

Abstract
Sjögren‐Larsson syndrome (SLS) is a rare inborn error of lipid metabolism. The syndrome is caused by mutations in the ALDH3A2 gene, resulting in a deficiency of fatty aldehyde dehydrogenase. Most patients have a clearly recognizable severe phenotype, with congenital ichthyosis, intellectual disability, and spastic diplegia. In this study, we describe two patients with a remarkably mild phenotype. In both patients, males with actual ages of 45 and 61 years, the diagnosis was only established at an adult age. Their skin had been moderately affected from childhood onward, and both men remained ambulant with mild spasticity of their legs. Cognitive development, as reflected by school performance and professional career, had been unremarkable. Magnetic resonance spectroscopy of the first patient was lacking the characteristic lipid peak. We performed a literature search to identify additional SLS patients with a mild phenotype. We compared the clinical, radiologic, and molecular features of the mildly affected patients with the classical phenotype. We found 10 cases in the literature with a molecular proven diagnosis and a mild phenotype. Neither a genotype‐phenotype correlation nor an alternative explanation for the strikingly mild phenotypes was found. New biochemical techniques to study the underlying metabolic defect in SLS, like lipidomics, may in the future help to unravel the reasons for the exceptionally mild phenotypes. In the meantime, it is important to recognize these mildly affected patients to provide them with appropriate care and genetic counseling, and to increase our insights in the true disease spectrum of SLS.
Keywords: ALDH3A2, FALDH, Sjögren‐Larsson syndrome, spastic diplegia
SYNOPSIS.
Sjögren‐Larsson syndrome (OMIM #270200) is a recognizable clinical disorder that almost always leads to a (classic) severe phenotype; in this study, we learn that mild phenotypes exist, with the same genetic and biochemical abnormalities, and normal intelligence but otherwise essentially the same—but far less severe—neurocutaneous syndrome.
1. INTRODUCTION
In 1957, Sjögren and Larsson described a syndrome with congenital ichthyosis, intellectual disability, and spastic diplegia, in a series of 28 Swedish patients with a rather homogeneous clinical phenotype, enabling the recognition of the disorder that has since been called Sjögren‐Larsson syndrome (SLS).1
In 1988, it was found that abnormalities in the lipid metabolism were responsible for the clinical features of SLS.2 The enzyme involved, fatty aldehyde dehydrogenase (FALDH), is part of the fatty alcohol nicotinamide adenine dinucleotide oxidoreductase complex and catalyses oxidation of many different medium‐ and long‐chain fatty aldehydes, derived from fatty alcohols, into fatty acids. FALDH deficiency results in the accumulation of fatty aldehydes and fatty alcohols, which is considered the principal causative disease mechanism in SLS. In 1996, the ALDH3A2 gene was discovered to be coding for the FALDH enzyme. ALDH3A2 mutations have been identified in all patients with SLS.3 To date, more than 100 mutations in the ALDH3A2 gene have been described,4 and all patients, except some extremely rare cases, appear to suffer from essentially the same, aforementioned clinical triad of symptoms, with only minor variations in severity.5, 6, 7, 8, 9
Our group has several decades of experience with patient care for children and adults with SLS, as well as clinical SLS research. We have learned to know SLS in its common, severe form, and had—among more than 30 patients—recognized only one sib pair with a milder phenotype in the past (cases H1 and H2 of SLS10 and cases 18 and 19 of SLS7). Recently, a third patient with a remarkably mild SLS phenotype was referred to our clinic.
Here we describe two of the three mildly affected Dutch patients (one patient did not give informed consent). Additionally, we performed a literature study to identify other mild cases of SLS. We aim to increase awareness and expand the phenotypic spectrum of SLS. By making comparisons between the mild vs classical presentation, we hope to identify the factors involved in determining the clinical outcome and understand the natural history, thus improving our insights into the underlying disease mechanism.
2. METHODS
2.1. Patient studies
We performed an observational study on two unrelated, male, adult patients from the Netherlands, with a mild SLS phenotype. The study was performed according to the tenets of the declaration of Helsinki (2013 revision), and was approved by the Regional Committee on Research Involving Human Subjects. Informed consent was obtained from both participants prior to inclusion in the study.
The patients were seen in our outpatient clinic in 2018. Full physical and neurological examinations were performed. Additionally, a detailed ophthalmologic examination was performed to study visual function and retinal morphology. Visual acuity was measured using Snellen charts. Slit‐lamp examination, ophthalmoscopy, optical coherence tomography (OCT), and fundus photography were performed. In patient 1, cerebral magnetic resonance imaging (MRI) and MR‐spectroscopy (MRS) were performed. Patient 2 underwent MR imaging in 1995, but did not give consent for a follow‐up scan.
2.2. Literature study
We searched in PubMed for papers in which individual cases of SLS were described in sufficient detail. We only included patients with a biochemically or genetically proven diagnosis. We searched on PubMed using “Sjögren‐Larsson syndrome” as search term. All papers on SLS, published from 1988 (first report about the enzyme defect) to September 1, 2019 (date of searching), were screened for descriptions of patients with “nonclassical” phenotypes. Only English papers were included. Cases were defined as being nonclassical, that is, mild, when one of the core symptoms (ichthyosis, intellectual disability, and spasticity) was missing or when at least two of the three core symptoms were described as being mild.
3. RESULTS
3.1. Case descriptions
3.1.1. Patient 1
This 45‐year‐old male patient presented at our hospital with a new diagnosis of SLS. He was born late preterm, with a dry skin. In the following years, ichthyosis developed, covering mainly his trunk and showing sharply demarcated affected areas (Figure 1A‐C). From the age of 30 years on, his ichthyosis became less evident (Figure 1D), but he still suffered from considerable pruritus. During childhood and early adulthood, his motor skills had been considered “clumsy,” but the patient neither experience stiffness nor needed adjusted shoes. Neurological examination after experiencing a traffic accident at the age of 30 years showed—for the first time—a clear (but mild) spastic gait. He required glasses from already a young age and experienced photophobia. A formal IQ test had never been performed but based on his school carrier (he finished high school), employment history (he worked in a stockroom), and general performance, his cognitive functions were considered normal. His speech was unremarkable. His parents were nonconsanguineous, and he had one healthy brother. His spastic gait had been attributed to his preterm birth until a neurologist put all the symptoms together and tested the patient for SLS at the age of 45 years.
Figure 1.

Skin abnormalities of patients 1 (A‐D) and 2 (E‐H). Skin photographs of patients 1 and 2 at different ages. A,E, Photography of the trunk of patient 1 at the age of 3 years (A) and of patient 2 at the age of 7 years (E). A sharply demarcated hyperpigmented, ichthyosiform plaque is seen. B,F, Close‐up photography of the right axilla of both patients. C,G, Photography of the back of both patients (C, patient 1 at the age of 17 years; G, patient 2 at the age of 7 years), with a sharply demarcated hyperpigmented ichthyosiform plaque. D,H, Photography of the trunk of patient 1 at the age of 45 years (D) and adult age of patient 2 (H). Sharply demarcated, partially hyperkeratotic plaques with lichenification and an ichthyosiform desquamation in the axillae and medial aspects of the arms and elbows are seen in patient 1. In patient 2, the abdomen, axillae, and medial aspects of the arms and elbows are affected and sharply demarcated
On examination, a good‐natured man was seen. In the axillae, groins, and the lumbar region, ichthyosis was present. Remarkably, the medial parts of the upper arms were affected, whereas the lateral parts were not (Figure 1B and D). On the legs a mild, diffuse ichthyosiform desquamation was seen; the face, neck, most of the trunk, and lateral sides of the arms were not affected. He showed a spastic paraplegia, but was able to walk without aids. Visual acuity was 0.5 and 0.4 for the right and left eye, respectively, and findings on ophthalmoscopy and OCT were compatible with SLS maculopathy (Figure 2 A and B).
Figure 2.

Retinal abnormalities of patients 1 and 2. A, Fundus photography of patient 1 at the age of 45 years, with a few perifoveal crystals visible around the macula lutea together with a lack of the expected physiological darkening. B, Macular OCT scan of patient 1, with a thinned macula, hyperreflective dots suggesting crystalline deposits and interruptions of the photoreceptor layer visible suitable with SLS maculopathy. Next to this, a mild central serous chorioretinopathy is seen. C, Fundus photography of patient 2 at the age of 61 years, with parafoveal crystals visible. D, Macular OCT scan of patient 2, with also a thinned macula and changes in the retinal pigment epithelium. OCT, optical coherence tomography
Cerebral MRI showed diffuse, subtle signal changes of the periventricular white matter without other abnormalities. MRS revealed normal spectra of the parieto‐occipital white and occipital gray matter, and (thus) did not show the typical “lipid peak” at 1.3 ppm (Figure 3).11 FALDH activity in lymphocytes was below detection limit of the enzyme assay. Sanger sequencing showed compound heterozygosity for two pathogenic variants in the ALDH3A2 gene, namely, c.682C>T (p.[Arg228Cys]) and c.943C>T (p.[Pro315Ser]).
Figure 3.
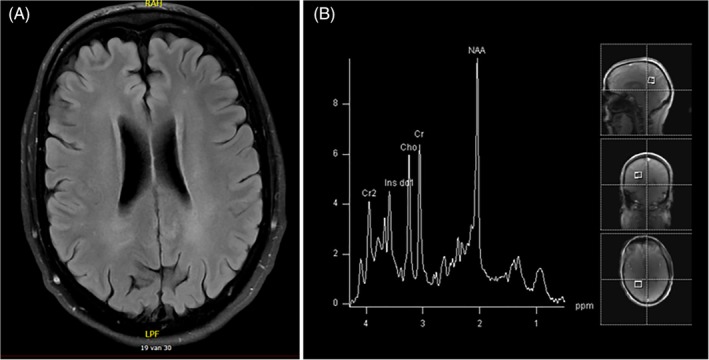
Cerebral magnetic resonance imaging and spectroscopy of patient 1. A; T2‐weightened MRI scan with subtle abnormalities in the periventricular white matter. B, MRS from the white matter, without the characteristic lipid peaks. MRI, magnetic resonance imaging; MRS, magnetic resonance spectroscopy
3.1.2. Patient 2
This 61‐year‐old male patient was already known for more than 25 years in our clinic, and we have described his phenotype previously.7, 10, 11, 12 He was diagnosed with SLS at the age of 34 years after clinical suspicion, using a targeted enzyme analysis. He was born at term. He was noted to have dry skin at the age of approximately 9 months, later described to be lamellar ichthyosis. A sharp demarcation of the skin abnormalities was seen (Figure 1, E‐G). Despite the use of acitretin, his skin showed a subtle ichthyosiform desquamation which was still pruritic (Figure 1H). At the age of 33 years, a mild spastic paraplegia was diagnosed after adolescent‐onset spastic paraplegia. The patient was able to walk without assistance, with adjusted shoes. He suffered from a mild visual impairment and photophobia. He had a (low) normal intelligence with an IQ of 83 and a mild speech impairment. He was married and had two healthy children. He has one affected sister with SLS and five unaffected healthy siblings. The affected sister had an overall phenotype that was even slightly milder than the phenotype of her brother.7, 10
On examination, a pleasant, mildly dysarthric man was seen. Mild ichthyosis was seen on the back, in the axillae and the back of his legs. The remainder of the skin appeared normal. He showed a mild spastic diplegia, with mild contractures of the ankles. Tendon reflexes were increased in the legs and he had a bilateral Babinski sign. His arms were neurologically unaffected. Best corrected visual acuity was 0.4 for both eyes. On slit‐lamp exam, a bilateral cortical and nuclear cataract was seen. Ophthalmoscopy revealed crystalline deposits around the macula, and the physiological retinal darkening caused by macular pigment was lacking (Figure 2C). Retinal imaging using OCT showed a thinned macula, with hyperreflective dots and an interrupted photoreceptor layer in both eyes (Figure 2D). In the left eye, foveal microcysts were seen. At the age of 38 years, an MRI of the brain had been performed, which showed mild periventricular hyperintense signals and mild ventricular enlargement. No MRS was performed. Biochemical and molecular analysis previously had shown FALDH deficiency in fibroblasts, and compound heterozygosity for two pathogenic variants in the ALDH3A2 gene, namely, c.551C>T (p.[Thr184Met]) and c.943C>T (p.[Pro315Ser]).
3.2. Case reports in literature
A review of literature, revealed six publications describing 10 non‐Dutch patients with a mild phenotype (Table 1).13, 14, 15, 16, 17, 18 Two additional case reports describing mildly affected patients were excluded due to insufficient biochemical or genetic information.19, 20
Table 1.
Case reports found in the literature describing patients with a mild phenotype of Sjögren‐Larsson syndrome
| Case number | Paper | Case in paper | Age | Sex | Skin | Eyes | Spasticity | Speech | Intellectual disability | Gestational age | FAO/FALDH activity | ALDH3A2 mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Nigro13 | A | 14 y | F | Ichthyosis on abdomen, axillae, extremities. Less pronounced lesions on back and face | No abnormalities on ophthalmologic evaluations | Gradually worsened spasticity, however able to walk without assistance | Normal | No evidence of mental retardation, but diagnosed as having a learning disability | ND | 6/not tested (pmol/min/mg protein; normal 75 ± 26) | ND |
| 2 | B | 12 y | M | Ichthyosis on legs, axillae, soles | No abnormalities on ophthalmologic evaluations | Gradually worsened spasticity, however, able to walk without assistance | Normal | No evidence of mental retardation, but diagnosed as having a learning disability | ND | 2/398 (pmol/min/mg protein; normal 75 ± 26/8540 ± 1158) | ND | |
| 3 | C | 7 y | F | No abnormalities | ND | Minimally abnormal gait | Normal | No evidence of mental retardation | ND | 3/257 (pmol/min/mg protein; normal 75 ± 26/8540 ± 1158) | ND | |
| 4 | Kawakami14 | 1 | 13 mo | M | Mild ichthyosis of lower abdomen and dorsal aspects of extremities | No abnormalities | No history of spastic diplegia, but he had club feet | ND | Developmental delay | Normal pregnancy and delivery | Not tested/694 ± 212 PMol/min/mg (mean ± SD; normal 2536 ± 649). | ND |
| 5 | 2 | 5 y | M | Mild ichthyosis on his lower abdomen and the dorsal aspects of his extremities | No abnormalities | Walk without assistance, no history of spastic diplegia | Normal | Learning disability, mental retardation gradually worsened | ND | Histochemical testing revealed reduced alcohol dehydrogenase activity in the ichthyotic skin, similar to that found in patient 1 in this paper | ND | |
| 6 | Carney15 | 1 | 4 y | ND | Ichthyosis+ | ND | +/Ambulatory | ND | Mild mental retardation | ND | FALDH activity 9% | c.286_296del/c.1268G>A |
| 7 | Didona16 | 1 | 12 y | F | Ichthyosis became evident during the first month of life | Pigmentary retinopathy was ruled out | Due to spasticity in the legs, the patient first walked at 24 months of age only with support, on her tip‐toes, and with adducted hips and flexed knees. She underwent surgical correction of leg contractures and started to walk independently with a spastic gait at 7 years of age. Since then, her motor disability has remained stable and currently, at 12 years of age, spasticity involves only the lower limbs | Normal language development | Moderate learning difficulties | ND | Not tested | c.769insA (homozygous) |
| 8 | 2 | 5 y | M | Ichthyosis present at birth | Pigmentary retinopathy was ruled out | Leg spasticity became evident in the subsequent few months, but the diplegia remained mild and surgical intervention has been avoided so far | ND | Mild mental retardation | ND | 935 pmol/min/mg (normal values 6750‐20 570) | c.1094C>T, c.471+2T>G | |
| 9 | Tachibana17 | 1 | 5 y | F | Ichthyosis on trunk, limbs | ND | Spasticity in both legs, MRI mild abnormalities, MRS lipid peak | ND | No deficiencies in intellectual development | 39 wk | ND | c.1339A>G, c.504_505insAG |
| 10 | Papathemeli18 | 1 | 3 y | F | Ichthyosis on axillae, neck, palms, soles, flexures | BCVA 0.5/0.3, fundoscopy and OCT normal | Bilateral spasticity of legs. MRI white matter lesions, MRS moderate increase lipid | ND | Normal (IQ 95) | 39 wk | ND | c.551C>T (homozygous) |
Abbreviations: BCVA, best corrected visual acuity; F, female; M, male; ND, not described.
The case reports included children from ages 1 to 14. The cases described different spectrum of severity for the three cardinal clinical symptoms. In three patients, the ichthyosis was described as being absent or mild. In seven patients, the spasticity was mild, for example, reflected by the fact that these patients were ambulatory. Intellectual development was normal in eight patients. Considering other prevalent symptoms, ophthalmological abnormalities were not seen in six out of seven patients in whom data were reported, and speech was normal in five patients (out of the five in whom it was reported explicitly). Gestational age was only described in three patients; all three were born at term. All five patients in whom mutation analysis had been performed had different ALDH3A2 gene mutations. In most of them, and in all remaining cases without a genetic diagnosis, enzyme deficiency had confirmed the clinical diagnosis of SLS.
4. DISCUSSION
We describe two adult patients with both biochemical and genetical confirmations of SLS, presenting with a milder spectrum of this disorder from the classical form. Both patients display a rather straight‐forward neurological disorder with normal cognitive functions, and adolescent‐ or adult‐onset spastic paraplegia with slow progression and preservation of ambulation during at least decades. Preterm birth is typically associated with the classical phenotype of SLS, and it has not been documented in mildly affected cases.21 The ichthyosis of patients 1 and 2 occurred early in life but did not follow the distinctive pattern seen in the classical phenotype. Interestingly, pruritus was a key complaint similar to the classical phenotype, and also normal appearing skin was felt to be itchy. Patients with “milder SLS phenotypes” thus appear to suffer from essentially the same clinical syndrome, although to a lesser degree. This is further confirmed by the observation that the retinal abnormalities of patients 1 and 2 were also similar to the classical SLS patients, showing a thinned retina with crystalloid depositions and interruptions of the photoreceptor layer.22
Applying MRS, lipid accumulation can be demonstrated in cerebral white matter of patients with classic SLS. Remarkably, the spectra of patient 1 did not show the typical resonances reflecting lipid accumulation. The other mildly affected Dutch patient known in our center, the sister of patient 2, did not demonstrate the typical lipid peak on MRS in previous studies.7, 10 The absence of lipid accumulation, or at least much lower brain tissue concentrations of abnormal lipids (below the detection limit of MRS), might be one of the explanations for the mild neurological phenotype.
The 10 clinical descriptions found in the literature of mildly affected patients with genetically or biochemically proven SLS are not uniform; the extent to which brain, skin, and eyes are affected varies between patients (Table 1). All patients were children. Patient 5 was only 13 months old at the time of reporting, thus may not express all the symptoms. It is well‐possible other adult patients with mild phenotype exist but are not diagnosed—and described in literature—yet.
We tried to understand why some patients show a milder phenotype, because this might contribute to a better understanding of the disease mechanisms underlying SLS. Enzyme activity measurements were not useful for this purpose because there is no clear correlation between residual enzyme activity and clinical phenotype.23 Because FALDH is a member of a large family of aldehyde dehydrogenases,24 most available enzyme assays are not 100% specific for FALDH activity. This hampers the ability to reliably determine small amounts of residual FALDH enzyme activity and consequently stands in the way of investigations studying a potential relationship between severity of the phenotype and residual FALDH activity.
We focused on the mutations of patients 1 and 2, and searched the mutations in Pubmed and the Leiden Open Variation Database (LOVD, https://databases.lovd.nl/shared/genes/ALDH3A2). The first variant in patient 1 (c.682C>T) was described in six siblings in a homozygous state and a variable phenotype.5 Although the patients in this cohort had slightly variable patterns of severity across symptoms, we think that they still display the clearly recognizable, common SLS phenotype. The second mutation of patient 1 (c.943C>T) is one of the most common mutations in the classical SLS population. Patient 2 has this same c.943C>T mutation. The second mutation of patient 2 (c.551C>T) is described in two other cases: the milder case from the report of Papathemeli et al18 (case 10 in Table 1) and a patient with a classical phenotype.25 The mutations described in the mild cases identified in our literature review were compared with the LOVD database as well and listed in Table 2. Altogether, we concluded that there is no clear genotype‐phenotype correlation to explain the mild phenotype of some patients with SLS. This lack of genotype‐phenotype correlation was also described in a recent paper by Abdel‐Hamid et al.31
Table 2.
ALDH3A2 gene mutations in mildly affected patients with Sjögren‐Larsson syndrome
| Casea | Mutation | Other cases found in LOVD database | ||
|---|---|---|---|---|
| Papers | Clinical features | FALDH activity | ||
| NL1 | c.682C>T | Lossos5 | Variable patterns of severity in SLS symptoms | 4% of normal |
| Shamriz26 | Classical SLS, coexistence with cytidine deaminase deficiency | Not described | ||
| Hidalgo27 | Classical SLS | Not described | ||
| NL1; NL2 | c.943C>T | Willemsen7 | Classical SLS | Reduced |
| Ganemo28 | Classical SLS | Not described | ||
| Sillen29 | Not described | Not described | ||
| Rizzo30 | Classical SLS | Reduced | ||
| NL2; 10 | c.551C>T | Jean‐Francois25 | Classical SLS | Not described |
| 6 | c.286_296del | No | Not applicable | Not applicable |
| c.1268G>A | No | Not applicable | Not applicable | |
| 7 | c.769insA | No | Not applicable | Not applicable |
| 8 | c.1094C>T | Sillen29 | Not described | Not described |
| Rizzo30 | Classical SLS | Reduced | ||
| c.471+2T>G | Rizzo30 | Classical SLS | Reduced | |
| 9 | c.1339A>G | No | Not applicable | Not applicable |
| c.504_505insAG | No | Not applicable | Not applicable | |
Abbreviations: LOVD, Leiden Open Variation Database; SLS, Sjögren‐Larsson syndrome.
Cases NL1 and NL2 are the described cases from the current paper; cases 7‐10 correspond with Table 1.
Apart from a (missing) genotype‐phenotype correlation, other mechanisms could potentially affect the severity of the phenotype in SLS. First, mildly affected patients may have “active alternative metabolic routes” to escape the metabolic defect, or may have “upstream” variants that decrease the flux of lipids through the pathways in which FALDH plays a role.
Second, mosaicism or revertant mosaicism of the genetic mutation might explain the peculiar distribution of the affected and nonaffected skin in patients 1 and 2.32, 33 However, the pattern of the ichthyosis in these patients (Figure 1) looks similar and does not fit any of the described mosaic distributions.32, 34, 35
Irrespective of the lack of understanding of the underlying mechanisms, we find it important to increase the awareness of the mild phenotype of SLS. Patients with this phenotype could present to neurologists, ophthalmologists, and dermatologists, and it is important to recognize this nonclassical phenotype of SLS. These patients should receive appropriate (genetic) counseling, and may possibly need different treatment approaches in the future. Studying these patients hopefully may increase our insights into the disease, both on an individual level (with a chance on better personalized care) and on a group level (with a more complete definition of the disease spectrum and better understanding of the underlying disease mechanisms).
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
AUTHOR CONTRIBUTIONS
P.S.: execution of the study, clinical assessment of patients, data interpretation, writing the manuscript. J.v.G.: clinical assessment of patients, correction of manuscript. P.v.D.: clinical assessment of patients, correction of manuscript. P.M.S.: clinical assessment of patients, correction of manuscript. S.F.: enzyme activity measurements, correction of manuscript. T.d.H.: correction of manuscript. M.M.B.S.: clinical assessment of patients, correction of manuscript. T.T.: clinical assessment of patients, correction of manuscript. M.A.A.P.W.: design of the study, clinical assessment of patients, data interpretation, correction of manuscript. M.A.A.P.W. is the guarantor for the article
INFORMED CONSENT
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2013. Informed consent was obtained from all patients for being included in the study.
DATA AVAILABILITY
Patient data are saved in the electronic patient records of our hospital.
Staps P, van Gaalen J, van Domburg P, et al. Sjögren‐Larsson syndrome: The mild end of the phenotypic spectrum. JIMD Reports. 2020;53:61–70. 10.1002/jmd2.12099
Communicating Editor: Areeg El‐Gharbawy
REFERENCES
- 1. Sjögren T, Larsson T. Oligophrenia in combination with congenital ichthyosis and spastic disorders; a clinical and genetic study. Acta Psychiatr Neurol Scand Suppl. 1957;113:1‐112. [PubMed] [Google Scholar]
- 2. Rizzo WB, Dammann AL, Craft DA. Sjögren‐Larsson syndrome. Impaired fatty alcohol oxidation in cultured fibroblasts due to deficient fatty alcohol:nicotinamide adenine dinucleotide oxidoreductase activity. J Clin Invest. 1988;81:738‐744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. De Laurenzi V, Rogers GR, Hamrock DJ, et al. Sjögren‐Larsson syndrome is caused by mutations in the fatty aldehyde dehydrogenase gene. Nat Genet. 1996;12:52‐57. [DOI] [PubMed] [Google Scholar]
- 4. Cho KH, Shim SH, Kim M. Clinical, biochemical, and genetic aspects of Sjogren‐Larsson syndrome. Clin Genet. 2018;93:721‐730. [DOI] [PubMed] [Google Scholar]
- 5. Lossos A, Khoury M, Rizzo WB, et al. Phenotypic variability among adult siblings with Sjogren‐Larsson syndrome. Arch Neurol. 2006;63:278‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davis K, Holden KR, S'Aulis D, Amador C, Matheus MG, Rizzo WB. Novel mutation in Sjogren‐Larsson syndrome is associated with divergent neurologic phenotypes. J Child Neurol. 2013;28:1259‐1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Willemsen MA, IJlst L, Steijlen PM, et al. Clinical, biochemical and molecular genetic characteristics of 19 patients with the Sjogren‐Larsson syndrome. Brain. 2001;124:1426‐1437. [DOI] [PubMed] [Google Scholar]
- 8. Weustenfeld M, Eidelpes R, Schmuth M, Rizzo WB, Zschocke J, Keller MA. A genotype‐based database for variants causing the Sjogren‐Larsson syndrome. Hum Mutat. 2019;40:177‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Amr K, El‐Bassyouni HT, Ismail S, et al. Genetic assessment of ten Egyptian patients with Sjogren‐Larsson syndrome: expanding the clinical spectrum and reporting a novel ALDH3A2 mutation. Arch Dermatol Res. 2019;311:721‐730. [DOI] [PubMed] [Google Scholar]
- 10. van Domburg PH, Willemsen MA, Rotteveel JJ, et al. Sjogren‐Larsson syndrome: clinical and MRI/MRS findings in FALDH‐deficient patients. Neurology. 1999;52:1345‐1352. [DOI] [PubMed] [Google Scholar]
- 11. Willemsen MA, Van Der Graaf M, Van Der Knaap MS, et al. MR imaging and proton MR spectroscopic studies in Sjogren‐Larsson syndrome: characterization of the leukoencephalopathy. Am J Neuroradiol. 2004;25:649‐657. [PMC free article] [PubMed] [Google Scholar]
- 12. Willemsen MA, Cruysberg JR, Rotteveel JJ, Aandekerk AL, Van Domburg PH, Deutman AF. Juvenile macular dystrophy associated with deficient activity of fatty aldehyde dehydrogenase in Sjögren‐Larsson syndrome. Am J Ophthalmol. 2000;130:782‐789. [DOI] [PubMed] [Google Scholar]
- 13. Nigro JF, Rizzo WB, Esterly NB. Redefining the Sjogren‐Larsson syndrome: atypical findings in three siblings and implications regarding diagnosis. J Am Acad Dermatol. 1996;35:678‐684. [DOI] [PubMed] [Google Scholar]
- 14. Kawakami T, Saito R, Fujikawa Y, et al. Incomplete Sjogren‐Larsson syndrome in two Japanese siblings. Dermatology. 1999;198:93‐96. [DOI] [PubMed] [Google Scholar]
- 15. Carney G, Wei S, Rizzo WB. Sjogren‐Larsson syndrome: seven novel mutations in the fatty aldehyde dehydrogenase gene ALDH3A2. Hum Mutat. 2004;24:186. [DOI] [PubMed] [Google Scholar]
- 16. Didona B, Codispoti A, Bertini E, et al. Novel and recurrent ALDH3A2 mutations in Italian patients with Sjogren‐Larsson syndrome. J Hum Genet. 2007;52:865‐870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tachibana Y, Aida N, Enomoto K, Iai M, Kurosawa K. A case of Sjogren‐Larsson syndrome with minimal MR imaging findings facilitated by proton spectroscopy. Pediatr Radiol. 2012;42:380‐382. [DOI] [PubMed] [Google Scholar]
- 18. Papathemeli D, Mataftsi A, Patsatsi A, et al. Atypical presentation of Sjogren‐Larsson syndrome. Case Rep Pediatr. 2017;2017:7981750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Balakumar B, Arora S, Palocaren T. Sjogren‐Larsson syndrome—unusual presentation with pathological femoral neck fracture: a case report. J Pediatr Orthop B. 2012;21:583‐586. [DOI] [PubMed] [Google Scholar]
- 20. Bhallil S, Chraibi F, Andalloussi IB, Tahri H. Optical coherence tomography aspect of crystalline macular dystrophy in Sjogren‐Larsson syndrome. Int Ophthalmol. 2012;32:495‐498. [DOI] [PubMed] [Google Scholar]
- 21. Staps P, Hogeveen M, Fuijkschot J, van Drongelen J, Willemsen M. Understanding fetal factors that contribute to preterm birth: Sjogren‐Larsson syndrome as a model. J Perinat Med. 2018;46:523‐529. [DOI] [PubMed] [Google Scholar]
- 22. Staps P, Cruysberg JRM, Roeleveld N, Willemsen MAAP, Theelen T. Retinal morphology in Sjögren‐Larsson syndrome on OCT: from metabolic crystalline maculopathy to early‐onset macular degeneration. Ophthalmol Retina. 2019;3:500‐509. [DOI] [PubMed] [Google Scholar]
- 23. Weustenfeld M, Eidelpes R, Schmuth M, Rizzo WB, Zschocke J, Keller MA. Genotype and phenotype variability in Sjogren‐Larsson syndrome. Hum Mutat. 2019;40:177‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vasiliou V, Nebert DW. Analysis and update of the human aldehyde dehydrogenase (ALDH) gene family. Hum Genomics. 2005;2:138‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jean‐Francois E, Low JY, Gonzales CR, Sarraf D. Sjogren‐larsson syndrome and crystalline maculopathy associated with a novel mutation. Arch Ophthalmol. 2007;125:1582‐1583. [DOI] [PubMed] [Google Scholar]
- 26. Shamriz O, Molho‐Pessach V, Shaag A, Daum H, Stepensky P. Coexistence of two rare autosomal recessive disorders: activation‐induced cytidine deaminase deficiency and Sjogren‐Larsson syndrome. Isr Med Assoc J. 2016;18:636‐638. [PubMed] [Google Scholar]
- 27. Hidalgo ET, Orillac C, Hersh A, Harter DH, Rizzo WB, Weiner HL. Intrathecal baclofen therapy for the treatment of spasticity in Sjogren‐Larsson syndrome. J Child Neurol. 2017;32:100‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ganemo A, Jagell S, Vahlquist A. Sjogren‐Larsson syndrome: a study of clinical symptoms and dermatological treatment in 34 Swedish patients. Acta Derm Venereol. 2009;89:68‐73. [DOI] [PubMed] [Google Scholar]
- 29. Sillen A, Anton‐Lamprecht I, Braun‐Quentin C, et al. Spectrum of mutations and sequence variants in the FALDH gene in patients with Sjogren‐Larsson syndrome. Hum Mutat. 1998;12:377‐384. [DOI] [PubMed] [Google Scholar]
- 30. Rizzo WB, Craft DA, Somer T, Carney G, Trafrova J, Simon M. Abnormal fatty alcohol metabolism in cultured keratinocytes from patients with Sjogren‐Larsson syndrome. J Lipid Res. 2008;49:410‐419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abdel‐Hamid MS, Issa MY, Elbendary HM, et al. Phenotypic and mutational spectrum of thirty‐five patients with Sjogren‐Larsson syndrome: identification of eleven novel ALDH3A2 mutations and founder effects. J Hum Genet. 2019;64:859‐865. [DOI] [PubMed] [Google Scholar]
- 32. Jackson R. The lines of Blaschko: a review and reconsideration: observations of the cause of certain unusual linear conditions of the skin. Br J Dermatol. 1976;95:349‐360. [DOI] [PubMed] [Google Scholar]
- 33. Gudmundsson S, Wilbe M, Ekvall S, et al. Revertant mosaicism repairs skin lesions in a patient with keratitis‐ichthyosis‐deafness syndrome by second‐site mutations in connexin 26. Hum Mol Genet. 2017;26:1070‐1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kromann AB, Ousager LB, Ali IKM, Aydemir N, Bygum A. Pigmentary mosaicism: a review of original literature and recommendations for future handling. Orphanet J Rare Dis. 2018;13:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Castori M, Tadini G. Discoveries and controversies in cutaneous mosaicism. G Ital Dermatol Venereol. 2016;151:251‐265. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Patient data are saved in the electronic patient records of our hospital.