate to measure trace amounts of residual ARSA activity as well. It is also possible to use generic sulfatase substrates to selectively assay ARSA after immuno-precipitation purification14, but this requires a polyclonal antiserum that is available in limited quantities, and also relies on the assumption that no off-target sulfatases are being pulled down during the process. The current LC-MS/MS-based ARSA assay was highly ARSA-specific due to the usage of the natural substrate and was highly precise and sensitive as trace amounts of residual activity could be detected with statistical significance (Figure 3). It should be noted that the methods described in this study are for research only and do not meet The Clinical Laboratory Improvement Amendments (CLIA) validation guidelines for the development of laboratory developed tests (LDTs). Furthermore, studies of a larger cohort of patients and normal controls are needed to define a reference range. The fluorometric and LC-MS/MS assays may both be adequate for diagnostic purposes, where measurement of nominally low enzymatic activity is potentially sufficient for diagnosis when the patient exhibits symptoms that are characteristic of the disease. However, due to its high performance, we believe this new ARSA leukocyte assay can be beneficial for evaluating potential patients who are identified by NBS and may be at risk to develop MLD but are so far asymptomatic. This is especially important in the case of MLD where there is a high frequency of pseudodeficiency variants14.
We were able to detect ARSA activity in DBS only if the enzyme is purified or partially purified from the matrix. Immunoprecipitation of ARSA was useful in this context, but to date we have not been able to find a monoclonal antibody that works in the enzymatic assay. Reliance on a commercially-available polyclonal anti-ARSA antiserum is problematic for long-term sustainability of the assay. Fortunately, we found that a simple size-exclusion procedure was sufficient to remove the inhibitor(s) and make ARSA detectable in the DBS matrix. Furthermore, the protocol was simple and inexpensive to carry out. With automation, it is also appropriate for high throughput screening including NBS (see below).
Currently, the ARSA enzymatic activity assay in DBS is implemented as the second-tier test for our ongoing large-scale de-identified pilot study for the NBS of MLD, where the ARSA enzymatic assay is performed on samples with abnormal sulfatide results. With this two-tier algorithm, the false positive rate is largely reduced and so is the accompanying anxiety to families in a real-world scenario. It should be noted that the ARSA activity in newborns reported here cannot be used as a normal range as these DBS were stored at room temperature for 1–2 months prior to analysis, and ARSA is known to be unstable under this storage condition. Since the ARSA enzymatic assay in DBS is conducted in a high throughput manner with a turnaround time of 2 min per sample, we are also exploring the option of using this assay as the first-tier screening test for MLD.
CONCLUSIONS
In summary, a novel ARSA leukocyte and DBS assay by LC-MS/MS was developed. Both assays used deuterated natural sulfatide as substrate, therefore are highly specific to ARSA. This is essential for accurately diagnosing MLD and MSD patients. Implementation of this new ARSA DBS assay as a second-tier test is crucial for our on-going de-identified pilot study for the NBS of MLD. This high throughput assay may also serve as the first-tier screening test for MLD.
Supplementary Material
ACKNOWLEDGMENT
The work was funded by a grant from the National Institutes of Health, R01 DK067859.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
D7-C18:0-galactosylceramide synthesis, ESI source parameters, MRM parameters, assay optimization, stability test. (docx)
REFERENCES
- 1.Sessa M; Lorioli L; Fumagalli F; Acquati S; Redaelli D; Baldoli C; Canale S; Lopez ID; Morena F; Calabria A, Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. The Lancet 2016, 388 (10043), 476–487. [DOI] [PubMed] [Google Scholar]
- 2.Biffi A; Montini E; Lorioli L; Cesani M; Fumagalli F; Plati T; Baldoli C; Martino S; Calabria A; Canale S, Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013, 341 (6148), 1233158. [DOI] [PubMed] [Google Scholar]
- 3.Spacil Z; Babu Kumar A; Liao HC; Auray-Blais C; Stark S; Suhr TR; Scott CR; Turecek F; Gelb MH, Sulfatide Analysis by Mass Spectrometry for Screening of Metachromatic Leukodystrophy in Dried Blood and Urine Samples. Clin Chem 2016, 62 (1), 279–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lorioli L; Cesani M; Regis S; Morena F; Grossi S; Fumagalli F; Acquati S; Redaelli D; Pini A; Sessa M; Martino S; Filocamo M; Biffi A, Critical issues for the proper diagnosis of Metachromatic Leukodystrophy. Gene 2014, 537 (2), 348–51. [DOI] [PubMed] [Google Scholar]
- 5.Doherty K; Frazier SB; Clark M; Childers A; Pruthi S; Wenger DA; Duis J, A closer look at ARSA activity in a patient with metachromatic leukodystrophy. Mol Genet Metab Rep 2019, 19, 100460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dierks T; Schmidt B; Borissenko LV; Peng J; Preusser A; Mariappan M; von Figura K, Multiple sulfatase deficiency is caused by mutations in the gene encoding the human Cα-formylglycine generating enzyme. Cell 2003, 113 (4), 435–444. [DOI] [PubMed] [Google Scholar]
- 7.Lin N; Huang J; Violante S; Orsini JJ; Caggana M; Hughes EE; Stevens C; DiAntonio L; Chieh Liao H; Hong X; Ghomashchi F; Babu Kumar A; Zhou H; Kornreich R; Wasserstein M; Gelb MH; Yu C, Liquid Chromatography-Tandem Mass Spectrometry Assay of Leukocyte Acid alpha-Glucosidase for Post-Newborn Screening Evaluation of Pompe Disease. Clin Chem 2017, 63 (4), 842–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han M; Jun SH; Song SH; Park HD; Park KU; Song J, Ultra-performance liquid chromatography-tandem mass spectrometry measurement of leukocyte arylsulfatase A activity using a natural substrate. Ann Lab Med 2015, 35 (1), 165–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cesani M; Lorioli L; Grossi S; Amico G; Fumagalli F; Spiga I; Filocamo M; Biffi A, Mutation Update of ARSA and PSAP Genes Causing Metachromatic Leukodystrophy. Hum Mutat 2016, 37 (1), 16–27. [DOI] [PubMed] [Google Scholar]
- 10.Jerfy A; Roy A, The sulphatase of ox liver. XVI. A comparison of the arylsulphatase and cerebroside sulphatase activities of sulphatase A. Biochimica et Biophysica Acta (BBA)-Enzymology 1973, 293 (1), 178–190. [DOI] [PubMed] [Google Scholar]
- 11.Fluharty AL; Edmond J, [58] Arylsulfatases A and B from human liver In Methods in enzymology, Elsevier: 1978; Vol. 50, pp 537–547. [DOI] [PubMed] [Google Scholar]
- 12.Morena F; di Girolamo I; Emiliani C; Gritti A; Biffi A; Martino S, A new analytical bench assay for the determination of arylsulfatase a activity toward galactosyl-3-sulfate ceramide: implication for metachromatic leukodystrophy diagnosis. Anal Chem 2014, 86 (1), 473–81. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y; Yi F; Kumar AB; Chennamaneni NK; Hong X; Scott CR; Gelb MH; Turecek F, Multiplex Tandem Mass Spectrometry Enzymatic Activity Assay for Newborn Screening of the Mucopolysaccharidoses and Type 2 Neuronal Ceroid Lipofuscinosis. Clin Chem 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan MA; Dean CJ; Hopwood JJ; Meikle PJ, Diagnosis of metachromatic leukodystrophy by immune quantification of arylsulphatase A protein and activity in dried blood spots. Clinical chemistry 2008, 54 (11), 1925–1927. [DOI] [PubMed] [Google Scholar]
- 15.Martino S; Consiglio A; Cavalieri C; Tiribuzi R; Costanzi E; Severini GM; Emiliani C; Bordignon C; Orlacchio A, Expression and purification of a human, soluble Arylsulfatase A for Metachromatic Leukodystrophy enzyme replacement therapy. J Biotechnol 2005, 117 (3), 243–51. [DOI] [PubMed] [Google Scholar]
- 16.Matzner U; Breiden B; Schwarzmann G; Yaghootfam A; Fluharty AL; Hasilik A; Sandhoff K; Gieselmann V, Saposin B-dependent reconstitution of arylsulfatase A activity in vitro and in cell culture models of metachromatic leukodystrophy. J Biol Chem 2009, 284 (14), 9372–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borremans B, Ammonium improves elution of fixed dried blood spots without affecting immunofluorescence assay quality. Trop Med Int Health 2014, 19 (4), 413–6. [DOI] [PubMed] [Google Scholar]
- 18.De Jesus VR; Zhang XK; Keutzer J; Bodamer OA; Muhl A; Orsini JJ; Caggana M; Vogt RF; Hannon WH, Development and evaluation of quality control dried blood spot materials in newborn screening for lysosomal storage disorders. Clin Chem 2009, 55 (1), 158–64. [DOI] [PubMed] [Google Scholar]
- 19.Liao HC; Spacil Z; Ghomashchi F; Escolar ML; Kurtzberg J; Orsini JJ; Turecek F; Scott CR; Gelb MH, Lymphocyte Galactocerebrosidase Activity by LC-MS/MS for Post-Newborn Screening Evaluation of Krabbe Disease. Clin Chem 2017, 63 (8), 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wenger DA; Sattler M; Clark C; McKelvey H, An improved method for the identification of patients and carriers of Krabbe’s disease. Clinica Chimica Acta 1974, 56 (2), 199–206. [DOI] [PubMed] [Google Scholar]
- 21.Chang PL; Rosa NE; Davidson RG, Differential assay of arylsulfatase A and B activities: a sensitive method for cultured human cells. Analytical biochemistry 1981, 117 (2), 382–389. [DOI] [PubMed] [Google Scholar]
- 22.Bostick W; Dinsmore S; Mrochek J; Waalkes T, Separation and analysis of arylsulfatase isoenzymes in body fluids of man. Clinical chemistry 1978, 24 (8), 1305–1316. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.