

Abstract
Motivation
Genome annotation is an important step for all in-depth bioinformatics analysis. It is imperative to augment quantity and diversity of genome-wide annotation data for the latest reference genome to promote its adoption by ongoing and future impactful studies.
Results
We developed a python toolkit AnnoGen, which at the first time, allows the annotation of three pragmatic genomic features for the GRCh38 genome in enormous base-wise quantities. The three features are chemical binding Energy, sequence information Entropy and Homology Score. The Homology Score is an exceptional feature that captures the genome-wide homology through single-base-offset tiling windows of 100 continual nucleotide bases. AnnoGen is capable of annotating the proprietary pragmatic features for variable user-interested genomic regions and optionally comparing two parallel sets of genomic regions. AnnoGen is characterized with simple utility modes and succinct HTML report of informative statistical tables and plots.
Availability and implementation
1 Introduction
The human reference genome plays a central role in today’s biology and translational medicine researches. The latest assembly GRCh38 was endorsed for ‘enduring quality’ (Schneider et al., 2017) and advantage on high throughput sequencing data analysis (Guo et al., 2017).
A plethora of genome annotation tools is existent to provide steady support for human genomics researches, especially in the scaffold of GRCh37. ANNOVAR (Wang et al., 2010) is an excellent example with rich annotation libraries and friendly user experience. With the focus set on the identification of annotated genomic regions and characterization with diverse features, ANNOVAR spares any numeric aggregation or quantitative comparison endeavors. GREAT (McLean et al., 2010) and GLANET (Otlu et al., 2017) come along with less comprehensive feature characterization, but they are equipped with statistical tests for enrichment summary and functional interpretation. The GLANET publication did a rigorous theoretical comparison of related tools in 2017.
Here, we propose a new toolkit for annotating three pragmatic features in the framework of GRCh38 for the first time. We performed intensive computation to obtain base-wise quantities for the entire whole genome of 3 billion base pairs. The curated novel genomic features include Energy, Entropy and Homology Score (HS). Additionally, GC content is provided alongside at base wise resolution. These valuable pre-calculated data formed the basis of the newly devised Python application AnnoGen, which achieves retrieval, aggregation and comparison of the diverse pragmatic features pertaining to user-designated genomic regions. As we demonstrate in the case studies, AnnoGen enables access and exploitation of these proprietary pragmatic feature values and paves the way for high-level inquiry of research entities in light of these pragmatic features.
2 Materials and methods
Three novel types of pragmatic features alongside with GC content were calculated for every single nucleotide position in each human chromosome of GRCh38, excluding the terminal stretches of 10 k∼10 m bases that roughly corresponded to telomere territories. As expounded below, all features are initially defined with respect to a sequence interval instead of a single position, so for operation’s sake, we extracted the immediate upstream 100-bp sequence window and attributed all feature scores derived within this window to the position in question. The 100-bp sequence window slides across the whole sequential chromosome nucleotide by nucleotide, thus giving rise to base-wise feature scores.
Block entropy of a DNA sequence on the basis of an n-block was calculated with Equation (Schmitt and Herzel, 1997), where designated the frequency of an n-mer sequence motif in the running 100-bp DNAsequence window. AnnoGen curated three entropy scores with variant block lengths, namely H2, H3 and H4.
Secondary structure was predicted for each running 100-bp genomic window and the corresponding folding energy was estimated based on thermodynamic parameters of DNA structure motifs (SantaLucia and Hicks, 2004). AnnoGen calculated two variants of Energy scores, delta_G37 and delta_H.
HS was defined as the number of perfect alignments of a DNA stretch to the entire genome. HS is conceptually similar to the so-called ‘mappability’ in other applications (Derrien et al., 2012), but our advantage lies in the ab initio exhaustive sequence alignment to obtain these base-wise HS values. ‘perfect alignment’ was technically defined as zero basepair mismatch.
In addition to the above three novel pragmatic features, base-wise GC content has also been curated alongside with the three above novel features. All four types of features constitute the crucial data library of AnnoGen, which is aimed primarily to annotate these proprietary features for variable user-interested genomic regions.
The primary input expected by AnnoGen consists of a set of chromosome intervals (DNA sequence stretch delimited by coordinate endpoints). In the Annotation utility mode, AnnoGen annotates each interval with summary scores for DNA energy, entropy, GC content and mapping uniqueness (HS). If supplied with two comparative sets of chromosome intervals, AnnoGen invokes the Comparison utility mode to conduct a Zero-Inflated Poisson analysis for the HS values and a Wilcoxon test for the other features. An HTML report will be generated to deliver three major results: (i) catalog of input intervals by genomic region type; (ii) statistical comparison result including statistics and P values; and (iii) comparative distributions of each type of feature scores between the two input channels.
3 Results
We conducted two case studies to exhibit AnnoGen’s pragmatic genomic features and demonstrate its potential to guide high-level inquiry of research entities.
First, we employed AnnoGen to examine 41 cataloged gene types in GRCh38. The diverse gene types possesses one (e.g. scRNA) to 709 025 (protein-coding) genomic intervals which consume 42 (TR_D_gene) to 232 224 598 (protein-coding) nucleotides. AnnoGen calculated aggregate feature scores (mean, median, quantiles, etc.) for each individual interval, upon which we further derived the median scores for each gene type (Fig. 1A). The landscape of GC content precisely echos our commonsense knowledge (Piovesan et al., 2019). As for HS, all but four gene types had median value of 1, indicating that a random 100-bp DNA sequence in most gene types are most likely unique in the whole genome. Macro_lncRNA, rRNA, scRNA and non_coding RNA are less unique in sequence homology, having median HS values of 8.3, 4, 2.5 and 1.5, respectively.
Fig. 1.
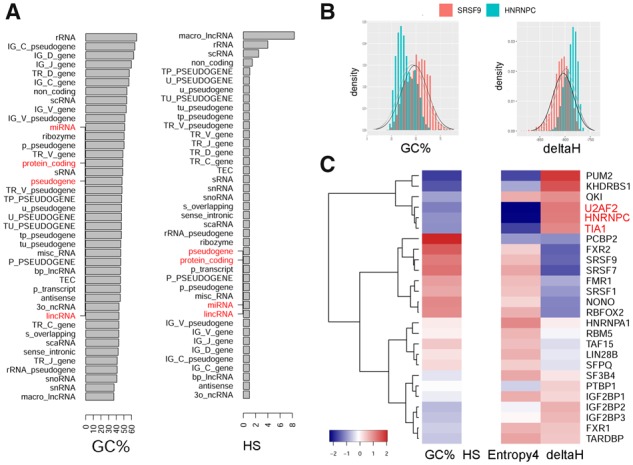
Case study results generated by AnnoGen. (A) Median GC content and Homology score of 41 gene types cataloged in GRCh38. Four major and common gene types are highlighted in red. (B) Representative comparative distribution plots outputted in the Comparison utility mode of AnnoGen. (C) Hierarchical clustering of 26 RNA-binding proteins according to summary progamatic feature scores of their respective binding target sequences. Each feature score (column) was scaled to standard z-scores. Three RNA-binding proteins with almost indiscriminable U-rich motifs are highlighted in red
Additionally, we employed AnnoGen to compare binding target sequences of different RNA-binding proteins. Highly probable binding segments in 3’-UTR for 26 RNA-binding proteins were downloaded (Yu et al., 2019) and aligned back to GRCh38 genome with NCBI’s blastn program. Pairwise comparison was conducted with the Comparison utility mode of AnnoGen. The example result (Fig. 1B) revealed that target regions of HNRNPC had much lower GC% yet rather higher energy than those of SRSF9. This is reasonable because HNRNPC favors a poly-U motif while SRSF9 identifies a GGA triplex core in its binding motif. In the resultant clustering dendrogram (Fig. 1C), HNRNPC clustered with TIA1 and U2AF2 unsurprisingly, because all three have almost indiscriminable U-rich motifs.
4 Conclusion
We developed a python toolkit AnnoGen with enormous base wise quantities of three novel pragmatic genomic features for GRCh38, including Energy, Entropy and HS. The HS is an exceptional feature solved through exhaustive alignment between running DNA windows against the entire genome. This led to a first-ever precise solution of the otherwise heuristically approximated mappability scores. In addition to the annotation capability with regards to proprietary pragmatic features, AnnoGen also enables appropriate statistical comparisons of these features between comparative sets of genomic regions and provides succinct HTML report of informative statistical tables and plots. The design allows additional feature tracks to be added to AnnoGen at ease. However, genomic features describing interaction, such as Hi-C data (Belton et al., 2012; Krietenstein et al., 2019), are not easily ported to AnnoGen without substantial modifications. Characterized with enormous amount of annotation data for pragmatic genomic features and streamlined statistical analysis and reporting, AnnoGen makes a noteworthy supplement to the growing family of GRCh38-tailored annotation toolboxes.
Funding
This work was supported by Cancer Center Support Grant from National Cancer Institute [P30CA118100].
Conflict of Interest: none declared.
References
- Belton J.M. et al. (2012) Hi-C: a comprehensive technique to capture the conformation of genomes. Methods, 58, 268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien T. et al. (2012) Fast computation and applications of genome mappability. PLoS One, 7, e30377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y. et al. (2017) Improvements and impacts of GRCh38 human reference on high throughput sequencing data analysis. Genomics, 109, 83–90. [DOI] [PubMed] [Google Scholar]
- Krietenstein N. et al. (2019) Ultrastructural details of mammalian chromosome architecture. bioRxiv639922. [DOI] [PMC free article] [PubMed]
- McLean C.Y. et al. (2010) GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol., 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otlu B. et al. (2017) GLANET: genomic loci annotation and enrichment tool. Bioinformatics, 33, 2818–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piovesan A. et al. (2019) On the length, weight and GC content of the human genome. BMC Res. Notes, 12, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SantaLucia J., Hicks D. (2004) The thermodynamics of DNA structural motifs. Annu. Rev. Biophys. Biomol. Struct., 33, 415–440. [DOI] [PubMed] [Google Scholar]
- Schmitt A.O., Herzel H. (1997) Estimating the entropy of DNA sequences. J. Theor. Biol., 188, 369–377. [DOI] [PubMed] [Google Scholar]
- Schneider V.A. et al. (2017) Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res., 27, 849–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K. et al. (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res., 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H. et al. (2019) beRBP: binding estimation for human RNA-binding proteins. Nucleic Acids Res., 47, e26. [DOI] [PMC free article] [PubMed] [Google Scholar]