

Abstract
Motivation
Two-dimensional [15N-1H] separated local field solid-state nuclear magnetic resonance (NMR) experiments of membrane proteins aligned in lipid bilayers provide tilt and rotation angles for α-helical segments using Polar Index Slant Angle (PISA)-wheel models. No integrated software has been made available for data analysis and visualization.
Results
We have developed the PISA-SPARKY plugin to seamlessly integrate PISA-wheel modeling into the NMRFAM-SPARKY platform. The plugin performs basic simulations, exhaustive fitting against experimental spectra, error analysis and dipolar and chemical shift wave plotting. The plugin also supports PyMOL integration and handling of parameters that describe variable alignment and dynamic scaling encountered with magnetically aligned media, ensuring optimal fitting and generation of restraints for structure calculation.
Availability and implementation
PISA-SPARKY is freely available in the latest version of NMRFAM-SPARKY from the National Magnetic Resonance Facility at Madison (http://pine.nmrfam.wisc.edu/download_packages.html), the NMRbox Project (https://nmrbox.org) and to subscribers of the SBGrid (https://sbgrid.org). The pisa.py script is available and documented on GitHub (https://github.com/weberdak/pisa.py) along with a tutorial video and sample data.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Oriented sample solid-state nuclear magnetic resonance (OS-ssNMR) spectroscopy enables the acquisition of highly resolved spectra of membrane proteins aligned in lipid bilayers (Opella and Marassi, 2004). In contrast to solution NMR and magic-angle spinning ssNMR, anisotropic contributions dominate chemical shifts and dipolar couplings of OS-ssNMR spectra, leading to enhanced spectral dispersion, especially for α-helices. These parameters provide invaluable topological restraints for structure determination and potentially provide highly sensitive probes that capture subtle signal transduction mechanisms that conventional structural techniques miss (Matthews et al., 2006).
Two-dimensional [15N-1H] separated local field (SLF) experiments (Hester et al., 1976) of uniformly 15N-labeled samples, such as PISEMA (Wu et al., 1994) and SAMPI4 (Nevzorov and Opella, 2007), provide residue-specific orientational restraints by correlating amide 15N chemical shifts and 15N-1H dipolar couplings. The introduction of magnetically aligned media, such as bicelles (Sanders and Landis, 1995) and macrodiscs (Park et al., 2011), has substantially improved the quality of these experiments. These lipid-mimetic systems provide high hydration levels and high lipid-protein ratios, which help stabilize membrane proteins structure and function (Dürr et al., 2012). These improvements yield resolution sufficient for studies of larger multi-spanning systems (Weber and Veglia, 2019). For α-helical proteins, SLF spectra produce circular patterns of the resonances, reflecting the periodic nature of secondary structures, described accurately by the Polar Index Slant Angle (PISA)-wheel model (Marassi and Opella, 2000; Wang et al., 2000). These phenotypical models predict cross-peak positions for each residue as a function of the tilt (or slant) and rotational angles of the overall helical segment (Denny et al., 2001), and they are commonly used in conjunction with selective labeling and unlabeling schemes for resonance assignments; while simultaneously determining descriptive topological parameters without complete structural calculations.
Although PISA models have been integral to OS-ssNMR for almost two decades, no software has been developed for widespread use by the ssNMR community for quantitative data analysis. We have, therefore, built the PISA-SPARKY plugin into NMRFAM-SPARKY (Lee et al., 2015) as part of our development of an integrative platform for biomolecular NMR research (Lee et al., 2016).
2 Materials and methods
The user loads the plugin Input Window by entering the two-letter code ‘PS’. This sets up simulation and fitting calculations for submission to the PISA-SPARKY webserver. The plugin was written in Python 2.7 using the Tkinter GUI module. The PISA simulation method is detailed in Supplementary Data and Supplementary Fig. S1 along with all parameters and their associated default values (Supplementary Table S1). Job ID is in date/time format YYMMDD_HHmmSS followed by a random number. Results are returned to the user’s SPARKYHOME/PISA/Job ID directory (The two-letter code ‘RD’ is used to set SPARKYHOME) and read using the Check button, which displays the Output Window (see Fig. 1 or Supplementary Fig. S2 for full screenshot and workflow) showing all information contained in the log file with visualization and export options. Simulated peaks and a line taken from interpolated PISA data points in the accompanying wave file are displayed in the Spectral Window by the Draw a Wheel button. The Plot Waves button, which utilizes the NDP-PLOT module (Lee et al., 2016), is used to prepare chemical shift and dipolar coupling wave plots. Peaks selected in the Spectral Window may be highlighted in PyMOL by the associated button. The Export DC button is used to export scaled dipolar coupling restraints as input into PONDEROSA-C/S (Lee et al., 2014) for automated structure calculation.
Fig. 1.
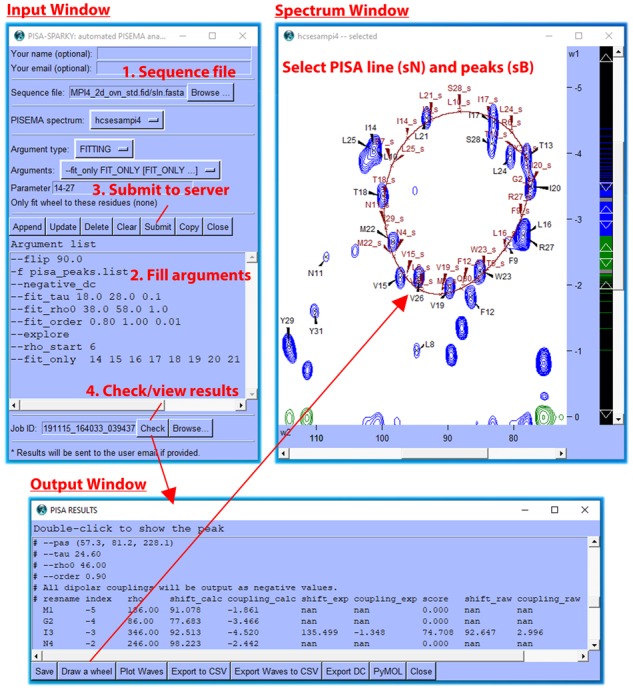
Partial screenshot and workflow of the PISA-SPARKY plugin
Fittings are executed by the pisa.py script, which, if desired, may be used as a standalone tool (see Supplementary Data for additional information) on a local machine in a shell environment. The webserver provides an installation-free environment for computation-intensive jobs. The webserver consists of an AMD 48-core Opteron 2.6 GHz, 128 GB RAM, running the 64-bit Linux CentOS and Python 3 virtual environment. The pisa.py script was written in Python 3.7 using the Numpy library for numerical calculations and the concurrent library to parallelize fitting routines over multiple cores. Interactive communication between the user’s local computer and the remote webserver is handled using HTML, CGI/Perl, Bash and SSH.
3 Results
The plugin is demonstrated in the Supplementary Data using an hcSE-SAMPI4 spectrum of sarcolipin reconstituted into an unflipped bicelle (Wang et al., 2019). Examples describe usage of assignment-free fitting against manually specified spectral boundaries (Supplementary Fig. S3), exhaustive fitting to assignments selected within NMRFAM-SPARKY (Supplementary Fig. S2), basic PISA-wheel simulation (Supplementary Fig. S4A), error analysis (Supplementary Fig. S4B) and PyMOL integration (Supplementary Fig. S5).
Supplementary Material
Acknowledgements
The authors thank Dimitri Maziuk for maintenance of the NMRFAM computing server, which hosts the PISA webserver.
Funding
This work has been supported the National Science Foundation [DBI 1902076 to J.L.M. and W.L.], and partially by the National Institutes of Health [R01 GM 64742 and HL 144130 to G.V.; P41 GM 103399 to J.L.M.] and the American Heart Association [19POST34420009 to D.K.W.]. Part of this work was carried out using hardware provided by the University of Minnesota Supercomputing Institute.
Conflict of Interest: none declared.
References
- Denny J.K. et al. (2001) PISEMA powder patterns and PISA wheels. J. Magn. Reson., 152, 217–226. [DOI] [PubMed] [Google Scholar]
- Dürr U.H.N. et al. (2012) The magic of bicelles lights up membrane protein structure. Chem. Rev., 112, 6054–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hester R.K. et al. (1976) Separated local field spectra in NMR: determination of structure of solids. Phys. Rev. Lett., 36, 1081–1083. [Google Scholar]
- Lee W. et al. (2014) PONDEROSA-C/S: client-server based software package for automated protein 3D structure determination. J. Biomol. NMR, 60, 73–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W. et al. (2015) NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics, 31, 1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W. et al. (2016) Integrative NMR for biomolecular research. J. Biomol. NMR, 64, 307–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marassi F.M., Opella S.J. (2000) A solid-state NMR index of helical membrane protein structure and topology. J. Magn. Reson., 144, 150–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews E.E. et al. (2006) Dynamic helix interactions in transmembrane signaling. Cell, 127, 447–450. [DOI] [PubMed] [Google Scholar]
- Nevzorov A.A., Opella S.J. (2007) Selective averaging for high-resolution solid-state NMR spectroscopy of aligned samples. J. Magn. Reson., 185, 59–70. [DOI] [PubMed] [Google Scholar]
- Opella S.J., Marassi F.M. (2004) Structure determination of membrane proteins by NMR spectroscopy. Chem. Rev., 104, 3587–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.H. et al. (2011) Nanodiscs versus macrodiscs for NMR of membrane proteins. Biochemistry, 50, 8983–8985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders C.R., Landis G.C. (1995) Reconstitution of membrane proteins into lipid-rich bilayered mixed micelles for NMR studies. Biochemistry, 34, 4030–4040. [DOI] [PubMed] [Google Scholar]
- Wang J. et al. (2000) Imaging membrane protein helical wheels. J. Magn. Reson., 144, 162–167. [DOI] [PubMed] [Google Scholar]
- Wang S. et al. (2019) Improving the quality of oriented membrane protein spectra using heat-compensated separated local field experiments. J. Biomol. NMR, 73, 617–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber D.K., Veglia G. (2019) A theoretical assessment of the structure determination of multi-span membrane proteins by oriented sample solid-state NMR spectroscopy. Aust. J. Chem., doi: 10.1071/CH19307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.H. et al. (1994) High-resolution heteronuclear dipolar solid-state NMR spectroscopy. J. Magn. Reson., 109, 270–272. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.