

Abstract
Analyses of 19 amino acids, 38 acylcarnitines, and 3 creatine analogues (https://clir.mayo.edu) were implemented to test the hypothesis that succinic semialdehyde dehydrogenase deficiency (SSADHD) could be identified in dried bloodspots (DBS) using currently available newborn screening methodology. The study population included 17 post‐newborn SSADHD DBS (age range 0.8‐38 years; median, 8.2 years; 10 M; controls, 129‐353 age‐matched individuals, mixed gender) and 10 newborn SSADHD DBS (including first and second screens from 3 of 7 patients). Low (informative) markers in post‐newborn DBS included C2‐ and C4‐OH carnitines, ornithine, histidine and creatine, with no gender differences. For newborn DBS, informative markers included C2‐, C3‐, C4‐ and C4‐OH carnitines, creatine and ornithine. Of these, only creatine demonstrated a significant change with age, revealing an approximate 4‐fold decrease. We conclude that quantitation of short‐chain acylcarnitines, creatine, and ornithine provides a newborn DBS profile with potential as a first tier screening tool for early detection of SSADHD. This first tier evaluation can be readily verified using a previously described second tier liquid chromatography‐tandem mass spectrometry method for γ‐hydroxybutyric acid in the same DBS. More extensive evaluation of this first/second tier screening approach is needed in a larger population.
Keywords: acylcarnitines, amino acids, creatine, dried bloodspots, newborn screening, succinic semialdehyde dehydrogenase deficiency
Abbreviations
- C2‐carnitine
acetyl‐carnitine
- C4‐OH‐carnitine
3‐hydroxybutyryl‐carnitine
- F
female
- M
male
Synopsis.
Quantitation of ornithine, short‐chain acylcarnitines, and creatine in newborn dried bloodspots may provide a metabolomic profile with potential as an first tier screen for succinic semialdehyde dehydrogenase deficiency.
1. INTRODUCTION
Succinic semialdehyde dehydrogenase deficiency (SSADHD) is a rare genetic disease associated with mutations of the ALDH5A1 gene, tissue accumulation of neuromodulators including γ‐aminobutyric acid (GABA) and γ‐hydroxybutyric acid (GHB), and tissue depletion of glutamine (gln), the precursor of GABA and glutamic acid (glu). SSADHD presents with nonspecific mild to moderate developmental delay, intellectual deficiency, severe expressive language impairment, neuropsychiatric problems (ADHD, obsessive compulsive disorder, autistic behavior), and variable epilepsy.1, 2, 3, 4, 5, 6, 7, 8 There have been reports of sudden unexplained death of epilepsy9, 10 (Gibson, unpublished).
Current approaches and major gaps to patient identification and treatment are summarized in Figure 1. Since SSADHD was first described in 1981, research has focused on identifying the spectrum of pathogenic ALDH5A1 mutations, understanding the molecular and biochemical basis of disease presentation, and testing promising therapeutics. The development of an animal model11 resulted in significant strides in elucidation of pathomechanisms and development of novel preclinical therapeutics, including a recently completed interventional trial with the GABABR antagonist SGS‐742 (http://www.clinicaltrials.gov; NCT02019667), as well as a completed open‐label trial of taurine which failed to demonstrate efficacy.12 Treatment for SSADHD remains symptomatic, yet an expanding therapeutic preclinical pipeline strongly suggests that targeted and effective therapies (in addition to vigabatrin, which directly targets GABA metabolism) for SSADHD are on the horizon.13
Figure 1.
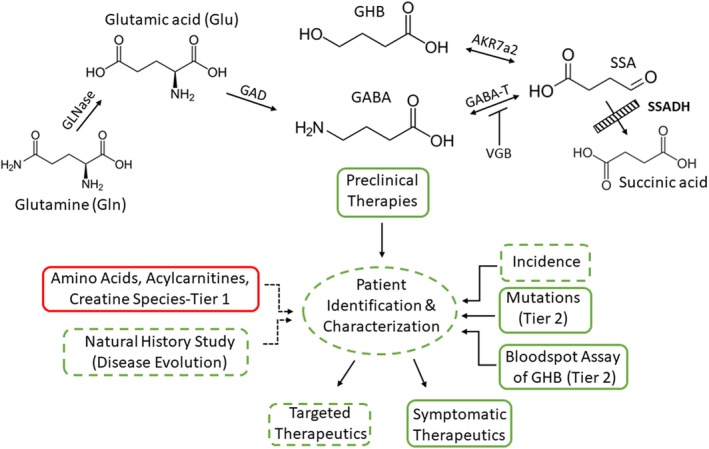
The GABA metabolic pathway (top) and overview of approaches toward patient identification and treatment (bottom). Metabolites elevated in patients with succinic semialdehyde dehydrogenase (SSADH) deficiency (SSADHD) include GABA, SSA (succinic semialdehyde), and GHB (γ‐hydroxybutyric acid); conversely, glutamine (gln) appears decreased. For the diagram, green indicates procedures or measures either achieved (solid line) or partially achieved/in progress (broken line). A tier 1 (tier 2 = confirmation of initial screen) bloodspot assay that is amenable to current NBS platforms is currently required (red). Additional abbreviations: GLNase, glutaminase; GAD, glutamic acid decarboxylase; GABA‐T, GABA‐transaminase (also referred to as ABAT, or aminobutyrate aminotransferase); SSADH, succinic semialdehyde dehydrogenase; AKR7a2, aldo‐keto reductase 7a2. VGB (vigabatrin; γ‐vinylGABA; Sabril) represents an irreversible inhibitor of GABA‐T
Implementation of newborn screening (NBS) for SSADHD is clinically important for a number of reasons. Expanded NBS would maximize the therapeutic benefit of targeted therapeutics, facilitate family planning, and remove the potential off‐target effects of commonly used symptomatic agents, effectively constituting “treatment by omission.” Further, NBS could synergize with a recently undertaken natural history study of SSADHD through significant expansion of the span of longitudinal assessment of the disorder. NBS would detect patients who will develop milder forms of the disease, or patients with clinical presentations so subtle that they may be misdiagnosed. The potential for delayed diagnosis is highlighted in the postmortem identification of an adult male in the fifth decade.5 Early patient diagnosis would further serve to refine disease prevalence estimates, increase the pool of patients available for a natural history study and future clinical trials, and provide insight on disease pathogenesis and the genetic factors that track with milder disease presentation.
The incidence of SSADHD has been estimated at 1 × 106 based upon mutation (allele) frequency (Dr Nilah Monnier, Stanford‐personal communication). Mutation analysis has been available for many years.14 Detection of SSADHD has continued to expand with the addition of ALDH5A1 gene analysis to several commercial epilepsy and intellectual disability panels; however, only ~50% of SSADHD patients have seizures, suggesting that many patients remain undiagnosed. On the other hand, methodology for GHB quantitation in dried bloodspots (DBS) has been presented.15 That method utilized liquid chromatography‐tandem mass spectrometry (LC‐MS/MS). At least in the United States, only a very limited number of states employ LC/MS‐MS for metabolite quantification, primarily because it is insufficiently high‐throughput (run time 2‐3 minutes per DBS vs 1 minute with MS/MS alone). States with smaller populations, and an often correspondingly lower birthrate, have the capacity to employ LC‐MS/MS which can expand their metabolic screening “menu,” but states with larger populations (New York, California, and Texas) may only have the capacity to employ LC‐MS/MS as a second tier screen, or for very specialized analytical needs. Nonetheless, GHB quantitation in DBS represents a very attractive second tier test for confirmation of SSADHD.
Recent surveys of SSADHD families worldwide indicate that the median age at disease onset is 1 year whereas the median age at diagnosis is 3 years, that is, a 2‐year delay after the first clinical symptoms. Under current circumstances (with a median age at diagnosis of 3 years), any natural history study of SSADHD would not include patients diagnosed in the early newborn period, representing a major confound from the neurodevelopmental perspective of a disease manifesting a prominent neurological phenotype. Moreover, these survey data provide a measurable perspective on the disease burden for SSADHD families who have to wait 3 years to achieve a diagnosis, as well as the public health and societal impact of delayed diagnosis. To address these unmet healthcare needs, we examined the hypothesis that an integrated screen of 19 amino acids, 38 acylcarnitines, and 3 creatine analogues (https://clir.mayo.edu) could be used to potentially identify a metabolomic pattern that could be used as an first‐tier screening tool for SSADHD using DBS, an approach that has been successful in a number of inborn errors of metabolism.16, 17, 18
2. MATERIALS AND METHODS
2.1. Dried bloodspots
DBS from post‐newborn SSADHD patients were collected with informed consent (WSU IRB 15901). Seventeen post‐newborn DBS included: 10 M/7F, ages 0.8‐38 years (median, 8.2), and 4 sibships (total, 8 patients), representing ~10% of published cases.19 SSADHD was previously confirmed through a combination of GHB measurement (urine, DBS), ALDH5A1 molecular analyses and expression, and assay of SSADH in white cells for older patients (Table 1). DBS were obtained using standard finger lance and blood collected onto 903 five spot blood cards (Eastern Business Cards, Greenville, South Carolina). Reference DBS encompassed an archival collection in the Mayo Clinical Laboratories (n = 129‐353, age range 0.5‐87.9 years; mixed gender).
Table 1.
Characteristics of patients from whom post‐newborn dried bloodspots were obtained
| Patient | Age (year) | Gender | Mutation 1 | Mutation 2 | Zygosity | GHB (physiol. fluid) | Notes on alleles |
|---|---|---|---|---|---|---|---|
| 1 | 10 | F | p.W204* | p.R425* | CH | Elevated | |
| 2 | 8 | M | p.W204* | p.R425* | CH | Elevated | |
| 3 | 24 | M | p.W204* | p.W204* | HZ | Elevated | <10% of nl in white cells |
| 4 | 40 | M | p.W204* | c.1054‐2A>Ca | CH | Elevated | Undetectable in white cells |
| 5 | M | NA | NA | ||||
| 6 | 1.5 | M | p.G176R | p.G409D | CH | 843 mmol/mol (urine; nl < 9) | Both alleles tested in HEK293 overexpression: 0% residual activity14 |
| 7 | 26 | F | p.G196D | p.G196D | HZ | CSF, 594; sera, 265 μmol/L (nl < 3, both fluids) | Allele p.G196D: 9% residual activity in HEK293 overexpression (Pop et al, unpublished) |
| 8 | 11 | M | See legend | See legend | 325 mmol/mol (urine; nl < 9) | ||
| 9 | 7 | F | p.T233M | p.T233M | HZ | Elevated | Allele tested in HEK293 overexpression: 4% residual activity14 |
| 10 | 15 | F | p.T233 M | p.T233M | HZ | Elevated | See above |
| 11 | 7 | M | p.C93F | p.C531Y | CH (parents untested) | 477 mmol/mol creatinine (<10) | Allele C93F in HEK293 overexpression: 3% residual activity14; C531Y 1% (Pop et al, unpublished) |
| 12 | 10 | M | p.C93F | p.C531Y | CH (parents untested) | 114 mmol/mol creatinine (<10) | See above |
| 13 | 18 | F | c.621delC | c.621delC | HZ | Elevated | |
| 14 | 18 | M | p.C93F | p.C93F | HZ | 119 mmol/mol (urine; nl < 9) | See above20 |
| 15 | 2 | F | p.G252C | p.G252C | HZ | Elevated | Allele p.G252C: 6% in HEK293 overexpression (Pop et al, unpublished) |
| 16 | 5 | F | p.G252C | p.G252C | HZ | Elevated | See above |
| 17 | 8 | M | p.G252C | p.G252C | HZ | Elevated | See above |
Note: Sibships: patients 2 and 3; 9 and 10; 11 and 12; and 15‐17; all confirmed by sequencing of the parents. For patient 8, Sanger sequencing of ALDH5A1 gene, exon 1 could not be amplified, suggesting a homozygous deletion (MLPA confirmation pending). Control range (nl) for SSADH activity in extracts of white cells, 1.9‐3.9 nmol/min/mg.
Abbreviations: CH, compound heterozygous; HZ, homozygous; NA, not available.
Splice variant at the canonical splice site which is considered pathogenic.
Newborn DBS were obtained with parental and State Newborn Screening Laboratory consents from seven patients (10 DBS), including three patients providing both first and second screens (approximate age at collection, 48 and 340 hours) (Table 2). Conditions of DBS storage included three first screens stored at 4°C, with the remainder kept at room temperature. Patient overlap between newborn and post‐newborn samples included a single sibship (first, second, and post‐newborn DBS from one sibling, and a second screen [first screen unavailable] with post‐newborn DBS from the older sibling).
Table 2.
Characteristics of patients from whom newborn dried bloodspots were obtained
| Patient | Current age (year) | Gender | Screen 1 | Screen 2 | Mutation 1 | Mutation 2 | Zygosity | GHB (urine) |
| 1 | 10 | F | x | p.W204* | p.R425* | CH | Elevated comparable to other patients | |
| 2 | 8 | M | x | p.W204* | p.R425* | CH | Elevated comparable to other patients | |
| 2 | 8 | M | x | p.W204* | p.R425* | CH | Elevated comparable to other patients | |
| 3 | 4 | M | x | p.G533R | c.1015‐2A>Ca | CH | Elevated comparable to other patients | |
| 3 | 4 | M | x | p.G533R | c.1015‐2A>Ca | CH | Elevated comparable to other patients | |
| 4 | 27 | M | x | NA | NA | NA | ||
| 5 | 8 | F | x | p.M445L | c.610‐2A>G | CH | “Marked elevation”b | |
| 6 | 9 | M | x | p.W204* | p.G441Rc | CH | 79‐156 mmol/mol | |
| 7 | 5 | F | x | c.104_127del p.Ser35* | c.1054‐2A>C | CH | “Marked elevation” | |
| 7 | 5 | F | x | c.104_127del p.Ser35* | c.1054‐2A>C | CH | “Marked elevation” |
Note: Patients 1 and 2 identical to patients 1 and 2 in Table 1; for urine GHB, control values are <9 mmol/mol creatinine.
Abbreviations: CH, compound heterozygous; HZ, homozygous.
In addition to elevated 4‐hydroxybutyric acid, the urine organic acids revealed elevations of 4,5‐dihydroxyhexanoic, glutaric, adipic, glycolic, 3‐hydroxypropionic, and 2‐hydroxyglutaric acids, all hallmarks of SSADH deficiency, in two unique urine samples.22, 23
PolyPhen characterization of p.G441R indicated a strong likelihood of pathogenicity.
2.2. Metabolic measurements
Analyses of amino acids, acylcarnitines (saturated and unsaturated), and creatine derivatives were performed using tandem mass spectrometry as previously described.18, 24 A single 3 mm punch from one DBS was used. At this time, we do not have insights into specific metabolic profile changes due to prematurity, drug treatment, alimentation, or other external factors which might influence the screening results. This will need to await implemented and expanded NBS for SSADH deficiency.
2.3. Collaborative Laboratory Integrated Reports
Collaborative Laboratory Integrated Reports (CLIR 2.12; https://clir.mayo.edu) is a web application that maintains an interactive database of laboratory results from multiple sites and provides on demand clinical decision support for their integrated interpretation.25 CLIR replace conventional reference intervals with continuous, covariate‐adjusted moving percentiles,26 as well as replacement of analyte decision limits (ie, cutoff values) with a condition‐specific degree of overlap between reference and disease ranges. Further, an additional feature of CLIR incorporates integration of primary markers and unbiased biomarker discovery by automated calculation of all possible permutations of ratios (A/B) plus manual selection of complex ratios and equations, which can be performed simultaneously for all conditions that can be diagnosed from the available laboratory measurements.
2.4. Data and statistical analyses
Metabolic measures were integrated within CLIR, facilitating comparison with anonymized DBS data from multiple NBS centers throughout the world. Analyte‐covariate analysis employed age as covariate. Gender was known for reference and patient values, but was only assessed as covariate for informative markers in the post‐newborn DBS samples of SSADHD. Statistical analysis included one‐way analysis of variance or two‐tailed t test using GraphPad Prism 8.0 (San Diego, California).
3. RESULTS
3.1. Informative biomarkers in newborn vs post‐newborn DBS
Informative biomarkers are shown in Figure 2 (A, newborn DBS; B, post‐newborn DBS). The y‐axis depicts metabolite abbreviations in units of μM. Reference data are shown as green boxes, and depict data falling between the 1%ile and the 99%ile of the reference population for that marker. Patient values are depicted in box and whisker format in quartile ranges. Error bars on the box represent the 1%ile (lowest) and 99%ile (highest) error bar. The bottom of the box represents the 10%ile while the top of the box represents the 90%ile, with the median represented by the horizontal line. The x‐axis depicts multiples of reference median in logarithmic scale for newborn reference data (A) and in exponential scale for post‐newborn samples (B). Informative markers (blue) for newborn DBS included C2‐, C3‐, C4‐ and C4‐OH carnitines, ornithine, and creatine (Figure 2A). For post‐newborn DBS, informative markers (blue) included C2‐ and C4‐OH carnitines, ornithine, histidine, and creatine.
Figure 2.
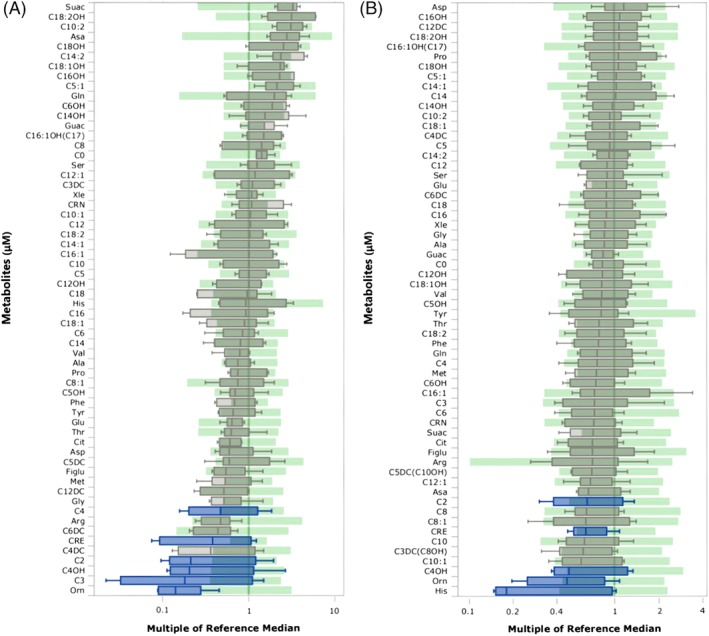
Comparison of multiple analytes against reference intervals for (A) newborn SSADHD DBS and (B) post‐newborn SSADHD DBS. Data for patients is presented as box and whisker (quartiles; 1%ile, 10%ile, median, 90%ile, 99%ile, whereas the reference range (green) lacks whiskers and presents data from 1%ile to 99%ile of reference for that marker. For (A), informative markers (blue) included ornithine, C2‐, C3‐, C4‐ and C4‐OH carnitines, and creatine; for (B), informative markers (blue) included histidine, ornithine, C2‐ and C4‐OH carnitines, and creatine. X‐axis values depict multiple of the reference median, shown for (A) in log scale and for (B) in exponential scale. Amino acids are shown in standard three letter code (eg, his = histidine). Xle represents the sum of leucine and isoleucine, isobaric species. Additional abbreviations: guac, guanidinoacetic acid; C0‐, free carnitine; crn, creatine; suac, succinylacetone; figlu, formiminoglutamic acid. Acylcarnitine metabolites are depicted as chain‐length (eg, C12 = dodecanoylcarnitine), as a monounsaturated (eg, C8:1) or diunsatured (C14:2) species, as the hydroxylated species (eg, C16OH), or as the unsaturated, hydroxylated species (C18:1OH). In selected instances (C16:1OH(C17), C5DC(C10OH), C3DC(C8OH)), identical molecular ions are produced such that the value represents the sum of the two isobaric species shown. DC represents dicarboxylic acid carnitine species (eg, C3DC, malonylcarnitine)
The individual data points for Figure 2A,B are comprehensively described in Table 3 (newborn DBS, corresponding to Figure 2A) and Table 4 (post‐newborn DBS, corresponding to Figure 2B). Summary data highlight the mean and SEM (SE of the mean) for the 10 newborn (Table 3) and 17 post‐newborn DBS (Table 4), as well as showing the summary characteristics of data for parallel control DBS available from the CLIR database. For control newborn DBS, shown are the 1st, 50th and 99th centiles for individual control data points ranging from n = 9840 for creatine to up to n = 5 089 207 data points for C3‐carnitine. For control post‐newborn DBS, shown are the 1st and 99th centiles, as well as the median, for individual control DBS data points representing n = 129 for creatine up to n = 353 data points for ornithine.
Table 3.
Summary of informative markers from newborn dried bloodspots of SSADH‐deficient patients in comparison to control percentiles
| Patients | Age (h) | Sex | Ornithine | C2 | C3 | C4 | C4‐OH | CRE |
|---|---|---|---|---|---|---|---|---|
| 336 | F | 12.3 | 2.7 | 0.06 | 0.03 | 0.033 | 39.9 | |
| 48 | M | 6.7 | 3.2 | 0.08 | 0.045 | 0.02 | 101.4 | |
| 360 | M | 6.6 | 2.1 | 0.04 | 0.048 | 0.027 | 30.9 | |
| 48 | M | 10.8 | 4.9 | 0.29 | 0.067 | 0.047 | 161.4 | |
| 312 | M | 10.4 | 3.1 | 0.19 | 0.059 | 0.022 | 53.3 | |
| 48 | M | 9.4 | 6.8 | 0.61 | 0.196 | 0.054 | 216.5 | |
| 48 | F | 19.2 | 45.2 | 2.56 | 0.26 | 0.50 | 438.5 | |
| 48 | M | 35.4 | 24.6 | 1.76 | 0.41 | 0.168 | 521.0 | |
| 336 | F | 15.6 | 5.4 | 0.33 | 0.188 | 0.040 | 159.2 | |
| 48 | F | 9.1 | 4.7 | 0.42 | 0.139 | 0.033 | 209.9 | |
| Summary | Mean | 13.6 | 10.3 | 0.63 | 0.14 | 0.09 | 193.2 | |
| SEM | 2.7 | 4.4 | 0.27 | 0.04 | 0.05 | 52.5 | ||
| Controls | Count | 2 379 498 | 2 935 441 | 5 089 207 | 2 939 759 | 1 065 684 | 9840 | |
| 1%ile | 20.4 | 8.5 | 0.6 | 0.10 | 0.06 | 254 | ||
| 50%ile | 72 | 22.9 | 1.7 | 0.22 | 0.18 | 425 | ||
| 99%ile | 235 | 47.8 | 4.1 | 0.58 | 0.46 | 684 |
Ages (h) are approximate. C2‐, C3‐, C4‐, and C4‐OH represent acylcarnitine species (corresponding to Figure 2A). Additional abbreviations: Count represents the number of newborn data points available from CLIR; CRE, creatine; ile, percentile; SEM, SE of the mean.
Table 4.
Summary of informative markers in post‐newborn dried bloodspots from SSADH‐deficient patients in comparison to control
| Patients | Age (year) | Sex | Ornithine | Histidine | C2 | C4‐OH | CRE |
|---|---|---|---|---|---|---|---|
| 22.7 | M | 28.9 | 26.3 | 5.9 | 0.06 | 147.8 | |
| 8.7 | F | 8.8 | 25.4 | 4.7 | 0.04 | 138.3 | |
| 6.8 | M | 6.3 | 21.0 | 4.3 | 0.06 | 126.9 | |
| 38.2 | M | 27.1 | 29.4 | 12.1 | 0.07 | 151.4 | |
| 4.1 | M | 9.4 | 25.6 | 7.5 | 0.05 | 154.4 | |
| 0.9 | M | 14.2 | 20.6 | 17.3 | 0.15 | 134.1 | |
| 24.3 | F | 15.7 | 25.4 | 7.7 | 0.05 | 164.9 | |
| 9.6 | M | 12.1 | 25.3 | 6.2 | 0.07 | 176.8 | |
| 5.8 | F | 21.9 | 49.4 | 9.2 | 0.06 | 166.6 | |
| 13.5 | F | 19.1 | 66.4 | 10.2 | 0.05 | 251.5 | |
| 5.6 | M | 20.8 | 141.1 | 9.3 | 0.05 | 133.1 | |
| 8.2 | M | 27.1 | 129.0 | 9.1 | 0.05 | 137.9 | |
| 16.6 | F | 36.9 | 144.2 | 12.9 | 0.14 | 240.5 | |
| 7.1 | M | 12.5 | 21.7 | 9.6 | 0.04 | 121.8 | |
| 4.2 | F | 8.8 | 22.8 | 9.2 | 0.05 | 119.4 | |
| 0.8 | F | 7.6 | 21.7 | 15.4 | 0.11 | 108.7 | |
| 18.4 | M | 27.6 | 113.3 | 19.7 | 0.15 | 182.0 | |
| Summary | Mean | 17.9 | 53.5 | 10.0 | 0.07 | 156.2 | |
| SEM | 2.2 | 11.3 | 1.0 | 0.01 | 9.6 | ||
| Controls | Count | 353 | 129 | 350 | 347 | 343 | |
| Min | 13.3 | 41.0 | 6.0 | 0.04 | 115.2 | ||
| Max | 88.2 | 320.3 | 40.1 | 0.40 | 475.2 | ||
| Mean | 35.3 | 131.0 | 15.4 | 0.13 | 243.2 | ||
| 1%ile | 14.4 | 54.8 | 6.9 | 0.05 | 127.8 | ||
| Median | 33.4 | 121.5 | 14.4 | 0.12 | 234.7 | ||
| 99%ile | 72.3 | 309.6 | 33.9 | 0.34 | 444.8 |
Control age range, 0.5‐87.9 years, mean 38.3 years. C2‐ and C4‐OH represent informative acylcarnitines (refer to Figure 2B). Additional abbreviations: Count represents the number of control data points available from CLIR; CRE, creatine; ile, percentile; SEM, SE of the mean; min, minimum; max, maximum.
3.2. Comparison of gender and screen number for informative markers
Sufficient samples were only available for post‐newborn DBS, and for the informative markers noted above there were no significant differences with respect to gender. For informative markers in newborn DBS, only creatine demonstrated a significant change with age (first screen (~48 hours post birth), [275 ± 68 μM (range 101‐521, n = 6)]; second screen (~312‐360 hours post birth), [71 ± 30 μM (range 31‐159, n = 4); P < .05, two tailed t test]). This was not unexpected given the inverse age relationship for creatine noted above.
4. DISCUSSION
4.1. Comparison of metabolic panels to detect SSADHD in DBS
4.1.1. Informative amino acid markers
Based upon the known metabolic correlations between glutamine, glutamate, and GABA,27, 28, 29 our initial prediction was glutamic acid and glutamine would serve as informative markers for SSADHD in DBS. These amino acids were, however, noninformative, whereas ornithine and histidine were. Indeed, the most consistent amino acid dysregulation was that of ornithine, both in post‐newborn and newborn DBS. Low ornithine has hitherto not been reported in plasma amino acid analysis of patients with SSADHD. Conversely, ornithine has been implicated in the ocular toxicity associated with vigabatrin, an antiepileptic whose mode of action encompasses irreversible inactivation of GABA‐transaminase (Figure 1) with concomitant elevation of GABA, a finding analogous to that of SSADHD.30 Shank and Campbell31 demonstrated that both orn and gln can serve to replenish glu and GABA pools, although gln has a more prominent role in this process.
Of interest, histidine was only informative for post‐newborn DBS and not newborn DBS. It is noteworthy that histidine is conjugated with GABA in CNS to derive the dipeptide homocarnosine, an osmoregulator that is also increased in cerebrospinal fluid of SSADHD patients.32, 33 Nevertheless, we could not document the presence of homocarnosine in post‐newborn SSADHD DBS, perhaps indicating that the enzyme required for GABA‐histidine conjugation is not active in the newborn period.
4.1.2. Informative acylcarnitine markers
Short‐chain acylcarnitine species were informative in both post‐newborn and newborn SSADHD DBS, encompassing C2‐ and C4‐OH carnitine in the post‐newborn samples and C2‐, C3‐, C4‐, and C4‐OH in newborn SSADHD DBS. This may not be surprising in view of the accumulation of both GHB and succinic semialdehyde in SSADHD,13, 34 which may interfere with short chain fatty acid metabolism. This observation is further supported by the early reports of dicarboxylic aciduria and unusual tetronic acid derivatives in SSADHD.22, 35 Depleted levels of acetyl‐carnitine further suggest reduced mitochondrial function, which we and others have observed both in SSADHD and other disorders of fat oxidation.36, 37 Low levels of 3‐hydroxybutyryl‐carnitine may also provide insight into the success of the ketogenic diet in aldh5a1 −/− mice.38 Administration of the ketogenic diet to these animals significantly elevated blood levels of 3‐hydroxybutyrate while significantly improving the phenotype of seizures and runted growth in this model.
4.1.3. Creatine as an informative marker for SSADHD DBS
Creatine was an informative marker in both post‐newborn and newborn SSADHD DBS. Previously, we had documented the presence of 4‐guanidinobutyrate in tissue and physiological fluids of both aldh5a1 −/− mice and patients with SSADHD.39, 40 This species is predicted to derive from conjugation of GABA (in lieu of glycine) in the arginine:glycine amidinotransferase reaction of the creatine biosynthetic pathway. On the other hand, if this pathway is engaged, we might expect depletion of arginine in DBS, which was not an informative amino acid marker in either post‐newborn or newborn SSADHD DBS.
4.1.4. Utility of metabolomic profiles as a potential 1st tier screen for SSADH deficiency
For both newborn and post‐newborn SSADHD DBS, there was a degree of overlap with reference ranges. However, the individual markers taken together may provide a metabolomic profile with a high degree of probability for accurate detection of SSADH deficiency. For example, it is informative to compare the mean values of biomarkers for patients (n = 10; Table 3) in comparison to the 1%ile of the control data range (n = 9840‐5 089 207) (the appropriate data are italicized in Table 3). These results highlight the fact that the patient means are below this control percentile for both ornithine and creatine, and very close to this percentile for the four acylcarnitine species. Accordingly, the newborn profile of C2‐, C3‐, C4‐ and C4‐OH acylcarnitines, creatine, and ornithine in DBS may highlight an informative “biosignature” of SSADH deficiency.
For post‐newborn DBS (Table 4), it is similarly informative to compare the mean of patient DBS (n = 17) to the 1%ile of the control DBS (n = 129‐353) for the five biomarkers shown, including orn, his, C2‐ and C4‐OH acylcarnitines, and creatine. For histidine, the mean patient DBS data were below the 1%ile of parallel DBS control data, while the mean C4‐OH for patient DBS is proximal to the parallel 1%ile control DBS data (pertinent data sets in Table 4 are italicized), while the mean for all five biomarkers resides well below the median of control DBS data. The fact that four of six biomarkers show parallel patterns between newborn and post‐newborn SSADHD DBS (ornithine, C2‐ and C4‐OH‐acylcarnitines, and creatine) lends further credence to the idea that this “biosignature” of biomarkers is informative for SSADH deficiency. At this time, and in the absence of specific funding for those studies, retrospective “in silico” analyses of metabolic patients in archival newborn DBS has not been undertaken, but such data will provide key sensitivity and specificity data, as well as predictive value, for the metabolomic profile results we have observed.
5. LIMITATIONS AND FUTURE STUDIES
A primary limitation with this study is the low number of both post‐newborn and newborn SSADHD DBS, especially the latter. This issue is compounded by the fact that different states within the USA retain archival NBS DBS for variable times, with some states discarding samples at the end of 1 year, others at 6‐8 years, and still others retaining their samples for decades or indefinitely. This provides inherent challenges in accruing substantial numbers of newborn SSADHD DBS. Moreover, conditions of storage are often variable, with some states storing archival DBS at room temperature, while others may keep these samples at 4°C. For post‐newborn DBS, an additional confound is medication (we did not have information on medication intake for the current study), and the potential influence of medications on DBS metabolites. We plan to significantly expand the number of newborn SSADHD DBS for further evaluation.
It remains to be determined if GABA (free, or esterified) can be quantified in human DBS, but at present this analyte is not present on US NBS panels. Moreover, it would be worthwhile to evaluate the current SSADHD DBS for the presence of 4‐guanidinobutyrate, which may be a potentially relevant biomarker for SSADHD, as well as further characterizing the presence/absence of homocarnosine and carnosine (the latter the dipeptide of his and β‐alanine). These might be pertinent to the development of a sensitive first‐tier screen for SSADHD using DBS. A second‐tier screen already exists that can quantify GHB in DBS (see Introduction15, 41) or via mutation analysis.14 Finally, any methodology pertinent to the newborn detection of SSADHD may have relevance to the identification of GABA‐transaminase deficiency,42 as more patients are being identified. With regard the latter, our prediction is that ornithine will remain an informative marker in DBS.
6. CONCLUSIONS
Quantitation of short‐chain acylcarnitines, creatine, and ornithine provides a newborn DBS profile with potential as a first tier screening tool for early detection of SSADHD. This first tier evaluation can be readily verified using a previously described second tier LC‐MS/MS method for GHB in the same DBS. Our approach awaits more extensive evaluation in a larger population.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
AUTHOR CONTRIBUTIONS
Brown, Turgeon, Rinaldo‐data derivation, drafting of manuscript, editing of manuscript.
Pop, Salomons‐data derivation, statistical analyses, manuscript editing, data analysis.
Roullet and Gibson‐preparation of initial manuscript draft, editing, data and statistical analyses.
ETHICAL APPROVAL STATEMENT
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all families and State Newborn Screening Programs for study inclusion.
ACKNOWLEDGMENTS
This study was supported by R01 HD91142 from the National Institutes of Health. We gratefully acknowledge the assistance of the Biochemical Genetics Laboratory of the Mayo Clinic, Rochester, Minnesota, for assistance in the development of this work using the Collaborative Laboratory Integrated Reports (https://clir.mayo.edu). The assistance of Drs. Yilmaz Yildiz, Francois Feillet, Johannes Haberle, Gabriela Horvath, Ellen Crushell, and Saadet Andrews in provision of dried bloodspots and clinical details from screening for succinic semialdehyde dehydrogenase deficiency patients is gratefully acknowledged. We thank the parents of patients, and the patients themselves, for their support of this study.
Brown M, Turgeon C, Rinaldo P, et al. Longitudinal metabolomics in dried bloodspots yields profiles informing newborn screening for succinic semialdehyde dehydrogenase deficiency. JIMD Reports. 2020;53:29–38. 10.1002/jmd2.12075
Madalyn Brown and Coleman Turgeon contributed equally to this study.
Funding information National Institutes of Health, Grant/Award Number: HD091142
REFERENCES
- 1. Gibson KM, Gupta M, Pearl PL, et al. Significant behavioral disturbances in succinic semialdehyde dehydrogenase (SSADH) deficiency (gamma‐hydroxybutyric aciduria). Biol Psychiatry. 2003;54:763‐768. [DOI] [PubMed] [Google Scholar]
- 2. Gogou M, Spilioti M, Tramma D, Papadopoulou‐Alataki E, Evangeliou A. Succinic Semialdehyde dehydrogenase deficiency presenting as autism Spectrum disorder. Indian J Pediatr. 2016;83:1036‐1037. [DOI] [PubMed] [Google Scholar]
- 3. Knerr I, Gibson KM, Jakobs C, Pearl PL. Neuropsychiatric morbidity in adolescent and adult succinic semialdehyde dehydrogenase deficiency patients. CNS Spectr. 2008;13:598‐605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Knerr I, Gibson KM, Murdoch G, et al. Neuropathology in succinic semialdehyde dehydrogenase deficiency. Pediatr Neurol. 2010;42:255‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lapalme‐Remis S, Lewis EC, De Meulemeester C, et al. Natural history of succinic semialdehyde dehydrogenase deficiency through adulthood. Neurology. 2015;85:861‐865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Parviz M, Vogel K, Gibson KM, Pearl PL. Disorders of GABA metabolism: SSADH and GABA‐transaminase deficiencies. J Pediatr Epilepsy. 2014;3:217‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pearl PL, Parviz M, Vogel K, Schreiber J, Theodore WH, Gibson KM. Inherited disorders of gamma‐aminobutyric acid metabolism and advances in ALDH5A1 mutation identification. Dev Med Child Neurol. 2014;57:611‐617. 10.1111/dmcn.12668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vogel KR, Pearl PL, Theodore WH, McCarter RC, Jakobs C, Gibson KM. Thirty years beyond discovery – clinical trials in succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism. J Inherit Metab Dis. 2013;36:401‐410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Horino A, Kawawaki H, Fukuoka M, et al. A case of succinic semialdehyde dehydrogenase deficiency with status epilepticus and rapid regression. Brain Dev. 2016;38:866‐870. [DOI] [PubMed] [Google Scholar]
- 10. Pearl PL, Shukla L, Theodore WH, Jakobs C, Gibson KM. Epilepsy in succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism. Brain Dev. 2011;33:796‐805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hogema BM, Gupta M, Senephansiri H, et al. Pharmacologic rescue of lethal seizures in mice deficient in succinate semialdehyde dehydrogenase. Nat Genet. 2001;29:212‐216. [DOI] [PubMed] [Google Scholar]
- 12. Schreiber JM, Pearl PL, Dustin I, et al. Biomarkers in a Taurine trial for succinic Semialdehyde dehydrogenase deficiency. JIMD Rep. 2016;30:81‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Malaspina P, Roullet J‐B, Pearl PL, Ainslie GR, Vogel KR, Gibson KM. Succinic semialdehyde dehydrogenase deficiency (SSADHD): pathophysiological complexity and multifactorial trait associations in a rare monogenic disorder of GABA metabolism. Neurochem Int. 2016;99:72‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Akaboshi S, Hogema BM, Novelletto A, et al. Mutational spectrum of the succinate semialdehyde dehydrogenase (ALDH5A1) gene and functional analysis of 27 novel disease‐causing mutations in patients with SSADH deficiency. Hum Mutat. 2003;22:442‐450. [DOI] [PubMed] [Google Scholar]
- 15. Forni S, Pearl PL, Gibson KM, Yu Y, Sweetman L. Quantitation of gamma‐hydroxybutyric acid in dried blood spots: feasibility assessment for newborn screening of succinic semialdehyde dehydrogenase (SSADH) deficiency. Mol Genet Metab. 2013;109:255‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de Sain‐van der Velden MG, Rinaldo P, Elvers B, et al. The proline/citrulline ratio as a biomarker for OAT deficiency in early infancy. JIMD Rep. 2012;6:95‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rinaldo P, Lim JS, Tortorelli S, Gavrilov D, Matern D. Newborn screening of metabolic disorders: recent progress and future developments. Nestle Nutr Workshop Ser Pediatr Program. 2008;2:81‐93. [DOI] [PubMed] [Google Scholar]
- 18. Turgeon C, Magera MJ, Allard P, et al. Combined newborn screening for succinylacetone, amino acids, and acylcarnitines in dried blood spots. Clin Chem. 2008;54:657‐664. [DOI] [PubMed] [Google Scholar]
- 19. Attri SV, Singhi P, Wiwattanadittakul N, et al. Incidence and geographic distribution of succinic semialdehyde dehydrogenase (SSADH) deficiency. JIMD Rep. 2017;34:111‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cordeiro D, Bullivant G, Cohn RD, Raiman J, Mercimek‐Andrews S. Outcome of patients with inherited neurotransmitter disorders. Can J Neurol Sci. 2018;45:571‐576. [DOI] [PubMed] [Google Scholar]
- 21. Xiong HY, Alipanahi B, Lee LJ, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015;347(6218):1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brown GK, Cromby CH, Manning NJ, Pollitt RJ. Urinary organic acids in succinic semialdehyde dehydrogenase deficiency: evidence of alpha‐oxidation of 4‐hydroxybutyric acid, interaction of succinic semialdehyde with pyruvate dehydrogenase and possible secondary inhibition of mitochondrial beta‐oxidation. J Inherit Metab Dis. 1987;10:367‐375. [DOI] [PubMed] [Google Scholar]
- 23. Yamakawa Y, Nakazawa T, Ishida A, et al. A boy with a severe phenotype of succinic semialdehyde dehydrogenase deficiency. Brain Dev. 2012;34:107‐112. [DOI] [PubMed] [Google Scholar]
- 24. Tortorelli S, Eckerman JS, Orsini JJ, et al. Moonlighting newborn screening markers: the incidental discovery of a second‐tier test for Pompe disease. Genet Med. 2018;20:840‐846. [DOI] [PubMed] [Google Scholar]
- 25. Hall PL, Marquardt G, McHugh DMS, et al. Post‐analytical tools improve performance of newborn screening by tandem mass spectrometry. Genet Med. 2014;16:889‐895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mørkrid L, Rowe AD, Elgstoen KBP, et al. Continuous age‐ and gender‐adjusted reference intervals of urinary markers for cerebral creatine deficiency syndromes: a novel approach to the definition of reference intervals. Clin Chem. 2015;61:760‐768. [DOI] [PubMed] [Google Scholar]
- 27. Chowdhury GM, Gupta M, Gibson KM, Patel AB, Behar KL. Altered cerebral glucose and acetate metabolism in succinic semialdehyde dehydrogenase‐deficient mice: evidence for glial dysfunction and reduced glutamate/glutamine cycling. J Neurochem. 2007;103:2077‐2091. [DOI] [PubMed] [Google Scholar]
- 28. Cooper AJ, Jeitner TM. Central role of glutamate metabolism in the maintenance of nitrogen homeostasis in normal and hyperammonemic brain. Biomolecules. 2016;6(2). 10.3390/biom6020016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang J, Shen J. Elevated endogenous GABA concentration attenuates glutamate‐glutamine cycling between neurons and astroglia. J Neural Transm (Vienna). 2009;116:291‐300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vogel KR, Ainslie GR, Walters DC, et al. Succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism: an update on pharmacological and enzyme‐replacement therapeutic strategies. J Inherit Metab Dis. 2018;41:699‐708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shank RP, Campbell GL. Ornithine as a precursor of glutamate and GABA: uptake and metabolism by neuronal and glial enriched cellular material. J Neurosci Res. 1983;9:47‐57. [DOI] [PubMed] [Google Scholar]
- 32. Gupta M, Polinsky M, Senephansiri H, et al. Seizure evolution and amino acid imbalances in murine succinate semialdehyde dehydrogenase (SSADH) deficiency. Neurobiol Dis. 2004;16:556‐562. [DOI] [PubMed] [Google Scholar]
- 33. Jansen EE, Gibson KM, Shigematsu Y, Jakobs C, Verhoeven NM. A novel, quantitative assay for homocarnosine in cerebrospinal fluid using stable‐isotope dilution liquid chromatography‐tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;830:196‐200. [DOI] [PubMed] [Google Scholar]
- 34. Struys EA, Jansen EE, Gibson KM, Jakobs C. Determination of the GABA analogue succinic semialdehyde in urine and cerebrospinal fluid by dinitrophenylhydrazine derivatization and liquid chromatography‐tandem mass spectrometry: application to SSADH deficiency. J Inherit Metab Dis. 2005;28:913‐920. [DOI] [PubMed] [Google Scholar]
- 35. Gibson KM, Goodman SI, Frerman FE, Glasgow AM. Succinic semialdehyde dehydrogenase deficiency associated with combined 4‐hydroxybutyric and dicarboxylic acidurias: potential for clinical misdiagnosis based on urinary organic acid profiling. J Pediatr. 1989;114:607‐610. [DOI] [PubMed] [Google Scholar]
- 36. Ribel‐Madsen A, Ribel‐Madsen R, Brøns C, Newgard CB, Vaag AA, Hellgren LI. Plasma acylcarnitine profiling indicates increased fatty acid oxidation relative to tricarboxylic acid cycle capacity in young, healthy low birth weight men. Physiol Rep. 2016;4(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sauer SW, Kölker S, Hoffmann GF, et al. Enzymatic and metabolic evidence for a region specific mitochondrial dysfunction in brains of murine succinic semialdehyde dehydrogenase deficiency (aldh5a1 −/− mice). Neurochem Int. 2007;50:653‐659. [DOI] [PubMed] [Google Scholar]
- 38. Nylen K, Velazquez JL, Likhodii SS, et al. A ketogenic diet rescues the murine succinic semialdehyde dehydrogenase deficient phenotype. Exp Neurol. 2008;210:449‐457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jansen EE, Verhoeven NM, Jakobs C, et al. Increased guanidino species in murine and human succinate semialdehyde dehydrogenase (SSADH) deficiency. Biochim Biophys Acta. 2006;1762:494‐498. [DOI] [PubMed] [Google Scholar]
- 40. Jansen EE, Struys E, Jakobs C, Hager E, Snead OC, Gibson KM. Neurotransmitter alterations in embryonic succinate semialdehyde dehydrogenase (SSADH) deficiency suggest a heightened excitatory state during development. BMC Dev Biol. 2008;8:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brown M, Ashcraft P, Arning E, Bottiglieri T, Roullet J‐B, Gibson KM. Gamma‐hydroxybutyrate content in dried bloodspots facilitates newborn detection of succinic semialdehyde dehydrogenase deficiency. Mol Genet Metab. 2019. 10.1016/j.ymgme.2019.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Koenig MK, Hodgeman R, Riviello JJ, et al. Phenotype of GABA transaminase deficiency. Neurology. 2017;88:1919‐1924. [DOI] [PMC free article] [PubMed] [Google Scholar]