

Abstract
Motivation
To facilitate accurate estimation of statistical significance of sequence similarity in profile–profile searches, queries should ideally correspond to protein domains. For multidomain proteins, using domains as queries depends on delineation of domain borders, which may be unknown. Thus, proteins are commonly used as queries that complicate establishing homology for similarities close to cutoff levels of statistical significance.
Results
In this article, we describe an iterative approach, called LAMPA, LArge Multidomain Protein Annotator, that resolves the above conundrum by gradual expansion of hit coverage of multidomain proteins through re-evaluating statistical significance of hit similarity using ever smaller queries defined at each iteration. LAMPA employs TMHMM and HHsearch for recognition of transmembrane regions and homology, respectively. We used Pfam database for annotating 2985 multidomain proteins (polyproteins) composed of >1000 amino acid residues, which dominate proteomes of RNA viruses. Under strict cutoffs, LAMPA outperformed HHsearch-mediated runs using intact polyproteins as queries by three measures: number of and coverage by identified homologous regions, and number of hit Pfam profiles. Compared to HHsearch, LAMPA identified 507 extra homologous regions in 14.4% of polyproteins. This Pfam-based annotation of RNA virus polyproteins by LAMPA was also superior to RefSeq expert annotation by two measures, region number and annotated length, for 69.3% of RNA virus polyprotein entries. We rationalized the obtained results based on dependencies of HHsearch hit statistical significance for local alignment similarity score from lengths and diversities of query-target pairs in computational experiments.
Availability and implementation
LAMPA 1.0.0 R package is placed at github (https://github.com/Gorbalenya-Lab/LAMPA).
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Due to high-throughput next-generation sequencing, genomics is outpacing functional and structural characterization of proteins (Brister et al., 2015). This gap is especially pronounced and fast growing for viruses, whose discovery and characterization in diverse habitats has been driven by metagenomics over the last 10 years (Suttle, 2007; Zhang et al., 2019).
In genomics projects, conceptually translated open reading frames (ORFs) are functionally characterized by bioinformatics tools which use homology recognition for annotation. To improve accuracy of protein annotation, bioinformatics tools use iterative searches of databases of individual sequences [e.g. PSI-BLAST (Altschul et al., 1997) versus GenBank (Sayers et al., 2019)], search profile databases [e.g. HMMER (Finn et al., 2011) or HHsearch (Remmert et al., 2012; Söding, 2005) versus Pfam (El-Gebali et al., 2019) or HHblits (Remmert et al., 2012) versus Uniclust30 (Mirdita et al., 2017)], and may involve comparison of query and target secondary structure [e.g. HHsearch versus SCOP (Fox et al., 2014)]. Annotation pipelines favor selectivity over sensitivity by imposing stringent cutoffs on similarity between query and database entries. Scores of similarity are interpreted in statistical frameworks using either expectation values (default cutoff E = 0.001, BLAST, HMMER, HHsearch) or homology Probability (default cutoff P = 95%, HHsearch).
To recognize distant homologs, popular HHsearch was fine-tuned based on a subset of SCOP 1.63 database with less than 20% pairwise sequence identity of structural domains (Söding 2005), where mean sequence length is equal 178 aa (Fox et al., 2014; Fig. 1), typical of functional and structural domain (Wheelan et al., 2000). Its hit statistical significance increases with score of similarity between query and target, and it depends on sizes and diversities of query and target (Remmert, 2011). Specifically, large size increases likelihood of a hit score emerging by chance, while the opposite is true for small size. Notwithstanding HHsearch training on protein domains, it has been routinely used in analysis of proteins of unknown domain organization. For a single-domain protein, statistical significance of hit similarity must be applicable to its domain, since sizes of both are similar. On the other hand, for multidomain queries, statistical support of a hit associated with individual domain may be underestimated due to inflated search space that encompasses other domains of the query protein (Altschul et al., 1997; Söding, 2005).
Fig. 1.

Length distribution of proteins in datasets relevant to comparison of HHsearch and LAMPA. This plot depicts sizes of six protein datasets labeled from A to F and used or cited in this study. (A) 6271 SCOP domains used for HHsearch training (range: 21–1504 aa); (B) 2985 RefSeq virus polyproteins (range: 1001–8572 aa); (C) 431 RefSeq virus polyproteins which include 507 regions exclusively annotated by LAMPA (range: 1039–8572 aa); (D) 507 hit regions generated by LAMPA from 431 RefSeq polyproteins (range: 88–2172 aa); (E) 507 domains tentatively demarcated around LAMPA hits (range: 164–732 aa); and (F) 41 designed sizes of each of three proteins, 123 in total, tested in computational experiments (range: 10–100.000 aa)
The query size issue could be of little practical consequence for proteins having closely related homologs in sequence databases. However, for identification of distant relationships, accurate estimation of statistical significance could be impactful. The above problem may be particularly acute for RNA viruses (Baltimore, 1971), which typically encode large multidomain proteins (>1000 aa) (Das and Arnold, 2015). (Hereafter and for sake of simplicity, we will use polyprotein to refer to virus multidomain proteins). They are much larger than most proteins of cellular organisms, whose length distributions resemble lognormal, with a mean below 500 aa (Zhang, 2000). Human immunodeficiency virus, Ebola virus, severe acute respiratory syndrome coronavirus and poliovirus, and very many other eukaryotic viruses encode polyproteins (Dougherty and Semler, 1993; Gorbalenya and Snijder, 1996). These polyproteins mediate replication/transcription and promote virus particle formation in either the synthesized form or after being proteolytically processed. Furthermore, the already known proteomes of RNA viruses are exceptionally diverse due to high mutation rate of RNA viruses (Sanjuan et al., 2010), with many relationships in twilight and midnight zones of homology (Habermann, 2016; Kuchibhatla et al., 2014).
In our recent HH-suit-mediated analysis of the largest known polyprotein of RNA virus (PSCNV, 13 556 aa) (Saberi et al., 2018), we initially annotated only three regions by homology (polyprotein 7.1%). To check whether this result could be partially attributed to an underestimation of genuine statistical significance of the similarity between polyprotein domains and target protein profiles, we split the polyprotein using comparative genomics and, indeed, identified three other homologs with high confidence (Saberi et al., 2018).
The above positive experience led us to formalize this approach in R package, called LAMPA, LArge Multidomain Protein Annotator, that we describe in this article. Also, we present proof-of-the principle for LAMPA in study of homology between RNA virus polyproteins and pfamA_31.0 database. It was further supported and expanded by evaluation of dependences of HHsearch statistics for fixed similarity score from lengths and diversities of query and target in computational experiments.
2 Materials and methods
2.1 Databases and virus protein dataset
We used pfamA_31.0 database (El-Gebali et al., 2019), accompanying HH-suite (Remmert et al., 2012), as target database to identify homology by profile searches and transfer annotation. We were interested in annotating virus proteins and selected a subset of NCBI Viral Genomes Resource database (RefSeq) (Brister et al., 2015) to serve as queries in homology searches and the source of expert annotation (Supplementary Text S1.1). Only proteins of true RNA viruses that use RNA-dependent RNA polymerase (RdRp), positive and negative single-strand ed RNA viruses, (+)ssRNA and (−)ssRNA, respectively, and double-stranded RNA viruses, dsRNA, were included in the query protein dataset (Supplementary Fig. S1). Protein sequences were obtained from ‘translation’ qualifiers of ‘CDS’ features in RefSeq genome entries. The query database included all 2985 protein sequences of RNA virus genomes listed in ‘Viral genome browser’ table on 26 July 2018 (Supplementary Table S1), that were 1000 aa or longer (protein length ranged from 1001 aa to 8572 aa, median = 2081 aa; Fig. 1). It was further grouped into 884 clusters using MMseqs2 (Steinegger and Söding, 2017), following the authors recommendations for multidomain proteins and defining sequence identity rate (–cluster-mode 1 –min-seq-id 0.3 –alignment-mode 3) and local alignment coverage (–cov-mode 0 -c 0.8) (see Supplementary Text S1.2 and Table S1). Most of these proteins are encoded in a single ORF (Firth and Brierley, 2012). We parsed RefSeq entries corresponding to the analyzed proteins to extract region annotations from ‘Region’ features (O’Leary et al., 2016). Other annotation features, such as ‘CDS’, ‘Protein’ and ‘Site’, which were not taken into analysis, may overlap with the ‘Region’ or include extra information. For further details about polyprotein query dataset see Supplementary Text S1.1.
2.2 Comparative sequence analysis
Transmembrane (TM) helices in protein sequences were predicted by TMHMM 2.0c (Sonnhammer et al., 1998). Secondary structures (SS) of query sequences, regardless of their length, were derived from the predictions made for the respective entire polyproteins by script addss.pl from HH-suite 3.0.0 (15 March 2015) (Steinegger et al., 2019), which used PSIPRED 3.5 tool (Jones, 1999). Query profiles were built and compared to a database by programs HHmake and HHsearch from HH-suite 2.0.16, respectively (Söding, 2005; http://wwwuser.gwdg.de/~compbiol/data/hhsuite/releases/all/). In all analyses, parameters of HH-suite programs were left at default values, with the exception of HHmake parameter ‘-M first’, indicating that columns with residue in the first sequence of the FASTA file are considered match states, and HHsearch three parameters: ‘-p 0’, allowing hits with Probability as low as zero; ‘-norealign’, blocking realignment of reported hits using maximum accuracy algorithm; ‘-alt 10’, enabling reporting up to 10 significant alternative alignments between a query and a target profile (Söding, 2005) (Supplementary Text S1.3). To identify statistically significant hits and homologous regions, HHsearch hits were subjected to post-processing under three cutoffs: Probability >95%, E-value <10 and hit length of >50 aa of the query sequence. Hits satisfying these thresholds and overlapping on query were combined into a cluster, extreme N- and C-terminal residues of which defined boundaries of region in the query that was homologous to target(s). Statistics of the top-scoring hit in the cluster defined the entire cluster, and name of the top-scoring target profile in the cluster annotated the query region. Unless stated otherwise, all reported analyses used the hits post-processing. Also, we used HHblits v.3 (Remmert et al., 2012) for analysis of selected polyproteins as detailed in Supplementary Text S1.4. Analysis and visualization were performed using R 3.3.0 (R Core Team, 2018, https://www.R-project.org/).
2.3 Statistics
P-value of Wilcoxon signed-rank test (PW) was calculated using function ‘wilcox.test’ from R package ‘stats’, with arguments ‘paired’ and ‘alternative’ set to values ‘TRUE’ and ‘greater’, respectively (R Core Team, 2018, https://www.R-project.org/).
2.4 Calculation of HHsearch P-value and Probability dependence from lengths and diversities of query-target pair for fixed hit score
HHsearch uses extreme value distribution (EVD) model for estimating hit’s P-value, E-value, and Probability from query-target local alignment similarity score. P-value for a given score is defined as:
| (1) |
where λ and μ are the EVD parameters that optimally approximate the score distribution of false positives for a given pair of query and target profiles. E-value is defined as Pvalue(score)*NDB, where NDB is the number of searched target profiles in the database. For calculations of λ and μ, HHsearch uses ‘profile auto-calibration’ that employs two simple artificial neural networks (Remmert, 2011). This default procedure makes use of dependence of λ and μ on four characteristics: profile lengths and sequence diversities of both query and target. The parameters of the neural networks were derived by training on a set of profiles based on 6271 sequences of SCOP20 v1.73 database (minimal, median and maximal protein lengths = 21 aa, 142 aa and 1504 aa, respectively; 5-to-95% range = 48-to-392 aa) (Fig. 1). Estimation for Probability of detecting homologous relationship (true positives) is also based on the EVD distribution but involves correction by the SS alignment score.
To learn how HHsearch performs on queries of our study with sizes close to or exceeding the largest protein in the training SCOP database, we conducted computational experiments using the HHsearch procedure that generates EVD parameters by adapting corresponding C++ source code into a Python Jupyter notebook (https://github.com/Gorbalenya-Lab/hh-suite-notebooks/tree/LAMPA). We approximated P-value and Probability of hit for fixed local alignment similarity score (including also SS alignment score for Probability) in relation to lengths and/or diversities of the corresponding query and target profiles, one of which may have been set to vary in large range of values (see Supplementary Text S1.5).
3 Results
3.1 LAMPA, iterative approach for homology recognition and functional annotation of multidomain proteins
LAMPA approach is aimed at improving detection of remote homology in large multidomain proteins (queries). Its multistage iterative procedure includes prediction of TM regions in query by TMHMM at the pre-iteration Stage #0 and comparisons of query and its regions with HH-suite profile database(s) (targets) using HHsearch for iterations at Stages #1–#3 (Fig. 2). As query, intact protein is used for Stages #0 and #1, and various protein regions are used for Stages #2 and #3. Iteration is a single execution of a procedure involving protein regions demarcation and submission of regions to HHsearch-mediated homology searches to identify statistically significant hits (values of post-processing cutoffs, specified in Section 2.2, are default). The approach stages are detailed below:
Fig. 2.
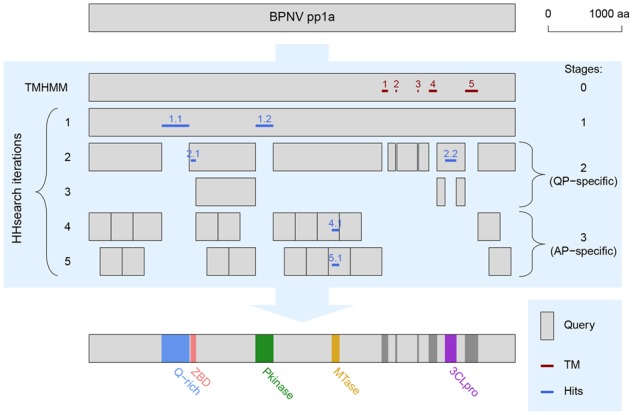
LAMPA workflow and its application to RNA virus polyprotein. Presented is outline of the LAMPA approach (blue background) applied to polyprotein 1a (pp1a) of BPNV. Gray bars, regions of BPNV pp1a that served as TMHMM or HHsearch queries. Iterations of the procedure and programs used are depicted on the left; stages are indicated on the right. Clusters of TM helices are depicted in dark red, clusters of hits—in dark blue. Hit double digits refer to iteration and hit position on polyprotein from left to right, respectively, except for hits at Stage #0 which are labelled with the position only. Hits and annotations obtained on Stage #1 represent output of conventional HHsearch. Q-rich, region rich in glutamine residue; ZBD, zinc-binding domain; Pkinase, protein kinase; MTase, methyltransferase; 3CLpro, 3C-like protease. For other details see text. (Color version of this figure is available at Bioinformatics online.)
Stage #0. Detection of TM regions in original query. TM region (domain) may include either single or few helices predicted by TMHMM. By default, more than one helix is included in a region if each helix is separated from its neighbor by <100 aa. Region boundaries are defined by either helix boundaries (single-helix region) or opposite boundaries of two respective terminal helices (multiple-helix region). TM regions are used to split original query into smaller regions (see Stage #2).
Stage #1. Detection of homology regions in original query. This is the first iteration of the annotation procedure that uses HHsearch-mediated homology search. Its input and output are the original query and hit-annotated regions, respectively.
Stage #2. Detection of homology regions in split query: query-protein-specific (QP-specific) iterations. To initiate this stage, the procedure selects regions of the original query that are flanked by either of the following: N- or C-terminus of the original query, TM regions and hits clusters identified at the Stages #0 and #1, respectively. These regions are used as input to HHsearch-mediated homology searches. Obtained hits are used for annotation and to demarcate flanking smaller non-annotated regions. The latter are used to initiate a new iteration in the manner described above. The iterations are repeated until no hits satisfying the cutoffs are identified.
Stage #3. Detection of homology regions in split query: average-protein-size-specific (AP-specific) iterations. Non-annotated regions after the Stage #2 are split into two overlapping sets of 300 aa queries (default). The most C-terminal queries of both sets are extended to include the remaining part of the respective region, if the remaining part is shorter than 300/2 = 150 aa (default) and if the extended query does not cover the entire region. The default 300 aa size is close to that of an average protein (AP), hence respective iterations are called AP-specific. Queries are defined starting from either the N-terminus (first AP-specific iteration) or 300/2 = 150 aa (default) downstream the N-terminus (second AP-specific iteration) of the non-annotated regions of Stage #2. They are run independently. During this stage, one and the same region of polyprotein may be found to have homolog and be annotated on both AP-specific iterations since two sets overlap.
3.2 LAMPA implementation
The above approach was realized as LAMPA 1.0.0 R package (see also Supplementary Text S1.6) that includes a single command ‘LAMPA’ with 15 arguments that allow user to specify a single protein query sequence, target database(s), information required to run HH-suit and TMHMM, and parameters of the LAMPA procedure, which are detailed in the package manual (see https://github.com/Gorbalenya-Lab/LAMPA/blob/master/LAMPA_manual.pdf). LAMPA package employs two external R packages: seqinr (Charif and Lobry, 2007) and IRanges (Lawrence et al., 2013). Output of the command is a directory, name of which is identical to the name of the file with query sequence by default. This directory contains a plot (similar to Fig. 2) and two tables summarizing TM predictions and homology annotations made for the query sequence (overlapping with Supplementary Table S2), as well as files with detailed information about hits constituting each cluster, and a folder with raw data (see package manual for details). Analysis of 2985 virus polyproteins against pfamA_31.0, detailed below, required 2000 min on 16 CPUs for LAMPA to complete (with 0.3–2.5 min per query, and approximately extra 1000 min compared to HHsearch). A separate script, not included in the LAMPA package, was used to automate analysis of multiple queries in this study.
3.3 Evaluation of LAMPA performance relative to HHsearch in analysis of RNA virus polyproteins
We evaluated LAMPA performance under default parameter values by querying pfamA_31.0 with 2985 RNA virus polyproteins (see Section 2.1; Fig. 1). This analysis documents dependence of HHsearch statistics on query size: split protein fragments or regions (‘LAMPA’) relative to intact proteins (‘HHsearch’). Only the most N-terminal cluster of hits was considered in 26 cases of overlapping clusters from the LAMPA AP-specific stage. For annotation-related statistics, we did not consider TM domains (LAMPA Stage #0, Fig. 2). The output of the LAMPA Stage #1 represented also output of the HHsearch run on intact proteins.
Additionally, HHsearch was also used for further statistical analyses of the difference between outputs of two tools. For these analyses, HHsearch output was not subject to post-processing (see Section 2.2) that allowed to analyse hits with Probability ≤95%, E-value ≥ 10 and size on query ≤50 aa (see below). This use of HHsearch was outside the LAMPA framework and required matching of hits obtained by LAMPA and HHsearch for evaluation. We restricted this matching to the top-scoring hits of LAMPA hit clusters and HHsearch that overlapped on query and targeted the same Pfam profile.
3.4 LAMPA outperforms HHsearch in recognizing homology and facilitating annotation of RNA virus polyproteins
Neither LAMPA nor HHsearch found homology between 163 proteins (5.5% of the dataset) and pfamA_31.0. For 2391 proteins (80.1%), LAMPA and HHsearch hit the same homologous regions, from 1 to 18. For 420 proteins (14.1%), LAMPA annotated from 1 to 3 extra regions on top of 1 to 15 found also by HHsearch (Fig. 3A). For each of the remaining 11 proteins (0.4%), a single region was hit by LAMPA only. Increase in number of annotated regions per protein by LAMPA was statistically significant (PW = 9.5e−86). By design of the procedure, HHsearch outperformed LAMPA for none of the polyproteins. For the three virus genome classes (2273 proteins in total), share of proteins, for which gain in number of annotated regions by LAMPA was observed, varied five-fold: (−)ssRNA viruses (3.1%), dsRNA viruses (10.2%) and (+)ssRNA viruses (15.9%). Among the 712 proteins with unknown virus genome class, LAMPA outperformed HHsearch for 22.2% of polyproteins. Increase in the number of annotated regions (Fig. 3D) was accompanied by the increase in the polyprotein coverage by annotations, which ranged from 1.0% to 25.5% of polyprotein length (Fig. 3B; PW = 1.18e−72).
Fig. 3.

Gain of homology recognition by LAMPA compared to HHsearch. Presented are four depictions of results of querying pfamA_31.0 with 2985 RNA virus proteins using LAMPA and HHsearch. (A) Number of regions (hit clusters) per query protein annotated by the two tools. Each protein is depicted by a transparent gray dot. Since multiple proteins may have the same or similar number of regions annotated by the two tools (x and y dot coordinates), dots may overlap. Gray density is proportional to the number of overlapping dots. Black line, diagonal. (B) Share of protein length (%) annotated by the two tools. For other details, see panel A. (C) Overlap between Pfam profiles that were linked to RNA virus proteins by the two tools. (D) Overlap between RNA virus polyprotein regions annotated by the two tools
Also, we compared lists of Pfam profiles hit by LAMPA and HHsearch and were used for region annotation (Fig. 3C, Supplementary Table S2). Both tools selected 173 profiles to annotate 5737 virus regions, and extra 67 profiles were used to annotate 5508 and 5947 virus regions by HHsearch and LAMPA, respectively. Also, additional 35 profiles were solely used by LAMPA to annotate 68 virus regions. Key enzymes of RNA viruses (RdRp, helicases, proteases and methyltransferases) dominated the shared part of the LAMPA and HHsearch Pfam profile lists (Supplementary Fig. S2A). In contrast, the LAMPA-restricted profiles did not include RdRp but included types of enzymes and non-enzymatic proteins not found in the shared list, e.g. seven kinase profiles (Supplementary Fig. S2B and Table S2). Many protein regions exclusively annotated by LAMPA were from most divergent RNA viruses (Shi et al., 2016).
3.5 Both QP- and AP-specific stages of LAMPA procedure contributed to gain of annotation
Gain of annotation by LAMPA compared to HHsearch is fully attributed to QP- and AP-specific stages. The gain was observed for 431 polyproteins, with the share of regions exclusively annotated by LAMPA varying from 6.2% to 100.0% (mean = 27.2%) of all recognized regions. Mean percentage of regions annotated in these proteins during the Stages #1–#3 were 72.8%, 17.1% and 10.2%, respectively (Fig. 4). During QP- and AP-specific stages, regions were identified in 322 proteins (10.8% of the whole dataset) and 126 proteins (4.2%), respectively.
Fig. 4.
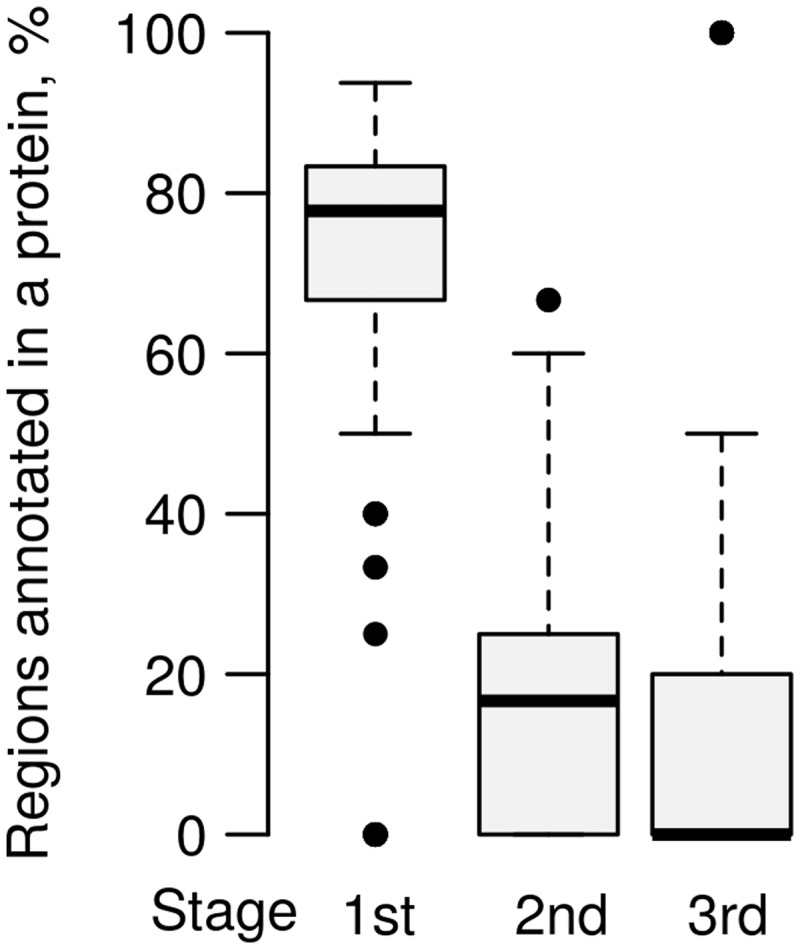
Contribution of different stages of LAMPA procedure to protein annotation. Contribution of three LAMPA stages to annotation of 431 proteins, including regions exclusively annotated by LAMPA, was measured by percentage of regions annotated in each protein. Total number of regions annotated in each protein was considered 100%, regardless of their actual number and share in the protein. The box plots, lower and upper limits of the box delimit the first (25%) and third (75%) quartiles, midline limit of the box—median, whiskers extend to the most extreme data point which is no >1.5 times the interquartile range from the box, data beyond that distance are represented by points
3.6 Increase of hit statistical significance by LAMPA compared to HHsearch is modest but common
LAMPA identified 507 clusters of hits on 431 proteins, HHsearch counterparts of which were removed by post-processing under the used thresholds (see Section 2.2; Fig. 3D). We used the top-scoring hits in these clusters to estimate the gain of statistical significance (Probability and E-value) by LAMPA compared to HHsearch and represent clusters in all analyses described below. We identified matching HHsearch hits for all 507 LAMPA hits (Supplementary Table S2) with 437 hits (86.2%) having identical coordinates on query. In each pair of hits, LAMPA hit was characterized by higher Probability and lower E-value (Fig. 5A and B). Probability increase by LAMPA compared to HHsearch was in the range from 0.5% to 37.6%, with mean 5.3% (Fig. 5A). Decimal logarithm of LAMPA to HHsearch E-values ratio ranged from −3.4 to −0.2 with mean −1.5 (Fig. 5B). Positive correlation between Probability and −logE-value was accompanied by E-value variation around two orders of magnitude for most Probabilities before and after they were elevated above the cutoff by LAMPA (Supplementary Fig. S3). Likewise, for E-values around 10−1, Probability varied approximately ±5%, illustrating that choice of statistic in addition to significance cutoff may affect output.
Fig. 5.

Gain of hit statistical significance by LAMPA compared to HHsearch. LAMPA hits to region queries, obtained during the QP-specific and AP-specific stages of LAMPA procedure, are compared with matching HHsearch hits to polyprotein queries, in respect to hit Probability (A) and E-value (B); and with matching HHsearch hits to putative domain queries (operational definition, see text for details), in respect to hit Probability (C) and E-value (D). Analyzed HHsearch hits were not subject to post-processing
3.7 LAMPA-demarcated regions may approximate authentic domains for purpose of homology detection
The LAMPA region queries may still be (much) larger than the actual domains, natural borders of which remain unknown. Because of this uncertainty, we reasoned that the gain of statistical significance by LAMPA compared to HHsearch might provide only a lower estimate for the actual difference between Probabilities and E-values of the respective hits obtained for the polyprotein and expected for its domains. To improve understanding about how close the obtained LAMPA Probabilities and E-values for protein regions may be to those of the actual domains, we adopted an operational definition of polyprotein domain in relation to homology hit and used it to approximate borders of the actual domains; in total 507 hits on 431 polyproteins (see above) were considered for this purpose. Operational domain was demarcated as LAMPA hit that was extended by 100 aa to the N- and C-terminus; if distance to the polyprotein terminus was <100 aa, extension was adjusted accordingly (which was used in 48 of 507 cases). The demarcated domain sizes ranged from 164 to 732 aa (mean = 315 aa) that was close to dominant domain size in public databases and narrower compared to the range of 88–2172 aa (mean = 479 aa) of region queries that produced the original LAMPA hits (Fig. 1). For each of 507 hits, we then compared Probability and E-value values, assigned by LAMPA, to those obtained by HHsearch for a matching hit in a separate analysis that used demarcated domains as queries and involved no hits post-processing (see Section 2.2; Supplementary Table S2).
We obtained data for all 507 hits, with 457 hits (90.1%) having identical coordinates on query in LAMPA and HHsearch analyses. The difference between the two Probability values ranged from −1.8% to 4.6% with mean and median close to zero (both were equal −0.2%); absolute value of the difference did not exceed 2% in 99.8% of cases (Fig. 5C). Decimal logarithm of the E-values ratio ranged from −1.3 to 1.8, mean 0.2 (Fig. 5D). These differences were evenly distributed and much smaller than those observed in comparison of LAMPA hits to region queries and HHsearch hits to polyprotein queries (Fig. 5A and B). Based on these results, we concluded that sizes of queries used by LAMPA during iterative stages may be close to those of the respective authentic domains for the purpose of statistical evaluation of homology and annotation transfer under the employed cutoff.
3.8 Increase of statistical significance of hits by LAMPA compared to HHsearch is proportional to respective decrease of query length
We then asked how LAMPA-based increase of statistical significance in 507 hits of 431 proteins in 504 pairs of polyprotein and Pfam profile depended on lengths of polyprotein (original query, varied between 1039 aa and 8572 aa) and its fragments (queries varied between 88 aa and 2172 aa at LAMPA Stages #2 and #3) (Fig. 1). We observed steady but highly uneven increase of Probability gain for polyproteins in the size range between 1001 aa and ∼3000 aa which then leveled (Fig. 6A). That positive dependence was stronger and more common when Probability gain was plotted against relative length decrease in queries of LAMPA compared to HHsearch, which varied in the range from 1× to 45.3×, with 68.2% of the decreases of query length being in the 1–10× range (Fig. 6B). Accordingly, Probability gain fall steeply with increase of the LAMPA query length up to 2172 aa; it was below 10% and 5% for LAMPA queries including >448 aa and 747 aa, respectively (Fig. 6C).
Fig. 6.

Relationship between Probability gain by LAMPA and query lengths. Difference between Probabilities of hit to region query (LAMPA Stages #2 or #3) versus polyprotein query (HHsearch without hits post-processing) (empty circle), is compared with difference between the respective approximated Probabilities for the matching hit in computational experiments (cross) at the y axis, for 507 hits in total. These values are plotted against values of three characteristics of respective queries at the x axis: (A) polyprotein length (Stage #1), (B) ratio of polyprotein to query region length (Stage #1 versus Stage #2/3) and (C) query region length (Stage #2/3)
3.9 Estimation of hits Probability by LAMPA may be approximated in computational experiment
Non-uniform dependence of Probability gain from query length (Fig. 6A and C) implied other characteristics are involved. Indeed, besides query length, target length and diversities of query and target are used by HHsearch for the calculation of λ and μ that affect hit score P-value (see Section 2.4). Accordingly, we analyzed the relationship between estimates of hit statistical significance and possible lengths of the corresponding query and target profiles systematically using computational experiments. They used local alignment similarity score of HHsearch hit of full-length query-target pair for approximating hit Probability on queries of other observed and computationally generated sizes, assuming that hit score may not change with query size. This assumption proved to be accurate within a margin of error (see below).
We used the HHsearch neural networks to generate EVD parameters, followed by calculation of Probability, as well as P-value, of hit to polyprotein region from local alignment similarity score of this hit in every full-length query-target pair for which hit Probability gain was observed (in total 507 hits; Figs 3D and 6; for details see https://github.com/Gorbalenya-Lab/hh-suite-notebooks/tree/LAMPA). First, we noted good agreement between gains of Probabilities obtained in computational experiments and LAMPA runs (Fig. 6). They are within of +0.7%/−0.4% deviation of Probability gain estimation by LAMPA for the 95 percentile of hit scores in the dataset (Supplementary Fig. S4A). The modest difference between the two values is explained by respective deviation of the underlying similarity score of the pairwise HHsearch hit alignment for polyprotein, which was fixed in computational experiments, from region-specific score that is calculated for actual query and target profiles by LAMPA. Thus, by default, the same hit alignment involving polyprotein and its part as queries might have slightly different scores and also coordinates, further contributing to difference between the respective Probabilities (and P-values, Supplementary Fig. S4B) in computational experiments.
3.10 P-value and Probability of HHsearch hits depend non-linearly on the lengths and diversities of query and target profiles in computational experiments
The increase of the hit Probability during QP- and AP-specific iterations (Fig. 6) is likely explained by the use of query length in the auto-calibration procedure of HHsearch (see Section 2.4). We then conducted four computational experiments for three selected query-target pairs (Supplementary Text S1.5) that were characterized by the largest Probability gain of LAMPA hit at Stages #2 (37.6%) and #3 (25.8%), respectively, and associated with the largest decrease of query size (47 fold) (Fig. 7, Supplementary Fig. S5 and Table S3). They also represent considerable ranges of hit scores (40.2, 41.1 and 67.2 for three pairs) and target diversities (6.7, 11.5 and 7.7). Forty-one computationally designed lengths of each of three queries were tested (Fig. 1 and Supplementary Text S1.5).
Fig. 7.

Relationship between hit statistical significance and profile lengths in computational experiments. HHsearch hit P-value (A–C) and Probability (D–F) were estimated for 41 designed lengths of query or target, each of which was equidistant from its immediate neighbor on base 10 logarithmic scale (see Supplementary Text S1). The 41 pairs of values were plotted to reveal relationship between two characteristics. These plots used hit score values of three query-target pairs, which are specified at the bottom of the figure and whose respective hit statistics values at the Stage #1 (HHsearch), and Stage #2 or #3 (LAMPA) are also depicted
In the three query-target pairs, both P-value and Probability showed strong non-linear dependence on designed sizes of query and target (Fig. 7) (hereafter we use ‘designed’ to distinguish computational experiment from LAMPA). Specifically, P-value changed steeply, with curves of designed queries and targets running in parallel relative to each other (Fig. 7A–C). In the designed length range from 100 aa to 10 000 aa, which encompasses most queries and targets of this study, P-value increased by approximately four orders of magnitude for queries of three pairs. This increase was limited to two orders of magnitude for the three selected queries illustrating LAMPA gain versus HHsearch. In contrast, dependence of Probability on length of designed queries and targets followed inverted logistic curve and differed between target and query as well as between the three pairs (Fig. 7D–F). Dependence of Probability on designed query size was most noticeable only below the 95% threshold, where it followed growth phase of logistic. The selected LAMPA and HHsearch queries were at different places of this growth phase in two query-target pairs (Fig. 7D and E) and outside the growth phase in third pair (Fig. 7F) which explained different Probability gains of LAMPA hit in these pairs. Hit score and target diversity contributed to variable Probability gain in three pairs (Supplementary Text S1.5).
3.11 LAMPA can significantly expand RefSeq expert annotation of RNA virus polyproteins
Finally, we compared annotations of the RNA virus polyproteins by LAMPA and HHsearch versus RefSeq experts (Fig. 8 andSupplementary Fig. S6). Concerning the number of annotated regions per polyprotein, LAMPA and HHsearch were as good as RefSeq for 38.8 and 41.4% of polyproteins, respectively, while RefSeq expert or LAMPA/HHsearch outperformed the other for 23.3/27.0% and 37.9/31.6% of polyproteins, respectively (Fig. 8A andSupplementary Fig. S6A). Notably, LAMPA and HHsearch annotated regions in 298 and 291 out of 426 polyproteins with no RefSeq annotation and increased the number of annotated region(s) for further 833 and 652 polyproteins. Increase in the number of annotated regions per protein by LAMPA but not HHsearch was statistically significant (PW = 3.11e−08 and 0.752, respectively). LAMPA and HHsearch annotations covered larger share of polyprotein (mean region length was 312 aa, 321 aa and 265 aa for LAMPA, HHsearch and RefSeq annotation, respectively). This coverage increase was observed for 78.7% and 77.5% proteins, respectively, (Fig. 8B andSupplementary Fig. S6B) and was statistically significant (PW = 1.07e−291 and 3.81e−273). We note that the above numbers apply to annotation in the ‘Region’ fields of RefSeq entries. Other fields may record non-redundant annotation which is particularly likely for RefSeq entries with zero regions annotated in the ‘Region’ field. These entries are in minority in the dataset. In summary, LAMPA expands further HHsearch annotation that may already improve RefSeq annotation of RNA virus polyproteins.
Fig. 8.

Summary statistic of annotation coverage by LAMPA and RefSeq experts. Comparison of the number of regions per protein (A) or percentage of protein length (protein coverage) (B) annotated by LAMPA (Stages #1–3) and RefSeq experts, based on analysis 2985 RNA virus proteins. Each protein is represented by a transparent gray dot; dot density is proportional to the number of proteins with identical characteristics. Black line, diagonal
4 Discussion
In this article, we present an iterative LAMPA pipeline for advanced homology detection in large multidomain proteins and proof-of-the-principle for LAMPA in its application to RNA virus polyproteins. Statistical apparatus of HHsearch, used in LAMPA, was trained on a dataset of structurally defined domains with the median size of 142 aa to ascertain high sensitivity and selectivity, although HHsearch is used for annotation of proteins, regardless of their domain composition and size. This expand ed application of HHsearch is due to two factors: (i) in contrast to sequence diversity of query (profile) (see HHblits), domain composition of query received relatively little attention in relation to HHsearch sensitivity; (ii) considerable complexity and uncertainty of domain delineation in protein sequences. We have addressed both aspects in this study and offer a practical solution to the detection of distant homology in multidomain proteins using conventional profile-based tools in the LAMPA pipeline, which could be particularly useful in the on-going exploration of the Virosphere (Saberi et al., 2018; Suttle, 2007; Zhang et al., 2019).
Length along with diversity are the two characteristics of query and target that determine hits Probability and P-value in HHsearch profiles’ auto-calibration procedure (Remmert, 2011). We employed this procedure in computational experiments of high accuracy to plot the dependence of hits Probability and P-value from designed query/target lengths of several query-target pairs over a large size range that was beyond those used for tuning the auto-calibration procedure (12–1504 aa) and this study (1001–8572 aa) (Fig. 1). The produced plots revealed constrained statistic-specific shape of considerable variation for the two statistics characterizing a hit score in relation to query size (Fig. 7). Due to training of the auto-calibration procedure on the domain dataset, this variation informs about hit score statistics in application to single-domain proteins. When applied to multidomain proteins, like those used in this study, it illustrates how statistical significance of hit scores may be underappreciated depending on difference of sizes of the intact protein and its domains. This underappreciation is realized regardless of multidomain protein size, although it may be consistently considerable only for large proteins.
In line with the Formula 1 (see Section 2.4), the computational experiments revealed also complex dependencies of statistical significance of HHsearch hits on designed target length and profile diversities of query and target (Fig. 7 and Supplementary Fig. S5). These dependencies explained variable gains of hit statistical significance by LAMPA compared to HHsearch in different query-target pairs. They also provide theoretical foundation for further efforts of improving the homology recognition by LAMPA through enriching queries using HHblits and targeting several databases, as is discussed below.
For queries including single domain or larger, false-positive rate of LAMPA may not be different from that of HHsearch (Remmert et al., 2012; Söding, 2005), which is used for calculation of hit statistical significance. Our results were obtained with Probability cutoff of 95%, which was chosen to ascertain homology detection and suppress false positives. The user may use E-value instead of Probability or lower the cutoff that will trade confidence in homology detection for increasing polyprotein coverage. We expect LAMPA to outperform HHsearch at these lower cutoffs as well. Due to logistic dependence between Probability and query length (Fig. 7D–F), Probability gains with under 95% cutoffs could be bigger than reported here.
We used TMHMM and HHsearch to functionally annotate polyproteins on structural grounds and by homology, respectively; they were used by LAMPA to delimit uncharacterized polyprotein regions that queried Pfam 31.0 further. (As discussed in Supplementary Text S1.3, the use of HHsearch in the LAMPA framework was adjusted for analysis of RNA virus polyproteins). Once this iterative query-specific characterization at the QP-stage was exhausted, we used average protein domain size to delimit the remaining non-annotated regions during further database searches. This AP-stage has elements of arbitrariness which were partially addressed ad hoc by using two alternative starting points for query delimitation.
This aspect and the entire pipeline may be advanced further. At the Stage #0, other programs in addition to TMHMM may assist with functional annotation, e.g. mapping disordered regions, or regions anomalously enriched with certain amino acid residues, or cleavage sites for particular proteases like it was demonstrated in our recent study (Saberi et al., 2018). In that study, HHsearch was used to scan several databases, and this provision is also available in the LAMPA 1.0.0 package. Also, iterative profile programs, e.g. PSI-BLAST or HHblits, could be incorporated in the LAMPA to enrich query and improve homology recognition by targeting proteins that are not part of curated profile databases. These improvements could increase relative share of the QP-stage in homology detection and region annotation. In theory, the LAMPA may identify all domains at the #1 and QP-stage, with the AP-stage generating no hits, either due to the lack of queries or homology. Notwithstand ing future advances, the current LAMPA version may already complement HHblits, the current top homology search tool. Indeed, under the 95% Probability cutoff HHblits failed to annotate 195 of 507 regions that LAMPA but not HHsearch annotated in 431 polyproteins of this study (Supplementary Table S2 and Text S1.4).
The reported gain of hit statistical significance by LAMPA compared to HHsearch was modest but sufficient to elevate many hits above the Probability 95% cutoff. It improved homology detection and hit coverage in 14.4% of polyproteins which were enriched with sequences that share not >30% identity with others in the dataset. Thus, gain of hit statistical significance by LAMPA compared to HHsearch could be larger for viruses that prototype genera or higher rank taxa rather than species dominating our dataset (see Supplementary Text S1.2).
LAMPA annotation was most frequent for (+)ssRNA viruses, which correlates with their abundance and expand ed diversity relative to dsRNA and (−)ssRNA viruses. Most newly detected homologs may already be known in other related viruses, which is evident from names and descriptions of hit Pfam profiles that often refer to viruses and their proteins (Supplementary Table S2). However, they also include those not reported in literature, e.g. ZBD and MTase domains in pp1a (YP_009052476.1) of BPNV, python tobanivirus (Fig. 2 and Supplementary Table S2). The detection of the MTase domain, which is apparently conserved in the distantly related fish WBV (YP_803214.1) in this genome location, is particularly intriguing. These viruses and other nidoviruses with genomes >20 kb are known to encode one or two MTases far downstream in the pp1b part of the pp1ab polyprotein (Saberi et al., 2018; Schutze et al., 2006; Stenglein et al., 2014) that were implicated in the 5’-end mRNA cap formation (Decroly et al., 2012). These and other functional assignments (Supplementary Table S2) could be used to direct experimental research and in reconstruction of evolution of RNA viruses.
LAMPA facilitates homology detection and may be used to improve annotation coverage by other tools and experts in genomic projects, as well as in curated databases, including RefSeq. However, other factors besides detection of homology may affect quality of annotation (Punta and Ofran, 2008; Radivojac et al., 2013) and they were outside the scope of this study.
Supplementary Material
Acknowledgements
The authors thank Andrey M. Leontovich and Igor A. Sidorov for discussions and assistance.
Funding
This work was supported by the EU Horizon2020 EVAg 653316 project and the LUMC MoBiLe program; A.E.G. was a Leiden University Fund (LUF) Professor.
Conflict of Interest: none declared.
References
- Altschul S.F et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltimore D. (1971) Expression of animal virus genomes. Bacteriol. Rev., 35, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brister J.R. et al. (2015) NCBI viral genomes resource. Nucleic Acids Res., 43, D571–D577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charif D., Lobry J.R., (2007) SeqinR 1.0-2: a contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysis In: Bastolla Uet al. (eds.) Structural Approaches to Sequence Evolution: Molecules, Networks, Populations. Springer Berlin Heidelberg, Berlin, Heidelberg, pp. 207–232. [Google Scholar]
- Das K., Arnold E. (2015) Negative-strand RNA virus L proteins: one machine, many activities. Cell, 162, 239–241. [DOI] [PubMed] [Google Scholar]
- Decroly E. et al. (2012) Conventional and unconventional mechanisms for capping viral mRNA. Nat. Rev. Microbiol., 10, 51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty W.G., Semler B.L. (1993) Expression of virus-encoded proteinases: functional and structural similarities with cellular enzymes. Microbiol. Rev., 57, 781–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Gebali S. et al. (2019) The Pfam protein families database in 2019. Nucleic Acids Res., 47, D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R.D. et al. (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res., 39, W29–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth A.E., Brierley I. (2012) Non-canonical translation in RNA viruses. J. Gen. Virol., 93, 1385–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox N.K. et al. (2014) SCOPe: structural classification of proteins–extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Res., 42, D304–D309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Snijder E.J. (1996) Viral cysteine proteinases. Perspect. Drug Discovery Des., 6, 64–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habermann B.H. (2016) Oh Brother, where art thou? Finding orthologs in the twilight and midnight zones of sequence similarity In: Pontarotti P. (ed.) Evolutionary Biology: Convergent Evolution, Evolution of Complex Traits, Concepts and Methods. Springer International Publishing, Cham, pp. 393–419. [Google Scholar]
- Jones D.T. (1999) Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol., 292, 195–202. [DOI] [PubMed] [Google Scholar]
- Kuchibhatla D.B. et al. (2014) Powerful sequence similarity search methods and in-depth manual analyses can identify remote homologs in many apparently “orphan” viral proteins. J. Virol., 88, 10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M. et al. (2013) Software for computing and annotating genomic ranges. PLoS Comput. Biol., 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirdita M. et al. (2017) Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res., 45, D170–D176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary N.A. et al. (2016) Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res., 44, D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punta M., Ofran Y. (2008) The rough guide to in silico function prediction, or how to use sequence and structure information to predict protein function. PLoS Comput. Biol., 4, e1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. (2018) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria.
- Radivojac P. et al. (2013) A large-scale evaluation of computational protein function prediction. Nat. Methods, 10, 221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmert M. (2011) Fast, sensitive protein sequence searches using iterative pairwise comparison of hidden Markov models. Doctoral Dissertation, Ludwig Maximilian University, Munich.
- Remmert M. et al. (2012) HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods, 9, 173–175. [DOI] [PubMed] [Google Scholar]
- Saberi A. et al. (2018) A planarian nidovirus expand s the limits of RNA genome size. PLoS Pathog., 14, e1007314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuan R. et al. (2010) Viral mutation rates. J. Virol., 84, 9733–9748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers E.W. et al. (2019) GenBank. Nucleic Acids Res., 47, D94–D99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutze H. et al. (2006) Characterization of White bream virus reveals a novel genetic cluster of nidoviruses. J. Virol., 80, 11598–11609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M. et al. (2016) Redefining the invertebrate RNA virosphere. Nature, 540, 539–543. [DOI] [PubMed] [Google Scholar]
- Söding J. (2005) Protein homology detection by HMM-HMM comparison. Bioinformatics, 21, 951–960. [DOI] [PubMed] [Google Scholar]
- Sonnhammer E.L. et al. (1998) A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol., 6, 175–182. [PubMed] [Google Scholar]
- Steinegger M., Söding J. (2017) MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol., 35, 1026–1028. [DOI] [PubMed] [Google Scholar]
- Steinegger M. et al. (2019) HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics, 20, 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenglein M.D. et al. (2014) Ball python nidovirus: a cand idate etiologic agent for severe respiratory disease in Python regius. mBio, 5, e01484–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttle C.A. (2007) Marine viruses—major players in the global ecosystem. Nat. Rev. Microbiol., 5, 801–812. [DOI] [PubMed] [Google Scholar]
- Wheelan S.J. et al. (2000) Domain size distributions can predict domain boundaries. Bioinformatics, 16, 613–618. [DOI] [PubMed] [Google Scholar]
- Zhang J. (2000) Protein-length distributions for the three domains of life. Trends Genet., 16, 107–109. [DOI] [PubMed] [Google Scholar]
- Zhang Y.Z. et al. (2019) Expanding the RNA virosphere by unbiased metagenomics. Annu. Rev. Virol., 6, 119–139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.