

Abstract
Analogs of cancer-associated Lewis Y (Ley) antigen with varying structures at the reducing end were synthesized by a highly efficient strategy involving one-pot preactivation-based iterative glycosylation to obtain the key tetra-/pentasaccharide intermediates, which was followed by stereoselective fucosylation. After global deprotection, these oligosaccharides were coupled with carrier protein keyhole limpet hemocyanin. The resultant glycan-protein conjugates were subjected to immunological studies in mice. It was disclosed that the conjugate of the pentasaccharide analog of Lewis Y antigen was more immunogenic than that of the hexasaccharide analog but the antisera of both conjugates could indiscriminately recognize each carbohydrate hapten. These results suggested that the short Lewis Y analog may be utilized to develop functional conjugate cancer vaccines. More importantly, the results also proved that the reducing end glucose residue in the hexasaccharide analog of Lewis Y was probably not be involved in its interaction with the immune system, which discovery can have a broad impact on the design of new cancer vaccines.
Graphical Abstract
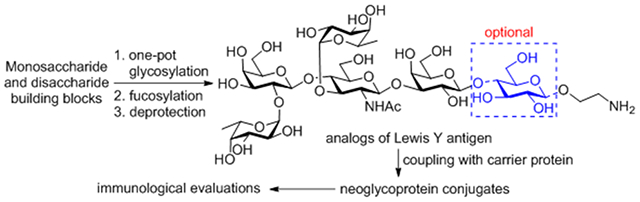
INTRODUCTION
Cell surfaces are covered by a thick layer of carbohydrates collectively known as cell glycocalyx, which is composed of glycans in the forms of glycoproteins, glycolipids, and other glycoconjugates. These glycoconjugates connect the inside and outside of cells to mediate cell recognition, proliferation, survival, and other cellular activities.1 The recognition and interaction between glycans and glycan-binding proteins are critical in cell biology and development of new therapies for various diseases. For example, cancer cells display unusual glycosylation patterns in the process of carcinogenesis, and the resultant abnormal glycans on the cancer cell surface are called tumor-associated carbohydrate antigens (TACAs).2 TACAs are promising targets for the development of cancer vaccines and immunotherapies which aim at stimulating the immune system to recognize TACA and to subsequently kill cancer cell.3
To develop effective cancer vaccines or immunotherapies, it is essential to have a deep understanding of the interaction between TACAs and the immune system or TACA-antibody recognition, which is a difficult topic. On the one hand, the conformational flexibility of carbohydrates makes the study of their structures and interplay within the immune system very challenging. As a result, structure-activity relationship analysis of TACAs using proper derivatives has become a useful tool to gain necessary information indirectly.4 On the other hand, complex carbohydrates needed for the structure-activity relationship study are difficult to come by. Thus, the problem is essentially boiled down to facile access to proper TACA derivatives for immunological studies.
Our research program aims at the development of TACA-based cancer vaccines or immunotherapies and is especially interested in establishing efficient methods for the synthesis of complex TACAs and related derivatives to facilitate the analysis of their structure-immunogenicity relationships and their interactions with antibodies. The current research was focused on Lewis Y antigen, a TACA abundantly expressed by several carcinomas.5 Specifically, we were interested in the question whether all of the sugar residues in the natural homolog of Lewis Y are necessary for its immunogenicity and for the elicitation of Lewis Y-specific antibodies. To answer this question, we developed a facile method for the synthesis of the natural hexasaccharide homolog of Lewis Y 1 and its analog 2 missing the inner core glucose unit (Figure 1), both with an aminoethyl group as a linker at the glycan reducing end. These synthetic and structurally well-defined oligosaccharides were then coupled to a carrier protein via the linker to afford glycoproteins that were immunologically investigated.
Figure 1.
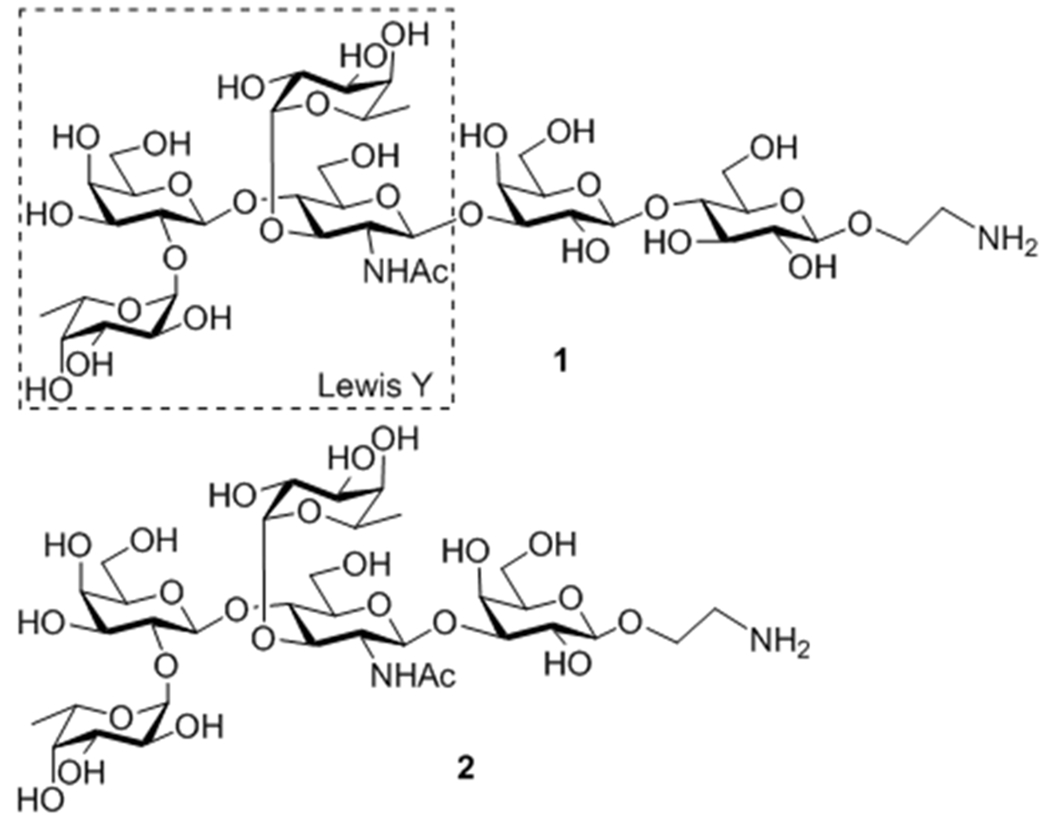
Structures of Lewis Y derivatives 1 and 2
RESULTS AND DISCUSSION
After Lewis Y was discovered as a TACA, it had attracted much attention in the 1990’s and several groups achieved its total synthesis.6 Encouraged by the therapeutic potentials of anti-Lewis Y monoclonal antibodies,7 a renaissance in its research has brought in more efficient synthetic strategies,8 highlighted by reactivity-based one-pot synthesis reported by the Wong group8d and solid phase synthesis reported by the Seeberger group.8c However, these strategies are either difficult to scale up or need tedious substrate manipulation to tune the reactivity for effective glycosylation reactions. Moreover, final global deprotection of complex protecting systems is always a challenge.
Here, we envisioned a practical and efficient synthesis for Lewis Y analogs based upon Huang and Ye’s one-pot glycosylation method.9 As outlined in Scheme 1, the key pentasaccharide intermediate 7 could be assembled from monosaccharide thioglycosides 3, 4, and 5 as glycosyl donors/acceptors and disaccharide 6 as an acceptor in a one-pot manner. Accordingly, 3 with the benzoyl group for temporary protection of its 2-O-position, which would facilitate neighboring group participation for stereoselective glycosylation, was activated with tolylsulfenyl chloride (TolSCl) and silver triflate (AgOTf) at −78 °C by Huang and Ye’s protocol. The reaction was completed in ca. 10 min as monitored by TLC showing the disappearance of starting materials. Thereafter, diol 4 was added to furnish the first glycosylation reaction, leading to a disaccharide product that was verified to have the desired β-1,4-linkage and a free 3-OH group. This reaction was both stereo- and regioselective probably because of the neighboring group participation effect (NGPE) and the higher reactivity of the 4-OH group as compared to the 3-OH in the glucosamine acceptor 4,9b, 10 respectively. At this point, fucosyl donor 5 was added, which was followed by adding TolSCl/AgOTf to complete the first stereoselective α-fucosylation of the resultant disaccharide. This reaction was chemoselective because under the given activation conditions armed thioglycoside 5 was much more reactive than the disarmed disaccharide intermediate with a phthalimido group at the 2-position. Subsequently, the same preactivation protocol was successfully employed for the glycosylation of disaccharide acceptor 6 by the resultant trisaccharide donor. The progress of all these reactions was monitored by TLC, which disclosed the consumption of starting materials and formation of products. In the end, the key intermediate 7 was purified through silica gel column chromatography to get a 43% overall yield after three one-pot glycosylation reactions and was fully characterized with NMR and HR MS. The stereochemistries of the glycosidic bonds were determined by coupling constants between H-1 and H-2, ranging from 7.2 Hz to 8.3 Hz for the β-linkages and 4.2 Hz for the α-linkage, and the detailed data are presented in the experimental section. Subsequently, the benzoyl and the phthalyl groups protecting the 2‴-O- and 2″-N-positions in 7 were removed simultaneously on treatment with hydrazine. Selective acetylation of the exposed amino group was readily achieved using acetic anhydride in methanol to give 8 in an excellent overall yield (75%). This enabled the introduction of the second fucosyl residue at the 2‴-O-position of 8 with 5 as the glycosyl donor and N-iodosuccinimide (NIS) and triflic acid (TfOH) as promoters to afford hexasaccharide 9. Global deprotection of 9 via Pd-catalyzed hydrogenolysis to remove all of the benzyl and benzylidene groups was straightforward to provide 1, which was finally purified by gel filtration column chromatography with Sephadex G-15 and fully characterized with NMR and HR MS data. Before purifying 1, triethylamine was added to neutralize acetic acid in the reaction mixture. However, we should note that it was very difficult to remove triethylamine later on. Therefore, we recommend here to directly condense the reaction mixture and subject it to gel filtration, and this procedure was adopted for the purification of 2.
Scheme 1.
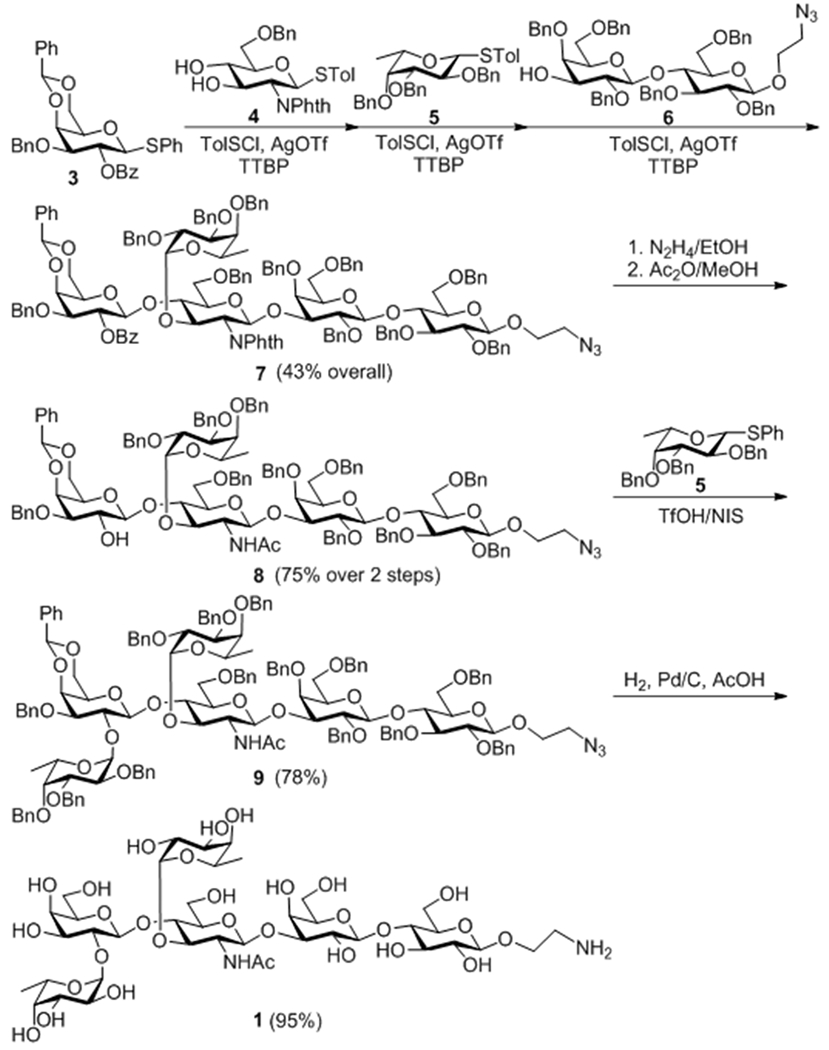
Synthesis of Lewis Y antigen analog 1
Throughout this synthesis, all of the glycosyl donors used were thioglycosides, which were effectively activated with an equal equivalent of tolylsulfenyl chloride. This combined with the one-pot protocol to save the tedious intermediate separation and characterization processes, as well as a one-step global deprotection protocol, made the synthesis especially efficient and scalable.
Lewis Y analog 2 was synthesized by the same strategy except for the replacement of disaccharide acceptor 6 with monosaccharide 10 (Scheme 2). These glycosylations gave comparable yields and the stereochemistry of all compounds was also confirmed by NMR data. In tetrasaccharide 11, the coupling constants are 8.4 Hz for H-1″ (4.74 ppm) and 8.5 Hz for H-1′ (5.32 ppm) for the newly generated β glycosidic bonds and 3.9 Hz for the α-fucose. In 13, the second fucosyl unit was confirmed to have α-configuration with a coupling constant of 3.9 Hz for its anomeric proton at 5.63 ppm.
Scheme 2.
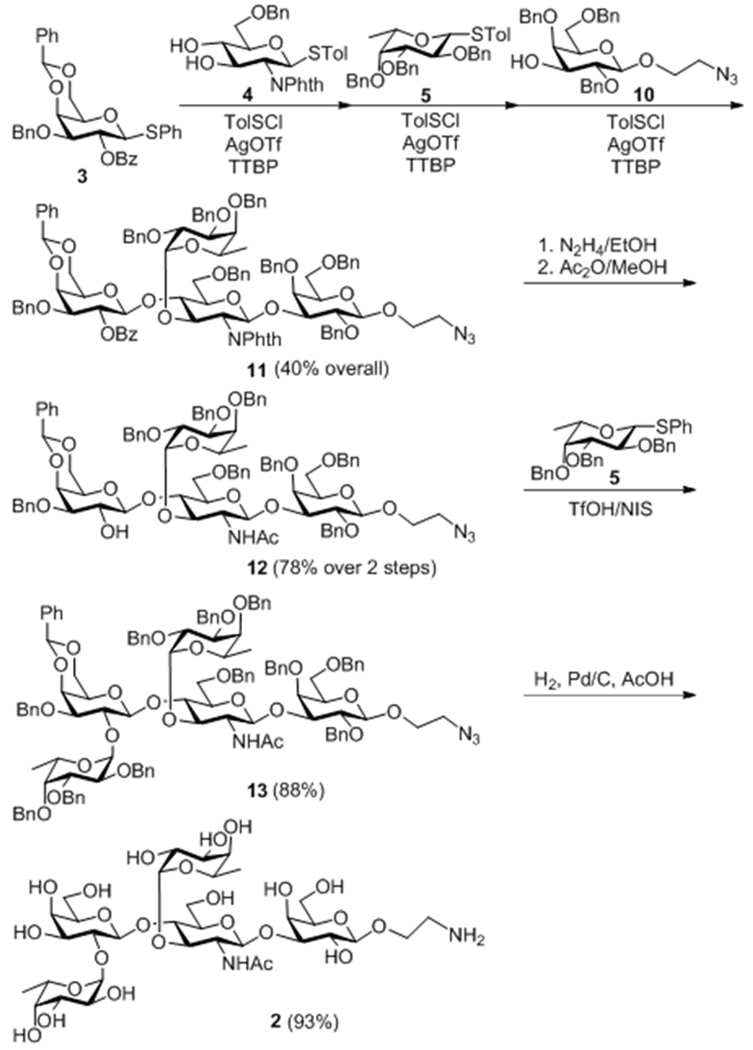
Synthesis of Lewis Y analog 2
Finally, synthetic Lewis Y analogs 1 and 2 were coupled with a carrier protein, keyhole limpet hemocyanin (KLH), to generate glycoconjugate vaccines 16a and 16b (Scheme 3), which were immunologically evaluated in mice. In the meantime, the corresponding human serum albumin (HSA) conjugates 17a and 17b were also prepared as the capture antigens to detect carbohydrate antigen-specific antibodies by enzyme-linked immunosorbent assay (ELISA). First, 1 and 2 were reacted with disuccinimidal glutarate 14 (15 equiv), a doubly activated ester of glutaric acid, to afford amides 15a and 15b, respectively, which still contained an activated ester. A large excess of 14 was applied to these reactions to avoid self-cross linkage of 1 or 2 into dimers. Thereafter, activated esters 15a and 15b were mixed with carrier proteins KLH and HSA, respectively, in a 0.1X PBS buffer, and the mixtures were stirred at room temperature for 3 days to furnish the conjugation reactions. The resultant conjugates 16a, 16b, 17a, and 17b were finally purified by size-exclusion column chromatography. The carbohydrate loadings of KLH conjugates 16a and 16b were determined by the reported phenol-sulfuric acid method,11 whereas the carbohydrate loadings of HSA conjugates 17a and 17b were determined by MS. Each type of conjugates had similar carbohydrate loadings (SI: 13.9 and 15.6% by weight or 65:1 and 84:1 by molar for KLH conjugates; 7.9 and 8.4% by weight or 6.0:1 and 7.6:1 by molar for HSA conjugates), which were all in the desired range. A glutaryl group was used as linker to couple haptens to KLH in conjugates 16a and 16b because it was verified to be nonimmunogenic and not elicit any antibody response.12
Scheme 3.
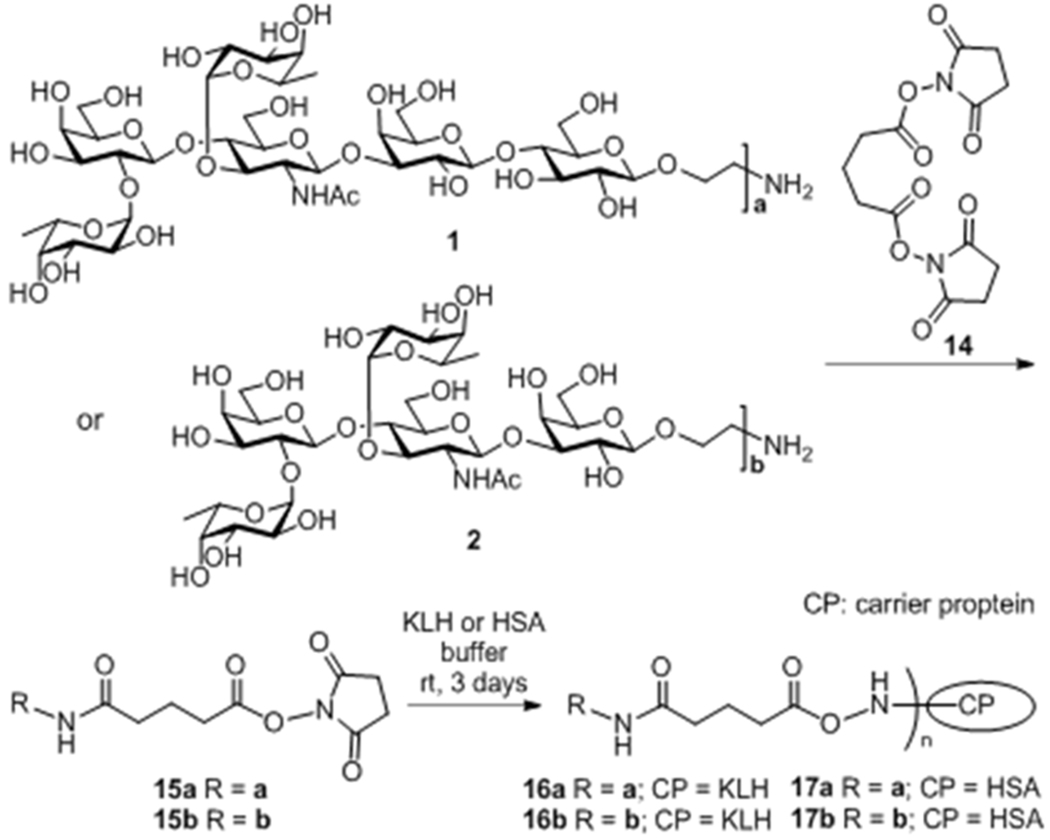
Conjugation of antigens with protein carriers
Conjugates 16a and 16b were subjected to immunological studies using female C57BL/6J mice (6-8 weeks of age). In this regard, an emulsion of each conjugate with the Freund’s adjuvant (FA) was injected subcutaneously (s.c.) on day 1 for the initial immunization (using complete FA, CFA) and injected intraperitoneally (i.p.) on day 8, 15 and 21 for boost immunizations (using incomplete FA, IFA) to a group of 5 mice, respectively. Blood samples were collected from these mice on day 0 before the initial immunization and on day 21 and 43 after boost immunizations and then subjected to antibody titer analysis by ELISA.
The results depicted in Figure 2A showed clearly that both 16a and 16b had already elicited the production of antigen-specific antibodies after the second boost immunization (day 21 antisera), but the total antibody titers of day 43 antisera, i.e., after the third boost immunization, were significantly higher than that of both day 0 and 21 antisera. These results disclosed the immunization dynamics and immune response reinforcement as a result of boost immunizations. Further analysis of the isotypes of induced antibodies revealed that the antibody response was mainly of IgG type (Figure 2B), while IgM antibody response was rather low in both cases (Figure 2C). These results were in agreement with literature reports that most glycoproteins induce mainly IgG antibody responses to carbohydrate antigens.13 The production of IgG antibodies indicates usually T-cell dependent immunity and antibody class switching and maturation as well as long-term immune memory,14 which is desirable for effective cancer immunotherapy.
Figure 2.
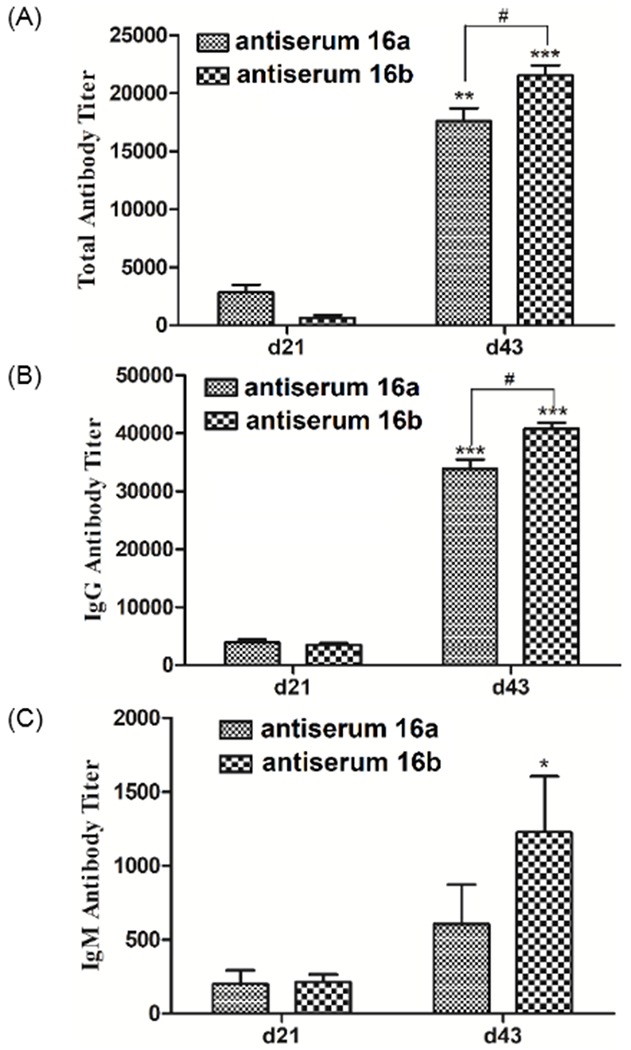
The total antibody (A), IgG antibody (B) and IgM antibody (C) titers of pooled day 21 and 43 sera from mice immunized with conjugates 16a and 16b. ELISA plates were coated with 17a and 17b to detect antibodies specific to the carbohydrate antigen in 16a and 16b, respectively. Antibody titers were calculated according to the method described in the experimental section. The mean value of three parallel experiments was shown for each sample, and the error bar showed the standard error of mean (SEM). *p < 0.05, **p < 0.01 and ***p < 0.001 as compared to normal serum; #p < 0.05 between the two groups.
Interestingly, conjugate 16b of the pentasaccharide analog of Lewis Y could elicit significantly stronger total and IgG antibody responses than conjugate 16a of the natural hexasaccharide homolog of Lewis Y (Figure 2A, Figure 2B). It should be noted that we applied conjugates 16a and 16b containing the same amount of carbohydrate haptens for the immunization of mice to avoid any potential influence of hapten dosages on the immune response. Even after taking into consideration of the potential impact of the different carbohydrate loadings in coating antigens 17a and 17b (7.9 vs 8.4%) for ELISA, the difference in IgG antibody titers of 16a and 16b antisera was still significant. Its reason is unclear but we can speculate. One reason may be that the small difference in hapten loading for 16a and 16b (13.9 vs 15.6%) had some impact on their immunogenicity. The other reason may be simply because the pentasaccharide analog of Lewis Y 2 was more immunogenic than its hexasaccharide analog 1. This should be reasonable because a branched pentasaccharide analog without the reducing end glucose is probably more conformationally rigid and is also more “unnatural” to the immune system, both of which may result in changes in immunological properties. Nonetheless, the results suggested that the pentasaccharide analog of Lewis Y may be employed to construct more effective conjugate vaccines for cancer immunotherapy, provided that the immune reactions induced by this analog could recognize Lewis Y and its natural hexasaccharide homolog on cancer cell surfaces.
To probe the option, we analyzed subsequently the cross-reactivity of each mouse antiserum from conjugates 16a and 16b with each carbohydrate hapten by ELISA. Accordingly, HSA conjugates 17a and 17b were used to coat the ELISA plates and detect antibodies that recognized the carbohydrate haptens in them. The results depicted in Figure 3 indicated clearly that the antibodies in these antisera could recognize and bind strongly to both carbohydrate haptens. Essentially, the two antisera had the same binding with both carbohydrate haptens, as the slightly higher antibody titers observed with conjugate 17b, as compared to that of 17a, were statistically insignificant and could be attributed to the slightly higher carbohydrate loading of 17b. These results have proved not only that the conjugates of pentasaccharide 2 may be used as effective vaccines to induce Lewis Y-recognizing antitumor immunities but also that the reducing end glucoside residue in Lewis Y may not be absolutely necessary for either its immunogenicity or its antibody recognition.
Figure 3.
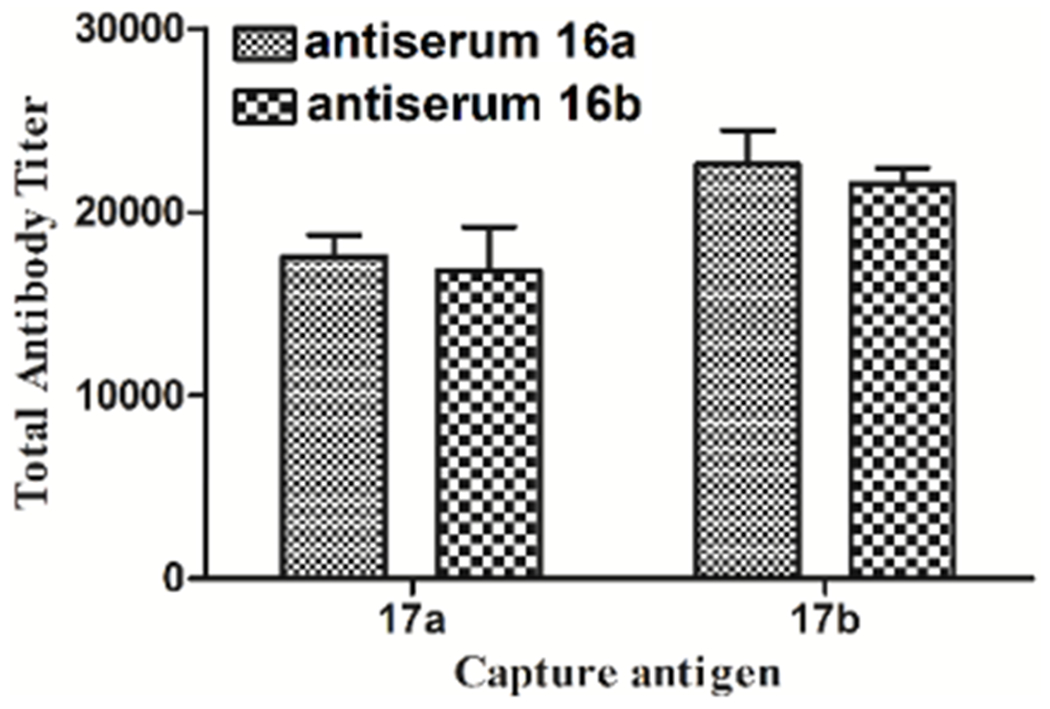
Results of cross-reactivity analysis. After ELISA plate was coated with capture antigen 17a or 17b, the titers of bound antibodies in the pooled day 43 antisera from mice immunized with both conjugates were examined. The mean of antibody titers of three parallel experiments was shown for each sample, and the error bar showed the SEM.
In conclusion, the hexasaccharide homolog of Lewis Y 1 and its pentasaccharide analog 2 were synthesized by the same effective and practical strategy, which is highlighted by preactivation-based regioselective and stereoselective one-pot assembly of a pentasaccharide or a tetrasaccharide as the key intermediate, use of glycosyl thioglycosides as the only glycosyl donors, and a one-step global deprotection protocol. This new strategy should be generally applicable for the synthesis of similar or related Lewis antigens. The synthesized oligosaccharides were covalently coupled with KLH to generate conjugate vaccines that were subjected to immunological evaluation in mice and enabled structure-immunogenicity relationship analysis. Our results of immunological studies had disclosed that the conjugate of the pentasaccharide analog of Lewis Y was at least similarly (probably more) immunogenic as the larger hexasaccharide analog. More importantly, antibodies elicited by the pentasaccharide hapten could indiscriminately recognize the hexasaccharide hapten, and vice versa. This has provided the opportunity to use the less complicated pentasaccharide for design and development of more potent glycoconjugate vaccines for cancer immunotherapy. Another important discovery of this work was that the reducing end glucose unit in Lewis Y hexasaccharide homolog may not be critical for its interaction with and recognition by the immune system. This discovery can be of broad importance since it indicates that for some complex TACAs the exposed non-reducing end structure may be sufficient for stimulating functional immune responses against cancer targets. We are currently preparing cross-reactive monoclonal antibodies to both Lewis Y analogs to gain more insights into the interaction between these antigens and their antibodies for the design of more effective vaccines.
EXPERIMENT SECTION
General Information.
Chemicals and materials were purchased from commercial sources and were used as received without further purification unless otherwise noted. molecular sieves 4Å were flame-dried under high vacuum and used immediately after cooling to rt under a N2 atmosphere. Analytical TLC was carried out on silica gel 60Å F254 plates with detection by a UV detector and/or by charring with 10% (v/v) H2SO4 in EtOH. Flash column chromatography was performed on silica gel 60 (230–400 Mesh). NMR spectra were acquired on an Agilent® or Varian® 400, 500 or 600 MHz machine with chemical shifts reported in ppm (δ) referenced to CHCl3 (1H NMR δ 7.26 ppm) or CDCl3 (13C NMR δ 77.0 ppm). Peak and coupling constant assignments are based on 1H NMR, 1H–1H COSY, and 1H–13C HMQC experiments.
Phenyl 2-O-benzoyl-3-O-benzyl-4,6-O-benzylidene-1-thio-β-D-galactopyranoside (3)
Prepared according to procedures described by Yu et al.15 1H NMR (300 MHz, CDCl3): δ 8.05-8.02 (m, 2H, ArH), 7.60-7.15 (m, 18H, ArH), 5.56 (t, J = 9.7 Hz, 1H), 5.49 (s, 1 H), 4.81 (d, J = 9.7 Hz, 1H), 4.63 (d, J = 12.8 Hz, 1H), 4.56 (d, J = 12.8 Hz, 1H), 4.39 (d, J = 12.5 Hz, 1H), 4.24 (d, J = 2.8 Hz, 1 H), 4.03 (d, J = 11.4 Hz, 1H), 3.77 (dd, J = 9.3, 3.2 Hz, 1H), 3.50 (s, 1H). 1H NMR data matches the literature report.
p-Tolyl 6-O-benzyl-2-deoxy-2-N-phthalimido-1-thio-β-D-glucopyranoside (4).
Prepared from glucosamine according to a literature procedure9b 1H-NMR (600 MHz, CDCl3): δ 7.79-6.96 (m, 13 H, ArH), 5.50 (d, J = 10.2 Hz), 4.56-4.51 (m, 2 H), 4.29 (t, 1 H, J = 10.2Hz), 4.15 (t, 1 H, J = 10.2 Hz), 3.77-3.69 (m, 2 H), 3.62-3.47 (m, 2 H), 3.09 (br, 1H), 2.50 (br, 1H), 2.25 (s, 3 H). 1H NMR data matches the literature report.
p-Tolyl 2,3,4-tri-O-benzyl-1-thio-β-L-fucopyranoside (5).
Prepared from L-Fucose according to a literature procedure.16 1H-NMR (400 MHz, CDCl3): δ 7.60-7.58 (m, 2 H, ArH), 7.40-7.28 (m, 15 H, ArH), 7.21-7.19 (m, 3 H, ArH), 5.01 (d, J = 11.5 Hz), 4.77 (dd, 2 H, J = 10.2, 4.2 Hz), 4.74 (s, 2 H), 4.15 (t, 1 H, J = 10.2 Hz), 4.67 (d, 1H, J = 11.6 Hz), 4.60 (d, 1 H, J = 9.6 Hz), 3.93 (t, 1 H, J = 9.5 Hz), 3.63 (dd, 1H, J = 2.8, 0.9 Hz), 3.59 (dd, 1H, J = 9.2, 2.8 Hz), 3.53 (q, 1H, J = 6.4 Hz), 1.27 (d, 3H, J = 6.4 Hz). 1H NMR data matches the literature report.
Azidoethyl (2,4,6-tri-O-benzyl-β-D-galactopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-β-D-glucopyranoside (6).
It was prepared from lactose according to a literature procedure.9b 1H NMR (600 MHz, CDCl3): δ 7.38-7.16 (m, 30H, ArH), 5.02 (d, J = 10.7 Hz, 1H), 4.92 (d, J = 11.1 Hz, 1H), 4.80 (d, J = 11.9 Hz, 1H), 4.77-4.74 (m, 4H), 4.69 (d, J = 11.3 Hz, 1 H), 4.61 (d, J = 11.9 Hz, 1 H), 4.56 (d, J = 11.9 Hz, 1 H), 4.44-4.38 (m, 4H), 4.28 (d, J = 11.9 Hz, 1 H), 4.06-4.03 (m, 1H), 3.96 (t, J = 9.7 Hz, 1 H), 3.84 (d, J = 2.9 Hz, 1 H), 3.80-3.69 (m, 3H), 3.58-3.40 (m, 10H), 2.18 (d, J = 6.5 Hz, 1 H). 13C NMR (150 MHz, CDCl3) δ.139.0, 138.7, 138.6, 138.4, 138.1, 138.0, 128.4, 128.37, 128.36, 128.3, 128.3, 128.04, 128.00, 127.99, 127.9, 127.74, 127.73, 127.7, 127.68, 127.62, 127.58, 127.55, 127.16, 103.6, 102.7, 82.7, 81.7, 80.6, 76.6, 75.9, 75.3, 75.2, 75.1, 75.0, 74.96, 74.01, 73.4, 73.2, 73.17, 68.2, 68.1, 68.0, 60.0, 50.1. 1H NMR data matches the literature report.
3-Azidoethyl (2-O-benzoyl-3-O-benzyl-4,6-O-benzylidene-β-D-galactopyranosyl)-(1→4)-[(2,3,4-tri-O-benzyl-α-L-fucopyranosyl)-(1→3)]-(6-O-benzyl-2-deoxy-2-N-phthalimido-β-D-glucopyranosyl)-(1→3)-(2,4,6-tri-O-benzyl-β-D-galactopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-β-D-glucopyranoside (7).
Galactosyl donor 3 (80 mg, 0.141mmol) and freshly activated molecular sieves MS 4 Å (1.0 g) in DCM (3 mL) were stirred at rt for 2 h. The mixture was cooled to −78 °C before silver triflate (109 mg, 0.42 mmol, 3.0 eq.) in acetonitrile (0.3 mL) was added. About 15 minutes later, p-TolSCl (22 μL, 0.141 mmol) was added. After complete activation of 3 was confirmed by TLC, a DCM solution (1 mL) of glucosamine acceptor 4 (62 mg, 0.123 mmol) and 2,4,6-Tri-tert-butylpyrimidine (TTBP) (35 mg, 0.141 mmol) was added into the reaction mixture. The reaction was monitored with TLC and when completion was confirmed (about 10 min), the fucosyl donor 5 (74 mg, 0.141 mmol) and TTBP (35 mg, 0.141 mmol) dissolved in DCM (1 mL) were added. The reaction mixture was stirred for an additional 10 min before p-TolSCl (22 μL, 0.141 mmol) was added. After the disaccharide acceptor resulted from the first step was completely consumed as indicated by TLC (in ca. 30 min), the lactosyl acceptor 6 (109 mg, 0.114 mmol) and TTBP (35 mg, 0.141 mmol) dissolved in DCM (2 mL) were added, which was followed by addition of silver triflate (36 mg, 0.141 mmol). After 10 min, the reaction mixture was charged with the last portion of p-TolSCl (22 μL, 0.141 mmol). The reaction progress was monitored by TLC. When the lactosyl acceptor was completely consumed, the reaction mixture was filtered through Celite. The solid was thoroughly washed with DCM and the filtrate was washed with saturated solution of NaHCO3. The organic layer was then dried over Na2SO4 and the solvent was removed in vacuum. The crude product was purified by silica gel column chromatography (hexanes/EtOAc = 1/1) to afford 7 as a white solid (107 mg, 43% yield). 1H NMR (600 MHz, CDCl3) δ 8.04 (d, 2H, J = 8.4 Hz), 7.62-6.78 (m, 67H), 5.62 (t, 1H, J = 9.8 Hz), 5.55 (s, 1H), 5.19 (d, 1H, J = 8.3 Hz, H-1″), 5.09 (d, 1H, J = 11.1 Hz), 4.88-4.83 (m, 2H), 4.80 (d, 1H, J = 7.9 Hz), 4.77 (d, 1H, J = 8.2 Hz, H-1‴), 4.78-4.75 (m, 1H), 4.71 (d, 1H, J = 13.2 Hz), 4.68 (d, 1H, J = 10.7Hz), 4.65 (d, 1H, J = 3.9 Hz), 4.59 (d, 1H, J = 4.2 Hz, H-1Fuc), 4.57-4.47 (m, 5H), 4.41-4.24 (m, 7H), 4.21 (d, 1H, J = 7.5Hz), 4.20 (d, 1H, J = 7.4 Hz, H-1), 4.18 (d, 1H, J = 8.2 Hz, H-1′), 4.17-4.13 (m, 4H), 4.11 (d, 1H, J = 8.0 Hz), 4.02-3.95 (m, 4H), 3.91-3.86 (m, 2H), 3.80-3.77 (m, 2H), 3.60-3.51 (m, 4H), 3.44-3.28 (m, 12H), 3.21 (d, 1H, J = 10.1Hz), 3.16 (s, 1H), 3.06 (s, 1H), 2.88 (d, 1H, J = 8.2 Hz), 1.23 (d, 3H, J = 6.4 Hz); 13C NMR (150 MHz, CDCl3) δ 164.5, 139.5, 139.49, 138.9, 138.6, 138.4, 138.35, 138.27, 138.2, 138.18, 137.83, 138.78, 133.6, 133.1, 129.8, 128.8, 128.5, 128.47, 128.37, 128.34, 128.24, 128.22, 128.17, 128.1, 128.04, 127.98, 127.96, 127.9, 127.83, 127.79, 127.76, 127.68, 127.67, 127.64, 127.62, 127.59, 127.53, 127.49, 127.42, 127.36, 127.1, 127.0, 126.9, 126.8, 126.7, 126.67, 126.2, 125.96, 103.5 (C-1), 102.6 (C-1′), 99.9 (C-1‴), 99.79 (PhCH), 99.76 (C-1″), 97.3 (C-1Fuc), 82.7, 82.3, 82.0, 81.5, 79.0, 78.9, 78.5, 76.6, 75.9, 75.4, 75.0, 74.74, 74.69, 74.65, 73.83, 73.79, 73.7, 73.2, 73.0, 72.8, 72.7, 71.4, 71.0, 70.7, 70.6, 70.1, 69.1, 68.3, 68.1, 68.0, 67.5, 66.6, 66.4, 57.1, 50.9, 30.1, 29.7, 16.2; HRMS [M + Na]+ m/z calcd for C131H132N4NaO27+ 2215.8971, found: 2215.9033.
3-Azidoethyl (3-O-benzyl-4,6-O-benzylidene-β-D-galactopyranosyl)-(1→4)-[(2,3,4-tri-O-benzyl-α-L-fucopyranosyl)-(1→3)]-(6-O-benzyl-2-deoxy-2-N-Acetyl-β-D-glucopyranosyl)-(1→3)-(2,4,6-tri-O-benzyl-β-D-galactopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-β-D-glucopyranoside (8).
Pentasacchride 7 (35mg, 0.016mmol) was dissolved in a mixture of ethanol (5 mL) and hydrazine hydrate (2 mL). The reaction mixture was refluxed overnight until TLC showed the complete removal of phthaly and acyl groups (product Rf = 0.4, DCM/MeOH = 20/1). The solvent was removed under vacuum. The residue was dissolved in DCM and the solution was thoroughly washed with water to remove excess of hydrazine. Removal of solvent in vacuum afforded the crude product as a yellowish solid, which was subjected to next step without further purification.
After the crude product was dissolved in methanol (5 mL), acetic anhydride (1 mL) was added. The resultant mixture was stirred at rt until TLC analysis indicated completion of the reaction in 1 h. The solvent was removed in vacuum and the residue was purified by silica gel flash chromatography (Hexanes/EtOAc = 1/1) to afford 8 as a white solid (24 mg, 75% overall yield). 1H NMR (600 MHz, CDCl3) δ 7.59 (d, 2H, J = 7.5 Hz), 7.50 (d, 2H, J = 7.2 Hz), 7.39-7.09 (m, 39H), 5.49 (s, 1H), 5.30 (d, 1H, J = 7.7 Hz), 5.29 (d, 1H, J = 7.9 Hz, H-1″), 5.12 (d, 1H, J = 11.7 Hz), 5.01 (d, 1H, J = 10.8 Hz), 4.93 (d, 1H, J = 3.1 Hz, H-1Fuc), 4.89 (d, 1H, J = 10.8 Hz), 4.81-4.59 (m, 13H), 4.53-4.49 (m, 2H), 4.48 (d, 1H, J = 7.3 Hz, H-1‴), 4.37 (d, 1H, J = 7.2 Hz, H-1′), 4.36 (d, 1H, J = 6.8 Hz, H-1), 4.36-4.30 (m, 4H), 4.26 (d, 1H, J = 11.1 Hz), 4.22-4.13 (m, 3H), 4.08-3.99 (m, 4H), 3.93-3.86 (m, 5H), 3.76 (d, 1H, J = 10.1 Hz), 3.70-3.66 (m, 3H), 3.60-3.55 (m, 4H), 3.50-3.20 (m, 13H), 2.87 (s, 1H), 2.52 (br, 1H), 1.23 (s, 3H), 1.01 (d, 3H, J = 6.7 Hz); 13C NMR (150 MHz, CDCl3) δ 169.9, 139.6, 139.4, 139.3, 139.1, 138.99, 138.94, 138.6, 138.3, 138.28, 138.2, 137.9, 137.8, 128.76, 128.5, 128.29, 128.28, 128.27, 128.25, 128.22, 128.15, 128.13, 128.09, 128.07, 128.02, 127.992, 127.987, 127.91, 127.90, 127.8, 127.6, 127.57, 127.53, 127.52, 127.50, 127.49, 127.43, 127.34, 127.32, 127.3, 127.1, 127.0, 126.96, 126.90, 125.8, 103.5 (C-1), 102.4 (C-1′), 101.4 (C-1‴), 100.7 (C-1″), 99.7 (PhCH), 97.5 (C-1Fuc), 82.8, 82.2, 81.6, 79.32, 79.30, 78.6, 76.5, 76.2, 75.7, 75.4, 75.0, 74.97, 74.9, 74.5, 74.3, 73.74, 73.68, 73.2, 73.03, 73.00, 72.98, 72.6, 71.4, 71.3, 70.1, 69.3, 68.5, 68.3, 68.0, 66.4, 66.0, 50.9, 22.9, 16.3; HRMS [M + Na]+ m/z calcd for C118H128N4NaO25+ 2023.8760, found: 2023.8866.
3-Azidoethyl (2,3,4-tri-O-benzyl-α-L-fucopyranosyl)-(1→2)-(3-O-benzyl-4,6-O-benzylidene-β-D-galactopyranosyl)-(1→4)-[(2,3,4-tri-O-benzyl-α-L-fucopyranosyl)-(1→3)]-(6-O-benzyl-2-deoxy-2-N-Acetyl-β-D-glucopyranosyl)-(1→3)-(2,4,6-tri-O-benzyl-β-D-galactopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-β-D-glucopyranoside (9).
After the mixture of fucosyl doner 5 (16 mg, 0.030 mmol), pentasaccharide intermediate 8 (12 mg, 0.006 mmol) and 4Å MS (1 g) in anhydrous DCM (5 mL) was stirred at rt under a N2 atmosphere for 0.5 h, it was cooled to −50 °C. NIS (7.5 mg, 0.033 mmol) and TfOH (1mg, 0.006 mmol) were added in one portion. The mixture was allowed to warm up to −20 °C when the reaction started as indicated by a color change. The reaction was maintained at tis temperature for 1 h, when TLC showed the completion of reaction. Saturated aq. NaHCO3 solution (5 mL) was added to quench the reaction and molecular sieves were removed by filtration. The aqueous phase was separated and extracted with DCM (3 × 10 mL). The organic phases were combined and washed with saturated aq. Na2SO3 solution, dried over Na2SO4, and condensed in vacuum. The crude product was purified by silica gel column chromatography (EA/hexanes = 2:3, v/v) to give 9 (12mg, 85%) as a white solid. 1H NMR (600 MHz, CDCl3) δ 7.55 (d, 2H, J = 7.6 Hz), 7.34-7.07 (m, 73H), 5.62 (d, 1H, J = 3.8 Hz, H-1″″), 5.41 (s, 1H), 5.39 (d, 1H, J = 8.8 Hz, H-1″), 5.38 (d, 1H, J = 7.9 Hz), 5.08 (d, 1H, J = 11.8 Hz), 4.97 (d, 1H, J = 10.9 Hz), 4.92 (d, 1H, J = 11.3 Hz), 4.87-4.85 (m, 2H), 4.76 (d, 1H, J = 3.1 Hz, H-1Fuc), 4.75-4.55 (m, 21H), 4.52 (d, 1H, J = 8.0 Hz, H-1‴), 4.51-4.46 (m, 4H), 4.43 (d, 1H, J = 7.6 Hz, H-1′), 4.42-4.36 (m, 2H), 4.33 (d, 1H, J = 7.6 Hz, H-1), 4.32-4.23 (m, 5H), 4.18 (d, 1H, J = 11.8 Hz), 4.10 (d, 1H, J = 11.6 Hz), 4.06-3.97 (m, 7H), 3.92-3.83 (m, 5H), 3.75-3.73 (m, 1H), 3.68-3.63 (m, 4H), 3.58-3.54 (m, 3H), 3.49-3.42 (m, 7H), 3.38-3.34 (m, 3H), 3.26 (d, 1H, J = 9.4 Hz), 3.17-3.14 (m, 1H), 3.11 (s, 1H), 3.08-3.04 (m, 1H), 2.88 (s, 1H), 1.25 (s, 3H), 1.26 (d, 3H, J = 7.3 Hz), 1.10 (d, 3H, J = 3.8 Hz); 13C NMR (150 MHz, CDCl3) δ 170.4, 139.6, 139.5, 139.3, 139.2, 139.0, 138.96, 138.88, 138.85, 138.84, 138.7, 138.4, 138.3, 138.24, 138.16, 138.0, 128.4, 128.34, 128.3, 128.27, 128.265, 128.2, 128.18, 128.14, 128.11, 128.09, 128.06, 128.05, 128.01, 128.0, 127.95, 127.94, 127.82, 127.79, 127.78, 127.65, 127.57, 127.52, 127.48, 127.42, 127.39, 127.34, 127.27, 127.23, 127.17, 127,13, 127.11, 127.0, 126.9, 126.82, 126.78, 103.5 (C-1), 102.4 (C-1′), 100.7 (C-1″), 99.8 (C-1‴), 99.7 (PhCH), 98.4 (C-1Fuc), 97.6 (C-1″″), 83.4, 82.9, 81.7, 81.1, 79.6, 78.9, 78.6, 78.5, 78.3, 76.6, 76.1, 75.8, 75.64, 75.63, 75.04, 57.02, 73.4, 73.2, 73.1, 73.0, 72.9, 72.6, 72.5, 72.3, 71.9, 71.2, 70.3, 68.3, 68.2, 68.1, 68.0, 66.9, 66.4, 66.3, 51.0, 29.7, 22.8, 16.3, 16.1; HRMS [M + Na]+ m/z calcd for C145H156N4NaO29+ 2440.0747, found: 2440.0657.
Azidoethyl 2,3,6-tri-O-benzyl-β-D-galactopyranoside (10).
It was prepared from galactose according to the same procedure as compound 6. 1H NMR (400 MHz, CDCl3): δ 7.40-7.29 (m, 15H, ArH), 4.98 (d, J = 10.7 Hz, 1H), 4.79 (d, J = 11.1 Hz, 1H), 4.68 (d, J = 11.1 Hz, 1H), 4.62 (d, J = 10.7 Hz, 1H), 4.44 (dd, J = 13.5, 11.3 Hz, 1 H), 4.61 (d, J = 8.5 Hz, 1 H), 4.08-4.01 (m, 1H), 3.85 (d, J = 1.8 Hz, 1 H), 3.70-3.58 (m, 6H), 3.54-3.45 (m, 1H), 3.42-3.38 (m, 1H), 2.22 (d, J = 3.5 Hz, 1 H).
2-Azidoethyl (2-O-benzoyl-3-O-benzyl-4,6-O-benzylidene-β-D-galactopyranosyl)-(1→4)-[(2,3,4-tri-O-benzyl-α-L-fucopyranosyl)-(1→3)]-(6-O-benzyl-2-deoxy-2-N-phthalimido-β-D-glucopyranosyl)-(1→3)-2,4,6-tri-O-benzyl-β-D-galactopyranoside (11).
After a mixture of galactosyl donor 3 (80 mg, 0.141mmol) and freshly activated molecular sieves MS 4 Å (1.0 g) in DCM (3 mL) was stirred at rt for 2 h, it was cooled to −78 °C and then silver triflate (109 mg, 0.42 mmol, 3.0 equiv) in acetonitrile (0.3 mL) was added. About 15 min later, p-TolSCl (22 μL, 0.141 mmol) was added. After complete activation of 3 as confirmed by TLC, a DCM solution (1 mL) of glucosamine acceptor 4 (62 mg, 0.123 mmol) and TTBP (35 mg, 0.141 mmol) was added. The reaction was monitored by TLC and when completion was confirmed (in about 10 min), the fucosyl donor 5 (74 mg, 0.141 mmol) and TTBP (35 mg, 0.141 mmol) in DCM (1 mL) were added. The reaction mixture was stirred for an additional 10 min before p-TolSCl (22 μL, 0.141 mmol) was added. After the complete consumption of the disaccharide acceptor resulted in the first step indicated by TLC (in about 30 min), the galactosyl acceptor 10 (59 mg, 0.114 mmol) and TTBP (35 mg, 0.141 mmol) dissolved in DCM (2 mL) was added to the reaction mixture followed by addition of silver triflate (36 mg, 0.141 mmol). After 10 min of stirring, the mixture was charged with the last portion of p-TolSCl (22 μL, 0.141 mmol), and the reaction was monitored with TLC. When the azidoethyl galactosyl acceptor was completely consumed, the reaction mixture was filtered through Celite. The solid was thoroughly washed with DCM and the filtrate was washed with saturated solution of NaHCO3. The organic layer was dried over Na2SO4 and the solvent was removed in vacuum followed by purification by silica gel column chromatography (hexanes/EtOAc = 1/1) to afford 11 as a white solid (80 mg, 40% yield. 1H NMR (600 MHz, CDCl3) δ 8.04 (d, 2H, J = 7.9 Hz), 7.66-6.97 (m, 50H), 6.87 (d, 2H, J = 7.9 Hz), 5.62 (t, 1H, J = 9.3 Hz), 5.56 (s, 1H), 5.32 (d, 1H, J = 8.5 Hz, H-1′), 4.93 (d, 1H, J = 11.7 Hz), 4.88-4.75 (m, 3H), 4.74 (d, 1H, J = 8.4 Hz, H-1″), 4.64 (d, 1H, J = 3.9 Hz, H-1Fuc), 4.59-4.49 (m, 5H), 4.44-4.18 (m, 8H), 4.16 (d, 1H, J = 7.6 Hz, H-1), 4.12 (d, 1H, J = 3.9 Hz), 4.02-3.82 (m, 7H), 3.67-3.56 (m, 3H), 3.50-3.40 (m, 5H), 3.35-3.32 (m, 1H), 3.28-3.23 (m, 2H), 3.18-3.16 (m, 2H), 2.99 (s, 1H), 1.25 (d, 3H, J = 6.6 Hz); 13C NMR (150 MHz, CDCl3) δ 164.5, 139.53, 139.5, 138.8, 138.5, 138.22, 138.2, 138.0, 137.8, 137.77, 133.8, 133.1, 129.8, 128.83, 128.78, 128.47, 128.46, 128.4, 128.3, 128.1, 128.03, 128.0, 127.97, 127.92, 127.8, 127.78, 127.74, 127.7, 127.67, 127.64, 127.62, 127.6, 127.5, 127.4, 127.3, 127.0, 126.9, 126.84, 126.82, 126.7, 126.0, 103.9 (C-1), 99.83 (C-1″), 99.8 (PhCH), 99.1 (C-1′), 97.4 (C-1Fuc), 80.4, 78.98, 78.96, 78.5, 77.1, 75.5, 74.7, 74.68, 74.6, 74.5, 73.79, 73.77, 73.39, 73.36, 72.7, 71.4, 71.2, 70.6, 70.57, 69.2, 69.1, 68.1, 67.7, 66.5, 66.4, 57.1, 50.7, 16.1; HRMS [M + Na]+ m/z calcd for C104H104N4NaO22+ 1783.7034, found: 1783.7068.
2-Azidoethyl (3-O-benzyl-4,6-O-benzylidene-β-D-galactopyranosyl)-(1→4)-[(2,3,4-tri-O-benzyl-α-L-fucopyranosyl)-(1→3)]-(6-O-benzyl-2-deoxy-2-N-acetamido-β-D-glucopyranosyl)-(1→3)-2,4,6-tri-O-benzyl-β-D-galactopyranoside (12)
After the solution of 11 (80 mg, 0.046 mmol) and N2H4·H2O (3.5 mL) in EtOH (10 mL) was refluxed overnight, MALDI TOF MS showed that all of the Phth and Bz groups were removed. The solvent was removed in vacuum, and the residue was dissolved in CH2Cl2 (10 mL). The resulting solution was thoroughly washed with 1N HCl, saturated NaHCO3, water and brine and then dried over Na2SO4. The solvent was removed in vacuum and dissolved in MeOH (5 mL) before anhydrous acetic anhydride (1 mL) was added at rt. The solution was stirred at rt for 1 h, and at this point, MALDI TOF MS showed the complete acetylation of the amino groups. The reaction mixture was concentrated in vacuum. The crude product was purified by flash column chromatography (CH2Cl2/MeOH = 40:1, v/v) to give 12 as a white solid (56 mg, 78%). 1H NMR (600 MHz, CDCl3) δ 7.57 (d, 2H, J = 7.6 Hz), 7.43 (d, 2H, J = 7.6 Hz), 7.38-7.17 (m, 39H), 7.12 (d, 2H, J = 7.0 Hz), 5.47 (s, 1H, PhCH), 5.24 (d, 1H, J = 7.1 Hz), 5.23 (d, 1H, J = 6.5 Hz, H-1′), 5.09 (d, 1H, J = 11.1 Hz), 5.00 (d, 1H, J = 11.6 Hz), 4.95 (d, 1H, J = 3.5 Hz, H-1Fuc), 4.86 (d, 1H, J = 11.6 Hz), 4.77-4.56 (m, 11H), 4.45 (d, 1H, J = 7.7 Hz, H-1″), 4.40-4.34 (m, 3H), 4.33 (d, 1H, J = 7.8 Hz, H-1), 4.27-4.20 (m, 3H), 4.13 (t, 1H, J = 9.5 Hz), 4.04-3.97 (m, 4H), 3.89-3.84 (m, 4H), 3.77-3.73 (m, 3H), 3.59-3.39 (m, 9H), 3.34-3.25 (m, 2H), 3.23 (s, 1H), 2.83 (s, 1H), 2.53 (br, 1H), 1.33 (s, 3H), 1.00 (d, 3H, J = 6.4 Hz); 13C NMR (150 MHz, CDCl3) δ 170.1, 139.5, 139.4, 139.1, 139.0, 138.9, 138.3, 138.0, 137.97, 137.9, 128.8, 128.7, 128.5, 128.34, 128.33, 128.30, 128.14, 128.11, 128.10, 128.0, 127.89, 127.86, 127.73, 127.66, 127.60, 127.53, 127.50, 127.49, 127.35, 127.30, 127.29, 127.25, 127.1, 126.9, 125.9, 103.9 (C-1), 101.4 (C-1″), 100.8 (C-1′), 99.7 (PhCH), 97.5 (C-1Fuc), 81.2, 79.3, 79.2, 78.9, 78.7, 75.7, 75.5, 74.9, 74.54, 74.51, 74.46, 73.8, 73.6, 73.5, 73.4, 72.9, 72.7, 71.5, 71.3, 70.1, 69.3, 69.2, 68.5, 67.8, 66.4, 66.1, 56.0, 51.0, 29.7, 23.0, 16.3; HRMS [M + Na]+ m/z calcd for C91H100N4NaO20+ 1591.6823, found: 1591.6061.
2-Azidoethyl (2,3,4-tri-O-benzyl-α-L-fucopyranosyl)-(1→2)-(3-O-benzyl-4,6-O-benzylidene-β-D-galactopyranosyl)-(1→4)-[(2,3,4-tri-O-benzyl-α-L-fucopyranosyl)-(1→3)]-(6-O-benzyl-2-deoxy-2-N-acetamido-β-D-glucopyranosyl)-(1→3)-2,4,6-tri-O-benzyl-β-D-galactopyranoside (13)
After the mixture of 12 (30 mg, 0.019 mmol), 5 (30 mg, 0.057 mmol) and 4Å MS (1 g) in CH2Cl2 (5 mL) was stirred at rt under a N2 atmosphere for 0.5 h, it was cooled to −50 °C. NIS (57 mg, 0.25 mmol) and AgOTf (18 mg, 0.07 mmol) were added in one portion. The mixture was allowed to warm up to −20 °C to start the reaction as indicated by the color change in solution. This temperature was maintained for 1 h, when TLC showed the completion of reaction. Saturated aq. NaHCO3 solution (5 mL) was added to quench the reaction and molecular sieves were removed by filtration. The aqueous phase was separated and extracted with CH2Cl2 (3 × 10 mL). The organic phases were combined and washed with saturated aq. Na2SO3 solution, dried over Na2SO4, and then concentrated in vacuum. The crude product was purified by silica gel column chromatography (EA/hexanes = 2:3, v/v) to give 13 (33 mg, 88%) as a white solid. 1H NMR (600 MHz, CDCl3) δ 7.56 (d, 2H, J = 7.5 Hz), 7.34-7.17 (m, 56H), 7.12 (d, 2H, J = 6.9 Hz), 5.64 (d, 2H, J = 3.4 Hz, H-1‴), 5.43 (s, 1H, PhCH), 5.37 (d, 1H, J = 7.7 Hz), 5.36 (d, 1H, J = 5.7 Hz, H-1′), 5.02 (d, 1H, J = 11.7 Hz), 4.94 (d, 1H, J = 11.4 Hz), 4.83 (d, 1H, J = 3.1 Hz, H-1Fuc), 4.83-4.67 (m, 17H), 4.51 (d, 1H, J = 8.0 Hz), 4.47 (d, 1H, J = 11.8 Hz, H-1″), 4.41-4.34 (m, 4H), 4.34 (d, 1H, J = 7.4 Hz, H-1), 4.27-4.25 (m, 3H), 4.12-3.84 (m, 13H), 3.77-3.70 (m, 4H), 3.49-3.26 (m, 7H), 3.21-3.18 (m, 1H), 3.12 (s, 1H), 2.87 (s, 1H), 2.72 (s, 1H), 1.27 (s, 3H), 1.12 (d, 3H, J = 6.9 Hz); 13C NMR (150 MHz, CDCl3) δ 170.4, 139.6, 139.4, 139.1, 139.07, 139.066, 138.9, 138.87, 138.8, 138.3, 138.2, 138.0, 137.9, 128.4, 128.38, 128.35, 128.30, 128.27, 128.2, 128.14, 128.12, 128.1, 128.08, 128.05, 128.0, 127.9, 127.8, 127.7, 127.6, 127.5, 127.49, 127.45, 127.42, 127.38, 127.37, 127.36, 127.34, 127.33, 127.32, 127.26, 127.25, 127.2, 127.1, 127.0, 126.9, 126.8, 125.8, 103.9 (C-1), 100.8 (C-1′), 99.9 (C-1″), 99.7 (PhCH), 98.3 (C-1Fuc), 97.5 (C-1‴), 79.5, 78.6, 78.4, 78.3, 75.6, 75.4, 74.9, 74.8, 74.7, 74.68, 74.5, 73.9, 73.85, 73.5, 73.1, 72.6, 72.5, 72.2, 71.9, 71.2, 70.2, 69.0, 68.3, 67.8, 66.9, 66.4, 66.3, 50.9, 29.7, 23.0, 16.3, 16.1; HRMS [M + Na]+ m/z calcd for C118H128N4NaO24+ 2007.8811, found: 2007.8770.
2-Azidoethyl α-L-fucopyranosyl-(1→2)-β-D-galactopyranosyl-(1→4)-[(α-L-fucopyranosyl)-(1→3)]-(2-deoxy-2-N-acetamido-β-D-glucopyranosyl)-(1→3)-β-D-galactopyranosyl-(1→4)-β-D-glucopyranoside (1).
The mixture of 9 (12 mg, 0.005mol) and 10% Pd/C (2 mg) in CH2Cl2/MeOH/H2O (3:3:1, 3.5 mL) and 3 drops of AcOH was stirred under an H2 atmosphere at 50 psi for 48 h. The catalyst was removed by filtration through a pad of Celite and the solid was washed with water. The combined filtrate was concentrated in vacuum and the resultant crude product was purified by gel filtration column chromatography using Sephadex G-15 with distilled water as the eluent, followed by lyophilization to afford 1 (5 mg, 95%) as a white solid. 1H NMR (600 MHz, D2O) δ 5.09 (d, 1H, J = 3.9 Hz, H-1Fuc), 4.93 (d, 1H, J = 6.4 Hz, H-1Fuc), 4.70 (overlap with solvent peak, H-5Fuc), 4.53 (d, 1H, J = 8.4 Hz, anomeric), 4.36 (d, 1H, J = 7.9 Hz, anomeric), 4.33 (d, 1H, J = 7.8 Hz, anomeric), 4.26 (d, 1H, J = 8.1 Hz, anomeric), 4.07 (d, 1H, J = 6.8 Hz), 3.98-3.93 (m, 3H), 3.82-3.36 (m, 38H), 3.29-3.17 (m, 3H), 3.10-3.08 (m, 2H), 1.85 (s, 3H), 1.08 (d, 3H, J = 6.6 Hz), 1.05 (d, 3H, J = 6.6 Hz); 13C NMR (150 MHz, D2O) δ 174.7, 102.8 (anomeric), 102.7 (anomeric), 101.9 (anomeric), 100.1 (anomeric), 99.3 (C-1Fuc), 98.5 (C-1Fuc), 81.9, 78.1, 76.3, 75.3, 74.8, 74.7, 74.1, 73.5, 73.0, 72.6, 71.9, 71.6, 69.9, 69.7, 69.1, 68.7, 68.2, 68.19, 67.6, 66.8, 66.7, 65.7, 61.4, 60.9, 60.8, 59.9, 46.6, 39.4, 39.3, 22.2, 15.4, 8.1; HRMS [M + H]+ m/z calcd for C40H71N2O29+ 1043.4137, found: 1043.4132.
2-Azidoethyl α-L-fucopyranosyl-(1→2)-β-D-galactopyranosyl-(1→4)-[(α-L-fucopyranosyl)-(1→3)]-(2-deoxy-2-N-acetamido-β-D-glucopyranosyl)-(1→3)-β-D-galactopyranoside (2).
Prepared from compound 13 with same procedure as for compound 1 (see experiment section in main text), and the yield was 93% (10 mg of compound 13 give 4 mg of compound 2 in presence of 2 mg of Pd/C). 1H NMR (600 MHz, D2O) δ 5.10 (d, 1H, J = 3.1 Hz, H-1Fuc), 4.94 (d, 1H, J = 3.5 Hz, H-1Fuc), 4.70 (m, overlap with solvent peak, H-5Fuc), 4.54 (d, 1H, J = 8.5 Hz, anomeric), 4.33 (d, 1H, J = 7.7 Hz, anomeric), 4.26 (d, 1H, J = 7.8 Hz, anomeric), 3.97 (d, 1H, J = 6.5 Hz), 3.97 (d, 1H, J = 3.3 Hz), 3.96-3.92 (m, 1H), 3.83-3.41 (m, 32H), 3.29-3.27 (m, 1H), 3.09-3.08 (m, 2H), 1.85 (s, 3H), 1.09 (d, 3H, J = 6.6 Hz), 1.06 (d, 3H, J = 6.6 Hz); 13C NMR (150 MHz, D2O) δ 174.7, 102.7 (anomeric), 102.4 (anomeric), 100.1 (anomeric), 99.4 (C-1Fuc), 98.5 (C-1Fuc), 82.0, 76.3, 75.3, 74.8, 74.6, 73.5, 73.0, 71.9, 71.6, 69.7, 69.66, 69.1, 68.7, 68.2, 67.6, 66.8, 66.7, 65.7, 61.4, 60.8, 60.0, 59.7, 39.4, 22.2, 19.6, 15.4; HRMS [M + H]+ m/z calcd for C34H61N2O24+ 881.3609, found: 881.3613.
Conjugation of oligosaccharides 1 and 2 with carrier proteins.
A mixture of 1 or 2 (6.0 mg) with disuccinimidal glutarate 14 (15 equiv) in DMF and PBS buffer (0.1 X) (4:1, 0.5 mL) was stirred at rt for 4 h. After the reaction solution was condensed in vacuum, EtOAc (9 volumes) was added. The precipitates were isolated and then washed with EtOAc 10 times and dried in vacuum to result in crude active ester 15a or 15b that was directly used for the next step. The crude product 15a or 15b was mixed with HSA or KLH in 0.1X PBS buffer (0.4 mL) at a molar ratio of 30:1 and 100:1 respectively. The solution was stirred at rt for 3 days and then applied to a Sephadex 200 column, which was eluted with 0.1X PBS buffer (I = 0.1, pH = 7.8) to separate the protein conjugate from remaining free oligosaccharide. Fractions containing the glycoprotein, which were characterized by the bicinchoninic acid (BCA) assay for proteins and also charring with 15% (v/v) H2SO4 in EtOH for carbohydrates, were combined and dialyzed against distilled water for 2 days, and finally lyophilized to afford the desirable glycoconjugate 16a, 16b, 17a or 17b as a white solid, respectively.
Analysis of the carbohydrate loadings of glycoconjugates.
The carbohydrate loadings were determined by literature methods,11 and the results are presented in SI. The sugar calibration curve was prepared using a standard solution of mixtures of fucose, galactose, N-acetylglucosamine, and glucose (1 mg/mL with molar ratios equal to the ratio in each antigen) in distilled water. Aliquots were transferred to 10 dry 10 ml tubes in 5 μL increments ranging from 5 to 50 μL. In another 10 mL test tube, accurately weighed samples of the glycoconjugates (final concentration should be in the range of the calibration curve derived from monosaccharide mixtures) to be analyzed were placed. At this point, all the tubes should contain between 5 to 50 μg of sugar, and one should contain an unknown amount of sugar in the conjugate to be determined. To all tubes were sequentially added 500 μL of 4% phenol and 2.5 mL of 96% sulfuric acid. The glycosyl linkages were cleaved and a colored complex was formed in this step. Solutions were transferred from the test tubes to cuvettes and measured at the wavelength of 490 nm. The calibration curve was obtained by plotting A490 absorbance against the weight (μg) of sugar in the standard samples. The amount of sugar present in each unknown sample was calculated based on the A490 of the unknown sample against the calibration curve, while the free protein KLH were used as blank control for conjugates. The carbohydrate loading of each KLH glycoconjugate was calculated according to the following equation.
The carbohydrate loadings of HSA conjugates were measured by MS (See Supporting Information) and calculated using the following equation.
Immunization of mice.
Twenty-five female C57BL/6J mice (6-8 weeks of age) were randomly separated into five groups for five mice in each group. On day 1, each mouse was immunized via subcutaneous injection of 0.1 mL of the CFA emulsion of a specific glycoconjugate containing 3 μg of a carbohydrate antigen. On day 8, 15 and 21, respectively, each mouse received intraperitoneal (i.p.) injection of the same glycoconjugate of the same dose as an IFA emulsion. Mice were bled by the saphenous vein on day 0 before the first injection and on day 21 and 43 after immunization. The blood samples were clotted and then centrifuged at 5000 rpm for 7 min at 4 °C to obtain sera which were stored at −80 °C before use. Note: The animal use protocol for this study (# 201609560) was approved by the Institutional Animal Care and Use Committee of University of Florida.
ELISA.
These assays were performed following previous protocols with minor modifications.17 ELISA plates were incubated with 100 μL/well of conjugate 1 or 2 solution (2 μg/mL) in coating buffer (0.1 M bicarbonate, pH 9.6) at 4 °C overnight and then at 37 °C for 1 h. After washing with PBST (PBS buffer containing 0.05% Tween-20) 3 times, the plates were treated with blocking buffer (1% BSA in PBST) at rt for 1 h. Subsequently, mouse serum was added to the plates (100 μL/well), and the plates were incubated at 37 °C for 2 h. For the detection of total, IgG and IgM antibodies, as well as the cross-reactivity analysis, the pooled mouse antiserum was diluted in a serial half-log manner from 1:300 to 1: 72900 in PBS. The plates were washed with PBST 3 times after the removal of serum, followed by incubating at rt for 1 h with AP-linked goat anti-mouse kappa (for total antibody), IgG or IgM antibody (100 μL/well) with dilutions at 1:1000 in PBS. After washing with PBST 3 times, the plates were incubated with p-nitrophenylphosphate (PNPP) solution (100 μL, 1.67 mg/mL in PNPP buffer) at rt for 30 min. The OD values of these plates were measured using a microplate reader at 405 nm wavelength. For antibody titer calculation, the OD values were plotted against the serum dilution numbers to obtain a best-fit logarithm line and the equation of this line was used to calculate the dilution number. The antibody titer is defined as the dilution number at which the OD value of 0.2 was achieved.
Statistical analysis.
Biological data were analyzed by independent t test using the SPSS software, where P < 0.05 was considered as statistically significant.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful for the financial supports from the S. and R. Scott Endowment, University of Florida and the National Institutes of Health (R01CA095142).
Footnotes
ASSOCIATED CONTENT
1H, 13C, and 2D NMR spectra, HRMS. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).(a) Stowell SR; Ju T; Cummings RD, Protein Glycosylation in Cancer. Annu. Rev. Pathol. Mech. Dis 2015, 10, 473–510; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ohtsubo K; Marth JD, Glycosylation in cellular mechanisms of health and disease. Cell (Cambridge, MA, U. S.) 2006, 126, 855–867; [DOI] [PubMed] [Google Scholar]; (c) Fuster MM; Esko JD, The sweet and sour of cancer: Glycans as novel therapeutic targets. Nat. Rev. Cancer 2005, 5, 526–542. [DOI] [PubMed] [Google Scholar]
- (2).(a) Lloyd KO, Humoral immune responses to tumor-associated carbohydrate antigens. Semin. Cancer Biol 1991, 2, 421–31; [PubMed] [Google Scholar]; (b) Feizi T, Carbohydrate antigens in human cancer. Cancer Surv 1985, 4, 245–69; [PubMed] [Google Scholar]; (c) Hakomori S, Aberrant glycosylation in cancer cell membranes as focused on glycolipids: overview and perspectives. Cancer Res. 1985, 45, 2405–14. [PubMed] [Google Scholar]
- (3).(a) Feng D; Shaikh AS; Wang F, Recent Advance in Tumor-associated Carbohydrate Antigens (TACAs)-based Antitumor Vaccines. ACS Chem. Biol 2016, 11, 850–863; [DOI] [PubMed] [Google Scholar]; (b) Ragupathi G; Deshpande PP; Coltart DM; Kim HM; Williams LJ; Danishefsky SJ; Livingston PO, Constructing an adenocarcinoma vaccine: Immunization of mice with synthetic KH-1 nonasaccharide stimulates anti-KH-1 and anti-Ley antibodies. Int. J. Cancer 2002, 99, 207–212. [DOI] [PubMed] [Google Scholar]
- (4).Lee YC; Lee RT, Carbohydrate-Protein Interactions: Basis of Glycobiology. Acc. Chem. Res 1995, 28, 321–7. [Google Scholar]
- (5).Nudelman E; Levery SB; Kaizu T; Hakomori S, Novel fucolipids of human adenocarcinoma: characterization of the major Ley antigen of human adenocarcinoma as trifucosylnonaosyl Ley glycolipid (III3FucV3FucVI2FucnLc6). J. Biol. Chem 1986, 261, 11247–53. [PubMed] [Google Scholar]
- (6).(a) Hummel G; Schmidt RR, Glycosylimidates. 79. A versatile preparation of the lactoneo-series antigens - preparation of sialyl dimer Lewis X and of dimer Lewis Y. Tetrahedron Lett. 1997, 38, 1173–1176; [Google Scholar]; (b) Windmueller R; Schmidt RR, Efficient synthesis of lactoneo series antigens H, Lewis X (Lex), and Lewis Y (Ley). Tetrahedron Lett. 1994, 35, 7927–30; [Google Scholar]; (c) Ball GE; O’Neill RA; Schultz JE; Lowe JB; Weston BW; Nagy JO; Brown EG; Hobbs CJ; Bednarski MD, Synthesis and structural analysis using 2-D NMR of sialyl Lewis X (SLex) and Lewis X (Lex) oligosaccharides: ligands related to E-selectin [ELAM-1] binding. J. Am. Chem. Soc 1992, 114, 5449–51; [Google Scholar]; (d) Behar V; Danishefsky SJ, Highly convergent synthesis of blood group determinant Lewisy in conjugate-forming form. Angew. Chem 1994, 106, 1536–8 (See also Angew Chem, Int Ed Engl, 1994, 33(14), 1468-70); [Google Scholar]; (e) Kinzy W; Loew A, Carbohydrate building blocks for tumor associated antigens. Synthesis of Lewis Y determinants. Carbohydr. Res 1993, 245, 193–218. [DOI] [PubMed] [Google Scholar]
- (7).(a) Smaletz O; Diz MDPE; do Carmo CC; Sabbaga J; Cunha-Junior GF; Azevedo SJ; Maluf FC; Barrios CH; Costa RL; Fontana AG; Madrigal V; Wainstein AJ; Yeda FP; Alves VA; Moro AM; Blasbalg R; Scott AM; Hoffman EW, A phase II trial with anti-Lewis-Y monoclonal antibody (hu3S193) for the treatment of platinum resistant/refractory ovarian, fallopian tube and primary peritoneal carcinoma. Gynecol. Oncol 2015, 138, 272–277; [DOI] [PubMed] [Google Scholar]; (b) Kudryashov V; Kim HM; Ragupathi G; Danishefsky SJ; Livingston PO; Lloyd KO, Immunogenicity of synthetic conjugates of Lewis y oligosaccharide with proteins in mice: towards the design of anticancer vaccines. Cancer Immunol Immunother 1998, 45, 281–286; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Scott AM; Geleick D; Rubira M; Clarke K; Nice EC; Smyth FE; Stockert E; Richards EC; Carr FJ; Harris WJ; Armour KL; Rood J; Kypridis A; Kronina V; Murphy R; Lee FT; Liu Z; Kitamura K; Ritter G; Laughton K; Hoffman E; Burgess AW; Old LJ, Construction, production, and characterization of humanized anti-Lewis Y monoclonal antibody 3S193 for targeted immunotherapy of solid tumors. Cancer Res. 2000, 60, 3254–3261. [PubMed] [Google Scholar]
- (8).(a) Mandal PK; Turnbull WB, Studies on the synthesis of Lewis-y oligosaccharides. Carbohydr. Res 2011, 346, 2113–2120; [DOI] [PubMed] [Google Scholar]; (b) Spassova MK; Bornmann WG; Ragupathi G; Sukenick G; Livingston PO; Danishefsky SJ, Synthesis of Selected LeY and KH-1 Analogues: A Medicinal Chemistry Approach to Vaccine Optimization. J. Org. Chem 2005, 70, 3383–3395; [DOI] [PubMed] [Google Scholar]; (c) Love KR; Seeberger PH, Automated solid-phase synthesis of protected tumor-associated antigen and blood group determinant oligosaccharides. Angew. Chem., Int. Ed 2004, 43, 602–605; [DOI] [PubMed] [Google Scholar]; (d) Mong K-KT; Wong C-H, Reactivity-based one-pot synthesis of a Lewis Y carbohydrate hapten: a colon-rectal cancer antigen determinant. Angew. Chem., Int. Ed 2002, 41, 4087–4090; [DOI] [PubMed] [Google Scholar]; (e) Buskas T; Li Y; Boons G-J, Synthesis of a dimeric lewis antigen and the evaluation of the epitope specificity of antibodies elicited in mice. Chem. - Eur. J 2005, 11, 5457–5467. [DOI] [PubMed] [Google Scholar]
- (9).(a) Huang X; Huang L; Wang H; Ye X-S, Iterative one-pot synthesis of oligosaccharides. Angew. Chem., Int. Ed 2004, 43, 5221–5224; [DOI] [PubMed] [Google Scholar]; (b) Miermont A; Zeng Y; Jing Y; Ye X-S; Huang X, Syntheses of LewisX and Dimeric LewisX: Construction of Branched Oligosaccharides by a Combination of Preactivation and Reactivity Based Chemoselective One-Pot Glycosylations. J. Org. Chem 2007, 72, 8958–8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Li Q; Guo Z, Pondering the Structural Factors that Affect 1,2-trans-Galactosylation: A Lesson Learnt from 3-O-β-Galactosylation of Galactosamine. J Carbohydr Chem 2018, 47, 347–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wrolstad ,RE; Acree TE; Decker EA; Penner MH; Reid DS; Schwartz SJ; Shoemaker CF; Smith D; Sporns P; Editors, Handbook of Food Analytical Chemistry: Pigments, Colorants, Flavors, Texture, and Bioactive Food Components. 2005; p 606 pp. [Google Scholar]
- (12).Wang Q; Ekanayaka SA; Wu J; Zhang J; Guo Z, Synthetic and immunological studies of 5’-N-phenylacetyl sTn to develop carbohydrate-based cancer vaccines and to explore the impacts of linkage between carbohydrate antigens and carrier proteins. Bioconjugate Chem. 2008, 19, 2060–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Zhou Z; Mandal SS; Liao G; Guo J; Guo Z, Synthesis and Evaluation of GM2-Monophosphoryl Lipid A Conjugate as a Fully Synthetic Self-Adjuvant Cancer Vaccine. Sci Rep 2017, 7, 11403; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang Q; Zhou Z; Tang S; Guo Z, Carbohydrate-monophosphoryl lipid a conjugates are fully synthetic self-adjuvanting cancer vaccines eliciting robust immune responses in the mouse. ACS Chem. Biol 2011, 7, 235–240; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ghazarian H; Idoni B; Oppenheimer SB, A glycobiology review: carbohydrates, lectins and implications in cancer therapeutics. Acta Histochem 2011, 113, 236–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Zhou Z; Liao G; Mandal SS; Suryawanshi S; Guo Z, A Fully Synthetic Self-Adjuvanting Globo H-Based Vaccine Elicited Strong T Cell-Mediated Antitumor Immunity. Chem Sci 2015, 6, 7112–7121; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Song C; Zheng X-J; Liu C-C; Zhou Y; Ye X-S, A cancer vaccine based on fluorine-modified sialyl-Tn induces robust immune responses in a murine model. Oncotarget 2017, 8, 47330; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kieber-Emmons T; Luo P; Qiu J; Chang TY; Insug O; Blaszczyk-Thurin M; Steplewski Z, Vaccination with carbohydrate peptide mimotopes promotes anti-tumor responses. Nat. Biotechnol 1999, 17, 660. [DOI] [PubMed] [Google Scholar]
- (15).Yu H; Williams DL; Ensley HE, 4-Acetoxy-2,2-dimethylbutanoate: a useful carbohydrate protecting group for the selective formation of β-(1→3)-D-glucans. Tetrahedron Lett. 2005, 46, 3417–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wang Z; Zhou L; El-Boubbou K; Ye X-S; Huang X, Multi-Component One-Pot Synthesis of the Tumor-Associated Carbohydrate Antigen Globo-H Based on Preactivation of Thioglycosyl Donors. J. Org. Chem 2007, 72, 6409–6420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zhou Z; Mondal M; Liao G; Guo Z, Synthesis and evaluation of monophosphoryl lipid A derivatives as fully synthetic self-adjuvanting glycoconjugate cancer vaccine carriers. Org. Biomol. Chem 2014, 12, 3238–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.