

Abstract
Motivation
The development of clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated protein 9 (Cas9) technology has provided a simple yet powerful system for targeted genome editing. In recent years, this system has been widely used for various gene editing applications. The CRISPR editing efficacy is mainly dependent on the single guide RNA (sgRNA), which guides Cas9 for genome cleavage. While there have been multiple attempts at improving sgRNA design, there is a pressing need for greater sgRNA potency and generalizability across various experimental conditions.
Results
We employed a unique plasmid library expressed in human cells to quantify the potency of thousands of CRISPR/Cas9 sgRNAs. Differential sequence and structural features among the most and least potent sgRNAs were then used to train a machine learning algorithm for assay design. Comparative analysis indicates that our new algorithm outperforms existing CRISPR/Cas9 sgRNA design tools.
Availability and implementation
The new sgRNA design tool is freely accessible as a web application, http://crispr.wustl.edu.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The clustered regularly interspaced short palindromic repeat (CRISPR)/Cas systems have provided an unprecedented opportunity for performing site-specific editing of a variety of genomes. In prokaryotes, CRISPRs are virus-derived DNA fragments which encode CRISPR RNA (crRNA) (Barrangou et al., 2007). The CRISPR/Cas systems fall into two classes: Class 1 systems use a complex consisting of multiple Cas protein subunits to degrade foreign nucleic acids and Class 2 systems use a single large Cas protein for the same purpose. The CRISPR/CRISPR-associated protein 9 (Cas9) system belongs to Class 2 systems and is the most widely used editing system due to its simplicity, high efficiency and low cost (Doudna and Charpentier, 2014). In conjunction with trans-activating crRNA (tracrRNA), the crRNA serves as a guide for Cas9 to bind and cleave foreign DNA (Deltcheva et al., 2011). In genome editing experiments, tracrRNA and crRNA are engineered into a combined single guide RNA (sgRNA) with a designed guide sequence complementary to the desired target (Jinek et al., 2012). Similar to the natural system, the sgRNA guides Cas9 to cleave the DNA at a specific genomic locus based on sequence match, resulting in a double-stranded DNA break. This break occurs precisely 3 nt upstream of an NGG protospacer adjacent motif (PAM) sequence (Chen et al., 2014). In mammalian cells, the DNA repair process often introduces indels, causing frameshift mutations and resulting in functional gene knockout. From this editing framework, advanced strategies have also been developed such as paired nicking for increased specificity (Ran et al., 2013) or inserting nucleotide sequences during the repair of double-strand break to generate knock-ins (Mali et al., 2013). The broad applicability of the CRISPR/Cas9 system stems from its ability to target DNA based on a synthetic sgRNA sequence, specifically the 20 nt guide sequence (gRNA) at the 5′ end of the sgRNA sequence.
It has been shown that the gRNA sequence is important for both targeting specificity and cleavage efficiency (Hsu et al., 2013; Jinek et al., 2013). Off-target Cas9 activity occurs when sequences similar to the gRNA occur elsewhere in the genome, potentially resulting in unintended knockout effects (Hsu et al., 2013). To address the off-target effects, various experimental approaches, mainly by altering the nuclease activity of Cas9 (resulting in high-fidelity Cas9) or gRNA design, have been established in recent years, resulting in significantly improved specificity for CRISPR/Cas9 targeting (Kleinstiver et al., 2016; Kocak et al., 2019; Ran et al., 2013; Slaymaker et al., 2016; Tycko et al., 2016). Further, recent studies have also developed bioinformatics methods to design sgRNA sequences with reduced off-target effects (Chuai et al., 2018; Doench et al., 2016; Hsu et al., 2013). However, these experimental innovations could still suffer from potential cleavage efficiency variations. More efficient Cas9 cleavage can potentially result in stronger knockout phenotypic changes. The importance of Cas9 cleavage efficiency is amplified when considering large-scale screening assays where many genes are to be knocked out using a genome-wide CRSIPR/Cas9 sgRNA library.
Several studies have approached the problem of sgRNA efficiency prediction, revealing sgRNA and target features that correlate with Cas9 cleavage efficiency (Chari et al., 2015; Chuai et al., 2018; Doench et al., 2014, 2016; Labuhn et al., 2018; Peng et al., 2018; Wong et al., 2015; Xu et al., 2015). Given the large number of features involved, machine learning methods are commonly employed for data modeling. To construct such computational models, a large number of sgRNAs need to be experimentally tested to build a robust training dataset for efficiency prediction. In order to do so, existing studies typically adopted biological enrichment schemes, in which gene editing events impact cell survival or other observable biological phenotypes. While these strategies avoid labor intensiveness on the experimental side, such indirect biological readouts could produce artifacts in the training data, as equally efficient Cas9 cleavage sites may not result in equal phenotypic changes or survival pressure. Furthermore, existing experimental studies were often focused on a small subset of genes and/or a single cell line, which limits the usefulness of the training data for general predictions. In our study, we generated a plasmid target library for experimental quantification of sgRNA efficiency in the CRISPR/Cas9 system. Using in silico designed target sites as presented from a large plasmid library, our large-scale training dataset reduces potential bias from specific experimental systems and is generalizable across other datasets. We performed comprehensive feature analysis of our sgRNA library and used the extracted features to train a machine learning model for sgRNA design.
Our final model, which we named sgDesigner, was developed by utilizing a stacked generalization framework to combine distinct models, resulting in more robust predictions (Wolpert, 1992). sgDesigner outperforms existing sgRNA design algorithms for sgRNA potency prediction and is publically accessible as a web application via http://crispr.wustl.edu.
2 Materials and methods
2.1 Cloning of sgRNA plasmid library
A pool of 12 472 oligonucleotides were synthesized by CustomArray, Inc. (Bothell, WA, USA). Each oligonucleotide contains a 20 nt gRNA sequence and paired 53 nt target sequence (including a NGG PAM), as presented in Supplementary Table S1. Among these oligos, 11 472 gRNA sequences were randomly selected from coding exons in humans. Most of these gRNAs (93%) cannot target the endogenous exon sites due to the lack of adjacent PAM domains in the genomic sequence. In these cases, a PAM domain was added next to the gRNA sequence in the plasmid to make the site targetable by Cas9. In addition, 1000 randomly shuffled gRNA sequences were included in the oligo pool to serve as negative control. Between the gRNA and target sequence, two BsmBI sites for Cas9 sgRNA scaffold cloning and a 12 nt unique molecular index (UMI) sequence for bioinformatics analysis were inserted. Two constant regions (20 nt each) at the 5′ and 3′ ends were added for PCR amplification of the oligonucleotides. The oligo pool was amplified by PCR with Phusion DNA polymerase (ThermoFisher) using primers ‘Cas9Lib_FP’ and ‘Cas9Lib_RP’ (Supplementary Table S5). Amplified DNA oligos were then gel purified using the QIAquick Gel Extraction kit (QIAGEN). Next, purified PCR products were assembled into the BsmBI-digested plasmid Lenti-gRNA-Puro (Addgene #84752) using the NEBuilder HiFi DNA Assembly kit. This plasmid was referred as the Library-1st plasmids in our study.
Cas9 sgRNA scaffold sequence was amplified from Lenti-CRISPR V2 (Addgene #52961) using the primers ‘scaffold RNA FP’ and ‘scaffold RNA RP’ (Supplementary Table S5). The Cas9 sgRNA scaffold PCR products were then gel purified. After BsmBI digestion of both library-1st plasmids and Cas9 sgRNA scaffold, the two fragments were ligated by T4 DNA ligase (Intact Genomics) to get the final library plasmids.
Following Gibson assembly or T4 DNA ligation, 2 µl of the reaction was transformed into 25 μl of ig™ 10B ElectroCompetent cells (Intact Genomics) by electroporation. To maximize library coverage, two electroporation reactions were performed. After transformation, cells were pooled and spread onto LB agar plates supplemented with 100 µg/ml ampicillin. All clones were harvested for plasmid DNA extraction by the PureYield Plasmid Midiprep kit (Promega). Throughout the cloning process, the transformation efficiency and library coverage were evaluated according to previously published guidelines (Joung et al., 2017). On average, there are about 300 colonies per sgRNA oligo in the plasmid library.
2.2 Lentivirus preparation
The infectious lentivirus particles were generated and packaged using 293 T cells (ATCC). In a 60 mm dish, 2 × 106 cells were seeded in 2.5 ml Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Gibco). About 2 µg of library plasmids or Cas9 expressing plasmids were mixed with 1.8 µg of psPAX2 (Addgene #12260) and 0.2 µg of pCMV-VSVG (Addgene #8454) in 250 µl of OPTI-MEM medium (ThermoFisher), while 12 µl of Lipofectamine 2000 (ThermoFisher) was diluted in 250 µl of OPTI-MEM medium. After 5 min of incubation at room temperature, the plasmid mixture and the diluted Lipofectamine 2000 were combined and incubated for 20 min at room temperature. After incubation, the 500 µl plasmid–lipofectamine mixture was dropped onto the 293 T cells. The transfection medium was replaced with regular cell culture medium 6 h post transfection. Virus was harvested at 40 h post transfection and filtered through a 0.45 µm Millex-HV membrane (Millipore).
2.3 Plasmid library delivery into HeLa/Cas9 cells
Adherent HeLa cells (ATCC) were cultured in DMEM medium supplemented with 10% FBS. Cells were cultured in a 37°C incubator supplied with 5% CO2. To establish the Cas9 stable expressing cells, HeLa cells (ATCC) were transduced with lentivirus containing Cas9 expressing transcripts from LentiCas9-Blast (Addgene #52962) at an MOI of 0.7. Two days after transduction, cells were selected with 10 µg/ml blasticidin for 4 days. The blasticidin-resistant cells were pooled and maintained in the presence of 10 µg/ml blasticidin. Cas9-expressing cells were transduced with the lentivirus expressing the plasmid library at an MOI of 0.3. Two days after transduction, cells were treated with 2 µg/ml puromycin for 3 days. Survived cells were harvested and genomic DNA was isolated for sequencing library construction.
2.4 Sequencing library construction
Genomic DNA was isolated using the GenElute Mammalian Genomic DNA Purification kit (SigmaMillipore). The sequencing library was constructed according to the methods described previously (Kim et al., 2017). In brief, the target sequence was first amplified and then the Illumina adaptor and barcode sequences were introduced by a second PCR. All primers used in these two PCRs were listed in Supplementary Table S5. The final PCR products were purified with AmpureXP beads (Beckman Coulter), quantified with the Quantifluor system (Promega) and then sequenced with MiSeq (Illumina).
2.5 Quantification of sgRNA efficiency
FASTQ raw sequencing data were de-multiplexed and ambiguous reads were filtered out. Each sequencing read was identified using its gRNA sequence and UMI and subsequently aligned to its reference sequence using Smith–Waterman alignment with affine gap penalty to detect editing (with parameters for match = 3, mismatch = −2, gap opening = −10 and gap extension = −1). In this step, plasmids that exhibited indels prior to exposure to Cas9 were excluded from further analysis. Proportion of reads edited was used to quantify sgRNA efficiency. sgRNAs with 100% of associated reads edited were considered of high efficiency, while those with ≤50% of associated reads edited were considered of low efficiency. A minimum read count of 10 per sgRNA was required for sgRNA inclusion in the analysis, and an additional criterion of at least two UMIs were required for each sgRNA included the high-efficiency group in order to maximize training data quality.
2.6 Computational tools
Data processing, sequence alignment and feature extraction were performed using custom Perl scripts. RNAfold (Hofacker, 2003) was used to compute sgRNA structural features. Features analysis was performed using MATLAB. Significance levels (P-values) were calculated using Student’s t-test for numerical features and χ2 test for binary features. Feature enrichment was determined by comparing functional sgRNAs with non-functional sgRNAs. Computational modeling and performance evaluation were performed using Python.
2.7 Independent testing datasets
A total of six testing datasets were gathered from published studies, namely: Wang, Koike-Yusa, FC, RES, Shalem and Chari (Chari et al., 2015; Doench et al., 2014, 2016; Koike-Yusa et al., 2014; Shalem et al., 2014; Wang et al., 2014). The Wang and Koike-Yusa datasets were downloaded from supplemental tables provided by Xu et al. (2015). We used the negative log2 fold change values for correlative analysis of sgRNA prediction. The FC and Shalem datasets were provided by Doench et al. (2014). The RES dataset was downloaded from the Azimuth website, which was implemented by Microsoft (Doench et al., 2016). The Chari dataset was directly retrieved from supplemental tables at the journal’s website (Chari et al., 2015). Additional details about these datasets can be found in Supplementary Table S4.
2.8 Model performance evaluation
We compared sgDesigner with three existing sgRNA design tools included RS2, Sequence Scan for CRISPR (SSC) and DeepCRISPR. RS2 prediction results were retrieved from Microsoft’s Azimuth 2.0 website, using predefined in vitro parameters (Doench et al., 2016). SSC prediction results were computed using the authors’ web-based implementation (Xu et al., 2015). DeepCRISPR predictions were generated using the command-line version with sequence features only (Chuai et al., 2018). Receiver operating characteristic (ROC) and Spearman correlation analyses were performed using the testing datasets to assess the consistency between experimentally determined sgRNA efficiencies and predicted efficiencies.
2.9 Availability of data
Our sgRNA design tool, sgDesigner, is freely accessible as a web application via http://crispr.wustl.edu. In addition, the source code and stand-alone version of sgDesigner are freely accessible at GitHub (https://github.com/wang-lab/sgDesigner). Additional supplementary data can be downloaded from journal’s website and Zenodo.org (http://doi.org/10.5281/zenodo.3572803).
3 Results
3.1 An sgRNA library for quantifying CRISPR/Cas9 editing efficiency
The overall experimental procedure is summarized in Figure 1a. In summary, we synthesized an sgRNA library and used it for cleavage efficiency quantification by high-throughput sequencing. Specifically, we designed a pool of oligonucleotides each containing a U6 promotor sequence, a gRNA sequence and a corresponding target sequence. The target sequence included a 20 nt gRNA-matching region, followed by an NGG PAM domain. In addition, to examine the potential impact of target flanking regions, we also included 30 distinct nt surrounding the target site, forming an extended target site of 53 nt. The oligos were cloned into plasmids by Gibson assembly, and then the sgRNA scaffold was inserted downstream of the gRNA sequence. In this way, each plasmid contains both an sgRNA expression cassette and a paired target sequence. This plasmid library was transduced into Cas9-expressing cells, and the editing of in silico designed target sequences was determined by sequencing. In this way, the potency of 7407 sgRNAs were quantified after filtering out low-quality reads. Overall, Cas9 activity was high, with 81.1% reads edited. We stratified the sgRNAs into high-efficiency (100% editing), medium-efficiency (51–99% editing) and low-efficiency (0–50% editing) groups and selected high-confidence sgRNAs for training (Fig. 1b). To emphasize the most predictive features affecting sgRNA efficiency, our strategy was to only consider the high- and low-efficiency groups for data modeling, resulting in a training set comprised of 746 functional sgRNAs and 563 non-functional sgRNAs (see Materials and methods, Supplementary Table S2).
Fig. 1.
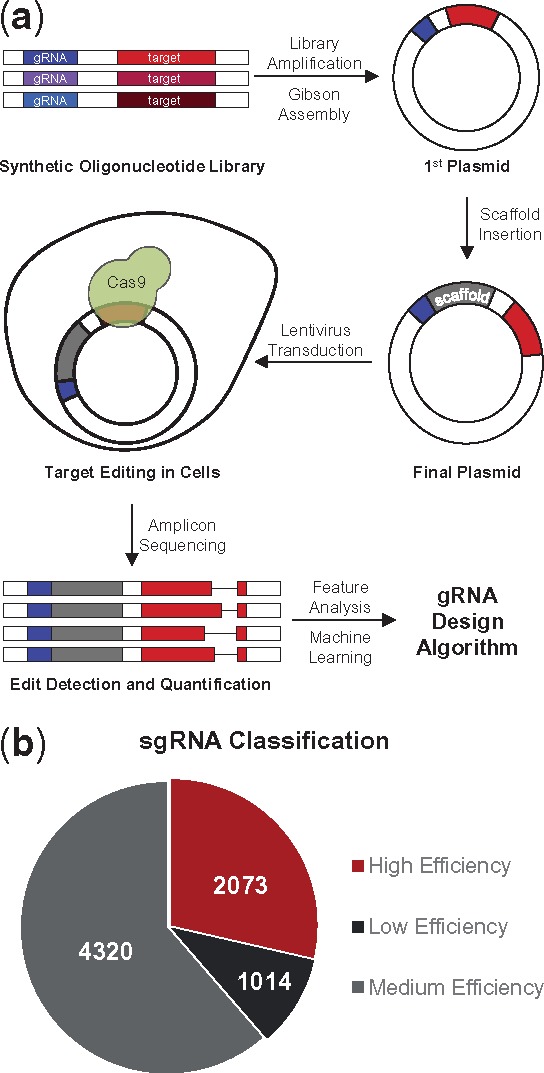
Determining sgRNA efficiencies using an in silico designed sgRNA library. (a) Experimental outline for construction of the sgRNA library and generation of training data for computational modeling. (b) Stratification of sgRNAs based on editing efficiency
3.2 sgRNA/target features
Previous studies have identified multiple sgRNA and target features contributing toward Cas9 activity, such as position-specific nucleotide composition and GC content (Chari et al., 2015; Chuai et al., 2018; Doench et al., 2014, 2016; Labuhn et al., 2018; Peng et al., 2018; Wong et al., 2015; Xu et al., 2015). However, feature comparison between published datasets reveals considerable discordance and further study is warranted to identify generalizable features affecting Cas9 efficiency. For example, Xu et al. (2015) demonstrated that guanines are preferred at positions −14 to −17 of the 20-mer gRNA sequence, whereas this was not observed by Doench et al. (2014). Our new dataset, with its direct cleavage quantification by employing a plasmid-based system for generation of target sites, provides a unique opportunity to isolate and quantify features that are intrinsically associated with Cas9 activity. We included a total of 26 nt for analysis, including the 20 nt gRNA-matching sequence, the NGG PAM and 3 nt downstream of the PAM (Fig. 2a). In total, 302 features were chosen as machine learning input (Supplementary Table S3). These features were extracted by a combination of sequence and structural analyses of the gRNA and target sites, as described in detail below. We also explored additional features of the 53 nt extended target sequence, including 15 nt and 12 nt flanking the gRNA-matching/PAM sequence at the 5′ end and 3′ end, respectively. However, none of the additional extended positions were statistically significant after correcting for multiple comparison, nor did they improve model performance (Supplementary Fig. S1). Thus, we excluded these additional nucleotide positions from further analysis.
Fig. 2.
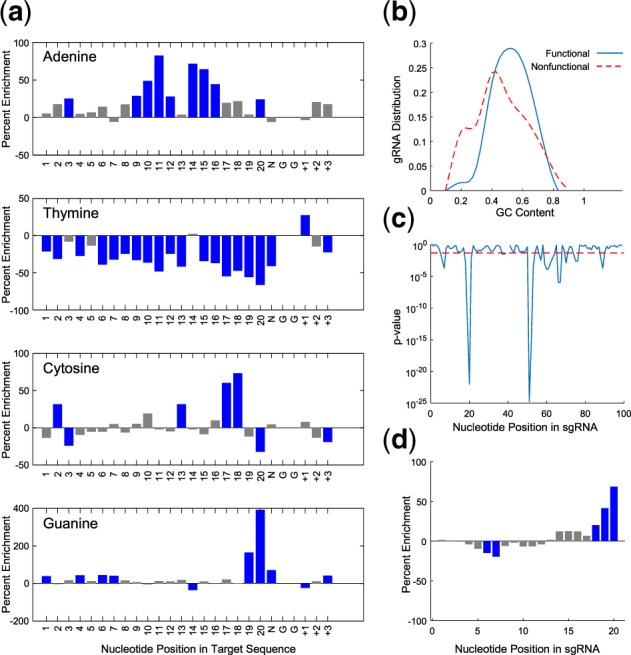
Feature analysis of efficient sgRNAs. (a) Position-specific nucleotide composition. A positive or negative value represents enrichment or depletion of the nucleotide, respectively. Statistically significant nucleotides are depicted in blue. (b) GC content of the gRNA. (c) Significance of nucleotide accessibility at each position in the sgRNA. The significance level (P = 0.05) is depicted with red dashes. (d) Percent enrichment of nucleotide accessibility at each position in the gRNA. Significant positions are depicted in blue. (Color version of this figure is available at Bioinformatics online.)
3.2.1 Nucleotide composition
Nucleotide composition at each target position is summarized in Figure 2a. Nucleotides at positions 1–20 are identical to those in the gRNA [with a thymine (T) to uracil (U) conversion], followed by the NGG PAM, which is a requirement for Cas9 targeting (Jinek et al., 2012). Positions +1, +2 and +3 represent the genomic context of the target sequence. Functional gRNAs were depleted in T throughout 19 of the 23 positions (P-values in the range of 2.4E−27–1.8E−02; average depletion of 38%), while position +1 was significantly enriched in T (P = 1.3E−02; enrichment of 28%; Fig. 2a). The overall depletion is in part due to transcriptional efficiency as opposed to an interaction involving Cas9. The U6 promotor used in the study recruits RNA polymerase III which recognizes a poly-T sequence as a termination signal (Nielsen et al., 2013). Consistent with this mechanism, none of the gRNAs in the high efficiency group contained a sequence of five or more contiguous T bases. However, this phenomenon does not account for the entirety of the depletion in T, since we still observed significant overall T depletion after excluding gRNAs with four or more contiguous Ts.
Nucleotides proximal to the PAM were the most predictive of Cas9 activity. Most significantly, functional gRNAs had strong enrichment in guanine (G) at positions 19, 20, and the N position of the PAM (P = 4.7E−19, 1.4E−36 and 5.9E−08; enrichment of 165, 392 and 71%, respectively). Adenines (A) were, however, more enriched toward the middle of the gRNA specifically at positions 9–12 and 14–16 (P-values in the range of 2.0E−08–1.8E−02; average enrichment of 53%). Cytosines (C) were most enriched at positions 17 and 18 (P = 1.2E−06 and 1.2E−07, enrichment of 60 and 73%, respectively).
3.2.2 GC content
We found decreased activity in gRNAs with extreme overall GC content. As shown in Figure 2b, the vast majority of gRNAs with GC content >80% and <30% were non-functional (depletion of 81 and 93%, respectively). These two features were significant and improved overall predictions (P = 1.8E−02 and 1.1E−09, respectively). In contrast, we did not observe model improvement using absolute GC content values.
3.2.3 Structural features
RNA molecules commonly form secondary structures through intramolecular interactions, resulting in differential accessibility for the nucleotides within the folded structure. This phenomenon can potentially result in unfavorable sgRNA structures affecting Cas9 efficiency. However, most sgRNA design tools did not consider sgRNA nucleotide accessibility for Cas9 editing prediction. Here, we investigated these structural features using RNAfold (Hofacker, 2003) for structure prediction. The present dataset showed that sgRNA nucleotide accessibility at positions 18–20 of the gRNA domain are crucial for efficient editing (P = 2.1E−3, 5.8E−13 and 9.9E−23, respectively; Fig. 2c). Functional sgRNAs tend to be accessible at these positions with enrichment values of 20, 41 and 68%, respectively (Fig. 2d). In the predicted sgRNA secondary structure, these three nucleotides proximal to the PAM align with the nucleotides in the scaffold at positions 51–53 due to a stem-loop formation at positions 21–50. Thus, interestingly, increased accessibility at 51–53 is also significantly correlated with high Cas9 efficiency (Fig. 2c). The sgRNA sequence at positions 51–53 is AAG, which would ideally bind to a CUU sequence at positions 18–20, or a UUU sequence when considering wobble base pairing. This may explain the observed depletion of U nucleotides at the 3′ end of the 20 nt gRNA. Our results suggest that there are more complex intramolecular interactions which may have been missed in other algorithms that do not consider structural features.
3.3 Assessment of modeling methods
Recently, several machine learning algorithms have been used to predict sgRNA efficiency. We summarize these algorithms into three categories: (i) regression models such as gradient boosting regression tree (Doench et al., 2016) and extreme gradient boost (XGBoost) (Peng et al., 2018), (ii) classification models, such as support vector machines (SVM) (Chari et al., 2015; Wong et al., 2015) and logistic regression (Doench et al., 2014) and (iii) emerging technologies or hybrid algorithms, such as deep learning technology (Chuai et al., 2018) and simple average of multiple models (Peng et al., 2018). Given the variety of potentially useful models, our strategy was to use a stacking framework in order to capture the advantages of multiple models. Our Stacking model was designed by stacking SVM and XGBoost using a logistic regression model as the combiner (Fig. 3). We compared Stacking performance against commonly used machine learning algorithms as well as models proposed in other sgRNA design studies.
Fig. 3.
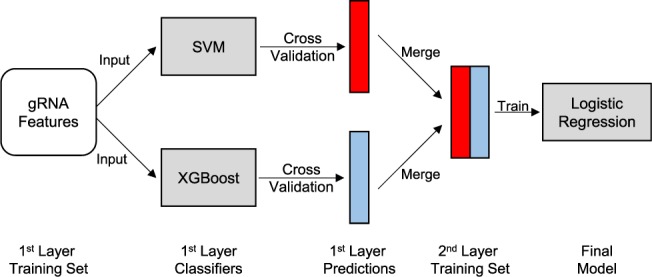
Stacking model framework. The features were first used as input to train first layer models (SVM and XGBoost). Fivefold cross-validation was performed for each individual model in the first layer and the predictions from each model were merged into a two-column feature set. The resulting feature set was then used to train the second layer model (logistic regression)
Using our training dataset comprised of 746 high-efficiency sgRNAs and 563 low-efficiency sgRNAs, we employed 7 different classification-based approaches and tested the performance of each model. The models tested include Stacking, XGBoost, L2-regularization logistic regression, SVM, adaboost, random forests and decision trees. Each of the model parameters were tuned based on the training dataset. We used ROC curve, Spearman correlation and predicted mean accuracy from cross-validation analysis to evaluate the performance of each model. Based on these assessment metrics, we conclude that the Stacking model had the best performance among all models included in the analysis (Fig. 4). The Stacking model developed using the training dataset was thus chosen to be the final model, which we named sgDesigner.
Fig. 4.
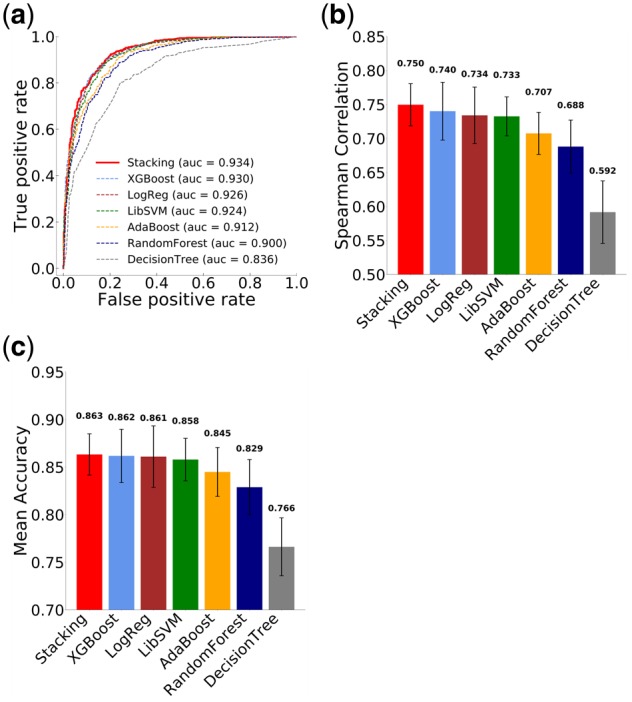
Comparison of different computational models. (a) ROC curve analysis. AUC values for individual models are shown in the legend. (b) Spearman correlation between experimentally determined efficiency and predicted efficiency score. Error bars indicate the SD. (c) Mean accuracy of sgRNA classification (high or low efficiency)
3.4 Validation of sgDesigner
To evaluate the general applicability of sgDesigner at predicting sgRNA efficiency, we curated six CRISPR/Cas9 sgRNA datasets from various cell lines (see Materials and methods for details; Supplementary Table S4). With these datasets, we compared sgDesigner with three existing state-of-the-art sgRNA design tools, including Doench Rule Set 2 (RS2) (Doench et al., 2016), SSC (Xu et al., 2015) and DeepCRISPR (Chuai et al., 2018). These existing tools were selected for comparison because they are currently widely used and freely accessible to the public. To avoid training bias, we only considered independent datasets that were not used to train respective models. The prediction results for these independent datasets were separately generated using each design tool. For each tool, prediction results for all independent datasets were combined for subsequent performance evaluation. Specifically, we performed ROC and Spearman correlation analyses and used true data labels to evaluate the performance of the design tools. The area under the curve (AUC)–ROC and correlation coefficient analysis results are summarized in Figure 5(a, b), with more detailed results for each dataset presented in Supplementary Figures S2 and S4. Further, we present detailed performance evaluation of sgDesigner across six individual datasets in Figure 5(c, d). Compared with other tools, sgDesigner had the best performance, as evaluated by ROC, precision-recall and correlation analyses. Specifically, sgDesigner consistently outperformed all competing tools across all six independent datasets (Supplementary Figs S2–S4). Overall, sgDesigner had consistently high performance across all testing datasets, with average ROC–AUC of 0.833 and a range of 0.765–0.899 (Fig. 5c). From these validation results, we conclude that sgDesigner has robust performance and consistently performs well across various experimental settings.
Fig. 5.
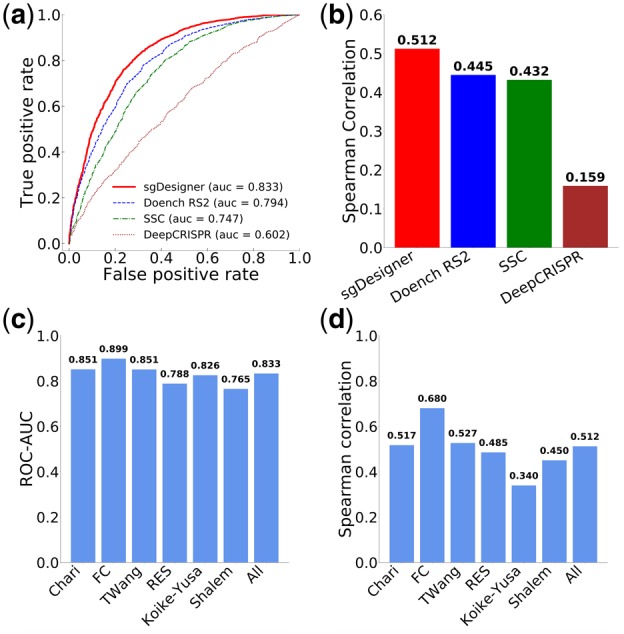
Comparison of sgDesigner with public sgRNA design tools. sgDesigner and three other algorithms were included in this analysis. Validation analysis was performed using six independent datasets, and the combined results are summarized here. Detailed results on each testing dataset are presented in Supplementary Figures S2 and S4. (a) ROC curve analysis. (b) Spearman correlations between experimentally determined efficiency and prediction score. (c) Summary of ROC–AUC values for sgDesigner on six independent datasets. (d) Summary of Spearman correlation coefficient values for sgDesigner on six independent datasets
3.5 Genome-wide sgRNA design database
Using the sgDesigner algorithm, we computed cleavage efficacy for CRISPR/Cas9 sgRNAs to target all human and mouse genes annotated in the NCBI RefSeq database. To reduce potential off-target editing, we also computed off-target scores for the sgRNAs and select those with greater specificity using our previously published algorithm (Wong et al., 2015). In brief, we performed both gRNA seed search and BLAST alignment to identify potential off-targets that share identical 13-mer seed sequence or with at least 85% overall sequence homology to the gRNA sequence. Of note, we focused on identifying off-targets from all known exons (for both coding and non-coding genes) instead of the entire genome space which contains other potentially important non-coding regions.
Our online database provides up to 20 sgRNA designs per gene in the human and mouse genomes. Our database and open-source custom sgRNA design tool are freely accessible at http://crispr.wustl.edu.
4 Discussion
As the CRISPR/Cas9 system has quickly become a ubiquitous gene editing tool in biological research, an increasingly pressing challenge is the design of efficient sgRNAs. Various bioinformatics tools have been developed to address this important issue. However, one major limitation of previous studies is related to the quality of the datasets used to train such tools. Most experimental methods are based on phenotypic screening and are not ideal at quantifying CRISPR/Cas9 editing efficiency. Successful gene editing is unlikely to produce consistent and precise phenotypic changes across all genes and target sites tested. Thus, such indirect methods introduce undesired noises in the datasets used to train machine learning algorithms, which could mask true features that are characteristic of sgRNA-guided Cas9 cleavage. Furthermore, sgRNAs tested in functional screens are typically designed for a subset of genes and tested in a single cell line. These restrictions may introduce biases specific to each experimental setting, such as those related to different levels of genomic accessibility, or different responses to DNA cleavage in a cell line or gene specific manner. All these factors may potentially reduce model generalizability. In the present study, we address these issues by using a new in silico designed plasmid library for sgRNA expression and target site presentation. We produced a new training dataset with precise and direct quantification of sgRNA efficiency, which was used to characterize general sgRNA features that are intrinsically associated with CRISPR/Cas9 cleavage. This new strategy was feasible due to a unique experimental design in which oligonucleotides were synthesized with both an sgRNA expression cassette and a corresponding target sequence in the same construct. Similar strategies were recently used to generate large-scale datasets for analysis of CRISPR/Cpf1 efficiency as well as for the analysis of CRISPR/Cas9 editing patterns and specificity (Allen et al., 2019; Kim et al., 2017; Shen et al., 2018; Tycko et al., 2018). Here, we demonstrate that an in silico designed Cas9 targeting system is useful at generating large-scale training data to characterize CRISPR/Cas9 cleavage efficiency. We were able to precisely quantify the efficiencies of a large number of sgRNAs within a single experiment, thus avoiding inconsistencies when merging datasets from heterogeneous experiments. Our final model, sgDesigner, had stable, high-quality performance across vastly different independent testing datasets in human and mouse experimental systems. However, it remains to be tested whether sgDesigner can be robustly applied to other biological systems, as the rules for CRISPR/Cas9 targeting could be different in other organisms not assessed in our study.
Equally important to training data quality, the choice of machine learning modeling methods also has great impact on the quality of predictions. Previous studies have not reached a consensus on the best modeling approach as seen in the variety of distinct frameworks proposed in sgRNA design studies. Most studies tested a single model or a small number of similar models, limiting the potential for model improvement. Thus, in the present study, we explored multiple vastly different frameworks to identify the best one at sgRNA efficiency prediction. Our final stacking model combined the advantages of multiple models and exhibited greater performance than individual models alone. In summary, through improvements in experimental design, data quality and computational modeling, we developed a new sgRNA design tool, which consistently outperformed competing tools under various experimental settings. Our tool is freely accessible as a web application via http://crispr.wustl.edu.
Funding
This work was supported by the National Institutes of Health [R01GM089784, R01DE026471 and R41GM126682 to X.W.].
Conflict of Interest: Weijun Liu was employed by Nawgen LLC.
Supplementary Material
References
- Allen F. et al. (2019) Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol., 37, 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R. et al. (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science, 315, 1709–1712. [DOI] [PubMed] [Google Scholar]
- Chari R. et al. (2015) Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat. Methods, 12, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. et al. (2014) Cut site selection by the two nuclease domains of the Cas9 RNA-guided endonuclease. J. Biol. Chem., 289, 13284–13294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuai G. et al. (2018) DeepCRISPR: optimized CRISPR guide RNA design by deep learning. Genome Biol., 19, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deltcheva E. et al. (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature, 471, 602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench J.G. et al. (2014) Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol., 32, 1262–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench J.G. et al. (2016) Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol., 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna J.A., Charpentier E. (2014) Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science, 346, 1258096. [DOI] [PubMed] [Google Scholar]
- Hofacker I.L. (2003) Vienna RNA secondary structure server. Nucleic Acids Res., 31, 3429–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P.D. et al. (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol., 31, 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M. et al. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science, 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M. et al. (2013) RNA-programmed genome editing in human cells. eLife, 2, e00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung J. et al. (2017) Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc., 12, 828–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.K. et al. (2017) In vivo high-throughput profiling of CRISPR-Cpf1 activity. Nat. Methods, 14, 153–159. [DOI] [PubMed] [Google Scholar]
- Kleinstiver B.P. et al. (2016) High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature, 529, 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocak D.D. et al. (2019) Increasing the specificity of CRISPR systems with engineered RNA secondary structures. Nat. Biotechnol., 37, 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike-Yusa H. et al. (2014) Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat. Biotechnol., 32, 267–273. [DOI] [PubMed] [Google Scholar]
- Labuhn M. et al. (2018) Refined sgRNA efficacy prediction improves large- and small-scale CRISPR-Cas9 applications. Nucleic Acids Res., 46, 1375–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P. et al. (2013) RNA-guided human genome engineering via Cas9. Science, 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen S. et al. (2013) Mechanism of eukaryotic RNA polymerase III transcription termination. Science, 340, 1577–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H. et al. (2018) CRISPR/Cas9 cleavage efficiency regression through boosting algorithms and Markov sequence profiling. Bioinformatics, 34, 3069–3077. [DOI] [PubMed] [Google Scholar]
- Ran F.A. et al. (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell, 154, 1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O. et al. (2014) Genome-scale CRISPR-Cas9 knockout screening in human cells. Science, 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M.W. et al. (2018) Predictable and precise template-free CRISPR editing of pathogenic variants. Nature, 563, 646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaymaker I.M. et al. (2016) Rationally engineered Cas9 nucleases with improved specificity. Science, 351, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko J. et al. (2016) Methods for optimizing CRISPR-Cas9 genome editing specificity. Mol. Cell, 63, 355–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko J. et al. (2018) Pairwise library screen systematically interrogates Staphylococcus aureus Cas9 specificity in human cells. Nat. Commun., 9, 2962–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T. et al. (2014) Genetic screens in human cells using the CRISPR-Cas9 system. Science, 343, 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpert D.H. (1992) Stacked generalization. Neural Netw., 5, 241–259. [Google Scholar]
- Wong N. et al. (2015) WU-CRISPR: characteristics of functional guide RNAs for the CRISPR/Cas9 system. Genome Biol., 16, 218–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H. et al. (2015) Sequence determinants of improved CRISPR sgRNA design. Genome Res., 25, 1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.