

Abstract
We report a specific and sensitive method to improve the coupling of propidium monoazide (PMA) with DNA derived from killed cells of Escherichia coli using UV light of 365 nm. UV light of three different intensities mainly 2.4 × 103, 4.8 × 103, and 7.2 × 103 μJ/cm2 was applied to E. coli cells each for 1, 3, and 5 min. PMA was found to be successfully cross-linked with the DNA from killed cells of E. coli at 4.8 × 103 μJ/cm2 in 3 min leading to the complete inhibition of PCR amplification of DNA derived from PMA-treated heat-killed cells. In spiked phosphate-buffered saline and potable water samples, the difference of the Cq values between PMA-treated viable cells and PMA-untreated viable cells ranged from −0.17 to 0.2, demonstrating that UV-induced PMA activation had a negligible effect on viable cells. In contrast, the difference of the Cq values between PMA-treated heat-killed cells and PMA-untreated heat-killed cells ranged from 8.9 to 9.99, indicating the ability of PMA to inhibit PCR amplification of DNA derived from killed cells to an equivalent as low as 100 CFU. In conclusion, this UV-coupled PMA-qPCR assay provided a rapid and sensitive methodology to selectively detect viable E. coli cells in spiked water samples within 4 h.
Electronic supplementary material
The online version of this article (10.1007/s42770-019-00161-8) contains supplementary material, which is available to authorized users.
Keywords: Viable, qPCR, Ultraviolet, Escherichia coli, Monitoring, Propidium monoazide
Water must be free from pathogens to become reliable for safe drinking purposes. Public health protection requires an immediate evaluation of microorganisms in drinking water to prevent outbreaks of microbial contamination. Children under the age of 5 years are more vulnerable to diarrheal diseases with about 800 child deaths per day [1, 2]. Escherichia coli is widely recognized not only as the indicator of fecal contamination of water resources but also as pathogen [3, 4]. However, culture-based methods and biochemical assays used for isolation and identification of E. coli strains require 72–96 h while giving little information about the type of the strain or the isolate. In order to prevent future outbreaks, it is necessary to develop rapid yet specific methods for the detection of E. coli. Thus, quantitative PCR (qPCR) has been effectively employed to overcome the shortcomings associated with culture-based methods mainly in terms of selectivity, specificity, and rapidity [5, 6]. However, persistence of DNA from dead or membrane-compromised cells in PCR reactions could cause false-positive results, hence, lead to overestimation of the viable cell counts in a water sample [7]. Such limitation actually restricts the effective use of PCR for determining microbiological safety of water samples. A propidium monoazide (PMA)-based method has been shown to selectively inhibit the PCR amplification of DNA derived from dead or membrane-compromised cells by treating samples with PMA before DNA extraction [8]. PMA can penetrate membrane-compromised cells and when exposed to an intense light source leads to covalent bond formation with the DNA strands [8]. This process inhibits subsequent PCR amplification [9]. Notably, PMA cannot penetrate viable cells with intact membranes; thus, only DNA from viable cells can be amplified by PCR [10]. This method has been successfully used for microbiological monitoring of pathogens in a number of previous studies [10–16]. Moreover, earlier studies employed high-wattage halogen lamps and LEDs for PMA light activation [8, 17].
In the present study, we have demonstrated PMA activation using UV light (365 nm). The CL-1000L UV Crosslinker (Analytik Jena, USA) is a multi-purpose ultraviolet exposure instrument (contains five mercury lamps) which utilizes a non-destructive 365 nm longwave UV radiation. Therefore, the purpose of the present study was (i) to improve the PMA treatment protocol using UV irradiation (365 nm) and (ii) to determine the effect of PMA on viable and heat-killed cells of E. coli in spiked potable water samples using the new protocol.
Escherichia coli MTCC 3221 cells were grown to logarithmic phase (optical density at 600 nm [OD600], 0.5 to 0.6) with constant shaking in 100 mL Luria Bertani (LB, HiMedia, Mumbai, India) broth at 37 °C. Aliquots of 1 mL of 0.5 OD600 (4 × 108 CFU/mL) viable cells of E. coli MTCC 3221 were added into autoclaved 1.5-mL microcentrifuge tubes and centrifuged at 11,000×g for 1 min to remove the media components. The pellet was washed twice with the sterile buffered saline to obtain final bacterial suspension of 400 μL. The bacterial cells contained in 400 μL aliquot were killed by using a boiling water bath for 5 min. Loss of viability of heat-killed bacterial cells was tested by spreading 400 μL of cell suspension on the LB (HiMedia) agar plates which yielded no growth of E. coli MTCC 3221 after incubation at 37 °C for 24 h. PMA treatment was performed according to Nocker et al. [9] with some modifications as described below. In brief, PMA (Biotium Inc., Hayward, CA, USA) was dissolved in 20% dimethyl sulfoxide (DMSO; Sigma-Aldrich, St. Louis, MO, USA) to obtain a stock concentration of 10 mM and stored at − 20 °C in the dark. An aliquot of 2.0 μL of 10 mM PMA was added to each 400 μL of viable cells, heat-killed cells, and mixtures of viable and killed cells to obtain a final concentration of 50 μM and incubated in dark for 10 min using opaque microcentrifuge tubes. Each PMA-treated cell suspension was then transferred to 5-mL sterile glass vials (Fisherbrand™, #03-338B, Indiana, PA, USA) in order to achieve maximum UV light treatment as mentioned below. PMA-treated cells were exposed to 365 nm UV light (8-W mercury lamp, #F8T5, HITACHI, Chiyoda, Tokyo, Japan) for 1, 3, and 5 min each at 2.4 × 103 microjoules per centimeter square (μJ/cm2), 4.8 × 103 μJ/cm2, and 7.2 × 103 μJ/cm2. UV treatment was given at a distance of 15 cm from the UV light source. To prevent the DNA degradation by UV irradiation, 5 mM MgCl2 (filter-sterilized) was added to killed and a mixture of killed and viable bacterial cells [18].
DNA from E. coli MTCC 3221 was extracted using a FastDNA™ Spin Kit (MP Biomedicals, Santa Ana, CA, USA) according to the manufacturer’s instructions. The concentration of DNA was determined by using a NanoDrop™ spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) which yielded 10.5 ng/μL with an A260/A280 ratio of 1.8. For PMA-qPCR, we used primers IEC-UP 5′-CAA TTTTCGTGTCCCCTTCG-3′ and IEC-DN 5′-GTTAATGATAGTGTGTCGAAAC-3′ [19] targeting distal and proximal conserved flanking regions of the 16S rRNA gene, the Internal Transcribed Spacer (ITS) region, and the 23S rRNA gene for detection of E. coli to amplify an expected size of 450 bp fragments. Oligonucleotide primers were synthesized by Integrated DNA Technologies (Coralville, IA, USA). The amplification reaction was carried out by using the FastStart Universal SYBR Green Master (Roche Diagnostics, Indianapolis, IN, USA) [6]. Five microliters of the template was transferred directly to a 15-μL PCR mixture containing 50 pmol/μL each of forward and reverse primer, 3.3 μg/μL BSA, 2× FastStart Universal SYBR Green Master, and the volume of PCR reaction mixture was adjusted using 2 μL of PCR grade water (Roche Diagnostics) [6]. In each experiment, 5 μL of PCR grade water (Roche Diagnostics) was added to PCR mixtures as a negative control. The qPCR mixtures were subjected to thermal cycling for 180 s at 95 °C and then 40 cycles of 30 s at 94 °C, 30 s at 58 °C, and 30 s at 72 °C. The amplification reaction was performed using the LightCycler® 96 Real-Time PCR system (Roche Diagnostics) in triplicate. The specificity of the assay was evaluated from melting profiles generated for the amplicons over a temperature range of 65 to 95 °C, where the melting temperature (Tm) was defined as the peak of fluorescence in the generated melting curves. The PCR products were further confirmed by 2% agarose gel electrophoresis and the gels were visualized on a UV transilluminator (E-Gel Imager System, Thermo Fisher Scientific). To evaluate the specificity of the amplification of the target gene, the amplicon obtained by qPCR was sequenced using the same set of primers at Eurofins Genomics (Bengaluru, India). The obtained sequences were analyzed using the BLAST software suite at the National Centre for Biotechnology Information (http://www.ncbi.nlm.nih.gov/blast) to determine the phylogenetic identity. A process control consisting of 500 Bacillus atrophaeus spores was incorporated in the qPCR reaction. The spores were prepared according to Picard et al. [20]. Primer sequence, F-5′-CACTTCATTTAGGCGACGATACT-3′ and R-5′-TTGTCTGTGAATCGGATCTTTCTC-3′, was used for detection of spores in qPCR reactions [20]. The standard curves were generated using purified DNA extracted from pure culture (OD600 = 0.5) of E. coli MTCC 3221 using dilutions in the range from 56 picograms to 0.007 picograms (1 × 103 CFU equivalents to 1 × 100 CFU equivalent), wherein the experiments were performed three times using three different DNA preparations. The Cq values for each set of reactions were plotted against log DNA quantities to obtain a standard curve.
For spiking experiments, E. coli MTCC 3221 was employed as a standard strain. The strain was grown to the logarithmic phase as described above. For spiking PBS (137 mM NaCl, 6.4 mM Na2HPO4, 2.7 mM KCl, 0.88 mM KH2PO4, pH 7.4), ten 400 μL aliquots were transferred each into 1.5-mL tubes and five were killed by heating as described above. Four each of the viable and heat-killed aliquots were spiked in 1 mL PBS. One heat-killed cell aliquot was plated onto LB agar which confirmed the complete loss of viability. For spiking potable water samples, ten potable water samples (total 1 L) were collected from Sancoale (Goa, India) in sterile screw-cap glass bottles and stored at 4–8 °C until used. Ten 400 μL aliquots of cell cultures were transferred into 1.5-mL tubes and five were killed by heating as described above. Four each of the viable and heat-killed aliquots were spiked in 1 mL double-autoclaved potable water samples. One heat-killed cell aliquot was spread plated on LB agar which confirmed the loss of cell viability of E. coli. Water samples were tested negative for the occurrence of naked environmental DNA prior to the spiking experiments. Each spiking experiment was performed in triplicate. B. atrophaeus spores were included in PMA-treated cell suspensions after the UV treatment to monitor the inhibition of PCR amplification by any inhibitory factors from the water samples. PMA treatment, UV irradiation, and DNA purification were performed as described above.
The statistical significance of differences in Cq values was assessed using a one-way analysis of variance (ANOVA) and a post hoc Tukey-Kramer multiple comparison test. All individual results were recorded and statistical significance was calculated using Microsoft Excel 2007 software (Microsoft Corporation; Redmond, WA, USA). To evaluate the effect of PMA on qPCR-amplifiable DNA from viable and killed cell samples, ΔCq (Cq value for PMA-treated cells − Cq value for untreated cells) values were calculated. When the (two-sided) p value was less than 0.05, we concluded that there was a significant difference between PMA treatment of viable and killed cells using UV radiation.
A novel strategy has been developed for real-time differentiation of DNA from viable and dead or membrane-compromised cells using PMA treatment as described previously [8]. This method involves the treatment of cells with PMA and subsequent PMA activation using high-intensity light. Many PMA-based studies have employed high-wattage halogen lamps, which produce heat during PMA treatment that may render viable cells to become membrane-compromised and in turn susceptible to PMA cross-linking [8, 17]. As an alternative to halogen lamps, the use of 460 nm LEDs was demonstrated by Hellein et al. [17]. However, improvements are required for PMA light activation. Therefore, the purpose of our study was aimed at discriminating between viable and killed E. coli cells by UV-induced PMA activation. PMA-qPCR assay was successfully applied to detect the target genes, namely, 16S rRNA gene, the ITS region, and the 23S rRNA gene of E. coli using IEC primer set [19] (Fig. 1a). Non-specific annealing and primer-dimer formation were not observed. The melting curve analyses demonstrated that the PMA-qPCR assay was highly specific. Such analyses also demonstrated the presence of accurate and reproducible melting peaks devoid of any non-specific products (Fig. 1b).
Fig. 1.
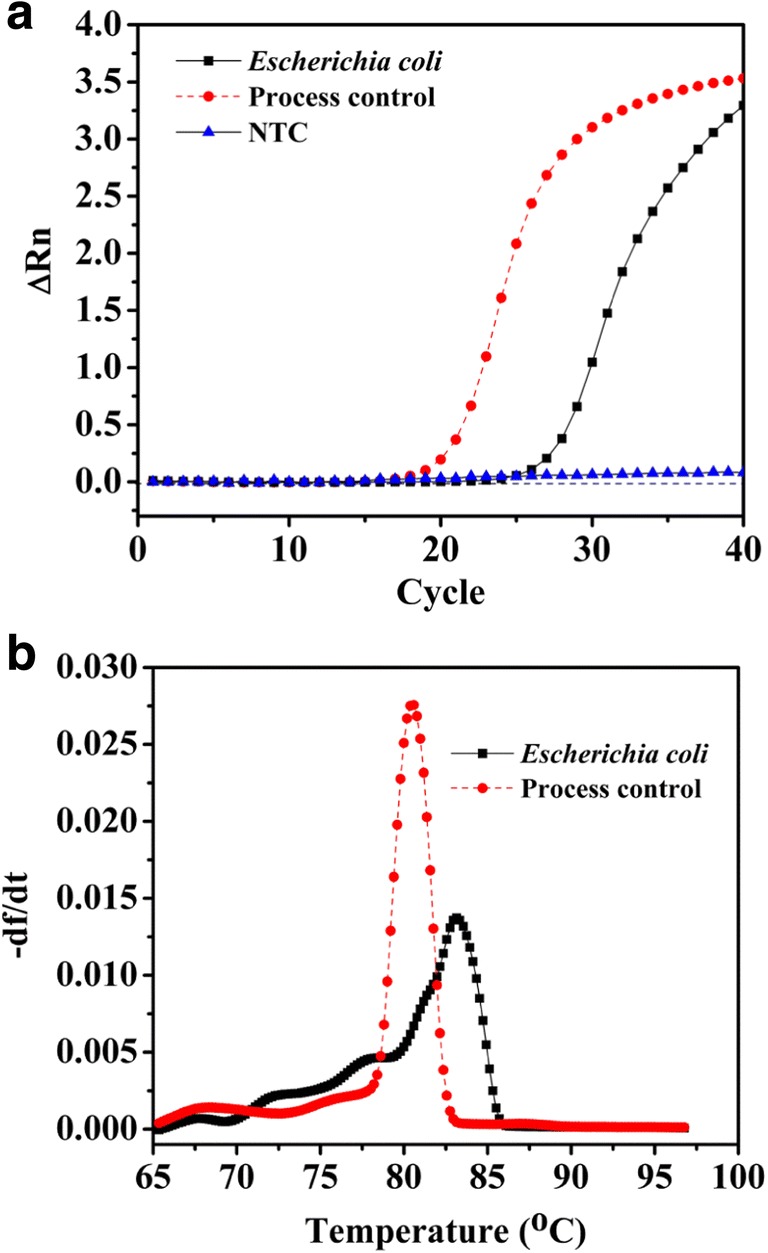
Amplification of DNA from Escherichia coli cells. Amplification signals (a) and typical melting curves (b) showing detection of DNA from pure cultures of Escherichia coli MTCC 3221. Bacillus atrophaeus spores were used as process control in qPCR reactions using specific primers targeting the atpD gene [20]. ∆Rn, fluorescence intensity change; − df/dt, fluorescence per unit temperature; NTC, no template control
PCR assays of PMA-treated cells could differentiate viable cells from a mixture of viable and killed cells of E. coli MTCC 3221 for UV radiation exposure at 4.8 × 103 μJ/cm2 for 3 min as shown in Fig. 2a. UV radiation exposures for 1 and 5 min did not yield any differentiation between viable and mixture of killed and viable cells (Supplementary Information, Table S1). The UV radiation treatment of PMA-treated cells showed that the cycle of quantitation (Cq) values of PMA-treated killed cells was higher than the Cq values of the mixture of viable and killed cells for E. coli MTCC 3221 (Fig. 2b). In contrast, PMA-qPCR assay could not completely distinguish the presence of viable cells from a mixture of viable and killed cells of E. coli MTCC 3221 when exposed to UV light at 2.4 × 103 μJ/cm2 and 7.2 × 103 μJ/cm2 either for 1, 3, or 5 min. UV irradiation of PMA-treated cells did not reveal significant changes in Cq values of PMA-treated viable cells when compared with the Cq values of the mixture of viable and killed cells for E. coli MTCC 3221 as illustrated in Figs. 3 and 4. In the case of UV light at 2.4 × 103 μJ/cm2 and 7.2 × 103 μJ/cm2, the covalent coupling of PMA with DNA derived from killed cells of E. coli MTCC 3221 could not be achieved when exposed either for 1, 3, or 5 min. The reason for these results is unknown; however, it could possibly be either due to the penetration of PMA into viable cells in the mixture of viable and killed cells as a result of UV irradiation or insufficient covalent binding of PMA to DNA from killed cells. However, in the present context, no supporting data are available for UV-induced cross-linking of PMA with the DNA from killed cells.
Fig. 2.
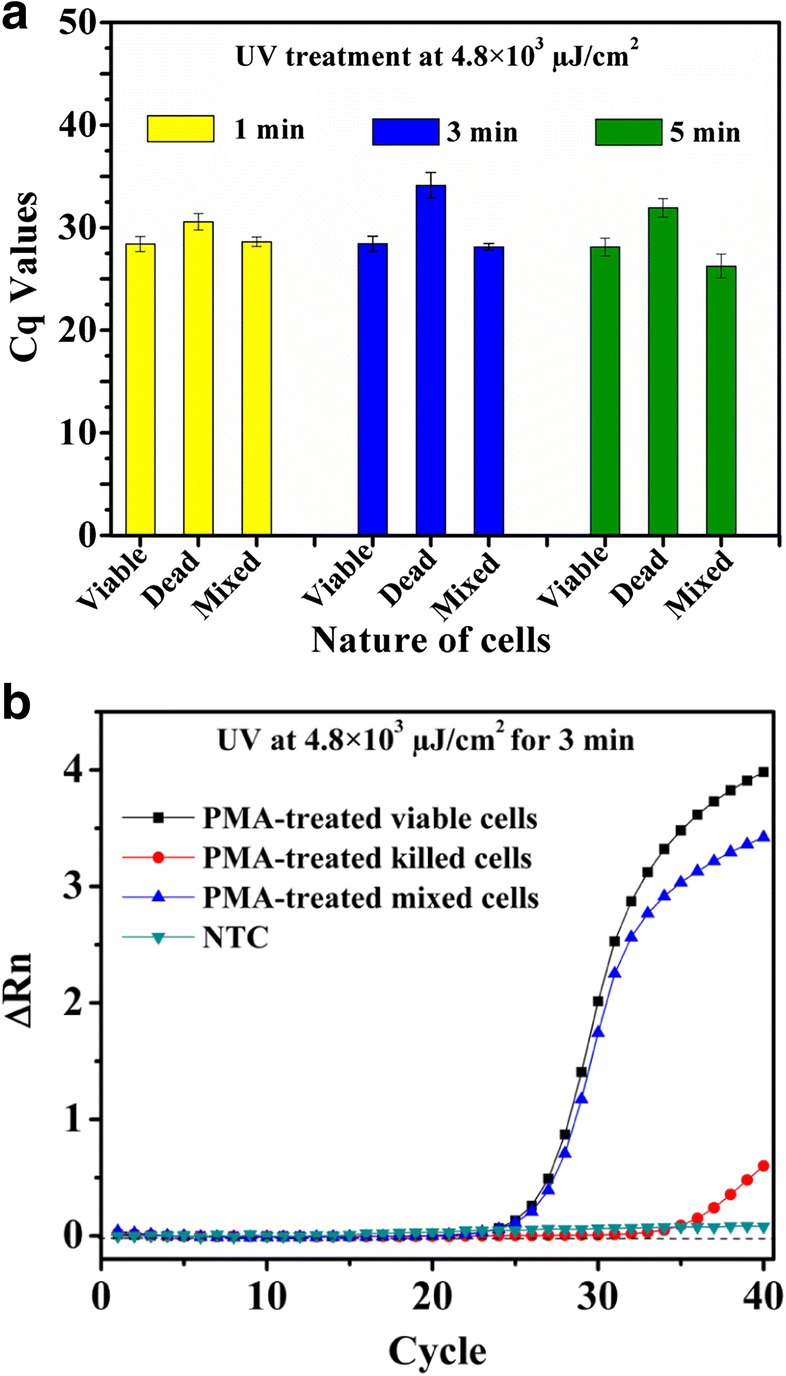
UV treatment at 4.8 × 103 μJ/cm2 to viable, killed, and a mixture of viable and killed cells of Escherichia coli MTCC 3221 for 3 min. UV treatment at 4.8 × 103 μJ/cm2 for 3 min resulted in complete inhibition of DNA amplification from killed cells by qPCR (a). Amplification curves for PMA-treated viable, killed, and a mixture of viable and killed cells of Escherichia coli MTCC 3221 at 4.8 × 103 μJ/cm2 for 3 min (b). Each bar represents the average Cq value of a triplicate experiment ± standard deviation. ∆Rn, fluorescence intensity change; NTC, no template control
Fig. 3.
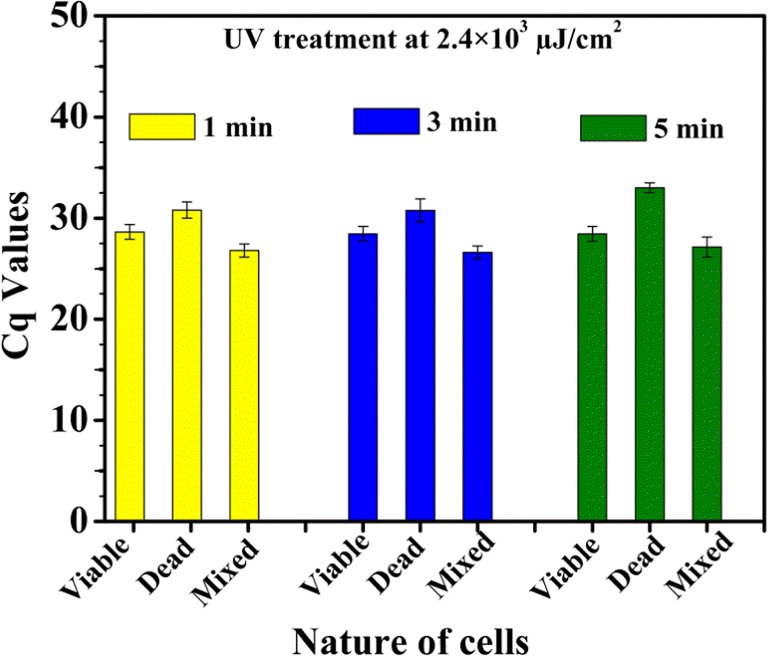
UV treatment of viable, killed, and a mixture of viable and killed Escherichia coli MTCC 3221 cells at 2.4 × 103 μJ/cm2 for 1, 3, and 5 min. Each bar represents the average Cq value of a triplicate experiment ± standard deviation
Fig. 4.
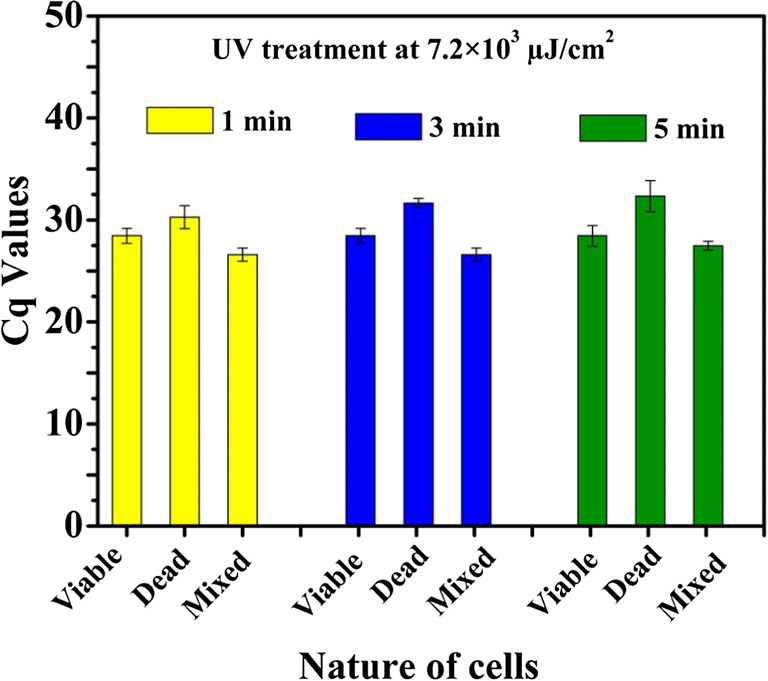
UV treatment at 7.2 × 103 μJ/cm2 to viable, killed, and mixture of viable and killed cells of Escherichia coli MTCC 3221 cells for 1, 3, and 5 min. Each bar represents the average Cq value of a triplicate experiment ± standard deviation
Applications of UV radiations for catalyzing the covalent attachment of nucleic acids to blotting membranes by activating interactions between thymines or uracils and the amine groups on the membrane matrix were reported previously [21]. Besides, the versatile wavelength of UV radiation is effective in many molecular biology applications such as restriction digestion of DNA and testing of recA mutations [18, 22]. In this study, PMA was coupled with the DNA derived from killed cells using UV light which resulted in complete inhibition of DNA amplification from killed cells. Due to UV irradiation, a declining tendency in Cq values of DNA from a mixture of viable and killed cells (average Cq, 28.13) was observed (Fig. 2a). The Cq values of mixed viable and killed cells extensively reflected the amount of DNA of viable cells (average Cq, 28.45) despite a large amount of DNA from killed cells in the mixture (average Cq, 34.14) (Fig. 2a, b). These observations indicated that PMA treatment differentiated between viable cells from a mixture of viable and killed cells of E. coli when exposed to UV radiation at 4.8 × 103 μJ/cm2 for 3 min. Herein, 1 and 5 min of UV exposure at 4.8 × 103 μJ/cm2 might be inadequate for covalent cross-linking of PMA to DNA derived from killed cells or PMA binding to DNA from killed cells might be hindered; however, this assumption requires a separate experimental validation. Nevertheless, 3 min of UV treatment was found to be optimum UV exposure time for PMA activation in the present study.
The sensitivity of the qPCR was determined by generating the standard curves (Supplementary Information, Fig. S2) using the optimized qPCR amplification assay conditions. The analysis of the standard curve revealed that the qPCR assay could detect as low as 6–7 femtograms of DNA per PCR reaction, which is equivalent to approximately 1 viable cell of E. coli [6, 23, 24]. The amplification efficiency of the PMA-qPCR was 90%. Agarose gel electrophoresis (2%) of the qPCR products also showed the desired quality of amplification devoid of any formation of primer-dimers (Supplementary Information, Fig. S3). The amplicon sequencing also confirmed the specificity of IEC primer set employed in UV-coupled PMA-qPCR assay for E. coli detection and quantification (Supplementary Information, Fig. S4).
PMA-qPCR of spiked water and PBS samples yielded the results in which PMA greatly reduced the amplifications of DNA derived from heat-killed samples. The ΔCq values for live cells ranged from − 0.17 to 0.2 (Table 1) indicating that UV-induced PMA activation had a negligible effect on viable cells (equivalent to 8 × 102 CFU; average Cq, 28.68). In contrast, ΔCq values for heat-killed cells ranged from 8.9 to 9.99 (Table 1), demonstrating the ability of PMA to inhibit PCR amplification of DNA derived from killed cells. These observations suggested that the amplification of DNA derived from PMA-treated heat-killed cells was reduced to an equivalent as low as 100 CFU (average Cq, 37.65). In addition, we observed significant differences between ΔCq values of PMA-treated heat-killed cells and all other sample types (PMA-treated viable cells, PMA-untreated viable cells, and PMA-untreated heat-killed cells; p < 0.03) as determined by paired t test and Tukey-Kramer procedure (Supplementary Information, Table S5). Subsequently, these results indicated that UV-induced PMA activation was sufficiently effective to inhibit PCR amplification of DNA derived from killed cells (Fig. 5). Consequently, these results showed that PMA coupled with UV light proved to be a good method to differentiate DNA of viable cells from that of killed cells. Moreover, setting and usage of halogen and LED light are cumbersome, whereas the use of CL-1000L UV Crosslinker is hassle-free, time-efficient, and effective for cross-linking PMA.
Table 1.
Average ΔCq values (Cq value for PMA-treated cells − Cq value for PMA-untreated cells) and standard deviations for amplification of DNA from Escherichia coli cells spiked in PBS and water samples
| Sample typen | Viable cells* | Heat-killed cells* |
|---|---|---|
| Escherichia coli cells–spiked in PBS | ||
| Sample 1 | − 0.17 ± 0.4 | 9.34 ± 0.36 |
| Sample 2 | − 0.32 ± 0.27 | 9.99 ± 1.18 |
| Escherichia coli cells–spiked in potable water | ||
| Sample 1 | 0.20 ± 0.13 | 8.9 ± 1.8 |
| Sample 2 | − 1.215 ± 0.94 | 9.965 ± 0.7 |
n = 3
*Mean ΔCq ± standard deviation
Fig. 5.
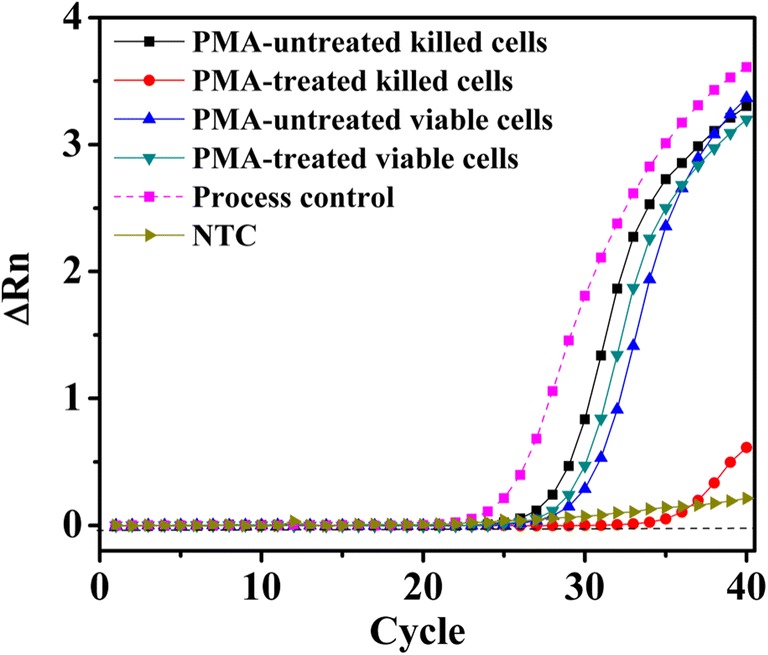
Amplification plot for comparison between PMA-treated and PMA-untreated viable and killed cells using UV light for 3 min at 4.8 × 103 μJ/cm2. ∆Rn, fluorescence intensity change; NTC, no template control
In the present study, the assay represented a significant improvement for selective detection of viable E. coli cells by UV-irradiated PMA-qPCR. This could be due to our modified PMA treatment process, as indicated by the significant differences in ∆Cq values between viable cells and killed cells resulting from PMA cross-linking to DNA by UV light (Table 1) and the selectivity of PMA in inhibiting DNA amplification from dead cells [25]. These improvements appear to be simple as they impart various advantages on the PMA-qPCR assay such as it becomes convenient to achieve a uniform cross-linking effect, which would contribute to the improved sensitivity of PMA-qPCR. It is also easy to place a large number of samples in CL-1000L UV Crosslinker and irradiate equal intensity of UV light exposure for cross-linking. Besides this, there is no heat generation during UV treatment, thereby, the killing of viable cells may be circumvented which otherwise renders viable cells susceptible to PMA treatment.
In conclusion, PMA-qPCR has been found to be specific and sensitive for the detection and quantification of viable E. coli using UV irradiation at 4.8 × 103 μJ/cm2 for 3 min. This assay has also been adapted for the detection of viable E. coli cells from spiked samples and, hence, this strategy may, therefore, be effectively used for accurate microbiological monitoring of water quality. However, further studies may be required in order to validate the time-dependent exposure of UV light for the activation of PMA to corroborate its equivalence to the already existing PMA treatment protocols.
Electronic supplementary material
(DOCX 686 kb)
Acknowledgments
We immensely thank the Indian Council of Medical Research (ICMR), Govt. of India, for awarding senior research fellowship (SRF-8/6/5/ITR-F/2018) to Mr. Rehan Deshmukh. Mr. Arun Kumar Prusty’s kind assistance in setting up and usage of CL-1000L UV Crosslinker is highly acknowledged.
Funding information
This research was supported by a grant from Centre for Research Excellence in Waste Water and Energy Management (CORE WWEM) Project of Birla Institute of Technology and Science—Pilani, K. K. Birla Goa Campus.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Gorchev HG, Ozolins G (2011) WHO guidelines for drinking-water quality. Vol 38. WHO Press 10.1016/S1462-0758(00)00006-6
- 2.Unicef. Water, sanitation and hygiene. http://www.unicef.org/wash/. Published 2015. Accessed August 19, 2019.
- 3.Graham N. Guidelines for drinking-water quality, 2nd edition, Addendum to volume 1 – recommendations, World Health Organisation, Geneva, 1998, 36 pages. Urban Water. 1999;1(2):183. doi: 10.1016/S1462-0758(00)00006-6. [DOI] [Google Scholar]
- 4.Deshmukh RA, Joshi K, Bhand S, Roy U. Recent developments in detection and enumeration of waterborne bacteria: a retrospective minireview. Microbiology Open. 2016;5(6):901–922. doi: 10.1002/mbo3.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maheux AF, Bissonnette L, Boissinot M, et al. Method for rapid and sensitive detection of Enterococcus sp. and Enterococcus faecalis/faecium cells in potable water samples. Water Res. 2011;45(6):2342–2354. doi: 10.1016/j.watres.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 6.Deshmukh RA, Bhand S, Roy U. A novel molecular quantitative method for rapid and sensitive detection of Escherichia coli from roof-harvested rainwater. Anal Methods. 2019;11(25):3155–3167. doi: 10.1039/c9ay00587k. [DOI] [Google Scholar]
- 7.Wang S, Levin RE. Discrimination of viable Vibrio vulnificus cells from dead cells in real-time PCR. J Microbiol Methods. 2006;64(1):1–8. doi: 10.1016/j.mimet.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 8.Nocker A, Cheung CY, Camper AK. Comparison of propidium monoazide with ethidium monoazide for differentiation of live vs. dead bacteria by selective removal of DNA from dead cells. J Microbiol Methods. 2006;67(2):310–320. doi: 10.1016/j.mimet.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 9.Nocker A, Sossa KE, Camper AK. Molecular monitoring of disinfection efficacy using propidium monoazide in combination with quantitative PCR. J Microbiol Methods. 2007;70(2):252–260. doi: 10.1016/j.mimet.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 10.Slimani S, Robyns A, Jarraud S, et al. (2012) Evaluation of Propidium Monoazide (PMA) Treatment directly on membrane filter for the enumeration of viable but non cultivable legionella by QPCR. Vol 88. 10.1016/j.mimet.2011.12.010 [DOI] [PubMed]
- 11.Nocker A, Sossa-Fernandez P, Burr MD, Camper AK. Use of propidium monoazide for live/dead distinction in microbial ecology. Appl Environ Microbiol. 2007;73(16):5111–5117. doi: 10.1128/AEM.02987-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taskin B, Gozen AG, Duran M. Selective quantification of viable Escherichia coli bacteria in biosolids by quantitative PCR with propidium monoazide modification. Appl Environ Microbiol. 2011;77(13):4329–4335. doi: 10.1128/AEM.02895-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Àlvarez G, González M, Isabal S, Blanc V, León R. Method to quantify live and dead cells in multi-species oral biofilm by real-time PCR with propidium monoazide. AMB Express. 2013;3(1):1. doi: 10.1186/2191-0855-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brescia CC, Griffin SM, Ware MW, Varughese EA, Egorov AI, Villegas EN. Cryptosporidium propidium monoazide-PCR, a molecular biology-based technique for genotyping of viable Cryptosporidium oocysts. Appl Environ Microbiol. 2009;75(21):6856–6863. doi: 10.1128/AEM.00540-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yáñez MA, Nocker A, Soria-Soria E, Múrtula R, Martínez L, Catalán V. Quantification of viable Legionella pneumophila cells using propidium monoazide combined with quantitative PCR. J Microbiol Methods. 2011;85(2):124–130. doi: 10.1016/j.mimet.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 16.Yoon J, Sung-Kwon M, Choi C, Buom-Yong R, Lee S (2019) Detection of viable but nonculturable Vibrio parahaemolyticus induced by prolonged cold-starvation using propidium monoazide real-time polymerase chain reaction. Lett Appl Microbiol :lam.13157. 10.1111/lam.13157 [DOI] [PubMed]
- 17.Hellein KN, Kennedy EM, Harwood VJ, Gordon KV, Wang SY, Lepo JE. A filter-based propidium monoazide technique to distinguish live from membrane-compromised microorganisms using quantitative PCR. J Microbiol Methods. 2012;89(1):76–78. doi: 10.1016/j.mimet.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 18.Whittaker PA, Southern EM. Ultraviolet irradiation of DNA: a way of generating partial digests for rapid restriction site mapping. Gene. 1986;41(1):129–134. doi: 10.1016/0378-1119(86)90276-3. [DOI] [PubMed] [Google Scholar]
- 19.Khan IUH, Gannon V, Kent R, et al. Development of a rapid quantitative PCR assay for direct detection and quantification of culturable and non-culturable Escherichia coli from agriculture watersheds. J Microbiol Methods. 2007;69(3):480–488. doi: 10.1016/j.mimet.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 20.Picard FJ, Gagnon M, Bernier MR, et al. Internal control for nucleic acid testing based on the use of purified Bacillus atrophaeus subsp. globigii Spores. J Clin Microbiol. 2009;47(3):751–757. doi: 10.1128/jcm.01746-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reed KC, Mann DA. Rapid transfer of DNA from agarose gels to nylon membranes. Nucleic Acids Res. 1985;13(20):7207–7221. doi: 10.1093/nar/13.20.7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sambrook J, Russell DW, David W (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press. https://books.google.co.in/books/about/Molecular_Cloning.html?id=Bosc5JVxNpkC. Accessed March 7, 2018.
- 23.Maheux AF, Bissonnette L, Boissinot M, et al. Rapid concentration and molecular enrichment approach for sensitive detection of Escherichia coli and Shigella species in potable water samples. Appl Environ Microbiol. 2011;77(17):6199–6207. doi: 10.1128/AEM.02337-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luna GM, Dell’anno A, Pietrangeli B, Danovaro R. A new molecular approach based on qPCR for the quantification of fecal bacteria in contaminated marine sediments. J Biotechnol. 2011;157:446–453. doi: 10.1016/j.jbiotec.2011.07.033. [DOI] [PubMed] [Google Scholar]
- 25.Nocker A, Camper AK. Selective removal of DNA from dead cells of mixed bacterial communities by use of ethidium monoazide. Appl Environ Microbiol. 2006;72(3):1997–2004. doi: 10.1128/AEM.72.3.1997-2004.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 686 kb)