

Abstract

DNA gyrase and topoisomerase IV are well-validated pharmacological targets, and quinolone antibacterial drugs are marketed as their representative inhibitors. However, in recent years, resistance to these existing drugs has become a problem, and new chemical classes of antibiotics that can combat resistant strains of bacteria are strongly needed. In this study, we applied our hit-to-lead (H2L) chemistry for the identification of a new chemical class of GyrB/ParE inhibitors by efficient use of thermodynamic parameters. Investigation of the core fragments obtained by fragmentation of high-throughput screening hit compounds and subsequent expansion of the hit fragment was performed using isothermal titration calorimetry (ITC). The 8-(methylamino)-2-oxo-1,2-dihydroquinoline derivative 13e showed potent activity against Escherichia coli DNA gyrase with an IC50 value of 0.0017 μM. In this study, we demonstrated the use of ITC for primary fragment screening, followed by structural optimization to obtain lead compounds, which advanced into further optimization for creating novel antibacterial agents.
Introduction
Recently, much research has been devoted to the development of novel antimicrobial agents against Gram-positive and Gram-negative bacteria that are resistant to the major antibiotics available at present.1−4 Among them, especially DNA gyrase and topoisomerase IV, which are the two types of type II topoisomerases present in bacteria, have attracted attention. These enzymes are involved in DNA replication, repair, and decatenation.5−7 DNA gyrase occurs as a heterodimer consisting of two subunits called GyrA and GyrB. GyrA is involved in DNA cleavage and recombination, whereas GyrB has ATPase activity, which provides the energy necessary for DNA cleavage and recombination.8 On the other hand, topoisomerase IV, which also has two subunits called ParC and ParE, is involved in decatenation of DNA and relaxation of supercoiled DNA.8,9 The fluoroquinolone antibacterial agents, such as ciprofloxacin, currently available in the market are DNA gyrase and topoisomerase IV inhibitors, and they exert their actions by interfering with DNA replication via stabilizing the cleavable complex formed by the enzyme, quinolone, and DNA.10 However, drug resistance to the fluoroquinolone antibacterial agents has become a critical clinical problem.11,12 In contrast, aminocoumarin antibiotics, such as novobiocin,13−15 are known to act through inhibiting GyrB/ParE, unlike the fluoroquinolone antibacterial agents. Regretfully, novobiocin could not be successfully launched in the market because of safety and tolerance problems (Figure 1).9,16
Figure 1.

Structures of ciprofloxacin and novobiocin.
Many research groups have been focusing their effort on the identification of potent GyrB/ParE inhibitors as novel antibacterial agents, in order to potentially overcome the drug resistance problem described above.17−19 Research and development on GyrB/ParE inhibitors has been performed through various drug discovery approaches, such as not only the deployment of natural products such as novobiocin,13−15 clorobiocin,20 cyclothialidine,21 and RU7911522 but also by implementation of hit-to-lead (H2L) optimization from high-throughput screening (HTS), for example, SPR719 (formerly VXc-486)23 and fragment-based screening, for example, AZD509924,25 and GP-4.26 However, none of these inhibitors have been launched in the market yet (Figure 2).9,16
Figure 2.

Some reported examples of GyrB/ParE inhibitors.
In this paper, we describe the synthesis and biological assay results of 2-oxo-1,2-dihydroquinoline-3-carboxamide derivatives for the identification of novel GyrB/ParE inhibitors, which eventually afforded dominant leads. We first performed enzyme-based HTS27 (full-length Escherichia coli DNA gyrase) of our compound library and found several micromolar potency HTS hit compounds that exhibited DNA gyrase- and topoisomerase IV-inhibitory activity. Then, by utilizing these hit compounds, we performed a distinctive H2L drug discovery, in which H2L was effectively implemented in combination with fragment-based drug discovery (FBDD) and structure-based drug discovery (SBDD). More precisely, the X-ray cocrystal structure of the HTS hit compound 1 in E. coli truncated GyrB (residues 1–220) was analyzed, and subsequently, the FBDD approach was applied to the core fragment 2a, which was obtained by fragmentation28,29 of the HTS hit structure 1 (Figure 3).
Figure 3.

Fragmentation of HTS hit 1.
In the FBDD approach, we focused on determination of the thermodynamic parameters by isothermal titration calorimetry (ITC) to identify 8-(methylamino)-quinolin-2(1H)-one 2d as a potent fragment. Among biophysical assays, ITC has attracted a lot of attention30−34 because it provides information regarding the thermodynamics and stoichiometry between a target protein and a ligand. ITC allows each thermodynamic parameter to be determined, including the dissociation constant (KD), enthalpy change (ΔH), and entropy change (−TΔS), directly by a single measurement. According to these thermodynamic parameters, the intermolecular interactions can be divided into the enthalpy-driven type (strong ΔH contribution) or entropy-driven type (strong −TΔS contribution). A ligand with strong ΔH contribution indicates that noncovalent interactions, such as hydrogen bonds, are efficiently formed at the protein binding site.35 Ideally, enthalpy-driven intermolecular interactions that are specific for a target molecule are desired for drug design.36,37 After identifying hit fragment 2d which showed desirable thermodynamic profiles, we performed SBDD based on X-ray cocrystal information to acquire highly active compounds. The SAR studies were guided by obtaining X-ray cocrystals of several expanded fragments and comparing their binding modes. Compound 13e interacted with the target protein GyrB in an enthalpy-driven manner and in addition showed antibacterial activity and high kinase selectivity. Herein, we report this rational H2L approach and creation of GyrB/ParE lead compounds based on the 8-(methylamino)-quinolin-2(1H)-one scaffold.
Results and Discussion
Chemistry
Fragments having a molecular weight of 250 or less were purchased or synthesized as indicated below. The representative core fragment 2d was prepared from 1,3-dichloro-2-nitrobenzene 3 as a starting material. Methylamine was introduced into 3 under microwave irradiation, followed by Heck reaction using Pd(tert-Bu3P)2 as a catalyst to yield 5. After the nitro group of 5 was reduced, the resulting amine 6 was cyclized by treating with NaOMe to yield 2d (Scheme 1).
Scheme 1. Synthesis of Core Fragment 2d.

Reagents and conditions: (a) MeNH2 in MeOH, DBU, 125 °C, microwave, 32%; (b) ethyl acrylate, Pd(tert-Bu3P)2, DIPEA, 160 °C, microwave, 64%; (c) Fe, NH4Cl, EtOH, H2O, 75 °C; (d) NaOMe, MeOH, 85 °C, 11% over two steps. See the Supporting Information for other synthetic fragments.
For the preparation of compounds 8, 8a, and 8c, DMT-MM or 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) was used as a coupling reagent for the amidation of 2e with the corresponding amines (7, 7a, and 7c). In contrast, compounds 8e and 8i were synthesized using anilines (7e and 7i), which were coupled with acid chlorides of 2e treated with thionyl chloride (Scheme 2).
Scheme 2. Synthesis of 8-Nonsubstituted 2-Quinolinone Derivatives 8 and 8a–i,

Reagents and conditions: (a) 7 or 7a, DMT-MM, DIPEA, EtOH, rt, 21% (compound 8); (b) 7c, HATU, DIPEA, DMF, rt; (c) (i) SOCl2, reflux, (ii) 7e or 7i, THF, 0 °C; (d) 1 N NaOH, THF, rt, 20–38% over two steps.
N-cyclohexyl-N-methylaminomethyl
Compounds 13 and 13a–h were also obtained in the same manner as described in Scheme 2 using carboxylic acid 2f which was synthesized by cyclization of aldehyde 11 with diethyl malonate, followed by hydrolysis under basic conditions (Scheme 3).
Scheme 3. Synthesis of 8-Methylamino-2-Quinolinone Derivatives 13 and 13a–h.

Reagents and conditions: (a) tributyl(vinyl)tin, Pd(tert-Bu3P)2, CsF, toluene, 140 °C, microwave, 91%; (b) OsO4, NaIO4, 1,4-dioxane, H2O, rt, 70%; (c) Fe, NH4Cl, EtOH, H2O, 75 °C, 46%; (d) diethyl malonate, piperidine, EtOH, 75 °C; (e) 1 N NaOH, THF, rt, 63% over two steps; (f) 7 or 7a–h, HATU, DIPEA, DMF, rt, or 60 °C; (g) 1 N NaOH, THF, rt, 6–58% over two steps.
Identification of HTS Hit Compounds
Initially, HTS using the E. coli DNA gyrase enzyme was performed on our universal compound library combining commercially available and in-house proprietary compounds. As a result, several tens of HTS hit compounds with an IC50 value of less than 20 μM were identified. For these HTS hits, various biophysical assays,36,38 including X-ray cocrystal structure analysis, ITC, thermal shift assay (TSA), and surface plasmon resonance (SPR), were utilized to identify compounds for which H2L could be performed more efficiently. As a result, compound 1, which was detected by TSA, SPR (data not shown), and ITC (Figure S1 in the Supporting Information), and for which we successfully obtained the X-ray cocrystal structure in E. coli truncated GyrB (residues 1–220), was selected as a reliable hit (Figure 4).
Figure 4.

X-ray structure analysis of HTS hit 1 in the ATPase domain of E. coli GyrB. (a) X-ray crystal structure of 1 in E. coli GyrB (residues 1–220, PDB code: 6KZV). Residues involved in key interactions (Asp73, Glu50, Arg76, and Arg136; E. coli GyrB numbering) are represented in a stick format. A conserved water molecule is displayed as a red ball. (b) Identical view on the two-dimensional (2D) plot showing the relevant interactions, indicated with dashed lines, and the distances are given in Angstrom units.
The X-ray cocrystal structure of compound 1 with E. coli GyrB truncated (residues 1–220) enzyme (Figure 4) indicated that the 2-quinolinone ring of 1 was located in the hinge region at the ATP binding site on the deep side of the pocket. Here, compound 1 formed a hydrogen bonding network between the Asp73 residue and a conserved water molecule. The phenyl ring extends from the 3-position of 2-quinolinone via an amide bond, and π-cation interaction with the salt bridge of the Glu50–Arg76 residue was found. Meanwhile, the N-cyclohexyl-N-methylaminomethyl moiety of 1 was revealed to be located in the hydrophobic floor (allowable space) and showed no conventional interaction with the target GyrB.
SBDD Approach Starting from HTS Hit
Because HTS hit 1 was selected as the origin of H2L, SBDD was performed using information on the superposition of HTS hit 1 and novobiocin (PDB code: 1AJ6) to obtain new lead compounds directly (Figure 5).
Figure 5.
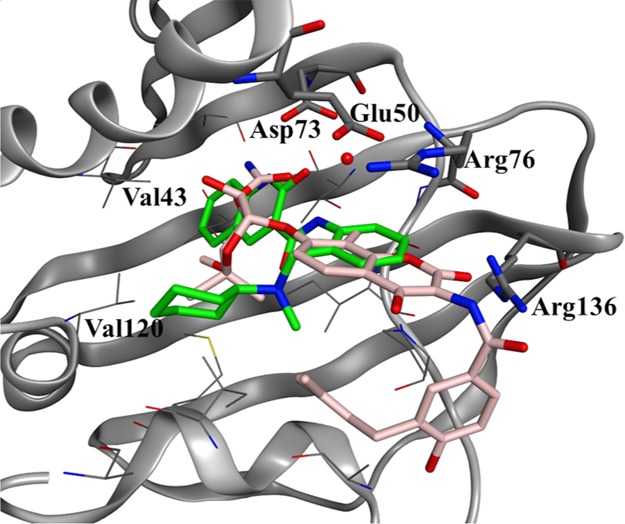
Overlap of novobiocin (light pink, PDB code: 1AJ6) and HTS hit 1 (green).
The X-ray cocrystal structure of novobiocin suggested the existence of a critical interaction of the 3-O-carbamoyl of the noviose sugar, the Asp73 residue, and the conserved water molecule (Figure 5).39 The carbamoyl moiety of novobiocin overlapped with the NHCO moiety of the 2-quinolinone part in compound 1. Furthermore, the hydrophobic pocket composed of residues such as Val43 and Val120 on the deep side was occupied by the phenyl portion of compound 1; therefore, it was expected that compound 1 would exhibit a stronger hydrophobic interaction than novobiocin. The coumarin moiety of novobiocin and the phenyl ring of compound 1 overlapped, both of which formed a π-stacking interaction with the salt bridge site of the Glu50–Arg76 residues. A hydrogen bond between the coumarin COO moiety of novobiocin and the Arg136 residue was observed at the entry of the ATP binding pocket, but no hydrogen bond was found at this moiety in compound 1 because of the absence of a suitable acceptor that interacts with Arg136. Therefore, it was inferred that a stronger interaction would be obtained by introducing an appropriate H-bond acceptor that interacted with the Arg136 residue (Figure 5).
We selected a carboxy group as the part corresponding to the COO site of novobiocin and tried to improve the activity by introducing a new proton acceptor into HTS hit 1 (Table 1). In regard to the position of introduction, we expected the same binding mode as novobiocin and prioritized the introduction to the meta position of the phenyl ring for HTS hit 1. As described above, because the N-cyclohexyl-N-methylaminomethyl site of HTS hit 1 does not directly interact with the amino acid residue of GyrB, a carboxy group was introduced to the derivatives from which the part had been removed. However, compounds 8i and 8a, in which the carboxy group had been introduced at the meta position, showed greatly decreased inhibitory activity. Besides, 8c was obtained by extending the alkyl chain of the linker, and 8e, with a carboxy group at the para position, exerted inhibitory activity equal to or less than that of HTS hit 1. This showed that the activity could not be improved by the introduction of the carboxy group in HTS hit 1 and derivatives without an N-cyclohexyl-N-methylaminomethyl moiety (Table 1).
Table 1. E. coli DNA Gyrase IC50 and Binding Thermodynamic Parameters of 2-Quinolinone Derivatives to GyrB.
![]()

Values are reported from the means of at least two independent experiments.
Reverse titration method was conducted, and value was the result of one experiment.
Investigation of the Core Fragment
Because previous attempts at improving the activity by introduction of a carboxy group into HTS hit 1 or its derivatives have failed, we redefined the strategy and tried to carry out FBDD based on the core fragment corresponding to the hinge region of the ATPase active site. In order to execute this approach reliably, we first focused on the fragments obtained from the fragmentation of HTS hit 1. As a biophysical assay, we envisioned that ITC screening would be advantageous as a fragment screening method to evaluate the formation of conventional hydrogen bonds, although its lower screening throughput was a demerit.28,38,40,41 Therefore, we assumed that the problem could be overcome by preparing a limited fragment set instead of the general fragment library often used in the FBDD method.42−44
First of all, HTS hit 1 was fragmented to obtain 2a corresponding to the hinge region. In the next step, in order to search for a core fragment more promising than the 2-quinolinone skeleton like 2a, approximately 160 fragments were selected that are structurally similar to core fragment 2a, including those shown in Figure 6 and Table 2 (see the Supporting Information for screened fragments). Among them, ITC screening was carried out for about 120 fragments that cleared the solubility test [dimethyl sulfoxide (DMSO) (40–200 mM) and assay buffer]. Unfortunately, the thermodynamic signals of these nonsubstituted fragments such as those shown in Figure 6 were very poor or not detected at all, suggesting that none of these fragments showed apparent enthalpy effects.
Figure 6.

Examples of evaluated fragments with quinolinone-like scaffold.
Table 2. ITC Evaluation of Representative Fragments with a Proton Donor at the C8 Position.

Not detected.
Values are the means of three separate experiments.
Taking the above results into consideration, we reviewed the X-ray crystallography data of the GyrB protein–HTS hit 1 complex. It was found that a relatively small hydrophobic pocket existed in the direction of the C8 position of the 2-quinolinone in the interaction with the Asp73 residue in the hinge region. Accordingly, we considered that installing an appropriate proton donor and a hydrophobic moiety would be expected to bring an enhancement of the interaction and filling of the space around the C8 position of fragment 2a (Figure 7). Therefore, we prepared these derivatives and performed the corresponding ITC assays (Table 2).
Figure 7.

(a) Enlarged view of the hinge region (surface caving: polar: purple, hydrophobic: green, and exposed: red). (b) Image of the proton donor and hydrophobic part introduced at the C8 position.
Some fragments related to 2-quinolinone had unfavorable solubility in the ITC buffer. Therefore, solubility tests were performed for each fragment as described above for ITC measurement, and only adequately soluble compounds were evaluated by ITC.45 The synthesis of 2-quinolinones with a substituent at the C8 position is relatively complicated. Hence, a limited number of hydrophilic or hydrophobic substituents were introduced at the C8 position. As shown in Figure 8, some fragments that are similar in structure to 2d, such as 8-(ethylamino) quinolin-2(1H)-one 2q and 8-aminoquinolin-2(1H)-one 2u, were included, but these fragments exhibited no exothermic effects. Most of these fragments had no effects. However, quite interestingly, 8-(methylamino)-quinolin-2(1H)-one 2d exhibited a clear exothermic signal (ΔH = −6.14 kcal/mol). These results suggested that the N-methylamino group at the C8 position of 2-quinolinone was suitable for appropriately filling the small hydrophobic pocket and enhancing the interaction with the hinge region of the target protein (Figure 8).
Figure 8.

Isothermal titration data for 2a and 2d with GyrB (2 mM ligand and 20 μM GyrB were used).
Hit Fragment Expansion
Having obtained the enthalpy-driven fragment 2d with high ligand efficiency (0.48),46 we adopted a fragment expansion approach based on the X-ray cocrystal information of 1. As before, a carboxy group was selected as the proton acceptor, and the resulting expansion derivatives with carbon linkers between the 3-carboxamide and N-phenyl ring, or between the N-phenyl ring and the carboxy group, were prepared. Moreover, the best substitution pattern (meta- and para-substitution) of each of the two substituents on the phenyl ring were explored to optimize their thermodynamic effects and in vitro potency (E. coli DNA gyrase IC50 values). The results are shown in Table 3.
Table 3. E. coli DNA Gyrase IC50 and Binding Thermodynamic Parameters of 8-Methylamino-2-Quinolinone Derivatives to GyrB.
![]()

IC50 values are reported from the means of at least two independent experiments.
Values are the means of at least three separate experiments.
IC50 ratio (8-H/8-NHMe).
Compound 13, in which the core fragment containing the N-methylamino moiety was adapted to simple compound 8, showed improved enzyme–inhibitory activity. However, because of the low solubility of compound 13, the thermodynamic parameters could not be obtained by ITC. Therefore, a carboxy group was introduced into compound 13 to try to improve both the solubility with a polar group and the activity by acquiring a new interaction. Compound 13a with a carboxy group introduced at the meta position exhibited a large thermodynamic effect (ΔH = −12.9 kcal/mol and KD = 0.023 μM), with a more than 470-fold increase of the activity as compared to 8a. As compared with 8c without the N-methylamino moiety, 13c (ΔH = −13.2 kcal/mol and KD = 0.023 μM) was confirmed to exhibit large exothermic heat and a more than 4500-fold improvement in activity. These thermodynamic profiles revealed a definite increase in enthalpy by the introduction of the N-methyl moiety at the C8 position. The results also suggested that expansion from specific core fragments such as 2d with promising thermodynamic profiles was very important.
On the other hand, when the phenyl ring of compound 13 was converted to meta-benzoic acid as in 13a, no improvement in the activity was observed, as shown in Table 1. The same was also true for compound 13b and the phenylacetic acid-type compound 13d, in which each linker was extended by one carbon atom. The results suggested that substitution of a carboxy group in the meta position in the above derivatives did not contribute to any enhancement of the interactions. Next, para-substituted derivatives were evaluated. 13e exhibited the largest exothermic signal among the tested compounds (ΔH = −13.7 kcal/mol and KD = 0.0019 μM) and was more potent (IC50 = 0.0017 μM) than the meta-substituted 13a (IC50 = 0.21 μM) against the E. coli DNA gyrase. An improvement by over 5200-fold was observed in the enzyme–inhibitory activity of 13e as compared to that of 8e, which had a proton at the C8 position of 2-quinolinone. Compound 13e showed a superior thermodynamic profile and enzyme–inhibitory activity to novobiocin. Unfortunately, linker optimization studies following 13e (i.e., 13f, 13g, and 13h) resulted in a substantial decrease of the activity (Table 3). X-ray cocrystallography analysis of 13a and 13e suggested that the terminal carboxylic acid of 13e interacted with the Arg136 residue of the target GyrB, whereas no interaction was detected between the carboxyl group of 13a and the Arg136 residue of GyrB (Figure 9). This observation can be invoked to explain the difference between the exothermic effects of 13a and 13e. Moreover, a cocrystal structure with GyrB (residues 1–220) was also obtained for 13d. Comparison of its thermodynamic parameters with 13a showed that both compounds had almost the same profile. On the other hand, comparison of the obtained cocrystal structure confirmed that 13d showed a binding mode different from that of 13a. Introduction of the carboxy group did not result in interaction with Arg136 in either 13a or 13d, but 13d formed a new hydrogen bond with Arg76. From this unique interaction, it was confirmed that Arg76 was closer to the carboxy group of 13d than that of 13a and 13e. The results also suggested that the π-cation interaction was weakened by the collapse of the salt bridge between Glu50–Arg76. Consequently, 13d had the same enzyme–inhibitory activity as 13a and greatly inferior enzyme–inhibitory activity as compared to 13e.
Figure 9.

Comparison of the binding modes of compounds 13a, 13e, and 13d. (a) X-ray crystal structures of 13a, 13e, and 13d in E. coli GyrB (residues 1–220, PDB code: 6KZX, 6KZZ, and 6L01). Residues involved in key interactions (Asp73, Glu50, Arg76, and Arg136) are represented in stick format. The conserved water molecules are displayed as red balls. (b) Identical view on the 2D plot showing relevant interactions, indicated with dashed lines; the distances are given in Angstrom units.
Antimicrobial Activity and Kinase Profiles
Finally, compounds 13c, 13e, and 13f were selected as potential leads with equal or better inhibitory activity of E. coli gyrase enzyme as compared to novobiocin (Table 3), and their biological activities were evaluated. Compounds 13c and 13f showed no antibacterial activity against both Gram-negative bacteria E. coli ATCC 25922 and Gram-positive bacteria Staphylococcus aureus ATCC 29213. The antibacterial activities of compound 13e against E. coli and S. aureus were 64 and 16 μg/mL, respectively (Table 4). The inhibitory activity of 13e against E. coli gyrase enzyme was more than 10-fold stronger than that of novobiocin. However, the antibacterial activity against E. coli was the same, and the activity against S. aureus was weak. This difference could be due to efflux mechanisms. In the presence of the efflux pump inhibitor phenylalanine–arginine β-naphthylamide (PAβN), the antibacterial activity of 13e against E. coli was ≤0.03 μg/mL. The ratio of the antibacterial activity of 13e against E. coli with and without PAβN was more than 2000-fold. The antibacterial activities of compounds 13c and 13f in the presence of PAβN were significantly stronger than the antibacterial activities without PAβN. Furthermore, compound 13e was evaluated for its inhibitory activity against topoisomerase IV and was confirmed to exhibit a similar degree of enzyme–inhibitory activity (IC50 = 0.98 μM) to that of novobiocin (IC50 = 0.82 μM). In addition, compound 13e showed an 8.4% inhibition at 50 μM for the human hepatocellular carcinoma (HepG2) cell, and the cytotoxicity was acceptable (Table S1 in the Supporting Information).
Table 4. Biological Profiles of Compounds 13c, 13e, and 13f.

DNA gyrase belongs to the GHKL family of ATPases,49 and its inhibitors sometimes cause cross-inhibition of human kinases; therefore, compound 13e was profiled using 96 kinases. It was confirmed to show a low inhibition against many kinds of kinases and had sufficient kinase specificity. An inhibition rate of 25% or more at 10 μM concentration was observed against only four kinds of kinases (CK1δ, CAMK2, DYRK1A, and HGK) (Table S2 in the Supporting Information).
Conclusions
During the course of an antibacterial project, we performed enzyme-based HTS (full-length E. coli DNA gyrase) of our compound library, which led to the identification of several micromolar potent hit compounds as GyrB/ParE inhibitors. To identify novel lead compounds from these HTS hits, we implemented the focused fragment screening combining an ITC-based direct measurement of thermodynamic parameters with X-ray cocrystallography analysis. As a result, we identified the potent fragment 2d with an N-methylamino moiety at the C8 position, in which the conventional interaction was further strengthened by slight structural transformation of the core fragment 2a in HTS hit 1. Subsequently, efficient fragment expansion led to the development of the novel lead compound 13e for DNA gyrase inhibitors. Compound 13e showed an over 3000-fold more potent inhibitory activity (IC50 = 0.0017 μM) against E. coli DNA gyrase as compared to HTS hit 1, and antibacterial activity against both E. coli ATCC 25922 and S. aureus ATCC 29213. Thus, SAR exploration utilizing the enthalpy-driven core fragment 2d led to successful development of the lead compound 13e, which is thermodynamically attractive and advantageous for further development. Currently, optimization studies on the lead compound to identify preclinical candidates are ongoing, and the results will be reported in due course.
Experimental Section
Chemistry
All reagents and solvents were of commercial quality and were used without further purification. Progress of the reactions was usually monitored by TLC using Merck silica gel 60 F254 plates or Fuji Silysia chromatorex NH plates. Purifications using silica gel column chromatography were performed on a Biotage Isolera One instrument using Biotage SNAP Ultra Cartridges (particle size: 25 μm sphere), BÜCHI Reveleris Flash Cartridges (particle size: 40 μm), or Biotage SNAP Isolute NH2 Cartridges (particle size: 50 μm). 1H NMR spectra were recorded at 400 MHz using a BRUKER AVANCE III HD 400 or 600 MHz using a JEOL ECA600 with TMS as the internal standard, and proton chemical shifts were expressed in parts per million (ppm) in the indicated solvent. Multiplicity was defined as s (singlet), d (doublet), t (triplet), q (quartet), dd (double doublet), m (multiplet), or br s (broad singlet). 13C NMR spectra were recorded at 125 MHz using a JEOL ECA500 with TMS as the internal standard, and carbon chemical shifts were expressed in ppm in DMSO-d6 solvent. High-resolution mass spectrometry (HRMS) was recorded on a Shimadzu LCMS-IT-TOF mass spectrometer with electrospray ionization (ESI)/atmospheric pressure chemical ionization (APCI) dual source. Microwave irradiation experiments were performed using a Biotage Initiator+ 60EXP with standard Pyrex vessels (capacity 2–20 mL). ESI mass spectra were recorded on an Agilent 6130 or 6150 Quadrupole LC/MS connected to an Agilent 1290 Infinity HPLC instrument under the following conditions: column, Waters Acquity CSH C18 (1.7 μm, 2.1 × 50 mm); mobile phase A, H2O containing 0.1% formic acid; mobile phase B, CH3CN containing 0.1% formic acid; gradient, 20% B to 99% over 1.2 min, followed by 99% B over 0.2 min; flow rate, 0.8 mL/min. The wavelengths of detection were 210 and 254 nm. All tested compounds possessed a purity of at least 95% as determined by LCMS analysis.
8-(Methylamino)quinolin-2(1H)-one (2d)
A mixture of 1,3-dichloro-2-nitrobenzene 3 (6.00 g, 31.3 mmol), 40% methylamine MeOH solution (4.44 mL, 43.5 mmol), and DBU (6.60 mL, 44.2 mmol) was stirred at 125 °C for 10 min under microwave irradiation. The reaction mixture was diluted with water and extracted with EtOAc, and the extract was washed with water and concentrated. The residue was purified by column chromatography on silica gel and eluted with 5% EtOAc/n-hexane to obtain 3-chloro-N-methyl-2-nitroaniline 4 as a purple solid (1.87 g, 32%). LCMS (ESI) m/z: 187 [M + H]+. A mixture of compound 4 (396 mg, 2.12 mmol), ethyl acrylate (2.34 mL, 21.5 mmol), bis(tri-tert-butylphosphine)palladium(0) (216 mg, 0.42 mmol), and DIPEA (12.0 mL, 68.7 mmol) was stirred at 160 °C for 15 min under microwave irradiation. The mixture was evaporated after cooling. The residue was purified by column chromatography on silica gel and eluted with 2–15% EtOAc/n-hexane to obtain ethyl (2E)-3-[3-(methylamino)-2-nitrophenyl]prop-2-enoate 5 as an orange oil (342 mg, 64%). LCMS (ESI) m/z: 251 [M + H]+. Ammonium chloride (37 mg, 0.69 mmol) and iron powder (305 mg, 5.46 mmol) were added to a solution of compound 5 (0.247 g, 0.987 mmol) in EtOH (6.5 mL) and water (0.72 mL). The mixture was stirred at 75 °C for 5 h. After cooling, the mixture was filtered on Celite with EtOAc wash. The filtrate was quenched with saturated aqueous NaHCO3 and extracted with EtOAc. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel and eluted with 7% EtOAc/n-hexane to obtain the crude product, which was mixed with 28% sodium methoxide methanol solution (197 mg, 1.02 mmol) in MeOH (4 mL). The mixture was stirred at 85 °C for 3 h. After cooling, the solvent was evaporated, followed by addition of CHCl3 and saturated aqueous NH4Cl. The aqueous layer was separated and extracted with CHCl3 using a phase separator. The combined organic layer was concentrated. The residue was purified by column chromatography on silica gel and eluted with CHCl3 to obtain 2d as a colorless solid (9 mg, 5.2% over two steps). 1H NMR (600 MHz, CDCl3): δ 12.93–12.52 (m, 1H), 7.85 (d, J = 9.1 Hz, 1H), 7.20–7.12 (m, 1H), 6.95 (d, J = 7.0 Hz, 1H), 6.76 (d, J = 7.8 Hz, 1H), 6.70 (d, J = 9.5 Hz, 1H), 3.03 (s, 3H). LCMS (ESI) m/z: 175 [M + H]+. Rt 0.618 min. HRMS (ESI/APCI dual) m/z: calcd for C10H10N2O [M + H]+, 175.0866; found, 175.0852.
8-(Methylamino)-2-oxo-1,2-dihydroquinoline-3-carboxylic Acid (2f)
Diethyl malonate (434 μL, 2.87 mmol) and piperidine (263 μL, 2.66 mmol) were added to a solution of 2-amino-3-(methylamino)benzaldehyde 11 (308 mg, 2.05 mmol) in EtOH (4 mL). The reaction mixture was stirred at 75 °C for 8 h. After cooling to room temperature, the resulting precipitates were collected by filtration and dried to obtain ethyl 8-(methylamino)-2-oxo-1,2-dihydroquinoline-3-carboxylate as a crude product, which was used without further purification. LCMS (ESI) m/z: 247 [M + H]+. The product was taken up in tetrahydrofuran (THF) (12 mL). To this suspension, a 1 M aqueous solution of NaOH (12 mL) was added, and the reaction mixture was stirred at room temperature for 8.5 h. A 1 M hydrochloric acid solution was added, and the resulting precipitates were collected by filtration, washed with water, and dried to obtain 2f as a yellow solid (281 mg, 63% over two steps). 1H NMR (600 MHz, DMSO-d6): δ 14.83 (br s, 1H), 12.26 (s, 1H), 8.87 (s, 1H), 7.30–7.22 (m, 2H), 6.87 (d, J = 5.8 Hz, 1H), 6.15 (d, J = 4.5 Hz, 1H), 2.86 (d, J = 4.5 Hz, 3H). LCMS (ESI) m/z: 219 [M + H]+. Rt 0.693 min. HRMS (ESI/APCI dual) m/z: calcd for C11H10N2O3 [M + H]+, 219.0764; found, 219.0769.
2-Oxo-N-phenyl-1,2-dihydroquinoline-3-carboxamide (8)
Aniline 7 (0.049 μL, 0.537 mmol), DMT-MM (163 mg, 0.589 mmol), and DIPEA (140 μL, 0.802 mmol) were added to a solution of 2e (102 mg, 0.539 mmol) in EtOH (2 mL). After stirring at room temperature for 6 h, more DMT-MM (80 mg, 0.289 mmol) and aniline (0.020 μL, 0.219 mmol) were added. After stirring at room temperature for 17 h, the mixture was concentrated. The residue was purified by column chromatography on NH-silica gel and eluted with 20–50% EtOAc/n-hexane and then with 0–20% MeOH/CHCl3 to obtain 8 as a colorless solid (30 mg, 21%). 1H NMR (600 MHz, DMSO-d6): δ 12.63 (br s, 1H), 12.16 (s, 1H), 8.98 (s, 1H), 8.03–7.99 (m, 1H), 7.76–7.72 (m, 2H), 7.72–7.69 (m, 1H), 7.51–7.47 (m, 1H), 7.42–7.37 (m, 2H), 7.36–7.32 (m, 1H), 7.16–7.11 (m, 1H). 13C NMR (125 MHz, DMSO-d6): δ 162.2, 161.0, 144.6, 139.5, 138.2, 133.1, 129.9, 129.1, 124.0, 123.1, 121.3, 119.8, 118.8, 115.5. LCMS (ESI) m/z: 265 [M + H]+. Rt 0.922 min. HRMS (ESI/APCI dual) m/z: calcd for C16H12N2O2 [M + H]+, 265.0972; found, 265.0968.
3-[(2-Oxo-1,2-dihydroquinoline-3-carbonyl)amino]benzoic Acid (8a)
Methyl 3-aminobenzoate 7a (200 mg, 1.32 mmol), DMT-MM (401 mg, 1.45 mmol), and DIPEA (346 μL, 1.98 mmol) were added to a solution of 2e (250 mg, 1.32 mmol) in EtOH (5 mL), and the mixture was stirred at room temperature for 21 h. The generated solid was collected, washed with MeOH and water, and dried to obtain 171 mg of methyl 3-(2-oxo-1,2-dihydroquinoline-3-carboxamido)benzoate. LCMS (ESI) m/z: 323 [M + H]+. The product (90 mg, 0.28 mmol) was taken up in THF (2.8 mL). A 1 M aqueous solution of NaOH (2.8 mL, 2.8 mmol) was added to this solution, and the reaction mixture was stirred at room temperature for 4 h. Then, a 1 M hydrochloric acid solution was added, and the resulting precipitates were collected by filtration, washed with water and n-hexane, and dried to obtain 8a as a brown solid (67 mg, 33% over two steps). 1H NMR (600 MHz, DMSO-d6): δ 13.16 (br s, 1H), 12.69 (s, 1H), 12.30 (s, 1H), 9.02–8.96 (m, 1H), 8.37–8.31 (m, 1H), 8.04–8.00 (m, 1H), 7.96–7.91 (m, 1H), 7.74–7.67 (m, 2H), 7.56–7.47 (m, 2H), 7.38–7.31 (m, 1H). 13C NMR (125 MHz, DMSO-d6): δ 167.0, 162.2, 161.3, 144.7, 139.5, 138.4, 133.2, 131.6, 130.0, 129.4, 124.7, 123.9, 123.1, 121.2, 120.4, 118.8, 115.5. LCMS (ESI) m/z: 307 [M – H]−. Rt 0.798 min. HRMS (ESI/APCI dual) m/z: calcd for C17H12N2O4 [M + H]+, 309.0870; found, 309.0855.
3-{2-[(2-Oxo-1,2-dihydroquinoline-3-carbonyl)amino]ethyl}benzoic Acid (8c)
Methyl 3-(2-aminoethyl)benzoate hydrochloride 7c (199 mg, 0.92 mmol), HATU (350 mg, 0.92 mmol), and DIPEA (0.438 mL, 2.51 mmol) were added to a solution of 2e (158 mg, 0.84 mmol) in DMF (3 mL). The mixture was stirred at room temperature for 5 h, diluted with EtOAc, and washed three times each with saturated aqueous NaHCO3 and water. The separated organic layer was dried over MgSO4 and concentrated. The residue was purified by column chromatography on NH-silica gel eluting with CHCl3 to obtain 157 mg of methyl 3-{2-[(2-oxo-1,2-dihydroquinoline-3-carbonyl)amino]ethyl}benzoate. LCMS (ESI) m/z: 351 [M + H]+. A 1 M aqueous solution of NaOH (2.0 mL, 2.0 mmol) was added to the solution of methyl 3-{2-[(2-oxo-1,2-dihydroquinoline-3-carbonyl)amino]ethyl}benzoate (30 mg, 0.0856 mmol) in THF (2.0 mL), and the reaction mixture was stirred at room temperature for 5 h and concentrated. Then, a 1 M hydrochloric acid solution was added, and the resulting precipitates were collected by filtration and washed with water to obtain 8c as a colorless solid (11 mg, 21% over two steps). 1H NMR (600 MHz, DMSO-d6): δ 12.87 (br s, 1H), 12.44 (s, 1H), 9.84 (t, J = 5.8 Hz, 1H), 8.83 (s, 1H), 7.97–7.92 (m, 1H), 7.87–7.84 (m, 1H), 7.81–7.78 (m, 1H), 7.65 (ddd, J = 8.4, 7.1, 1.4 Hz, 1H), 7.56–7.51 (m, 1H), 7.43 (t, J = 7.6 Hz, 1H), 7.41 (d, J = 8.3 Hz, 1H), 7.32–7.26 (m, 1H), 3.65–3.58 (m, 2H), 2.93 (t, J = 7.2 Hz, 2H). 13C NMR (125 MHz, DMSO-d6): δ 167.3, 162.7, 161.9, 143.7, 139.8, 139.4, 133.2, 132.6, 130.9, 129.7, 129.6, 128.6, 127.2, 122.8, 121.5, 118.6, 115.2, 40.2, 34.9. LCMS (ESI) m/z: 335 [M – H]−. Rt 0.756 min. HRMS (ESI/APCI dual) m/z: calcd for C19H16N2O4 [M + H]+, 337.1183; found, 337.1173.
4-[(2-Oxo-1,2-dihydroquinoline-3-carbonyl)amino]benzoic Acid (8e)
A mixture of 2e (111 mg, 0.587 mmol) and thionyl chloride (8.0 mL, 0.11 mol) was stirred at reflux temperature for 2 h. The reaction mixture was cooled to room temperature and concentrated to obtain the acid chloride. Methyl 4-aminobenzoate 7e (106 mg, 0.701 mmol) was added to a solution of the acid chloride in THF (4 mL) at 0 °C, and the reaction mixture was stirred at 0 °C for 1 h. The resulting precipitates were collected by filtration, washed with MeOH, and dried to obtain 101 mg of methyl 4-[(2-oxo-1,2-dihydroquinoline-3-carbonyl)amino]benzoate. LCMS (ESI) m/z: 323 [M + H]+. The product was taken up in THF (3 mL). A 1 M aqueous solution of NaOH (3 mL) was added to this suspension, and the reaction mixture was stirred at room temperature for 5 h. Then, a 1 M hydrochloric acid solution was added to this suspension, and the resulting precipitates were collected by filtration, washed with water, and dried to obtain 8e as a colorless solid (36 mg, 20% over two steps). 1H NMR (600 MHz, DMSO-d6): δ 12.78 (br s, 1H), 12.71 (s, 1H), 12.43 (s, 1H), 9.00 (s, 1H), 8.03 (d, J = 7.4 Hz, 1H), 7.97 (d, J = 8.7 Hz, 2H), 7.85 (d, J = 8.7 Hz, 2H), 7.78–7.68 (m, 1H), 7.49 (d, J = 8.3 Hz, 1H), 7.42–7.32 (m, 1H). 13C NMR (125 MHz, DMSO-d6): δ 166.8, 162.2, 161.4, 144.9, 142.1, 139.6, 133.3, 130.6, 130.0, 125.8, 123.1, 121.0, 119.2, 118.8, 115.5. LCMS (ESI) m/z: 309 [M + H]+. Rt 0.802 min. HRMS (ESI/APCI dual) m/z: calcd for C17H12N2O4 [M + H]+, 309.0870; found, 309.0850.
4-{[Cyclohexyl(methyl)amino]methyl}-3-[(2-oxo-1,2-dihydroquinoline-3-carbonyl)amino]benzoic Acid (8i)
Compound 8i was prepared from methyl 3-amino-4-{[cyclohexyl(methyl)amino]methyl}benzoate 7i in a similar manner to that described for compound 8e. 1H NMR (600 MHz, CD3OD): δ 9.00 (s, 1H), 8.49–8.46 (m, 1H), 7.91–7.86 (m, 2H), 7.73–7.69 (m, 1H), 7.55–7.52 (m, 1H), 7.47–7.44 (m, 1H), 7.39–7.35 (m, 1H), 4.32–4.26 (m, 2H), 3.28–3.22 (m, 1H), 2.66 (br s, 3H), 2.12–2.05 (m, 2H), 1.93–1.86 (m, 2H), 1.72–1.66 (m, 1H), 1.60–1.51 (m, 2H), 1.43–1.33 (m, 3H). 13C NMR (125 MHz, DMSO-d6): δ 167.3, 161.7, 161.4, 144.7, 139.6, 137.2, 134.4, 133.0, 130.1, 129.9, 124.6, 123.0, 122.9, 122.1, 118.6, 115.3, 61.1, 55.0, 36.1, 27.4, 25.9, 25.6. LCMS (ESI) m/z: 434 [M + H]+. Rt 0.542 min. HRMS (ESI/APCI dual) m/z: calcd for C25H27N3O4 [M + H]+, 434.2074; found, 434.2050.
8-(Methylamino)-2-oxo-N-phenyl-1,2-dihydroquinoline-3-carboxamide (13)
Aniline 7 (0.016 mL, 0.175 mmol), HATU (68 mg, 0.179 mmol), and DIPEA (0.127 mL, 0.727 mmol) were added to a solution of 2f (35 mg, 0.160 mmol) in DMF (2 mL). The mixture was stirred at room temperature for 17 h, diluted with EtOAc, and washed with saturated aqueous NaHCO3 and water. The separated organic layer was dried over MgSO4 and concentrated. The residue was purified by column chromatography on silica gel and eluted with 0–6% MeOH/CHCl3 to obtain 13 as a yellow solid (12 mg, 26%). 1H NMR (600 MHz, DMSO-d6): δ 12.23 (br s, 1H), 11.75 (br s, 1H), 8.88 (s, 1H), 7.73 (d, J = 7.8 Hz, 2H), 7.39 (t, J = 7.8 Hz, 2H), 7.24–7.22 (m, 1H), 7.21–7.18 (m, 1H), 7.13 (t, J = 7.2 Hz, 1H), 6.81 (d, J = 7.4 Hz, 1H), 6.17 (d, J = 4.5 Hz, 1H), 2.86 (d, J = 4.5 Hz, 3H). 13C NMR (125 MHz, DMSO-d6): δ 162.2, 161.0, 145.7, 138.3, 136.2, 129.1, 127.2, 123.9, 123.9, 120.5, 119.8, 119.0, 116.8, 111.3, 29.9. LCMS (ESI) m/z: 294 [M + H]+. Rt 1.014 min. HRMS (ESI/APCI dual) m/z: calcd for C17H15N3O2 [M + H]+, 294.1237; found, 294.1221.
3-{[8-(Methylamino)-2-oxo-1,2-dihydroquinoline-3-carbonyl]amino}benzoic Acid (13a)
Methyl 3-aminobenzoate 7a (30 mg, 0.20 mmol), HATU (76 mg, 0.20 mmol), and DIPEA (0.096 mL, 0.55 mmol) were added to a solution of 2f (40 mg, 0.18 mmol) in DMF (3 mL). The mixture was stirred at room temperature for 21 h and concentrated. The residue was suspended with water and stirred for 5 min, and the resulting precipitates were collected to obtain methyl 3-{[8-(methylamino)-2-oxo-1,2-dihydroquinoline-3-carbonyl]amino}benzoate as a crude product, which was used without further purification. LCMS (ESI) m/z: 350 [M – H]−. The product was taken up in THF (1.8 mL). A 1 M aqueous solution of NaOH (1.8 mL) was added to this solution, and the reaction mixture was stirred at room temperature for 4 h. Then, a 1 M hydrochloric acid solution was added, and the resulting precipitates were collected by filtration, washed with water, and dried to obtain 13a as a yellow solid (36 mg, 58% over two steps). 1H NMR (600 MHz, DMSO-d6): δ 13.06 (br s, 1H), 12.30 (s, 1H), 11.77 (s, 1H), 8.90 (s, 1H), 8.33 (t, J = 1.9 Hz, 1H), 7.95 (dd, J = 7.8, 1.9 Hz, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.52 (t, J = 7.8 Hz, 1H), 7.29–7.24 (m, 1H), 7.24–7.19 (m, 1H), 6.83 (d, J = 7.8 Hz, 1H), 6.18 (d, J = 4.5 Hz, 1H), 2.87 (d, J = 4.5 Hz, 3H). 13C NMR (125 MHz, DMSO-d6): δ 167.0, 162.2, 161.3, 145.8, 138.5, 136.2, 131.6, 129.4, 127.2, 124.7, 123.9, 120.4, 120.4, 119.0, 116.9, 111.3, 29.9. LCMS (ESI) m/z: 338 [M + H]+. Rt 0.885 min. HRMS (ESI/APCI dual) m/z: calcd for C18H15N3O4 [M + H]+, 338.1135; found, 338.1116.
3-({[8-(Methylamino)-2-oxo-1,2-dihydroquinoline-3-carbonyl]amino}methyl)benzoic Acid (13b)
Compound 13b was prepared from methyl 3-(aminomethyl)benzoate hydrochloride 7b in a similar manner to that described for compound 13a. 1H NMR (600 MHz, DMSO-d6): δ 12.98 (br s, 1H), 11.56 (br s, 1H), 10.22 (t, J = 6.1 Hz, 1H), 8.79 (s, 1H), 7.93 (s, 1H), 7.84 (d, J = 7.8 Hz, 1H), 7.60 (d, J = 7.8 Hz, 1H), 7.47 (t, J = 7.8 Hz, 1H), 7.21–7.14 (m, 2H), 6.79 (dd, J = 7.2, 1.9 Hz, 1H), 6.12 (d, J = 4.5 Hz, 1H), 4.64 (d, J = 6.1 Hz, 2H), 2.84 (d, J = 4.5 Hz, 3H). 13C NMR (125 MHz, DMSO-d6): δ 167.2, 163.0, 161.9, 145.3, 139.9, 136.1, 131.9, 130.9, 128.7, 128.2, 127.9, 127.1, 123.6, 120.6, 118.8, 116.7, 111.0, 42.1, 29.9. LCMS (ESI) m/z: 352 [M + H]+. Rt 0.806 min. HRMS (ESI/APCI dual) m/z: calcd for C19H17N3O4 [M + H]+, 352.1292; found, 352.1291.
3-(2-{[8-(Methylamino)-2-oxo-1,2-dihydroquinoline-3-carbonyl]amino}ethyl)benzoic Acid (13c)
Compound 13c was prepared from methyl 3-(2-aminoethyl)benzoate hydrochloride 7c in a similar manner to that described for compound 13a. 1H NMR (600 MHz, DMSO-d6): δ 11.53 (br s, 1H), 9.83 (t, J = 5.6 Hz, 1H), 8.73 (s, 1H), 7.85 (s, 1H), 7.79 (d, J = 7.8 Hz, 1H), 7.51 (d, J = 7.8 Hz, 1H), 7.41 (t, J = 7.8 Hz, 1H), 7.20–7.13 (m, 2H), 6.77 (dd, J = 6.4, 2.7 Hz, 1H), 6.07 (d, J = 4.5 Hz, 1H), 3.64–3.58 (m, 2H), 2.92 (t, J = 7.2 Hz, 2H), 2.83 (d, J = 4.5 Hz, 3H). 13C NMR (125 MHz, DMSO-d6): δ 167.3, 162.7, 161.9, 144.9, 139.8, 136.1, 133.3, 130.9, 129.6, 128.6, 127.2, 127.0, 123.6, 120.7, 118.7, 116.7, 110.9, 40.2, 34.9, 29.9. LCMS (ESI) m/z: 366 [M + H]+. Rt 0.914 min. HRMS (ESI/APCI dual) m/z: calcd for C20H19N3O4 [M + H]+, 366.1448; found, 366.1449.
(3-{[8-(Methylamino)-2-oxo-1,2-dihydroquinoline-3-carbonyl]amino}phenyl)acetic Acid (13d)
Compound 13d was prepared from methyl (3-aminophenyl)acetate 7d in a similar manner to that described for compound 13a. 1H NMR (600 MHz, DMSO-d6): δ 12.35 (br s, 1H), 12.13 (s, 1H), 11.73 (s, 1H), 8.88 (s, 1H), 7.70 (dd, J = 7.4, 1.2 Hz, 1H), 7.58 (s, 1H), 7.33 (t, J = 7.8 Hz, 1H), 7.28–7.18 (m, 2H), 7.03 (d, J = 7.4 Hz, 1H), 6.82 (dd, J = 7.4, 1.2 Hz, 1H), 6.18 (d, J = 4.5 Hz, 1H), 3.60 (s, 2H), 2.86 (d, J = 4.5 Hz, 3H). 13C NMR (125 MHz, DMSO-d6): δ 172.5, 162.2, 161.0, 145.6, 138.2, 136.2, 135.9, 128.9, 127.2, 125.0, 123.9, 120.7, 120.5, 119.0, 118.1, 116.8, 111.3, 40.7, 29.9. LCMS (ESI) m/z: 352 [M + H]+. Rt 0.856 min. HRMS (ESI/APCI dual) m/z: calcd for C19H17N3O4 [M + H]+, 352.1292; found, 352.1289.
4-{[8-(Methylamino)-2-oxo-1,2-dihydroquinoline-3-carbonyl]amino}benzoic Acid (13e)
Compound 13e was prepared from methyl 4-aminobenzoate 7e in a similar manner to that described for compound 13a. 1H NMR (400 MHz, DMSO-d6): δ 12.55 (br s, 1 H), 12.06 (br s, 1 H), 8.84 (s, 1H), 7.91 (d, J = 8.3 Hz, 2H), 7.77 (d, J = 8.3 Hz, 2H), 7.23–7.12 (m, 2H), 6.76 (d, J = 7.0 Hz, 1H), 6.13 (d, J = 4.5 Hz, 1H), 2.82 (d, J = 4.5 Hz, 3H). 13C NMR (125 MHz, DMSO-d6): δ 167.0, 162.3, 161.5, 145.8, 141.9, 136.4, 130.6, 127.5, 126.7, 123.8, 120.2, 119.1, 119.0, 116.8, 111.2, 29.9. LCMS (ESI) m/z: 338 [M + H]+. Rt 0.890 min. HRMS (ESI/APCI dual) m/z: calcd for C18H15N3O4 [M + H]+, 338.1135; found, 338.1137.
4-({[8-(Methylamino)-2-oxo-1,2-dihydroquinoline-3-carbonyl]amino}methyl)benzoic Acid (13f)
Compound 13f was prepared from methyl 4-(aminomethyl)benzoate hydrochloride 7f in a similar manner to that described for compound 13a. 1H NMR (600 MHz, DMSO-d6): δ 11.56 (br s, 1H), 10.22 (t, J = 6.0 Hz, 1H), 8.78 (s, 1H), 7.92 (d, J = 8.3 Hz, 2H), 7.45 (d, J = 8.3 Hz, 2H), 7.21–7.15 (m, 2H), 6.79 (dd, J = 7.0, 2.1 Hz, 1H), 6.16–6.08 (m, 1H), 4.65 (d, J = 6.0 Hz, 2H), 2.84 (d, J = 4.5 Hz, 3H). 13C NMR (125 MHz, DMSO-d6): δ 167.1, 163.0, 162.0, 145.3, 144.4, 136.1, 129.5, 127.3, 127.1, 123.6, 120.6, 118.8, 116.7, 111.0, 42.2, 29.9. LCMS (ESI) m/z: 352 [M + H]+. Rt 0.784 min. HRMS (ESI/APCI dual) m/z: calcd for C19H17N3O4 [M + H]+, 352.1292; found, 352.1286.
4-(2-{[8-(Methylamino)-2-oxo-1,2-dihydroquinoline-3-carbonyl]amino}ethyl)benzoic Acid (13g)
Compound 13g was prepared from methyl 4-(2-aminoethyl)benzoate hydrochloride 7g in a similar manner to that described for compound 13a. 1H NMR (600 MHz, DMSO-d6): δ 12.83 (br s, 1H), 11.53 (s, 1H), 9.83 (t, J = 5.8 Hz, 1H), 8.74 (s, 1H), 7.88 (d, J = 8.3 Hz, 2H), 7.41 (d, J = 8.3 Hz, 2H), 7.20–7.12 (m, 2H), 6.77 (dd, J = 6.8, 1.9 Hz, 1H), 6.06 (d, J = 4.5 Hz, 1H), 3.67–3.57 (m, 2H), 2.94 (t, J = 7.0 Hz, 2H), 2.83 (d, J = 4.5 Hz, 3H). 13C NMR (125 MHz, DMSO-d6): δ 167.2, 162.7, 161.9, 144.9, 144.7, 136.1, 129.4, 128.9, 128.8, 127.0, 123.6, 120.7, 118.7, 116.7, 110.9, 35.1, 29.9. LCMS (ESI) m/z: 366 [M + H]+. Rt 0.814 min. HRMS (ESI/APCI dual) m/z: calcd for C20H19N3O4 [M + H]+, 366.1448; found, 366.1446.
(4-{[8-(Methylamino)-2-oxo-1,2-dihydroquinoline-3-carbonyl]amino}phenyl)acetic Acid (13h)
Compound 13h was prepared from ethyl (4-aminophenyl)acetate 7h in a similar manner to that described for compound 13a. 1H NMR (600 MHz, DMSO-d6): δ 12.12 (s, 1H), 11.72 (br s, 1H), 8.88 (s, 1H), 7.67 (d, J = 8.7 Hz, 2H), 7.27 (d, J = 8.7 Hz, 2H), 7.25–7.18 (m, 2H), 6.82 (d, J = 7.8 Hz, 1H), 6.19 (d, J = 4.5 Hz, 1H), 3.55 (s, 2H), 2.86 (d, J = 4.5 Hz, 3H). 13C NMR (125 MHz, DMSO-d6): δ 172.7, 162.2, 160.9, 145.6, 136.8, 136.2, 130.6, 130.0, 127.2, 123.9, 120.5, 119.7, 119.0, 116.8, 111.2, 40.1, 29.9. LCMS (ESI) m/z: 352 [M + H]+. Rt 0.853 min. HRMS (ESI/APCI dual) m/z: calcd for C19H17N3O4 [M + H]+, 352.1292; found, 352.1293.
Protein Production
The gene fragment encoding residues 2–220 of GyrB was amplified from the genomic DNA of E. coli by PCR using primers with NdeI and BamHI sites and subcloned into pET21a vectors. The protein expression in E. coli BL21(DE3) cells was induced with 0.4 mM isopropyl β-thiogalactopyranoside at OD600 = 0.5 for 3 h at 37 °C. The protein was purified using a sepharose-novobiocin column,50 dialyzed against 20 mM Tris-hydrochloric acid, pH 7.5, and concentrated to 10 mg/mL. The purity of the protein was determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis to be >95%.
Crystallization, Data Collection, and Processing
Complexes with inhibitors were crystallized by the sitting drop vapor diffusion method at 20 °C. Crystals were flash-frozen and measured at a temperature of 100 K. The diffraction data were collected in-house using a Rigaku R-AXIS VII image-plate detector and a MicroMax-007 generator operating with a copper target. The data were processed using the CrystalClear package. The structure was determined by molecular replacement with MolRep51 using the structure of the PDB code of 1AJ6 as the search model. The structure was refined with Refmac5.52 The data collection and refinement statistics are summarized in Table 5.
Table 5. X-ray Data Collection and Refinement Statistics.
| 1 | 13a | 13e | 13d | |
|---|---|---|---|---|
| Data Collection | ||||
| space group | P41 | P212121 | P212121 | P212121 |
| Cell Dimensions | ||||
| a (Å) | 53.44 | 37.87 | 41.30 | 41.58 |
| b (Å) | 53.44 | 68.57 | 67.08 | 67.07 |
| c (Å) | 67.88 | 68.80 | 68.29 | 68.25 |
| α (deg) | 90.00 | 90.00 | 90.00 | 90.00 |
| β (deg) | 90.00 | 90.00 | 90.00 | 90.00 |
| γ (deg) | 90.00 | 90.00 | 90.00 | 90.00 |
| resolution (Å) | 2.40 (2.49–2.40) | 2.10 (2.17–2.10) | 2.00 (2.07–2.00) | 2.60 (2.69–2.60) |
| Rmerge (%) | 8.1 (34.8) | 9.4 (33.5) | 11.2 (28.9) | 14.9 (32.5) |
| I/σI | 12.6 (4.2) | 9.7 (4.2) | 10.2 (4.5) | 7.8 (4.1) |
| completeness (%) | 99.3 (98.7) | 99.7 (100.0) | 98.7 (97.5) | 100.0 (100.0) |
| redundancy | 4.90 (4.94) | 4.77 (4.73) | 6.09 (6.18) | 5.72 (5.91) |
| Refinement | ||||
| resolution (Å) | 53.45–2.40 | 48.56–2.10 | 47.84–2.00 | 47.84–2.60 |
| no. of reflections (work/free) | 7120/343 | 10360/523 | 12495/644 | 5930/287 |
| Rwork/Rfree | 19.6/26.0 | 19.8/27.0 | 19.0/23.8 | 23.0/28.7 |
| rms deviation | ||||
| bond lengths (Å) | 0.012 | 0.015 | 0.017 | 0.012 |
| bond angles (deg) | 1.57 | 1.70 | 1.69 | 1.63 |
Isothermal Titration Calorimetry
All the ITC experiments were conducted on an Auto iTC200 (Malvern Instruments). The experiments were performed at 25 °C in ITC buffer (10 mM Tris, pH 7.5, 1–5% (v/v) DMSO). GyrB (1–220) at 3–75 μM was titrated with compounds at 0.03–2 mM. The titrations were carried out with 120–240 s intervals between the 1.5–2 μL injections. The first injection for each sample was excluded from the data fitting. Analysis of the data was performed with ORIGIN7 using a one-site binding model. The ITC experiments were run at least in duplicate, except for the reversible experiment and analyzed independently, and the average thermodynamic values were calculated.
Evaluation of the Inhibitory Activities on E. coli DNA Gyrase and Topoisomerase IV
The DNA supercoiling inhibition activity of purified E. coli DNA gyrase and the DNA—relaxation activity of E. coli topoisomerase IV were determined by reference to previous reports.53,54 The DNA gyrase reaction mixture contained 20 mM Tris-hydrochloric acid (pH 8.0), 35 mM CH3CO2NH4, 8 mM MgCl2, 1 mM dithiothreitol, 0.5 mM ATP, 4.6% glycerol, 0.005% Brij35, and 1 ng/mL relaxed pBR322. The topoisomerase IV reaction mixture contained 20 mM Tris-hydrochloric acid (pH 8.0), 35 mM CH3CO2NH4, 8 mM MgCl2, 1 mM dithiothreitol, 0.5 mM ATP, 4.6% glycerol, 0.005% Brij35, and 4 ng/mL of supercoiled pBR322. The reaction mixture was incubated at room temperature for 60 min for E. coli DNA gyrase and 30 min for E. coli topoisomerase IV. After incubation, freshly prepared H19 dye solution (ProFoldin, MA, USA) was added to each well, followed by incubation for 15 min, and the fluorescence was measured at Ex 485 nm/Em 535 nm. The IC50 was defined as the compound concentration that reduced the enzymatic activity observed in compound-free controls by 50%.
Evaluation of the Antibacterial Activity
The minimum inhibitory concentration was determined using the Clinical and Laboratory Standards Institute (CLSI) methodology, described in a CLSI document.55,56S. aureus ATCC 29213 and E. coli ATCC 25922 were obtained from the American Type Culture Collection.
Cytotoxicity Testing
Subconfluent HepG2 (ATCC HB-8065) cells were incubated with 8-(methylamino)-2-oxo-1,2-dihydroquinoline derivatives at 10 and 50 μM for 48 h at 37 °C. Cell proliferation was evaluated by using a Cell Counting Kit-8 (CCK-8) (Dojindo Molecular Technologies, Kumamoto, Japan) according to the manufacturer’s instructions, and percentage inhibitions of cells are calculated.
Kinase Selectivity Assay
Microfluidics-based technology was used in this assay for kinase profiling. The base components of the screening were a LabChip EZ Reader (PerkinElmer) and a biochemical assay using the ProfilerPro Kinase Selectivity Assay Kit (PerkinElmer). The assay was carried out in a final volume of 25 μL containing the fluorescence-labeled peptide substrate, the enzyme, ATP, and the test compound. This technology also uses the charge or shift in electrophoretic mobility of the labeled substrates upon enzymatic conversion to its product. As a result, this assay system eliminated the need for the use of radioactive reagents or other secondary reagents, such as antibodies.
Briefly, the recombinant enzyme was preincubated with or without the test compounds (final concentration 10 μM) at 28 °C for 15 min in 100 mM HEPES (pH 7.5) containing 10 mM MgCl2, 4% DMSO, 0.003% Brij 35, 0.004% Tween 20, and 1 mM dithiothreitol. Fluorescence-labeled peptide substrate (final concentration, 1.5 μM) and ATP (at the ATP Km apparent) were added, followed by incubation at 28 °C for 90 min. The kinase reaction was terminated by the addition of 3 mM ethylenediaminetetraacetic acid. The phosphorylated peptide was separated from the substrate peptide and quantified using the LabChip EZ Reader and then directly used to quantify the product conversion rate.
Acknowledgments
The authors thank Dr. Takashi Yoshizumi for his contributions to the project, Asami Fujii and Ayumi Kita for recording the isothermal titration calorimetry, Yunoshin Tamura and Dr. Junya Yamagishi for the in silico support, Reiko Wada for performing the kinase assay, Akira Higuchi for recording the high-resolution mass spectra, and Dr. Atsushi Okada and Dr. Yuta Oki for recording the nuclear magnetic resonance spectra.
Glossary
Abbreviations
- APCI
atmospheric pressure chemical ionization
- ATCC
American Type Culture Collection
- CAMK2
calcium/calmodulin-dependent protein kinase-II
- CK1δ
casein kinase 1δ
- CLSI
Clinical and Laboratory Standards Institute
- DIPEA
N,N-diisopropylethylamine
- DMT-MM
4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride
- DYRK1A
dual specificity tyrosine phosphorylation regulated kinase 1A
- GyrA
A-subunit of DNA gyrase
- GyrB
B-subunit of DNA gyrase
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- HepG2
human hepatocellular carcinoma
- HGK
hematopoietic progenitor kinase/germinal center kinase-like kinase
- H2L
hit-to-lead
- ITC
isothermal titration calorimetry
- PAβN
phenylalanine–arginine β-naphthylamide
- ParC
C-subunit of topoisomerase IV
- ParE
E-subunit of topoisomerase IV
- SPR
surface plasmon resonance
- TSA
thermal shift assay
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c00865.
Accession Codes
PDB codes for structures of GyrB(1–220):compound complexes are the following: 6KZV (GyrB:1); 6KZX (GyrB:13a); 6L01 (GyrB:13d); and 6KZZ (GyrB:13e). Authors will release the atomic coordinates upon article publication.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Silver L. L. Challenges of Antibacterial Discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. 10.1128/cmr.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher H. W.; Talbot G. H.; Bradley J. S.; Edwards J. E.; Gilbert D.; Rice L. B.; Scheld M.; Spellberg B.; Bartlett J. Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- Fischbach M. A.; Walsh C. T. Antibiotics for Emerging Pathogens. Science 2009, 325, 1089–1093. 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwynn M. N.; Portnoy A.; Rittenhouse S. F.; Payne D. J. Challenges of antibacterial discovery revisited. Ann. N.Y. Acad. Sci. 2010, 1213, 5–19. 10.1111/j.1749-6632.2010.05828.x. [DOI] [PubMed] [Google Scholar]
- Champoux J. J. DNA Topoisomerases: Structure, Function, and Mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- Maxwell A.; Lawson D. The ATP-Binding Site of Type II Topoisomerases as a Target for Antibacterial Drugs. Curr. Top. Med. Chem. 2003, 3, 283–303. 10.2174/1568026033452500. [DOI] [PubMed] [Google Scholar]
- Collin F.; Karkare S.; Maxwell A. Exploiting bacterial DNA gyrase as a drug target: current state and perspectives. Appl. Microbiol. Biotechnol. 2011, 92, 479–497. 10.1007/s00253-011-3557-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champoux J. J. DNA TOPOISOMERASES: Structure, Function, and Mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- Mayer C.; Janin Y. L. Non-quinolone Inhibitors of Bacterial Type IIA Topoisomerases: A Feat of Bioisosterism. Chem. Rev. 2014, 114, 2313–2342. 10.1021/cr4003984. [DOI] [PubMed] [Google Scholar]
- Aldred K. J.; Kerns R. J.; Osheroff N. Mechanism of Quinolone Action and Resistance. Biochemistry 2014, 53, 1565–1574. 10.1021/bi5000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalhoff A. Resistance surveillance studies: a multifaceted problem-the fluoroquinolone example. Infection 2012, 40, 239–262. 10.1007/s15010-012-0257-2. [DOI] [PubMed] [Google Scholar]
- Blair J. M. A.; Webber M. A.; Baylay A. J.; Ogbolu D. O.; Piddock L. J. V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. 10.1038/nrmicro3380. [DOI] [PubMed] [Google Scholar]
- Fairbrother R. W.; Williams B. L. Two New Antibiotics. Lancet 1956, 268, 1177–1179. 10.1016/s0140-6736(56)90052-6. [DOI] [PubMed] [Google Scholar]
- Hinman J. W.; Caron E. L.; Hoeksema H. The Structure of Novobiocin1,2. J. Am. Chem. Soc. 1957, 79, 3789–3800. 10.1021/ja01571a047. [DOI] [Google Scholar]
- Alt S.; Mitchenall L. A.; Maxwell A.; Heide L. Inhibition of DNA gyrase and DNA topoisomerase IV of Staphylococcus aureus and Escherichia coli by aminocoumarin antibiotics. J. Antimicrob. Chemother. 2011, 66, 2061–2069. 10.1093/jac/dkr247. [DOI] [PubMed] [Google Scholar]
- Bisacchi G. S.; Manchester J. I. A New-Class Antibacterial-Almost. Lessons in Drug Discovery and Development: A Critical Analysis of More than 50 Years of Effort toward ATPase Inhibitors of DNA Gyrase and Topoisomerase IV. ACS Infect. Dis. 2015, 1, 4–41. 10.1021/id500013t. [DOI] [PubMed] [Google Scholar]
- Azam M. A.; Thathan J.; Jubie S. Dual targeting DNA gyrase B (GyrB) and topoisomerse IV (ParE) inhibitors: A review. Bioorg. Chem. 2015, 62, 41–63. 10.1016/j.bioorg.2015.07.004. [DOI] [PubMed] [Google Scholar]
- Oblak M.; Kotnik M.; Solmajer T. Discovery and Development of ATPase Inhibitors of DNA Gyrase as Antibacterial Agents. Curr. Med. Chem. 2007, 14, 2033–2047. 10.2174/092986707781368414. [DOI] [PubMed] [Google Scholar]
- Durcik M.; Tomašič T.; Zidar N.; Zega A.; Kikelj D.; Mašič L. P.; Ilaš J. ATP-competitive DNA gyrase and topoisomerase IV inhibitors as antibacterial agents. Expert Opin. Ther. Pat. 2019, 29, 171–180. 10.1080/13543776.2019.1575362. [DOI] [PubMed] [Google Scholar]
- Hooper D. C.; Wolfson J. S.; McHugh G. L.; Winters M. B.; Swartz M. N. Effects of Novobiocin, Coumermycin A1, Clorobiocin, and Their Analogs on Escherichia coli DNA Gyrase and Bacterial Growth. Antimicrob. Agents Chemother. 1982, 22, 662–671. 10.1128/aac.22.4.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetschi E.; Angehrn P.; Gmuender H.; Hebeisen P.; Link H.; Masciadri R.; Nielsen J. Cyclothialidine and its congeners: A new class of DNA gyrase inhibitors. Pharmacol. Ther. 1993, 60, 367–380. 10.1016/0163-7258(93)90017-8. [DOI] [PubMed] [Google Scholar]
- Musicki B.; Periers A.-M.; Laurin P.; Ferroud D.; Benedetti Y.; Lachaud S.; Chatreaux F.; Haesslein J.-L.; Iltis A.; Pierre C.; Khider J.; Tessot N.; Airault M.; Demassey J.; Dupuis-Hamelin C.; Lassaigne P.; Bonnefoy A.; Vicat P.; Klich M. Improved antibacterial activities of coumarin antibiotics bearing 5′,5′-dialkylnoviose: biological activity of RU79115. Bioorg. Med. Chem. Lett. 2000, 10, 1695–1699. 10.1016/s0960-894x(00)00304-8. [DOI] [PubMed] [Google Scholar]
- Brown-Elliott B. A.; Rubio A.; Wallace R. J. Jr. In VitroSusceptibility Testing of a Novel Benzimidazole, SPR719, against Nontuberculous Mycobacteria. Antimicrob. Agents Chemother. 2018, 62, e01503–18. 10.1128/aac.01503-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basarab G. S.; Hill P. J.; Garner C. E.; Hull K.; Green O.; Sherer B. A.; Dangel P. B.; Manchester J. I.; Bist S.; Hauck S.; Zhou F.; Uria-Nickelsen M.; Illingworth R.; Alm R.; Rooney M.; Eakin A. E. Optimization of Pyrrolamide Topoisomerase II Inhibitors Toward Identification of an Antibacterial Clinical Candidate (AZD5099). J. Med. Chem. 2014, 57, 6060–6082. 10.1021/jm500462x. [DOI] [PubMed] [Google Scholar]
- Eakin A. E.; Green O.; Hales N.; Walkup G. K.; Bist S.; Singh A.; Mullen G.; Bryant J.; Embrey K.; Gao N.; Breeze A.; Timms D.; Andrews B.; Uria-Nickelsen M.; Demeritt J.; Loch J. T.; Hull K.; Blodgett A.; Illingworth R. N.; Prince B.; Boriack-Sjodin P. A.; Hauck S.; MacPherson L. J.; Ni H.; Sherer B. Pyrrolamide DNA Gyrase Inhibitors: Fragment-Based Nuclear Magnetic Resonance Screening To Identify Antibacterial Agents. Antimicrob. Agents Chemother. 2012, 56, 1240–1246. 10.1128/aac.05485-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tari L. W.; Li X.; Trzoss M.; Bensen D. C.; Chen Z.; Lam T.; Zhang J.; Lee S. J.; Hough G.; Phillipson D.; Akers-Rodriguez S.; Cunningham M. L.; Kwan B. P.; Nelson K. J.; Castellano A.; Locke J. B.; Brown-Driver V.; Murphy T. M.; Ong V. S.; Pillar C. M.; Shinabarger D. L.; Nix J.; Lightstone F. C.; Wong S. E.; Nguyen T. B.; Shaw K. J.; Finn J. Tricyclic GyrB/ParE (TriBE) Inhibitors: A New Class of Broad-Spectrum Dual-Targeting Antibacterial Agents. PLoS One 2013, 8, e84409. 10.1371/journal.pone.0084409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi Y.; Azuma Y.; Kizaki H. An Assay Method for DNA Topoisomerase Activity Based on Separation of Relaxed DNA from Supercoiled DNA Using High-Performance Liquid Chromatography. Anal. Biochem. 1993, 210, 63–68. 10.1006/abio.1993.1151. [DOI] [PubMed] [Google Scholar]
- Chen H.; Zhou X.; Wang A.; Zheng Y.; Gao Y.; Zhou J. Evolutions in fragment-based drug design: the deconstruction-reconstruction approach. Drug Discovery Today 2015, 20, 105–113. 10.1016/j.drudis.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Zhang J.; Yu Z.; Zhang H.; Wang Y.; Lingel A.; Qi W.; Gu J.; Zhao K.; Shultz M. D.; Wang L.; Fu X.; Sun Y.; Zhang Q.; Jiang X.; Zhang J.; Zhang C.; Li L.; Zeng J.; Feng L.; Zhang C.; Liu Y.; Zhang M.; Zhang L.; Zhao M.; Gao Z.; Liu X.; Fang D.; Guo H.; Mi Y.; Gabriel T.; Dillon M. P.; Atadja P.; Oyang C. Discovery of First-in-Class, Potent, and Orally Bioavailable Embryonic Ectoderm Development (EED) Inhibitor with Robust Anticancer Efficacy. J. Med. Chem. 2017, 60, 2215–2226. 10.1021/acs.jmedchem.6b01576. [DOI] [PubMed] [Google Scholar]
- Freire E. Do Enthalpy and Entropy Distinguish First in Class from Best in Class?. Drug Discovery Today 2008, 13, 869–874. 10.1016/j.drudis.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferenczy G. G.; Keserű G. M. Thermodynamics guided lead discovery and optimization. Drug Discovery Today 2010, 15, 919–932. 10.1016/j.drudis.2010.08.013. [DOI] [PubMed] [Google Scholar]
- Reynolds C. H.; Holloway M. K. Thermodynamics of Ligand Binding and Efficiency. ACS Med. Chem. Lett. 2011, 2, 433–437. 10.1021/ml200010k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rühmann E.; Betz M.; Heine A.; Klebe G. Fragment Binding Can Be Either More Enthalpy-Driven or Entropy-Driven: Crystal Structures and Residual Hydration Patterns Suggest Why. J. Med. Chem. 2015, 58, 6960–6971. 10.1021/acs.jmedchem.5b00812. [DOI] [PubMed] [Google Scholar]
- Folmer R. H. A. Integrating biophysics with HTS-driven drug discovery projects. Drug Discovery Today 2016, 21, 491–498. 10.1016/j.drudis.2016.01.011. [DOI] [PubMed] [Google Scholar]
- Ladbury J. E.; Klebe G.; Freire E. Adding calorimetric data to decision making in lead discovery: a hot tip. Nat. Rev. Drug Discovery 2010, 9, 23–27. 10.1038/nrd3054. [DOI] [PubMed] [Google Scholar]
- Keserű G. M.; Makara G. M. Hit discovery and hit-to-lead approaches. Drug Discovery Today 2006, 11, 741–748. 10.1016/j.drudis.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Klebe G. Applying thermodynamic profiling in lead finding and optimization. Nat. Rev. Drug Discovery 2015, 14, 95–110. 10.1038/nrd4486. [DOI] [PubMed] [Google Scholar]
- Renaud J.-P.; Chung C.-w.; Danielson U. H.; Egner U.; Hennig M.; Hubbard R. E.; Nar H. Biophysics in drug discovery: impact, challenges and opportunities. Nat. Rev. Drug Discovery 2016, 15, 679–698. 10.1038/nrd.2016.123. [DOI] [PubMed] [Google Scholar]
- Holdgate G. A.; Tunnicliffe A.; Ward W. H. J.; Weston S. A.; Rosenbrock G.; Barth P. T.; Taylor I. W. F.; Pauptit R. A.; Timms D. The Entropic Penalty of Ordered Water Accounts for Weaker Binding of the Antibiotic Novobiocin to a Resistant Mutant of DNA Gyrase: A Thermodynamic and Crystallographic Study. Biochemistry 1997, 36, 9663–9673. 10.1021/bi970294+. [DOI] [PubMed] [Google Scholar]
- Mashalidis E. H.; Śledź P.; Lang S.; Abell C. A three-stage biophysical screening cascade for fragment-based drug discovery. Nat. Protoc. 2013, 8, 2309–2324. 10.1038/nprot.2013.130. [DOI] [PubMed] [Google Scholar]
- Meiby E.; Simmonite H.; le Strat L.; Davis B.; Matassova N.; Moore J. D.; Mrosek M.; Murray J.; Hubbard R. E.; Ohlson S. Fragment screening by weak affinity chromatography: comparison with established techniques for screening against HSP90. Anal. Chem. 2013, 85, 6756–6766. 10.1021/ac400715t. [DOI] [PubMed] [Google Scholar]
- Lamoree B.; Hubbard R. E. Current perspectives in fragment-based lead discovery (FBLD). Essays Biochem. 2017, 61, 453–464. 10.1042/ebc20170028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keserű G. M.; Erlanson D. A.; Ferenczy G. G.; Hann M. M.; Murray C. W.; Pickett S. D. Design Principles for Fragment Libraries: Maximizing the Value of Learnings from Pharma Fragment-Based Drug Discovery (FBDD) Programs for Use in Academia. J. Med. Chem. 2016, 59, 8189–8206. 10.1021/acs.jmedchem.6b00197. [DOI] [PubMed] [Google Scholar]
- Williams G.; Ferenczy G. G.; Ulander J.; Keserű G. M. Binding thermodynamics discriminates fragments from druglike compounds: a thermodynamic description of fragment-based drug discovery. Drug Discovery Today 2017, 22, 681–689. 10.1016/j.drudis.2016.11.019. [DOI] [PubMed] [Google Scholar]
- Rühmann E.; Betz M.; Fricke M.; Heine A.; Schäfer M.; Klebe G. Thermodynamic signatures of fragment binding: Validation of direct versus displacement ITC titrations. Biochim. Biophys. Acta, Gen. Subj. 2015, 1850, 647–656. 10.1016/j.bbagen.2014.12.007. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. 10.1016/s1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- Mani N.; Gross C. H.; Parsons J. D.; Hanzelka B.; Müh U.; Mullin S.; Liao Y.; Grillot A.-L.; Stamos D.; Charifson P. S.; Grossman T. H. In vitro characterization of the antibacterial spectrum of novel bacterial type II topoisomerase inhibitors of the aminobenzimidazole class. Antimicrob. Agents Chemother. 2006, 50, 1228–1237. 10.1128/aac.50.4.1228-1237.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman T. H.; Bartels D. J.; Mullin S.; Gross C. H.; Parsons J. D.; Liao Y.; Grillot A.-L.; Stamos D.; Olson E. R.; Charifson P. S.; Mani N. Dual Targeting of GyrB and ParE by a Novel Aminobenzimidazole Class of Antibacterial Compounds. Antimicrob. Agents Chemother. 2007, 51, 657–666. 10.1128/aac.00596-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta R.; Inouye M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem. Sci. 2000, 25, 24–28. 10.1016/s0968-0004(99)01503-0. [DOI] [PubMed] [Google Scholar]
- Staudenbauer W. L.; Orr E. DNA gyrase: affinity chromatography on novobiocin-Sepharose and catalytic properties. Nucleic Acids Res. 1981, 9, 3589–3603. 10.1093/nar/9.15.3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. MOLREP: an Automated Program for Molecular Replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. 10.1107/s0021889897006766. [DOI] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53, 240–255. 10.1107/s0907444996012255. [DOI] [PubMed] [Google Scholar]
- Asha M. K.; Debraj D.; Prashanth D. s.; Edwin J. R.; Srikanth H. S.; Muruganantham N.; Dethe S. M.; Anirban B.; Jaya B.; Deepak M.; Agarwal A. In vitro anti-Helicobacter pylori activity of a flavonoid rich extract of Glycyrrhiza glabra and its probable mechanisms of action. J. Ethnopharmacol. 2013, 145, 581–586. 10.1016/j.jep.2012.11.033. [DOI] [PubMed] [Google Scholar]
- Peng H.; Marians K. J. Escherichia coli topoisomerase IV. Purification, characterization, subunit structure, and subunit interactions. J. Biol. Chem. 1993, 268, 24481–90. [PubMed] [Google Scholar]
- CLSI . Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard—Tenth Edition. CLSI document M07-A10; Clinical and Laboratory Standards Institute: Wayne, PA, 2015.
- CLSI . Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria; Approved Standard—Eighth Edition. CLSI document M11-A8; Clinical and Laboratory Standards Institute: Wayne, PA, 2012.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.