

Abstract
Background
The intracellular ROCK signaling pathway is an important modulator of blood pressure and of cardiovascular and renal remodeling when Rho-kinase activity is increased. Besides, in preclinical models of diabetes, ROCK activation has also a role in abnormal glucose metabolism as well as in subsequent vascular and myocardial dysfunction. In humans, there are a few data assessing ROCK activation in patients with type 2 diabetes mellitus (T2D) and no studies assessing upstream/downstream components of the ROCK pathway. We assessed here levels of ROCK activation and some of the RhoA/ROCK cascade molecules in peripheral blood mononuclear cells (PBMCs) in T2D patients under current treatment.
Methods
Cross-sectional observational study comparing 28 T2D patients under current antidiabetic treatment with 31 consecutive healthy subjects, matched by age and gender. Circulating levels of malondialdehyde, angiotensin II and inflammatory cytokines IL-6 and IL-8 were determined in all subjects. ROCK activation in PMBCs, upstream and downstream cascade proteins, and levels of the proinflammatory molecules VCAM, ICAM-1 and IL-8 were determined in their PMBCs by Western blot.
Results
Compared to healthy controls, ROCK activation in T2D patients measured by 2 direct ROCK targets in PBMCs was increased by 420 and 570% (p < 0001) and it correlated significantly with serum glucose levels. p38 MAPK phosphorylation (downstream from ROCK) and JAK-2 (upstream from ROCK) were significantly higher in the T2D patients by 580% and 220%, respectively. In T2D patients, significantly increased PBMC levels of the proinflammatory molecules VCAM-1, ICAM-1 and IL-8 were observed compared to control subjects (by 180%, 360% and 260%, respectively). Circulating levels of Ang II and MDA were significantly higher in T2D patients by 29 and 63%, respectively.
Conclusions
T2D patients under treatment with glucose-lowering drugs, antihypertensive treatment as well as with statins have significantly increased ROCK activation in their circulating leukocytes along with higher phosphorylation of downstream cascade proteins despite pharmacologic treatment, along with increased plasma angiotensin II and MDA levels. ROCK inhibition might have an additional role in the prevention and treatment of T2D.
Keywords: Diabetes, Metabolic syndrome, Rho kinase, ROCK, MYPT-1, ERM, Fasudil, JAK, p38 MAPK, Metformin
Background
Upregulated Rho kinase (ROCK) activity is involved in the pathogenesis of all aspects of the metabolic syndrome [1, 2]. The ROCK pathway or cascade is an intracellular mechanism that triggers cardiovascular remodeling with important roles in blood pressure regulation, vascular smooth muscle contraction, and cardiovascular and renal remodeling. The small RhoA protein is activated by agonists of receptors coupled to the cell membrane G protein, such as angiotensin II, noradrenaline, growth factors or by cytokines [3–5]. Once RhoA is activated, it translocates to the cell membrane where it activates ROCK. Activated ROCK plays a significant role in vascular smooth muscle cell contraction and regulates several cellular functions involved in cardiovascular remodeling (such as actin cytoskeleton organization, adhesion and motility, proliferation, differentiation, apoptosis and cell survival). ROCK also mediates upregulation of several proinflammatory, thrombogenic and fibrogenic molecules and downregulation of endothelial nitric oxide synthase [3, 4]. In this way, RhoA and ROCK activation have important effects on several cardiovascular diseases [3–6]. In humans with metabolic syndrome, ROCK activation determined in circulating leukocytes has been observed in 2 studies [2, 7].
Besides, in experimental models of metabolic syndrome and type 2 diabetes mellitus (T2D), ROCK activation has a role both in abnormal glucose metabolism pathophysiology as well as in subsequent vascular and myocardial dysfunction. Pharmacological ROCK inhibition increases the number of L cells in the gut, plasma levels of Glucagon-like peptide 1 (GLP1) and insulin and reduces blood glucose levels in mice [8]. Additionally, in mice with insulin resistance, ROCK inhibition increases secretion of GLP1 and glucose tolerance compared with saline administration [8]. Increased ROCK activity has been found in the skeletal muscle and vasculature of insulin-resistant Zucker obese rats and ROCK inhibition prevents the development of various aspects of metabolic syndrome including hypertension, insulin resistance, dyslipidemia, and obesity [9]. In streptozotocin induced diabetic mice, enhanced ROCK activity and decreased nitric oxide bioavailability contribute to altered reactivity to acetylcholine and Ang II in basilar arteries [10]. Furthermore, long-term administration of the ROCK inhibitor fasudil to diabetic rats, significantly ameliorated diabetes-induced cardiac contractile dysfunction at cellular and whole organ levels, specifically fasudil improved calcium removal in diabetic cardiomyocytes, normalized the G/F-actin ratio and promoted F-actin organization [11].
However, in humans there are a few data assessing ROCK activation in patients with T2D. Besides, there are no human studies assessing other components of the ROCK cascade (upstream or downstream) in T2D patients. In Chinese T2D patients under treatment, Liu et al. recently found that ROCK activity was significantly increased in circulating leukocytes [12] compared to control subjects. In a pilot study in patients with T2D and preserved left ventricular ejection fraction, short term fasudil administration improved echocardiographic parameters of left ventricle (LV) diastolic function, but ROCK activation in those patients was not reported [13]. Thus it is relevant to assess in another T2D population, ROCK activation levels simultaneously with proteins of the RhoA/ROCK cascade and with LV function.
The aim of this study was to assess levels of ROCK activation and some of the RhoA/ROCK cascade molecules in peripheral blood mononuclear cells (PBMCs) in type 2 diabetic patients under current treatment and its relationship with LV function and levels of Angiotensin II.
Methods
Study population
This cross-sectional observational study included consecutive T2D patients (n = 28), under current treatment with glucose-lowering drugs not changed during the previous 12 weeks, nonobese (body mass index < 27 kg/m2) and serum creatinine < 1.4 mg/dl and. All of them were also hypertensive patients controlled by antihypertensive treatment. Their clinical characteristics and the current medical treatment are depicted in Tables 1 and 2, respectively. As controls, 31 consecutive healthy subjects (by medical history, symptoms, electrocardiogram, and echocardiogram), matched by age and gender, not taking antidiabetic or antihypertensive drugs, were included. Exclusion criteria were neoplastic disease in the last 4 years, active infection in the last 8 weeks, and chronic lung, liver or kidney disease.
Table 1.
Demographics, blood pressure and heart rate
| Control subjects (n = 31) | T2D patients (n = 28) | p | |
|---|---|---|---|
| Age (years) | 57.8 ± 5.5 | 60.7 ± 5.4 | 0.080 |
| Men (%) | 34 | 58 | 0.150 |
| Weight (Kg) | 68.4 ± 8.6 | 70.1 ± 10.7 | 0.500 |
| Body mass index (BMI, Kg/m2) | 25.3 ± 2.3 | 26.4 ± 2.1 | 0.070 |
| Systolic blood pressure (mm Hg) | 114.1 ± 12.0 | 123.8 ± 16.0 | 0.010 |
| Diastolic blood pressure (mm Hg) | 75.4 ± 7.7 | 81.0 ± 9.0 | 0.010 |
| Heart rate (bpm) | 65.1 ± 8.9 | 73.0 ± 9.0 | 0.001 |
Values are shown as mean ± SD
Table 2.
Current pharmacological treatment in the T2D patients (n = 28)
| Pharmacological treatment | |
|---|---|
| Metformin (%) | 89 |
| Only metformin (%) | 14 |
| Other oral hypoglicemic drug (%) | 43 |
| Metformin + Other oral hypoglicemic drug (%) | 38 |
| Insulin (%) | 57 |
| Only insulin (%) | 4 |
| Insulin + Metformin (%) | 50 |
| Insulin + any oral hypoglicemic drug (%) | 54 |
| ACE inhibitors (%) | 21 |
| Angiotensin receptor blockers (%) | 75 |
| Betablockers (%) | 36 |
| Thiazides (%) | 25 |
| Statins (%) | 71 |
| Aspirin (%) | 43 |
The study was approved by the Human Research Ethics Committee at the Pontificia Universidad Catolica de Chile, School of Medicine and adhered to the Declaration of Helsinki principles. Written informed consent was obtained from all participants prior to any procedure.
Biochemical parameters
Fasting concentrations of total cholesterol, triglycerides (TG), low-density lipoprotein cholesterol (LDL-C), high density lipoprotein cholesterol (HDL-C), plasma glucose, and glycated hemoglobin A1c (HbA1c) were determined at the Central Clinical Laboratory, Red Christus UC.
Echocardiographic measurements
Echocardiograms were obtained with a 2.5-MHz transducer at the time of blood sampling with a Phillips IE-33 instrument (Andover, MA) to evaluate LV function, geometry, and mass. All measurements were performed blindly as previously described [14] and according to the recommendations of the American Society of Echocardiography [15]. The following variables in the parasternal short axis were measured: left atria diameter, interventricular septal thickness and posterior wall thickness, end diastolic dimension and end-systolic dimension. Ejection fraction was determined by the Simpson method. Systolic pulmonary artery pressure was assessed by continuous wave Doppler of the tricuspid regurgitation trace, to measure the difference in pressures between the right ventricle and right atrium and using the simplified Bernoulli equation to calculate this pressure difference using peak tricuspid regurgitation velocity [16].
Plasma levels of malondialdehyde
As one parameter associated to oxidative stress, we measured circulating malondialdehyde levels as previously described by determining the content of thiobarbituric acid-reactive substances [17].
Protein extraction from peripheral blood mononuclear cells (PBMCs)
For isolating peripheral blood mononuclear cells (PBMCs), 5 mL of whole venous blood containing ethylenediaminetetraacetic acid (EDTA) were poured over 5 mL of density gradient cell separation medium (Ficoll and sodium diatrizoate, Histopaque-1077, Sigma Chemical Co., St Louis, MO) and centrifuged [14, 18]. White cells were separated, resuspended and washed in phosphate buffered saline (PBS). Upon isolation (4–80 × 106 viable cells, 95% viability), cells were resuspended in lysis buffer containing 150 mM NaCl, 1% NP40, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate (SDS) and 50 mM Tris. Lysis buffer was supplemented with a protease inhibitor cocktail (1 μg/ml aprotinin, 1 μg/ml leupeptin and 1 mM phenylmethylsulphonyl fluoride) [14, 18]. Protein content was determined by the Lowry assay. ROCK activity and ROCK cascade proteins were determined by Western blot (details as follows).
Western blot analysis to determine ROCK activity and ROCK cascade proteins in PBMCs
ROCK activity was measured as the relationship of phosphorylated to total myosin phosphatase target subunit 1 (MYPT-1) and also by the relationship of phosphorylated to total ezrin, radixin, moesin (ERM), two direct ROCK substrates.
Soluble protein fractions were heated 5 min at 95 °C with SDS sample buffer (375 mM Tris–HCl pH 6.8, 6% SDS, 48% glycerol, 9% 2-mercaptoethanol and 0.03% bromophenol blue). Equal amounts of protein were loaded and separated on a 5% stacking and 8, 18% resolving SDS-PAGE gel (80 V), and transferred into a nitrocellulose membrane at 400 A during 2 h on ice. Blocking was performed with 5% BSA at room temperature. Blots were incubated overnight at 4 °C with the primary antibody. The relative amount of protein was estimated by chemiluminescence (ECL plus kit, Perkin Elmer which contains the substrate for horseradish peroxidase, HRP). Digital images were obtained with a Syngene G-Box automated system and analyzed by densitometry using the software UN-SCAN-IT™ (Silk Scientific Corporation).
The blots were incubated overnight with the following primary antibodies: anti myosin phosphatase target subunit 1 (anti-MYPT-1 antibody, rabbit polyclonal, 1/500 cell signaling, Cat 2634); anti-p-MYPT-1 antibody (phospho-MYPT1-Thr 853 rabbit polyclonal, 1/500, cyclex, Cat CY-P1025) [14, 16]; anti ezrin, radixin, moesin (anti-ERM antibody, total-ERM, rabbit polyclonal, 1/700, Cell Signaling, Cat 3142); anti-p-ERM antibody (phospho-ERM, ezrin Thr567, radixin Thr564, moesin Thr558, rabbit polyclonal, 1/700, Cell Signaling, Cat 3141); anti p-38 mitogen-activated protein kinase (anti-p38 MAPK antibody, p-38, rabbit polyclonal, 1/1000, Cell Signaling, Cat 9212); anti-phospho-p38 MAPK antibody (Thr180/Tyr182) (phospho-p-38, rabbit monoclonal, 1/1000, Cell Signaling, Cat 4511); anti-ROCK-1 antibody (mouse monoclonal, 1/500, BD Bioscience, Cat 611136); anti-ROCK-2 antibody (mouse monoclonal, 1/2000, BD Bioscience, Cat 610623); anti-p65 nuclear factor κB antibody (NFκB, rabbit polyclonal, 1/1000, Cell Signaling, Cat 8242); anti-vascular cell adhesion molecule 1 antibody (VCAM-1, goat polyclonal, 1/1000, Santa Cruz, Cat sc1504); anti-intracellular adhesion molecule 1 antibody (ICAM-1, mouse monoclonal, 1/1000, Santa Cruz, Cat sc8439); anti-interleukin 6 antibody (IL-6, rabbit polyclonal, 1/500, Abcam, Cat ab6672); anti-interleukin 8 antibody (IL-8, rabbit polyclonal, 1/500, Abcam, Cat ab7747); anti-myosin light chain 2 antibody (MLC-2, rabbit polyclonal, 1/1000, Cell Signaling, Cat cs3672) and anti-phospho-MLC-2 antibody (Thr18/Ser19) (p-MLC-2, rabbit polyclonal, 1/500, Cell Signaling, Cat cs3674); anti Janus kinase (anti JAK antibody, JAK, rabbit polyclonal, 1:1000 Cell signaling Cat cs3230); anti-phospho JAK2 antibody (p-JAK, 1:500 Rabbit polyclonal Cell signaling Cat sc3776); anti JNK antibody (JNK, rabbit polyclonal, 1:1000 cell signaling cs9252) and anti phospho c-Jun N-terminal kinase (anti-phospho JNK antibody, p-JNK, 1:500 rabbit polyclonal Cell signaling cat cs9251). The blots were then washed and incubated with a secondary antibody horseradish peroxidase conjugated goat anti-rabbit IgG (1:7500, Thermo Scientific, Cat) or a goat anti-mouse IgG (1:10.000, Santa Cruz, Cat sc2005) for 2 h. As a protein loading control, β-actin (β-actin, mouse monoclonal, 1/10,000, Sigma, Cat A2228) was used.
Circulating levels of angiotensin II and inflammatory cytokines IL-6 and IL-8
Venous blood samples were taken and immediately cooled, centrifuged at 4 °C and then stored at − 80 °C until analysis. Angiotensin II (Ang II) levels were quantified using commercial ELISA kits (Sigma, US; Enzo Life Sciences) according to the manufacturer instructions. The Ang II ELISA detection limits were 4.6 pg/ml with intra-assay coefficient of variation (CV) between 4.7 and 7.3% and inter-assay CV between 6.0 and 15.9%. Serum levels of IL-6 and IL-8 were also determined using the Abcam ELISA kit according to the manufacturer’s instructions.
Statistical analysis
Data are presented as mean ± SD. Differences between mean values were compared using t-test. Correlation analyses were performed by using Pearson correlation. p Values ≤ 0.05 were considered statistically significant.
Results
Clinical characteristics and laboratory tests
Twenty-eight consecutive patients with T2D with a mean diabetic history of 133 months and a mean HbA1C 7.9 ± 1.1% were included in the study. In the control group 31 consecutive healthy subjects without heart disease assessed by medical history, symptoms, electrocardiogram and echocardiogram were included. Age, weight, BMI and gender distribution were similar in both groups (Table 1). Blood pressure and heart rate were slightly higher in the T2D patients (Table 1). 96% of the T2D patients were receiving oral glucose-lowering drugs (most of them metformin) and 54% were treated with an oral glucose-lowering drug plus insulin (Table 2).
Compared to the control group, serum creatinine and potassium, triglycerides and SGOT levels were similar in T2D patients, but most of blood chemistry tests as well as levels of malondialdehyde were significantly different (p < 0.05) (Table 3).
Table 3.
Blood chemistry and plasma oxidative stress levels
| Control subjects (n = 31) | T2D patients (n = 28) | p | |
|---|---|---|---|
| Hematocrit (%) | 42.9 ± 3.4 | 40.8 ± 3.4 | 0.020 |
| Hemoglobin (mmol/L) | 14.5 ± 1.4 | 13.8 ± 1.3 | 0.040 |
| WBC/Ml | 6003.2 ± 1461.4 | 7465.9 ± 1733.8 | 0.020 |
| Plasma creatinine (mEq/L) | 0.8 ± 0.2 | 0.8 ± 0.2 | 0.690 |
| ESR | 6.5 ± 5.9 | 13.0 ± 15.9 | 0.030 |
| BUN (mg/L) | 13.2 ± 4.8 | 16.7 ± 4.1 | 0.030 |
| Potassium, serum (mEq/L) | 4.3 ± 0.7 | 4.3 ± 0.4 | 0.680 |
| Glucose, serum (mg/dL) | 89.2 ± 6.71 | 155.0 ± 59.2 | 0.001 |
| Uric acid (μmol/L) | 5.1 ± 1.2 | 4.4 ± 1.4 | 0.040 |
| BUN/plasma creatinine | 17.7 ± 3.4 | 22.2 ± 5.7 | 0.010 |
| Serum Cholesterol (mg/dL) | 204.5 ± 22.5 | 160.1 ± 42.3 | 0.001 |
| LDL Cholesterol (mg/dL) | 124.4 ± 23.9 | 87.2 ± 34.7 | 0.001 |
| HDL Cholesterol (mg/dL) | 55.7 ± 14.3 | 44.1 ± 9.4 | 0.001 |
| Triglicerydes (mg/dL) | 127.0 ± 63.3 | 144.1 ± 72.9 | 0.340 |
| SGOT (U/L) | 21.2 ± 6.3 | 19.7 ± 6.1 | 0.350 |
| MAU (ug/mL) | 2.2 ± 3.0 | 23.6 ± 40.77 | 0.010 |
| MAU/urine creatinine ratio (mg/g creat) | 4.9 ± 1.8 | 28.3 ± 50.6 | 0.030 |
| MDA (uM) | 1.1 ± 0.4 | 1.8 ± 1.1 | 0.010 |
Values shown as mean ± SD
NS nonsignificant, WBC white blood cells, BUN blood urea nitrogen, MDA malondialdehyde, HDL-C density lipoprotein cholesterol, MAU microalbuminuria
Echocardiographic LV dimensions and function
Compared with the control group, posterior wall thickness, septum thickness, and A wave velocity were significantly larger in the T2D patients by 8%, 12%, and 15%, respectively (Table 4). LV systolic function was normal in the T2D patients, whereas LV diastolic function assessed by the the E/A ratio was reduced (p < 0.02; Table 4).
Table 4.
Cardiac dimensions, LV systolic function and pulmonary artery systolic pressure by echocardiography
| Control subjects (n = 31) | T2D patients (n = 28) | p | |
|---|---|---|---|
| Posterior wall thickness (mm) | 8.7 ± 1.0 | 9.4 ± 1.3 | 0.020 |
| Septum thickness (mm) | 9.0 ± 1.0 | 10.1 ± 1.5 | 0.001 |
| LV end diastolic diameter (mm) | 48.8 ± 4.5 | 45.3 ± 4.7 | 0.001 |
| LV end systolic diameter (mm) | 30.1 ± 4.7 | 28.0 ± 4.5 | 0.090 |
| LV end diastolic diameter (mm)/BMI | 1.9 ± 0.4 | 1.7 ± 0.2 | 0.120 |
| LV end systolic diameter (mm)/BMI | 1.1 ± 0.3 | 1.1 ± 0.2 | 0.220 |
| LV end diastolic diameter (mm)/height (cms) | 0.3 ± 0.1 | 0.3 ± 0.0 | 0.930 |
| LV end sytolic diameter (mm)/height (cms) | 0.2 ± 0.0 | 0.2 ± 0.0 | 0.810 |
| Left atrial diameter (mm) | 37.2 ± 5.0 | 38.8 ± 4.1 | 0.150 |
| LV mass (g) | 189.0 ± 44.3 | 192.5 ± 48.9 | 0.780 |
| LV mass index (g/m2) | 103.5 ± 20.4 | 111.2 ± 24.6 | 0.210 |
| LV shortening fraction (%) | 38.1 ± 7.1 | 37.8 ± 7.2 | 0.810 |
| LV ejection fraction (%) | 67.4 ± 5.0 | 64.0 ± 7.3 | 0.070 |
| Pulmonary artery systolic pressure (mm Hg) | 25.4 ± 3.8 | 25.0 ± 4.2 | 0.770 |
| E wave velocity, cm/s | 70.1 ± 14.0 | 71.8 ± 10.8 | 0.600 |
| A wave velocity, cm/s | 69.1 ± 18.0 | 79.8 ± 15.4 | 0.020 |
| E/A | 1.0 ± 0.2 | 0.9 ± 0.2 | 0.010 |
Values shown as mean ± SD
LV left ventricle, E/A transmitral filling waves ratio
Rho-kinase activation in PBMC cells
In the T2D patients, the mean ratio between phosphorylated to total MYPT1 and ERM (MYPT1-P/T and ERM-P/T) in circulating leukocytes, was significantly increased by threefolds (Fig. 1, p < 0.001) and fivefolds (Fig. 2, p < 0.001), respectively, compared to control subjects. MYPT1-P/T and ERM-P/T ratios were directly correlated with serum glucose levels (r = 0.38, p < 0.02, Fig. 3a) and (r = 0.45, p < 0.004), respectively (Fig. 3b).
Fig. 1.
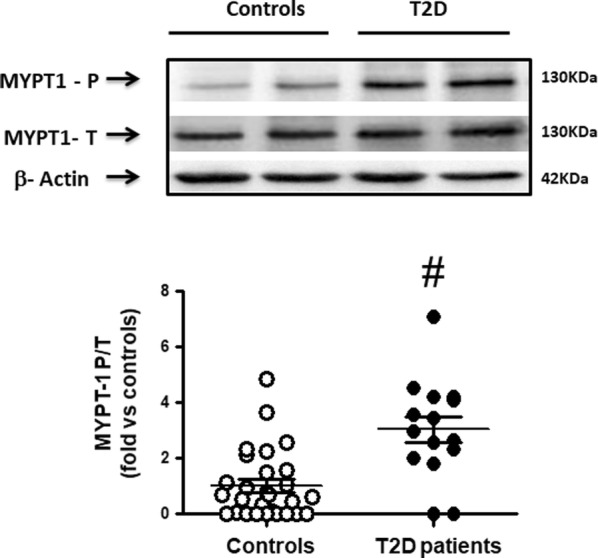
Increased ROCK activation in PBMCs assessed by MYPT-1 phosphorylation levels in T2D patients (Western blot). Upper panel. Representative western blots images for phosphorylated (MYPT1 P) and total MYPT1 (MYPT1 T) in PBMCs from 2 control subjects and 2 T2D patients. Lower Figure. Dot graph of phosphorylated/total MYPT1 ratios in PBMCs in Control subjects (n = 28, white circles) and in T2D patients (n = 15, black circles), data shown as mean ± SEM. Symbol: #p < 0,001 versus Controls
Fig. 2.

Increased ROCK activation in PBMCs assessed by ERM phosphorylation in T2D patients (Western blot). Upper panel. Representative western blots images for phosphorylated (ERM P) and total ERM (ERM T) in PBMCs from 2 control subjects and 2 T2D patients. Lower Figure. Dot graph of phosphorylated/total ERM ratios in PBMCs in Control subjects (n = 31, white circles) and in T2D patients (n = 18, black circles), data shown as mean ± SEM. Symbol: #p < 0,001 versus Controls
Fig. 3.

Linear relationship (Pearson coefficient) between serum glucose levels and ROCK activation in PBMCs assessed both by MYPT-1 (a), n = 39, and ERM phosphorylation levels (b), n = 41. White circles = Control subjects, black circles = T2D patients
Besides, ROCK1 levels in PBMCs were significantly higher in the T2D patients compared to healthy subjects (p < 0.002, Fig. 4), whereas similar levels of the ROCK 2 isoform were observed in both groups (Table 5).
Fig. 4.
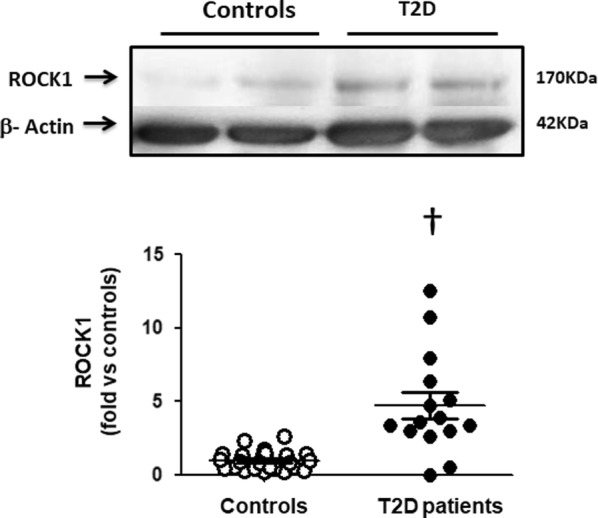
Increased ROCK1 isoform levels in PBMCs in T2D patients (Western blot). Upper panel. Representative western blots images of ROCK1 isoform in PBMCs from 2 control subjects and 2 T2D patients. Lower Figure. Dot graph comparing ROCK1 isoform levels in PBMCs in Control subjects (n = 25, white circles) and in T2D patients (n = 15, black circles), data shown as mean ± SEM. Symbol: †p < 0,002 versus controls
Table 5.
Components of the ROCK pathway in PBMCs (Western blot)
| Control subjects | T2D patients | p | |
|---|---|---|---|
| p38-MAPK P/T | 1.00 ± 0.90 | 5.81 ± 3.50 | 0.001 |
| p65-NFkB (UOD) | 1.00 ± 0.45 | 1.34 ± 1.18 | 0.240 |
| JAK2 P/T | 1.00 ± 0.68 | 2.28 ± 2.10 | 0.052 |
| ROCK2 (UOD) | 1.00 ± 0.57 | 0.68 ± 0.65 | 0.080 |
| JNK P/T | 1.00 ± 0.63 | 0.90 ± 1.16 | 0.960 |
| IL-6 (UOD) | 1.00 ± 1.00 | 1.03 ± 0.75 | 0.920 |
| MLC-2 P/T | 1.00 ± 0.63 | 0.98 ± 1.16 | 0.960 |
Values are shown as mean ± SD (fold vs control subjects) n = 22-27 Controls and 13 -18 T2D patients
p38-MAPK p38 mitogen-activated protein kinase, p65-NFkB Nuclear factor NF-kappa-B p65 subunit, JAK2 Janus kinase 2, ROCK2 Rho kinase isoform 2, JNK Jun amino-terminal kinase, IL-6 interleukin 6, MLC-2 myosin light chain 2, P/T phosphorylated/total, UOD units of optical density
Components of the ROCK cascade in circulating leukocytes
In the T2D patients JAK2 phosphorylation levels (upstream from ROCK) were increased by 2.2-fold and p38 MAPK phosphorylation levels (downstream from ROCK) were significantly increased by and 5.8-fold (Table 5) in their PBMCs.
Furthermore, a significant increase in the levels of VCAM-1 (by 1.8-fold, p < 0.05, Fig. 5), ICAM-1 (by 3.6-fold, p < 0.002, Fig. 6) and IL-8 (by 2.6-fold, p < 0.05, Fig. 7) were observed in the diabetic patients compared to control subjects.
Fig. 5.
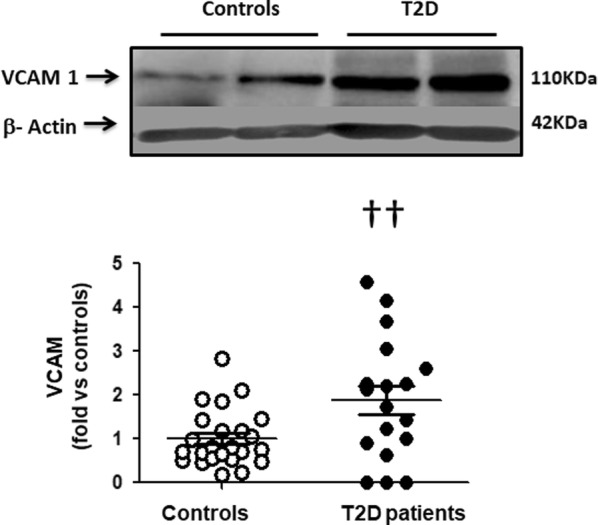
Higher VCAM-1 levels in PBMCs in T2D patients compared to control subjects (Western blot). Upper panel. Representative western blots images of VCAM in PBMCs from 2 control subjects and 2 T2D patients. Lower Figure. Dot graph comparing VCAM levels in PBMCs in Control subjects (n = 24, white circles) and in T2D patients (n = 18, black circles), data shown as mean ± SEM. Symbol: ††p < 0,05 versus controls
Fig. 6.
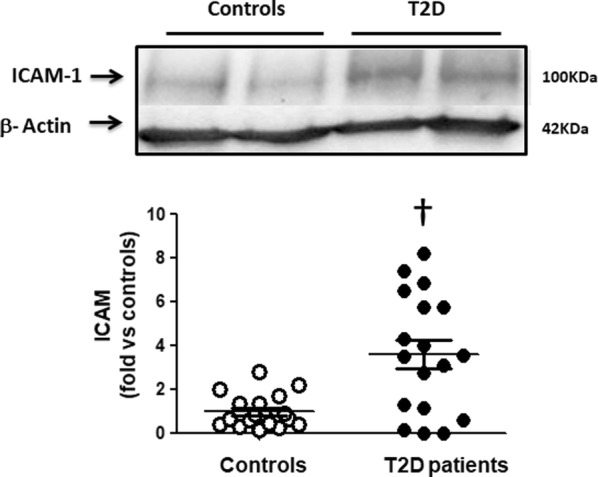
Increased ICAM-1 levels in PBMCs in T2D patients compared to control subjects (Western blot). Upper panel. Representative western blots images of ICAM in PBMCs from 2 control subjects and 2 T2D patients. Lower Figure. Dot graph comparing ICAM levels in PBMCs in Control subjects (n = 18, white circles) and in T2D patients (n = 18, black circles), data shown as mean ± SEM. Symbol: †p < 0,002 versus Controls
Fig. 7.
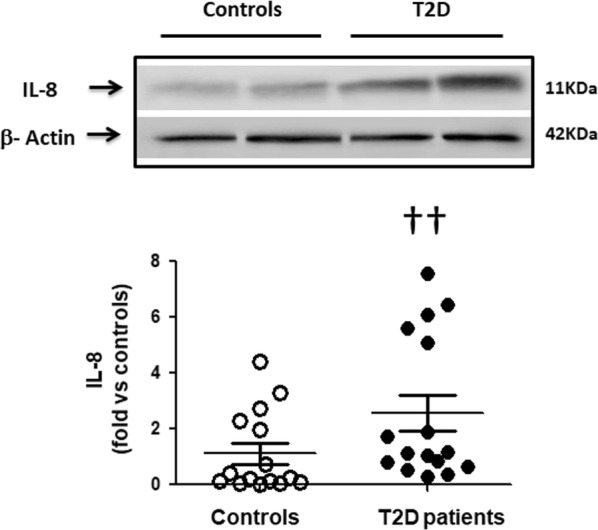
Increased IL-8 levels in PBMCs in T2D patients compared to control subjects (Western blot). Upper panel. Representative western blots images of IL-8 in PBMCs from 2 control subjects and 2 T2D patients. Lower Figure. Dot graph comparing IL-8 levels in PBMCs in Control subjects (n = 20, white circles) and in T2D patients (n = 16, black circles), data shown as mean ± SEM. Symbol: ††p < 0.05 versus Controls
Circulating levels of Angiotensin II, IL-8 and IL-6
Even though the T2D patients were receiving pharmacological treatment and all of them were clinically stable, circulating levels of Ang II were significantly higher in patients compared to controls by 29% (Table 6). Circulating levels of ILs 6 and 8 were similar in both groups (Table 6).
Table 6.
Circulating levels of interleukins 6 and 8 and angiotensin II
| Control subjects (n = 31) | T2D patients (n = 28) | p | |
|---|---|---|---|
| Interleukin-6 (pg/mL) | 0.95 ± 0.31 | 1.77 ± 0.52 | 0.180 |
| Interleukin-8 (pg/mL) | 4.89 ± 0.66 | 5.23 ± 1.08 | 0.780 |
| Angiotensin II (pg/mL) | 7.78 ± 2.80 | 10.08 ± 1.38 | 0.040 |
Values shown as mean ± SD; n = 10–30 Controls and 9–28 T2D patients
Discussion
The main findings in this study were that T2D patients under current glucose-lowering drugs compared with healthy control subjects have significant ROCK activation in their PBMCs as determined by increased phosphorylation of two ROCK direct targets (MYPT1 and ERM). Besides, increased activity of the ROCK cascade proteins JAK-2 (upstream) and p38-MAPK (downstream) were found. ROCK activation levels were here correlated with current blood glucose levels. In these patients, increased levels of the proinflammatory molecules VCAM, ICAM-1 and IL-8 and of the isoform ROCK1 were also found in their PMBCs despite pharmacologic treatment along with increased plasma angiotensin II and MDA levels (by 30% and 64%, respectively).
There is only one clinical study in T2D patients under treatment (26% of them were also hypertensives) observing significantly higher levels of ROCK activation in circulating leukocytes by 11% compared to controls [12] and our study is consistent with those observations, with larger significant differences in ROCK activation in our current study assessed by 2 different ROCK substrates phosphorylation. In both studies a significant relationship of ROCK activation in PBMCs with glucose levels has been observed.
Higher ROCK activation levels in the present study, were observed despite that 96% of the patients were receiving ACE inhibitors/ARBs and their blood pressure was normal but higher than in control subjects. In hypertensive patients, olmesartan [19, 20] reduces ROCK activation levels in PBMCs, as well as amlodipine [21] and eplerenone [22]. Consequently, it is possible to hypothesize that chronically elevated blood glucose levels may explain both higher ROCK activation levels and increased plasma angiotensin II levels in treated T2D patients (increased angiotensin levels II are associated with ROCK activation in PMBCs in normotensive rats with genetically induced high ACE levels and angiotensin II and in patients with heart failure) [23, 24].
The specific biological implication of ROCK activation in PMBCs in diabetic patients, as in patients with cardiovascular diseases, is not totally known. Our understanding of the pathophysiology of ROCK activation, downstream ROCK pathway and the related molecular mechanisms involved in its pathogenesis and how does activated ROCK in leukocytes affect each target organ is even less known [25]. In this regard, two preclinical studies assessing simultaneously ROCK activation in PMBCs, myocardium and in the aortic wall indicate a direct relationship between ROCK activation in PMBCs and in cardiovascular tissue, supporting the concept that ROCK activation in these circulating cells does reflect cardiovascular ROCK activation [23, 26].
In PBMCs, we observed here increased ROCK1 isoform levels in T2D patients and unchanged levels of the ROCK2 isoform. Patients with hypertension and patients with heart failure and reduced ejection fraction have similar ROCK isoform levels in their PBMCs [14, 18, 24]. There are no previous ROCK isoform measurements in humans with T2D but patients with metabolic syndrome have similar ROCK isoform levels compared to control subjects [7]. In non diabetic mice, double ROCK knockout promotes cardiac autophagy and fibrosis, whereas in ROCK2 knockout mice, autophagy is inhibited and increased cardiac fibrosis is observed [27]. At the same time loss of ROCK1 does not have an effect on autophagy [27]. In diabetic hearts from streptozotocin treated rats, ROCK activation is associated with over-expression of the ROCK2 isoform [28]. Cardiomyocytes from diabetic mice develop arrhythmic calcium transients in response to increased [Ca2+] which is attenuated in cardiomyocytes from diabetic ROCK2 mice [29] suggesting that ROCK2 contributes to diabetes-induced impaired cardiac calcium homeostasis. It is possible that ROCK2 may be a beneficially therapeutic target for heart protection in T2D [30], which needs further preclinical and clinical research as well as assessment of unwanted side effects [31].
Activation of the ROCK cascade has not been previously assessed in diabetic patients. Here, in the T2D patients receiving glucose-lowering drugs, both increased phosphorylation of the upstream molecule JAK-2 and of the downstream MAPK (p38 MAPK) were observed for the first time in this clinical context.
p38 MAPK plays important roles in regulating glucose metabolism in skeletal muscle and in adipose tissue [32]. Studies with cultured skeletal muscle cells indicate that p38 MAPK enhances insulin and exercise or AMPK-induced glucose uptake by activating GLUT4 [33, 34]. Besides, MAP kinases, such as the downstream ROCK cascade molecule p38-MAPK, have a role in the development of cardiac hypertrophy, remodeling, and contractile dysfunction (35 30). Increased cardiac p38-MAPK expression is associated with reduced contractility and the development of cardiomyopathy in mice [35]. In rats with cardiac remodeling induced by endurance exercise, the ROCK inhibitor fasudil reduces cardiac hypertrophy, remodeling and cardiac p38-MAPK levels [36]. Interestingly, PMBCs p38-MAPK phosphorylation levels and cardiac and aortic wall p38-MAPK phosphorylation levels secondary to ROCK activation have been observed in normotensive and in hypertensive rats [23, 26]. Even though LV systolic function assessed by LV ejection fraction was normal in the current T2D patients, this is the first observation of increased p38-MAPK phosphorylation in PMBCs in T2D patients, associated to ROCK activation, specifically in patients with high blood glucose levels, concentric remodeling and evidence of diastolic dysfunction (lower E/A ratio and increased A wave velocity).
In preclinical studies, the upstream JAK 2 protein has a known role in the development of diabetic nephropathy. High glucose levels augment ANG II-induced activation of JAK2 in renal glomerular mesangial cells and in vascular smooth vascular cells [37–39]. Interestingly, simvastatin modulates the detrimental effects of high glucose/ANG II in diabetic kidney glomeruli both in vitro and in vivo by preventing the high glucose and ANG II-induced geranylgeranyl-dependent activation of Rac or/and Rho and thereby blocking the activation of JAK2 and both STAT1 and STAT3 [40]. Besides, inflammation signaled by Janus kinases (JAKs) promotes progression of diabetic kidney disease [41]. In diabetic mice, enhanced expression of JAK2 selectively in glomerular podocytes increases pathological characteristics of diabetic kidney disease [42]. Recently, in a randomized clinical study in T2D patients at high risk for progressive kidney disease, baricitinib, an oral, reversible selective inhibitor of JAK1 and JAK2 reduced albuminuria [41]. In keeping with the abovementioned observations, in the current patients under treatment and with increased microalbuminuria, JAK activation (phosphorylation) compared to control subjects was observed.
Plasma MDA levels were measured here as an oxidative stress biomarker, more precisely a lipid peroxidation marker, and were higher in T2D patients compared to controls. Oxidative stress in diabetes exacerbates atrial structural and electrical remodeling, promotes Ca2+ mediated triggers and the initiation of atrial fibrillation [43]. MDA levels are elevated in patients with metabolic syndrome [44, 45]. Possibly, higher MDA levels in the T2D patients could originate from lower plasma antioxidant enzymes levels (not measured here) and to the over active leukocytes [44] secondary to ROCK activation.
Regarding T2D, Rho kinase activation and inflammation, T2D is a chronic low-grade inflammation state [46]. Several circulating inflammatory biomarkers are elevated in T2D patients under treatment compared to controls and are associated to a hypercoagulable state and vascular dysfunction [47]. Increased IL-6 (as well as IL-18) circulating levels in T2D patients under treatment could trigger an increased extracellular matrix proteolysis, which may modify vascular structure and function [48]. Interestingly, in the T2D patients under treatment (but still with hyperglycemia) we observed increased levels of ICAM-1, VCAM-1 and IL-8 in PBMCs associated to ROCK activation whereas their circulating of both IL-6 and IL-8 were similar to controls. The ROCK pathway plays a role in adhesion molecule expression and inflammatory cell infiltration in glomerular endothelial cells induced by advanced glycation end products [49]. Besides, ROCK activation mediates regulation of inflammatory signaling pathways in human retinal cells exposed to high-glucose as well as in diabetic mice [50].
Limitations
In this cross sectional study we used two groups, controls and T2D receiving glucose-lowering drugs (or insulin), antihypertensive drugs and some of them statins as well. It would be most interesting to assess ROCK levels in T2D patients without treatment. Nevertheless, in the only available study addressing ROCK activation in PBMCs in T2D patients 75.6% of them were receiving metformin, 58.5% gliclazide and 26.8% a DPP-4 inhibitor [12]. Besides, 63.7% were receiving ACE inhibitors or ARBs, 63.6% calcium-channel blockers and 68.4% statins [12]. Even though all these drugs may reduce ROCK activity, our results are consistent with those findings (compared with the control subjects, ROCK activity was significantly increased in T2D patients). Besides, since the T2D group was treated with glucose-lowering drugs (most of them with metformin) and some of them with insulin and they still had hyperglycemia, it would have been relevant to compare our current findings with a group of T2D patients with well controlled blood glucose levels.
Conclusions
T2D patients under current treatment with antidiabetic and antihypertensive drugs as well as with statins have significantly increased ROCK activation in their PMBCs, also with increased phosphorylation of upstream and downstream cascade proteins. ROCK activation levels were correlated here with current blood glucose levels. In these patients, increased levels of the proinflammatory molecules VCAM, ICAM-1 and IL-8 were also found in their PMBCs despite pharmacologic treatment along with increased plasma angiotensin II and MDA levels. It is possible to hypothesize that specific ROCK inhibition might have an additional role in the prevention and treatment of T2D.
Acknowledgements
To Ivonne Padillla, RN for her dedicated work with the patients.
Abbreviations
- EDTA
Ethylenediaminetetraacetic acid
- ERM
Ezrin, radixin, moesin
- GLP1
Glucagon-like peptide 1
- HbA1c
Glycated hemoglobin A1c
- ICAM-1
Intracellular adhesion molecule 1
- IL-6
Interleukin 6
- IL-8
Interleukin 8
- JAK
Janus kinase
- JNK
c-Jun N-terminal kinase
- LV
Left ventricle
- MAPK
Mitogen-activated protein kinase
- MLC-2
Myosin light chain 2
- MYPT-1
Myosin phosphatase target subunit 1
- NFκB
Nuclear factor κB
- PBMCs
Peripheral blood mononuclear cells
- ROCK
Rho kinase
- T2D
Type 2 diabetes mellitus
- VCAM-1
Vascular cell adhesion molecule 1
Authors’ contributions
MPO: study coordination, data analysis, manuscript writing. JEJ: study design and conduction, data analysis, final manuscript writing. PV: clinical work with the patients, data and manuscript discussion. JM: biochemical and molecular work, data analysis and discussion. LG: echocardiographic studies, data analysis, manuscript discussion. IG: echocardiographic studies, data analysis, manuscript discussion. SC: echocardiographic studies, data analysis, manuscript discussion. PM: echocardiographic studies, data analysis, manuscript discussion. LG: biochemical and molecular work, data analysis and discussion. LF: database analysis, manuscript discussion. All authors read and approved the final manuscript.
Funding
This work was supported by grants from the Agencia Nacional de Investigacion y Desarrollo (ANID, Chile): Fondecyt (Fondo Nacional de Desarrollo Cientiico y Tecnologico, Chile) 1161739 and 1121060 (to J.E.J), Anillo ACT192144 (to M.P.O and J.E.J) and FONDAP 15130011 (to L.G and M.P.O)
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
The study was approved by the Human Research Ethics Committee at the Pontificia Universidad Catolica de Chile, School of Medicine and a written informed consent was obtained from all participants prior to any study procedure.
Consent for publication
All the coauthors have agreed to publish this paper in the current version and format.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Jahani V, Kavousi A, Mehri S, Karimi G. Rho kinase, a potential target in the treatment of metabolic syndrome. Biomed Pharmacother. 2018;106:1024–1030. doi: 10.1016/j.biopha.2018.07.060. [DOI] [PubMed] [Google Scholar]
- 2.Leguina-Ruzzi A, Pereira J, Pereira-Flores K, Valderas JP, Mezzano D, et al. Increased RhoA/Rho-kinase activity and markers of endothelial dysfunction in young adult subjects with metabolic syndrome. Metab Syndr Relat Disord. 2015;13:373–380. doi: 10.1089/met.2015.0061. [DOI] [PubMed] [Google Scholar]
- 3.Shimokawa H, Takeshita A. Rho-kinase is an important therapeutic target in cardiovascular medicine. Arterioscler Thromb Vasc Biol. 2005;25:1767–1775. doi: 10.1161/01.ATV.0000176193.83629.c8. [DOI] [PubMed] [Google Scholar]
- 4.Jalil JE, Lavandero S, Chiong M, Ocaranza MP. Rho/Rho kinase signal transduction pathway in cardiovascular disease and cardiovascular remodeling. Rev Esp Cardiol. 2005;58:951–961. doi: 10.1157/13078132. [DOI] [PubMed] [Google Scholar]
- 5.Shimokawa H, Sunamura S, Satoh K. RhoA/Rho-kinase in the cardiovascular system. Circ Res. 2016;118:352–366. doi: 10.1161/CIRCRESAHA.115.306532. [DOI] [PubMed] [Google Scholar]
- 6.Dong M, Yan BP, Liao JK, Lam YY, Yip GW, Yu CM. Rho-kinase inhibition: a novel therapeutic target for the treatment of cardiovascular diseases. Drug Discov Today. 2010;15:622–629. doi: 10.1016/j.drudis.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu PY, Chen JH, Lin LJ, Liao JK. Increased Rho kinase activity in a Taiwanese population with metabolic syndrome. J Am Coll Cardiol. 2007;49:1619–1624. doi: 10.1016/j.jacc.2006.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petersen N, Frimurer TM, Pedersen MT, Egerod KL, Wewer Albrechtsen NJ, et al. Inhibiting RHOA signaling in mice increases glucose tolerance and numbers of enteroendocrine and other secretory cells in the intestine. Gastroenterology. 2018;155(1164–76):e2. doi: 10.1053/j.gastro.2018.06.039. [DOI] [PubMed] [Google Scholar]
- 9.Kanda T, Wakino S, Homma K, Yoshioka K, Tatematsu S, et al. Rho-kinase as a molecular target for insulin resistance and hypertension. FASEB J. 2006;20:169–171. doi: 10.1096/fj.05-4197fje. [DOI] [PubMed] [Google Scholar]
- 10.Islam MZ, Van Dao C, Miyamoto A, Shiraishi M. Rho-kinase and the nitric oxide pathway modulate basilar arterial reactivity to acetylcholine and angiotensin II in streptozotocin-induced diabetic mice. Naunyn Schmiedebergs Arch Pharmacol. 2017;390:929–938. doi: 10.1007/s00210-017-1396-x. [DOI] [PubMed] [Google Scholar]
- 11.Lai D, Gao J, Bi X, He H, Shi X, Weng S, et al. The Rho kinase inhibitor, fasudil, ameliorates diabetes-induced cardiac dysfunction by improving calcium clearance and actin remodeling. J Mol Med (Berl). 2017;95:155–165. doi: 10.1007/s00109-016-1469-1. [DOI] [PubMed] [Google Scholar]
- 12.Liu L, Tan L, Lai J, Li S, Wang DW. Enhanced Rho-kinase activity: pathophysiological relevance in type 2 diabetes. Clin Chim Acta. 2016;462:107–110. doi: 10.1016/j.cca.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 13.Guo R, Su Y, Yan J, Sun H, Wu J, et al. Fasudil improves short-term echocardiographic parameters of diastolic function in patients with type 2 diabetes with preserved left ventricular ejection fraction: a pilot study. Heart Vessels. 2015;30:89–97. doi: 10.1007/s00380-013-0458-3. [DOI] [PubMed] [Google Scholar]
- 14.Gabrielli L, Winter JL, Godoy I, McNab P, Padilla I, et al. Increased Rho-kinase activity in hypertensive patients with left ventricular hypertrophy. Am J Hypertens. 2014;27:838–845. doi: 10.1093/ajh/hpt234. [DOI] [PubMed] [Google Scholar]
- 15.Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, et al. Recommendations for chamber quantification: a report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr. 2005;18:1440–1463. doi: 10.1016/j.echo.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 16.Parasuraman S, Walker S, Loudon BL, Gollop ND, Wilson AM, et al. Assessment of pulmonary artery pressure by echocardiography—a comprehensive review. Int J Cardiol Heart Vasc. 2016;12:45–51. doi: 10.1016/j.ijcha.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pérez O, Castro P, Díaz-Araya G, Nettle D, Moraga F, et al. Persistence of oxidative stress after heart transplantation: a comparative study of patients with heart transplant versus chronic stable heart failure. Rev Esp Cardiol. 2002;55:831–837. doi: 10.1016/S0300-8932(02)76712-2. [DOI] [PubMed] [Google Scholar]
- 18.Ocaranza MP, Gabrielli L, Mora I, Garcia L, McNab P, et al. Markedly increased Rho-kinase activity on circulating leukocytes in patients with chronic heart failure. Am Heart J. 2011;161:931–937. doi: 10.1016/j.ahj.2011.01.024. [DOI] [PubMed] [Google Scholar]
- 19.Ravarotto V, Pagnin E, Maiolino G, Fragasso A, Carraro G, et al. The blocking of angiotensin II type 1 receptor and RhoA/Rho kinase activity in hypertensive patients: effect of olmesartan medoxomil and implication with cardiovascular-renal remodeling. J Renin Angiotensin Aldosterone Syst. 2015;16:1245–1250. doi: 10.1177/1470320315594324. [DOI] [PubMed] [Google Scholar]
- 20.Cantin C, Jalil JE, Bulnes JF, Novoa U, MacNab P, et al. Effect of early normotension with olmesartan on rho-kinase activity in hypertensive patients. Curr Vasc Pharmacol. 2020;18:87–91. doi: 10.2174/1570161117666190121103116. [DOI] [PubMed] [Google Scholar]
- 21.Hata T, Soga J, Hidaka T, Idei N, Fujii Y, et al. Calcium channel blocker and Rho-associated kinase activity in patients with hypertension. J Hypertens. 2011;29:373–379. doi: 10.1097/HJH.0b013e328340902d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fujimura N, Noma K, Hata T, Soga J, Hidaka T, et al. Mineralocorticoid receptor blocker eplerenone improves endothelial function and inhibits Rho-associated kinase activity in patients with hypertension. Clin Pharmacol Ther. 2012;91:289–297. doi: 10.1038/clpt.2011.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fierro C, Novoa U, González V, Ocaranza MP, Jalil JE. Simultaneous Rho kinase inhibition in circulating leukocytes and in cardiovascular tissue in rats with high angiotensin converting enzyme levels. Int J Cardiol. 2016;215:309–317. doi: 10.1016/j.ijcard.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 24.Ocaranza MP, Moya J, Jalil JE, Lavandero S, Kalergis AM, et al. Rho-kinase pathway activation and apoptosis in circulating leucocytes in patients with heart failure with reduced ejection fraction. J Cell Mol Med. 2019;24:1413–1427. doi: 10.1111/jcmm.14819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe H, Iino K, Ito H. Rho-kinase in leukocytes. An emerging biomarker for heart failure. Circ J. 2013;77:2471–2472. doi: 10.1253/circj.CJ-13-0935. [DOI] [PubMed] [Google Scholar]
- 26.Ocaranza MP, Fierro C, Jalil JE, Moya J, Gonzalez L, et al. Rho kinase activation in circulating leukocytes is related to hypertensive myocardial remodeling. Clin Sci (Lond). 2018;132:1837–1853. doi: 10.1042/CS20180312. [DOI] [PubMed] [Google Scholar]
- 27.Shi J, Surma M, Yang Y, Wei L. Disruption of both ROCK1 and ROCK2 genes in cardiomyocytes promotes autophagy and reduces cardiac fibrosis during aging. FASEB J. 2019;33:7348–7362. doi: 10.1096/fj.201802510R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin G, Craig GP, Zhang L, Yuen VG, Allard M, McNeill JH, MacLeod KM. Acute inhibition of Rho-kinase improves cardiac contractile function in streptozotocin-diabetic rats. Cardiovasc Res. 2007;75:51–58. doi: 10.1016/j.cardiores.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 29.Soliman H, Nyamandi V, Garcia-Patino M, Zhang PC, Lin E, Jia ZP, Tibbits GF, Hove-Madsen L, MacLeod KM. ROCK2 promotes ryanodine receptor phosphorylation and arrhythmic calcium release in diabetic cardiomyocytes. Int J Cardiol. 2019;281:90–98. doi: 10.1016/j.ijcard.2019.01.075. [DOI] [PubMed] [Google Scholar]
- 30.Tong S, Zhang L, Joseph J, Jiang X. A novel, potential therapeutic target in diabetic cardiomyocytes: ROCK2. Int J Cardiol. 2019;288:121. doi: 10.1016/j.ijcard.2019.03.045. [DOI] [PubMed] [Google Scholar]
- 31.Zhang BL, Yang DG. ROCK2: a novel, potential therapeutic target in diabetic cardiomyocytes? Int J Cardiol. 2020;298:119. doi: 10.1016/j.ijcard.2019.09.025. [DOI] [PubMed] [Google Scholar]
- 32.Nandipati KC, Subramanian S, Agrawal DK. Protein kinases: mechanisms and downstream targets in inflammation-mediated obesity and insulin resistance. Mol Cell Biochem. 2017;426(1–2):27–45. doi: 10.1007/s11010-016-2878-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGee SL, Hargreaves M. Exercise and skeletal muscle glucose transporter 4 expression: molecular mechanisms. Clin Exp Pharmacol Physiol. 2006;33:395–399. doi: 10.1111/j.1440-1681.2006.04362.x. [DOI] [PubMed] [Google Scholar]
- 34.Geiger PC, Wright DC, Han D-H, Holloszy JO. Activation of p38 MAP kinase enhances sensitivity of muscle glucose transport to insulin. Am J Physiol Endocrinol Metab. 2005;288:E782–E788. doi: 10.1152/ajpendo.00477.2004. [DOI] [PubMed] [Google Scholar]
- 35.Auger-Messier M, Accornero F, Goonasekera SA, Bueno OF, Lorenz JN, et al. Unrestrained p38 MAPK activation in Dusp1/4 double-null mice induces cardiomyopathy. Circ Res. 2013;112:48–56. doi: 10.1161/CIRCRESAHA.112.272963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ho TJ, Huang CC, Huang CY, Lin WT. Fasudil, a Rho-kinase inhibitor, protects against excessive endurance exercise training-induced cardiac hypertrophy, apoptosis and fibrosis in rats. Eur J Appl Physiol. 2012;112:2943–2955. doi: 10.1007/s00421-011-2270-z. [DOI] [PubMed] [Google Scholar]
- 37.Amiri F, Shaw S, Wang X, Tang J, Waller JL, et al. Angiotensin II activation of the JAK/STAT pathway in mesangial cells is altered by high glucose. Kidney Int. 2002;61:1605–1616. doi: 10.1046/j.1523-1755.2002.00311.x. [DOI] [PubMed] [Google Scholar]
- 38.Amiri F, Venema VJ, Wang X, Ju H, Venema RC, Marrero MB. Hyperglycemia enhances angiotensin II-induced janus-activated kinase/STAT signaling in vascular smooth muscle cells. J Biol Chem. 1999;274:32382–32386. doi: 10.1074/jbc.274.45.32382. [DOI] [PubMed] [Google Scholar]
- 39.Shaw S, Wang X, Redd H, Alexander GD, Isales CM, Marrero MB. High glucose augments the angiotensin II-induced activation of JAK2 in vascular smooth muscle cells via the polyol pathway. J Biol Chem. 2003;278:30634–30641. doi: 10.1074/jbc.M305008200. [DOI] [PubMed] [Google Scholar]
- 40.Banes-Berceli AK, Shaw S, Ma G, Brands M, Eaton DC, Stern DM, et al. Effect of simvastatin on high glucose- and angiotensin II-induced activation of the JAK/STAT pathway in mesangial cells. Am J Physiol Renal Physiol. 2006;291:F116–F121. doi: 10.1152/ajprenal.00502.2005. [DOI] [PubMed] [Google Scholar]
- 41.Tuttle KR, Brosius FC, 3rd, Adler SG, Kretzler M, Mehta RL, et al. JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: results from a Phase 2 randomized controlled clinical trial. Nephrol Dial Transplant. 2018;33:1950–1959. doi: 10.1093/ndt/gfx377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brosius FC, Tuttle KR, Kretzler M. JAK inhibition in the treatment of diabetic kidney disease. Diabetologia. 2016;59:1624–1627. doi: 10.1007/s00125-016-4021-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karam BS, Chavez-Moreno A, Koh W, Akar JG, Akar FG. Oxidative stress and inflammation as central mediators of atrial fibrillation in obesity and diabetes. Cardiovasc Diabetol. 2017;16(1):120. doi: 10.1186/s12933-017-0604-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Monserrat-Mesquida M, Quetglas-Llabrés M, Capó X, Bouzas C, Mateos D, Pons A, Tur JA, Sureda A. metabolic syndrome is associated with oxidative stress and proinflammatory state. Antioxidants (Basel). 2020;9(3):E236. doi: 10.3390/antiox9030236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen SJ, Yen CH, Huang YC, Lee BJ, Hsia S, Lin PT. Relationships between inflammation, adiponectin, and oxidative stress in metabolic syndrome. PLoS ONE. 2012;7:8–12. doi: 10.1371/journal.pone.0045693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X, Bao W, Liu J, Ouyang YY, Wang D, et al. Inflammatory markers and risk of type 2 diabetes: a systematic review and meta-analysis. Diabetes Care. 2013;36:166–175. doi: 10.2337/dc12-0702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Randeria SN, Thomson GJA, Nell TA, Roberts T, Pretorius E. Inflammatory cytokines in type 2 diabetes mellitus as facilitators of hypercoagulation and abnormal clot formation. Cardiovasc Diabetol. 2019;18(1):72. doi: 10.1186/s12933-019-0870-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kozakova M, Morizzo C, Goncalves I, Natali A, Nilsson J, Palombo C. Cardiovascular organ damage in type 2 diabetes mellitus: the role of lipids and inflammation. Cardiovasc Diabetol. 2019;18(1):61. doi: 10.1186/s12933-019-0865-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rao J, Ye Z, Tang H, Wang C, Peng H, et al. The RhoA/ROCK Pathway ameliorates adhesion and inflammatory infiltration induced by AGEs in glomerular endothelial cells. Sci Rep. 2017;7:39727. doi: 10.1038/srep39727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mohammad G, AlSharif HM, Siddiquei MM, Ahmad A, Alam K, Abu El-Asrar AM. Rho-associated protein kinase-1 mediates the regulation of inflammatory markers in diabetic retina and in retinal müller cells. Ann Clin Lab Sci. 2018;48:137–145. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.