

Abstract

A lignin-graft-poly(lactic-co-glycolic) acid (PLGA) biopolymer was synthesized with two types of lignin (LGN), alkaline lignin (ALGN) and sodium lignosulfonate (SLGN), at different (A/S)LGN/PLGA ratios (1:2, 1:4, and 1:6 w/w). 1H NMR and Fourier-transform infrared spectroscopy (FT-IR) confirmed the conjugation of PLGA to LGN. The (A/S)LGN-graft-PLGA biopolymers were used to form nanodelivery systems suitable for entrapment and delivery of drugs for disease treatment. The LGN-graft-PLGA NPs were generally small (100–200 nm), increased in size with the amount of PLGA added, monodisperse, and negatively charged (−48 to −60 mV). Small-angle scattering data showed that particles feature a relatively smooth surface and a compact spherical structure with a distinct core and a shell. The core size and shell thickness varied with the LGN/PLGA ratio, and at a 1:6 ratio, the particles deviated from the core–shell structure to a complex internal structure. The newly developed (A/S)LGN-graft-PLGA NPs are proposed as a potential delivery system for applications in biopharmaceutical, food, and agricultural sectors.
Introduction
Biofriendly materials with controlled properties and degradation profiles are desired for various drug delivery applications. The materials must be biodegradable and biocompatible, abundant, and affordable to be adopted by the pharmaceutical industry. In the quest for biofriendly materials, natural polymers such as cellulose, lignin, chitosan, alginic acid, zein, and polysaccharides were successfully assembled in the form of nanoparticles, microparticles, films, gels, and other delivery platforms.1,2 Also, synthetic polymers such as poly(caprolactone) (PCA), poly(d,l lactic-co-glycolic acid) (PLGA), and poly(ethylene glycol) (PEG) were used alone or in different combinations to obtain biofriendly materials capable to satisfy the needs of drug delivery.1,3,4
More recently, strides have been made on the synthesis of new biopolymers formed by chemically attaching natural polymers to synthetic polymers. Lignin (LGN), a byproduct of the paper and biofuel industry with limited industrial applications, was chosen mainly because of its biocompatibility, low toxicity, abundance, and low cost.5,6 An irregular polymer formed by a random combination of sinapyl, coumaryl, and coniferyl alcohols linked by ether links or condensed C–C bonds, lignin exhibits a hydrophilic behavior. The abundance of hydroxyl groups present in the aromatic and aryl chains provide potential for functionalization,7−9 and several studies reported the chemical modification of lignin by covalent attachment of synthetic polymers and oils. The chemically modified lignin was developed into suitable products such as new dispersants, adhesives, surfactants, films, capsules, and microparticles.8,10−14 Lignin was successfully modified by attaching poly(ε-caprolactone),15,16 urethane groups,17 poly(glycidyl methacrylate)-co-poly(ethylene glycol)methacrylate (PGEA-PEGMA),18 maleimido undecylenic acid,19,20 epoxy groups reacted with poly(ethylene glycol) diglycidyl ether,21 succinic anhydride or dodecyl-succinic anhydride (DSA),22 cyclic carbonates (ethylene carbonate, propylene carbonate, vinyl ethylene carbonate, and glycerol carbonate),23,24 poly(lactic acid),25 poly 2-(trimethylamino) ethyl methacrylate,26 and fatty acids.27 The chemically modified lignin showed better performance when compared to the blended lignin due to the contributions of the new attached moieties, which enhanced lignin solubility in organic solvents, stability in solution and UV light resistance, targeting, drug loading, and hydrophobic and hydrophilic balance.28−30
The application of modified lignin as a biomaterial suitable for the synthesis of delivery systems at the nanoscale has been accomplished by several groups.29 A modified lignin with acrylamide was synthesized by free radical polymerization to obtain a more stable lignin. The modified lignin formed aggregates with size ranging from 50 to 350 nm as a function of sodium chloride concentrations and time.31 A lignin-based delivery system was synthesized by cross-linking kraft lignin with different ratios of C6H12Br12 to obtain 95% conversion of phenolics into primary hydroxyalkyl ethers. The size of the nanospheres was in the range of 100–700 nm with a high polydispersity index based on dynamic light scattering (DLS). The ionic interactions between the sulfonic groups of lignin and the cationic surfactant were responsible for the structure formation.32,33
In another study, hollow nanocapsules were synthesized by interfacial polyaddition in inverse miniemulsion of toluene diisocyanate (TDI) to cross-link lignin. The nanocontainers were loaded with hydrophilic active components that were released by enzymes present in the environment. The size of the nanocapsules was around 311–390 nm in a buffer solution with a wall thickness of 10–20 nm, and the capsules were stable over months.34
A cancer treatment in the form of a polymeric nanoparticle delivery system was developed based on lignin nanoparticles loaded with the anticancer drug, hydroxycamptothecin. The nanoparticles were functionalized with folic acid and poly(ethylene glycol) (PEG). The mean nanoparticle size was around 150 nm with narrow size distribution. The drug loading was around 24% and the blood circulation time was increased around 7-fold compared to the free drug. After 24 days of the treatment, the lignin-based nanoparticles significantly reduced the tumor growth (tumor growth inhibition was 80.5 ± 33.4%) and increased survival time in rats.35
The aim of this study was to develop lignin-based, stable, biodegradable, and biocompatible polymeric nanodelivery systems in the 100 nm range without the aid of surfactants. Polymeric nanoparticle synthesis commonly requires the use of surfactants followed by a purification step necessary to remove excess surfactants. The purification step is not necessary when surfactants are not used, which results in a reduction of time and cost of the nanoparticle synthesis. To reach this aim, an amphiphilic novel biopolymer was synthesized by grafting hydrophilic natural polymer (lignin in two forms, alkaline lignin (ALGN) and lignosulfonate (SLGN)) on a hydrophobic synthetic polymer (PLGA) to be further assembled into polymeric nanoparticles. The synthetic polyester poly(lactic-co-glycolic acid) (PLGA) was selected due to its biocompatibility and biodegradability, important for biopharmaceutical applications. By covalently linking the hydrophilic natural polymer (lignin) with the hydrophobic synthetic polymer (PLGA), the new (A/S)LGN-graft-PLGA biopolymer was formed.
More specifically, the LGN-graft-PLGA biopolymer was formed from ALGN and SLGN linked to PLGA at three different LGN:PLGA mass ratios (1:2, 1:4, and 1:6 w/w). Following polymer characterization by NMR and Fourier-transform infrared spectroscopy (FT-IR), the LGN-graft-PLGA polymer was formed into nanoparticles. Nanoparticles were characterized by measuring size, size distribution, ζ-potential, stability, and morphology by DLS, transmission electron microscopy (TEM), and small-angle X-ray scattering/small-angle neutron scattering (SAXS/SANS).
Results and Discussion
LGN-graft-PLGA Biopolymer Structure
The approach for synthesizing surfactant-free lignin nanoparticles was to covalently link LGN with PLGA. The coupling was performed in a two-step reaction by the addition of oxalyl chloride. 1H NMR and FT-IR were used to confirm the covalent link between LGN and PLGA.
The 1H NMR plots for ALGN-graft-PLGA (Figure 1A) and SLGN-graft-PLGA (Figure 1B) show the characteristic peaks of PLGA at 5.1–5.2 and 4.6–4.8 ppm, corresponding to the lactide (CH groups) and glycolide (CH2 groups) monomers, respectively. The shifts in 5.6–5.75 ppm represent double-bond formation in the lignin moiety by excess of oxalyl chloride used in the first reaction. Also, methyl groups of lactide at approximately 1.5–1.7 ppm were observed (Figure 1A,B). All of the conjugate ratios synthesized with ALGN and SLGN showed the same pattern. The lignin polymer was observed at a range of 6.4–7.5 ppm that shows aromatic hydrogens. The −OCH3 groups are at 3.7–3.8 ppm, which appear in all conjugates formed with ALGN and SLGN. Also, −OCOCH3 was observed at 1.7–2.2 ppm in all of the synthesized graft polymers (Figure 1A,B). The water and dimethyl sulfoxide (DMSO) peaks were present in the samples at 3.3–3.4 and 2.65, respectively.
Figure 1.

H NMR of (A) ALGN-graft-PLGA and (B) SLGN-graft-PLGA graft polymers synthesized at 1:2, 1:4, and 1:6 mass ratios.
FT-IR showed typical lignin and PLGA bands for the conjugated biopolymer. The bands between 1660 and 1720 cm–1 were provided by the strong vibration of carbonyl groups present in PLGA (C=O). Typical vibration bands of lignin were at 1590–1596 and 1508–1510 cm–1, corresponding to the vibrations of the aromatic rings (α-carbonyl groups symmetric and asymmetric, respectively) that are not present in the PLGA polymer. Also, the absorption band at 1030–1050 cm–1 shows the primary and the secondary alcohol groups present in ALGN-graft-PLGA (Figure 2A) and SLGN-graft-PLGA (Figure 2B). The PLGA stretching bands provided by C–CO–O (symmetric and asymmetric) vibrations between 1300 and 1150 cm–1 were present in all LGN-graft-PLGA samples analyzed (Figure 2A,B).
Figure 2.

FT-IR spectrum for(A) PLGA 50:50, ALGN, ALGN-graft-PLGA 1:2 w/w, ALGN-graft-PLGA 1:4 w/w, and ALGN-graft-PLGA 1:6 w/w and (B) PLGA 50:50, SLGN, SLGN-graft-PLGA 1:2 w/w, SLGN 1:4 w/w, and SLGN-graft-PLGA 1: 6 w/w from bottom to top.
ALGN-graft-PLGA Nanoparticle Characterization
PLGA is currently formed into nanoparticles in combination with surfactants (anionic, cationic, nonionic, and zwitterionic) and the excess surfactants must be removed by a purification step (i.e., ultrafiltration, dialysis, or ultracentrifugation), thereby increasing the cost of the synthesis.36
The approach followed in the surfactant-free lignin nanoparticle synthesis was to covalently link the natural biopolymer lignin with PLGA to form a new biopolymer, (A/S)LGN-graft-PLGA. The ALGN-graft-PLGA nanoparticles showed a spherical shape with a narrow size distribution (Figure 3), even though no surfactant was used in the aqueous phase during nanoparticle synthesis. The biopolymer with more lignin (1:2 ALGN/PLGA w/w) produced smaller nanoparticles compared with the biopolymer with less lignin (1:4 and 1:6 ALGN/PLGA and SLGN/PLGA w/w, respectively) (Table 1). The TEM images indicated that particles had an outer layer (hydrophilic interface provided by lignin) with a gray ring surrounding the white PLGA hydrophobic core. The ALGN-graft-PLGA mean nanoparticle size was minimally affected when suspended in water at pH values ranging from 1.6 to 8.6 (Figure 4). The ζ-potential varried from −5 to −35 mV over the pH range studied.
Figure 3.

TEM pictures of (A) ALGN/PLGA 1:2 w/w, (B) ALGN/PLGA 1:4 w/w, (C) ALGN/PLGA 1:6 w/w, (D) SLGN/PLGA 1:2 w/w, (E) SLGN/PLGA 1:4 w/w, and (F) SLGN/PLGA 1:6 w/w. Uranyl acetate was used as a contrast agent in all samples. TEM images used for size analysis are included in the Supporting Information (SI).
Table 1. ALGN-Graft-PLGA and SLGN-Graft-PLGA Polymeric Nanoparticle Characteristics Based on DLS at pH 6.
| polymer (w/w) | hydrodynamic sizeDLS ± S.D.DLS | PDIDLS ± S.D.DLS | ζ-potential (mV) ± S.D. |
|---|---|---|---|
| ALGN-graft-PLGA 1:2 | 113.8 ± 3.4 | 0.31 ± 0.03 | –53.8 ± 6.9 |
| ALGN-graft-PLGA 1:4 | 144.1 ± 3.0 | 0.35 ± 0.04 | – 60.0 ± 0.8 |
| ALGN-graft-PLGA 1:6 | 195.5 ± 2.5 | 0.33 ± 0.01 | –52.2 ± 0.8 |
| SLGN-graft-PLGA 1:2 | 106.5 ± 2.7 | 0.26 ± 0.029 | –48.2 ± 1.8 |
| SLGN-graft-PLGA 1:4 | 120.8 ± 1.4 | 0.28 ± 0.022 | –53.3 ± 1.0 |
| SLGN-graft-PLGA 1:6 | 193.7 ± 3.7 | 0.28 ± 0.040 | –55.8 ± 0.3 |
Figure 4.

Effects of pH on LGN-graft-PLGA nanoparticle mean size and ζ-potential for particles made by (a) ALGN/PLGA (1:2, 1:4, 1:6 mass ratios) and (b) SLGN/PLGA (1:2, 1:4, 1:16 mass ratios). The thick red lines represent nanoparticle size (left y axis) and the fine blue dotted lines represent ζ-potential (right y axis).
Both ALGN and SLGN had a constant size over a wide range of pHs at 1:2 and 1:4 LGN/PLGA ratios. At SLGN/PLGA 1:6, the mean polymeric nanoparticle size decreased from 350 to 290 nm when pH increased from 2 to 10. The ζ-potential of ALGN-graft-PLGA and SLGN-graft-PLGA nanoparticles was negative across the pH range tested; for SLGN-graft-PLGA nanoparticles, it decreased from −40 to −70 mV with an increase in pH from 2 to 10 (Figure 4). In general, a higher ζ-potential (negative or positive) promotes repulsion between polymeric nanoparticles and is desired to reduce nanoparticle aggregation for better stability needed in drug delivery applications. Similarly charged particles can be formed from PLGA in the presence of sodium dodecyl sulfate as a surfactant or other negatively charged surfactants.37
The LGN-graft-PLGA nanoparticles synthesized herein with ALGN and SLGN did not require surfactants during synthesis for stabilization purposes, reducing processing cost and time. The purification step traditionally required to remove excess surfactant by dialysis, ultrafiltration, or ultracentrifugation was avoided. Moreover, the new biopolymer LGN-graft-PLGA was designed to minimize nanoparticle aggregation during freeze-drying, so the LGN-graft-PLGA nanoparticles can be resuspended in water or buffers with no significant changes in size (Figure 4).
Small-Angle Neutron Scattering (SANS)
Small-angle neutron scattering (SANS) is very sensitive to hydrogens and can help to expand our understanding of nanoparticles, by a more detailed insight, by distinguishing the core and the shell.38,39 Here, we concentrate on 1:2 ALGN-graft-PLGA particles with trehalose as a cryoprotectant. The freeze-dried particles were redissolved in D2O, and the neutron scattering profiles were obtained at two different concentrations, 0.2 and 1.25% w/w (Figure 5). The scattered intensity or, more precisely, the scattering cross section, dσ/dΩ, is plotted as a function of the momentum transfer, Q, for the two concentrations.
Figure 5.

SANS diffraction data for ALGN-graft-PLGA 1:2 particles in D2O (pH 6.8). (a) Sphere model: The solid lines are modeled using the sphere model with a polydispersity index (PDI) of 0.4. (b) Core–shell model: The solid lines are modeled using a modified core–shell model with a PDI of 0.5. Concentrations of the particles are 0.2% w/w (pale green) and 1.25% w/w (green). Data are shown using symbols and the model fits are shown by red and blue lines.
At small Q, we observe a transition
into a power-law
decay, I ∝ Q–4, which continues
for almost 2 orders of magnitude in Q. The transition
occurs at around q ≈ 0.003 Å–1. This corresponds to an associated length-scale of the order of
2π/Q≈200 nm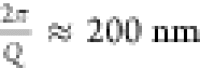 . This is almost twice as much as we would
expect from the hydrodynamic radius from dynamic light scattering
∼110 nm, and a more detailed discussion is required to explain
this apparent contradiction. However, we can clearly say that the
power-law belongs to a length-scale lesser than the particle size.
From experiments on colloids, we know that this slope refers to the
surface of the particles.40 More precisely,
the value of −4 indicates a surface fractal dimension of 2,
indicating a smooth surface at the length-scale of the experiments,
i.e., 5 × 10 –3 < Q/ Å–1 < 1 × 10–1 or the related
length-scales, 12 Å < size < 1200 Å, over 2 orders
of magnitude in length-scale.
. This is almost twice as much as we would
expect from the hydrodynamic radius from dynamic light scattering
∼110 nm, and a more detailed discussion is required to explain
this apparent contradiction. However, we can clearly say that the
power-law belongs to a length-scale lesser than the particle size.
From experiments on colloids, we know that this slope refers to the
surface of the particles.40 More precisely,
the value of −4 indicates a surface fractal dimension of 2,
indicating a smooth surface at the length-scale of the experiments,
i.e., 5 × 10 –3 < Q/ Å–1 < 1 × 10–1 or the related
length-scales, 12 Å < size < 1200 Å, over 2 orders
of magnitude in length-scale.
The simplest model that exists for spherical soft particles is a sphere form factor (Figure 5a). In a more realistic description, we may distinguish between the core and the shell (Figure 5b). At this point, it is important to elaborate on the important difference of a real sphere vs the sphere form factor. The sphere form factor assumes a constant scattering length density as a function of the distance from the center. Typically, a constant scattering length density is a good approximation for a core of nanoparticles, but not for polymer chains eventually in the chain or in a fuzzy surface. However, scattering experiments can show a core-only particle that equals the scattering of spheres for the case of a shell of negligible thickness or if the scattered intensity of the shell is much weaker than that of the core, e.g., if the contrast is weak. In other words, a sphere is a plausible first attempt to analyze the results. As a natural progression, we test the description by a core–shell form factor with the idea to explore the contribution of the shell and its thickness more in detail. Figure 5a,b tests these two simple models, sphere form factor41 and a modified core–shell model,42 respectively, as explained below. eqs 1–3.
The data in Figure 5a are modeled using a compact sphere form factor, P(Q,R)sphere,41 convoluted with a log-normal distribution to account for the size polydispersity, s(r). The scattering contribution is given by
| 1 |
With
 |
2 |
The hard-sphere radius from SANS is given by RHS, and the volume fraction is ϕ. The neutron scattering length density (NSLD) of the D2O solvent is given by ρsolv = 6.36 × 1010 cm–2. The NSLD for the particle ranges between ρparticle ≈ 2.12–2.14 × 1010 cm–2, due to the uncertainty in the overall molecular weight of the particle. So, we used an intensity prefactor, I0, in eq 1.
For the core–shell model in Figure 5b, the P(Q,R)sphere in eq 1 is replaced by the core–shell
form
factor, P(Q,R)core–shell. The scattering contribution similar to eqs 1 and 2 is given by  . The expression for F(Q)core is the same as that given by eq 2 for a compact hard-sphere core
of radius Rc with an NSLD ρc. The contribution from the shell is given by
. The expression for F(Q)core is the same as that given by eq 2 for a compact hard-sphere core
of radius Rc with an NSLD ρc. The contribution from the shell is given by
| 3 |
Here, Rp = Rc + Rshell, with the core radius and shell thickness given by Rc and Rshell, respectively. The neutron scattering length densities of the core and shell are given by ρc = 2.23 × 1010 cm–2 and ρshell = 1.89 × 1010 cm–2, respectively. The highly smeared blob scattering arising from the lignin tails can be described by an additional independent factor, AblobPblob, in eq 1. The blob scattering amplitude and the form factor is given by Ablob and Pblob, respectively, as was described in our previous work.42
Apparently, the structure of the nanoparticles is rather complex. At the same time, only a few features are visible in our data. Therefore, we constrained ourselves to a model that assumes two constant densities, one for the core and one for the shell. The sphere form factor approximately describes the experimental data with slight deviations at wide Q, but with substantial differences at a low Q. In other words, the fitting of a sphere form factor does not provide reliable information. Instead, the core–shell model describes the experimental intensities much better.
At this point, we need to be careful when discussing the data. Despite the fact that a better description by the core–shell model is in favor of a core–shell structure, the limited range toward lower Q does not allow to distinguish between size polydispersity, agglomeration of the polymeric nanoparticles, and a core–shell structure. In the case of agglomeration, we would observe an ongoing steep increase of the intensity with decreasing Q.40 It would also be possible that the polydispersity in size (around 30% from DLS) could cause a broader transition region from the Q–4 region (Porod region) to a plateau or, more precisely, the so-called Guinier region. As we do not observe this transition, we cannot reach a full conclusion. However, the better description of the data by the core–shell model may be indicative of a core–shell structure, which would imply a distinguishable core and shell.
From the fit of the model of a homogenous sphere, we obtain diameters of 58 ± 2 nm for both concentrations with a PDI of 0.4 ± 0.03. Apparently, experimental data are not well described at low Q, and the transition into the Guinier regions occurs at a higher Q, which implies that the real diameters are larger. This expectation is consistent with values from DLS (∼114 nm) and TEM. Description of experimental data using the diameter from DLS failed, though. In other words, the description by the sphere form factor gives a very rough estimate of a minimum particle size, but not any accurate value.
The core–shell model yields a particle diameter of 107 ± 4 nm and a PDI of 0.50 ± 0.04. The core diameter is 56 ± 2 nm. Within experimental uncertainty, these values agree to the approximation that the Q oscillation minimum from the scattering pattern is given by Q0 ≈ 2π/d for the particle diameter, d. The observed diameter is slightly less than the DLS value. However, given the experimental accuracy, we assume that SANS and DLS agree. At this point, a deeper discussion of these subtle differences is not attempted. It may simply be a consequence from the difference between the perimeter radius from SANS and the hydrodynamic radius from the diffusion coefficient from DLS. For more complex structures, typically, there is a numerical factor in between perimeter and hydrodynamic radius. However, it may also be the case that the DLS results are indicative of clustering, which we cannot explore further because of the limited Q range of the SANS experiment.
So far, our results point to a core–shell structure. To further explore the structure, we will utilize small-angle X-ray scattering and its advantage of a better Q-resolution. In the best case, this would reveal structural peaks, which are not visible in the case of SANS.
Small-Angle X-ray Scattering (SAXS)
To further expand our knowledge on the nanoparticles, we vary lignin (LGN), ALGN, and SLGN concentrations, and test different lignin/poly(lactic-co-glycolic) mass ratios, 1:2, 1:4, and 1:6. From SANS, we know that there is little, if any, concentration dependence; therefore, we concentrate on a single concentration of 1.25% w/w (12.5 mg/mL). Such a low concentration should minimize structure factor contributions from interparticle interactions and still have reasonable measurement times.
The data in Figure 6a,b for ALGN-graft-PLGA and SLGN-graft-PLGA, respectively, illustrate the differential scattering cross section, dσ/dΩ, as a function of the momentum transfer. In both subfigures, we see a substantial dependence on the LGN:PLGA ratios. In all of the cases, the intensity decreases with increasing Q. Apart from ALGN-graft-PLGA 1:2 and 1:4, there is no visible transition at low Q. As in the case of the SANS data, we observe a continuous decrease of the intensity with increasing Q. This is unfortunate because it makes accurate observations of the diameter of the colloids impossible. At first glance, this seems to contradict the SANS experiments. However, these measurements were only done for ALGN/PLGA 1:2. Thus, the observations match very well.
Figure 6.

Small-angle X-ray scattering (SAXS) profiles of (a) ALGN-graft-PLGA particles and (b) SLGN-graft-PLGA particles with LGN/PLGA mass ratios: 1:2, 1:4, and 1:6 in H2O (pH 7). The data are modeled by a modified core–shell model (red line).
Following eqs 1–3 from the SANS data, we know that a sphere form factor is not sufficient to model the scattering data. Therefore, we used a core–shell model as the simplest possible approach. The NSLD in eq 3 was replaced by the X-ray scattering length density (XSLD). For the core, it is given by 11.21 × 1010 cm–2, for the shell, 11.98 × 1010, and 9.44 × 1010 cm–2 for D2O. As a good initial estimate of the size, we chose the particle diameter for the Q ≈ 2π/Q0. The highly smeared nature of the scattering pattern increases the uncertainties in obtaining the final size. Despite this oversimplified approach, the fit yields a reasonable result, at least for ratios 1:2 and 1:4. However, a complete disagreement is more than evident for the 1:6 cases. We would like to emphasize that in 1:6 samples, the transitions from the flat Guinier plateau at low Q in the scattering pattern are not visible. Thus, the fit cannot determine the values. Instead, we used the smallest Q value, calculated the corresponding size, and tried to accomplish a reasonable agreement by keeping this value fixed in the model description. Obviously, this approach does not work for ALGN/PLGA 1:6 but it at least roughly reproduces the SLGN-graft-PLGA intensities. These observations point toward a core–shell structure that may get more complicated for certain ratios. We speculate that this is a consequence of the hydrophilic–hydrophobic balance (HLB) of the LGN and PLGA segments that are in interplay with each other and an attempt to minimize interfacial free energy change.43
The slightly higher intensities of the fit at low Q for ALGN-graft-PLGA 1:2 and 1:4 should be noted, which indicates that the fit could overestimate the size. On the other hand, in the case of SLGN-graft-PLGA, the transitions are barely visible and the values can have a systematic error that cannot be determined from the SAXS experiments (because the transition is not visible). Despite these obvious facts, the differences between the observations are surprisingly low. We do not dare to discuss this, as it might be purely coincidental.
We would also like to clarify the apparent disagreement of particle sizes from the three different techniques, DLS, SAXS, and TEM44 (provided in the SI). The hydrodynamic size obtained in the DLS first measures the diffusion coefficient and calculates size following the Stokes–Einstein equation. Since our particles are not homogenous spheres, the size of the particles in SAXS using a core–shell model, which gives the radius of gyration of the particles in solution, can have obvious differences. The large deviation in particle size indicates that the simple core−shell model cannot explain the data for samples ALGN-PLGA 1:6 and SLGN-PLGA 1:6, as can be seen from Figure 6. The outcome reported is not the size of the particles but rather the minimum size estimate obtainable by scattering intensity at low Q (Table 2). Contrary to both DLS and SAXS, the TEM data is obtained when the particles are in a dry state, stained with uranyl acetate. However, the initial values obtained for the core thickness were useful in refining the SAXS model.
Table 2. SAXS Modified Core–Shell Model Fit Results of ALGN-Graft-PLGA and SLGN-Graft-PLGA Polymeric Nanoparticles.
| polymer (w/w) | particle diameter (=core + 2*shell) | core diameter (nm) ± S.D. | shell thickness (nm) ± S.D. | polydispersity % |
|---|---|---|---|---|
| ALGN-graft-PLGA 1:2 | 82 ± 2 | 77 ± 2 | 2.2 ± 0.1 | 36 ± 3 |
| ALGN-graft-PLGA 1:4 | 86 ± 2 | 82 ± 2 | 1.8 ± 0.1 | 36 ± 4 |
| ALGN-graft-PLGA 1:6 | 412a | 252.0a | 80.0a | 30 ± 3 |
| SLGN-graft-PLGA 1:2 | 120 ± 3 | 78 ± 2 | 20.0 ± 0.3 | 40 ± 4 |
| SLGN-graft-PLGA 1:4 | 136 ± 4 | 124 ± 2 | 6.0 ± 1 | 30 ± 3 |
| SLGN-graft-PLGA 1:6 | 208a | 182.4a | 13a | 20 ± 2 |
Note: estimated minimum diameter from SAXS, as the particle diameter value cannot be determined.
Conclusions
Our approach was to covalently link a hydrophilic polymer (ALGN and SLGN) with PLGA to obtain a new biopolymer with amphiphilic behavior. The amphiphilic property of the new LGN-graft-PLGA biopolymer allows the synthesis of polymeric nanoparticles without the need for surfactants for stabilization purposes. As a result, there is no need for a purification step by dialysis, ultrafiltration, or ultracentrifugation, which are typically required to remove excess surfactants in other processes, thereby reducing the time and cost of nanoparticle synthesis. The (A/S)LGN-graft-PLGA NPs can be made in a wide range of sizes and surface charges by changing the LGN/PLGA ratio. The addition of more PLGA results in an increase in the nanoparticle diameter. The LGN-PLGA nanoparticles can be resuspended in water or buffers with no significant changes in size. Small-angle scattering data and TEM indicate that the particles have a spherical, core–shell structure with a relatively smooth surface, evident by the I–Q–4 relationship in the mid-Q region. SAXS analysis showed that particle core size increases with the addition of PLGA and corresponds with DLS data, where the overall particle size increased following a similar trend. Furthermore, SAXS revealed that ALGN and SLGN containing 1:6 ratio with PLGA deviate from the core–shell structure to yield much larger particles with a potentially complex structure, which needs further attention in future studies. Future studies will also attempt to entrap hydrophobic and hydrophilic drugs in the LGN-PLGA NPs. Codelivery of hydrophobic and hydrophilic drugs can be achieved with this novel nanoparticle by entrapping them either in the PLGA hydrophobic core or the lignin hydrophilic shell, controlling their release over time as a function of the degradation of the polymers LGN and PLGA, molecular weight of the polymer, and the lactide/glycolide ratio of PLGA.45 The versatility offered by the LGN-graft-PLGA biopolymer can open other avenues of applications including scaffolds, films, sensors, and packaging.
Experimental Procedures
Materials
PLGA 50:50 (38–54 kDa) was acquired from Sigma-Aldrich (St. Louis, MO). ALGN and SLGN were acquired from TCI Inc. (Portland, OR). Oxalyl chloride (purity ≥99%), dichloromethane (DCM, purity ≥99.9% extra dry), dimethyl sulfoxide (DMSO, purity ≥99.7%), trehalose (purity ≥99%), ethyl ether (purity ≥99%), ethanol, dimethylformamide (DMF, purity ≥99.5%), and ethyl acetate (purity ≥99.5%) were acquired from Fisher Scientific (Pittsburgh, PA).
Synthesis of the LGN-Graft-PLGA Biopolymer (ALGN and SLGN)
The coupling of lignin to PLGA was based on the acylation reaction for both types of lignin, (A/S)LGN (Scheme 1). Briefly, 2 g of PLGA was dissolved in 30 mL of DCM at room temperature in a three-neck round-bottom flask. A nitrogen flow was connected to a bubbler bottle with 1 M NaOH to neutralize HCl produced during the reaction. After complete dissolution of PLGA at room temperature, 5 equiv of oxalyl chloride was added dropwise with a glass syringe. The reaction was performed at room temperature with mild stirring for 4 h. The solution was concentrated with a rotavapor Buchi R-300 (Buchi Corporation, New Castle, DE). The polymer was precipitated with the addition of 150–200 mL of ethyl ether, and the white precipitate was washed at least three times to remove impurities. The solids were dried overnight under high vacuum. Next, the second reaction was performed with the dissolution of 1 g of dry PLGA-Cl in 20 mL of DMSO, which was added dropwise to 500 mg of (A/S)lignin dissolved in 20 mL of DMSO. The reaction was performed overnight at room temperature and nitrogen flow. The (A/S)LGN-graft-PLGA polymer was precipitated with the addition of 150–200 mL of ethyl ether; the washing step was repeated three times. The precipitated polymer was suspended in 20 mL of DCM and the organic phase was washed with water to remove unreacted lignin to obtain a clear supernatant. Finally, DCM was evaporated with a rotavapor Buchi R-300, and the polymer was dried under high vacuum for 3 days at 30 °C. The (A/S)LGN-graft-PLGA copolymers were stored at 2–4 °C for further characterization and use in nanoparticle synthesis.
Scheme 1. Lignin-PLGA Biopolymer Reaction.

SLGN and ALGN were used in the coupling reaction with PLGA (50:50) (R1, R2, R3, R4, and R5 are monomers (p-coumaryl alcohol, sinapyl alcohol, and coniferyl alcohol randomly distributed in lignin polymers)).
Synthesis of ALGN-Graft-PLGA Nanoparticles
The polymeric nanoparticles were synthesized by the emulsion evaporation technique with important modifications. No surfactants were added in the aqueous phase, and no purification steps were required. Briefly, 150–500 mg of PLGA–lignin was dissolved in 5 mL of ethyl acetate at room temperature under strong stirring. Next, the organic phase was added to the aqueous phase (50 mL of deionized water (DI) water). After 10 min of mixing, the suspension was homogenized with a microfluidizer (Microfluidics Corp., Westwood, MA) at 30 000 psi four times at 4 °C. Also, sonication can be used for small volumes. Next, the organic solvent was evaporated in a rotavapor R-300 (Buchi Corporation, New Castle, DE) at 32 °C under vacuum for at least 45 min. Finally, trehalose was added (1:1 mass ratio) as a cryoprotectant, and the samples were placed in a freeze-drier FreeZone 2.5 (Labconco Corporation, Kansas City, MO) for 2 days at −80 °C to remove water. Finally, the PLGA–lignin biopolymer nanoparticle samples were stored at −20 °C and the biopolymeric nanoparticle powders were resuspended in low-resistivity water before characterization.
Synthesis of SLGN-Graft-PLGA Nanoparticles
The synthesis of SLGN-graft-PLGA nanoparticles followed a similar emulsion evaporation technique. Two hundred milligrams of the SLGN-graft-PLGA biopolymer were dissolved in 5 mL of ethyl acetate under mild stirring for 1 h at room temperature. Next, the organic phase was added to 50 mL of low-resistivity water under stirring. The final emulsion was sonicated for 10 min with Vibra-Cell VC 750 (Sonics & Materials Inc., Newton, CT) at 38% amplitude (4 s “on” and 2 s “off”). The emulsion was placed in a round-bottom flask for solvent evaporation with a rotavapor Buchi R-300 (Buchi Corporation, New Castle, DE) at 33 °C for 1 h under vacuum. To the final clear suspension, trehalose was added in a 1:1 mass ratio for nanoparticle protection during freeze-drying. The samples were freeze-dried (Labconco Corp., Kansas City, MO) for 48 h at −80 °C. The particles were stored at −20 °C.
Biopolymer Characterization
The ALGN-graft-PLGA and SLGN-graft-PLGA conjugates were characterized with 1HNMR (Bruker 500, Billerica, MA) at 500 Hz in DMSO. The FT-IR spectrometer used was Bruker Tensor 27 (Bruker, Billerica, MA) where dry samples were placed on the detector.
Nanoparticle Characterization by Dynamic Light Scattering (DLS)
Size, polydispersity, and ζ-potential of the nanoparticles were measured by dynamic light scattering (DLS) using Malvern Zetasizer ZS (Malvern Panalytical, Westborough, MA). Briefly, 1 mL of the resuspended polymeric nanoparticle sample in low-resistivity water at concentrations of 0.2–0.4 mg/mL was placed in a cuvette or ζ-potential cell and analyzed at 25 °C.
Nanoparticle Characterization by Transmission Electron Microscopy (TEM)
The nanoparticle morphology was analyzed by transmission electron microscopy (TEM) JEM-1400 (Jeol USA Inc., Peabody, MA) as follows. One droplet of the resuspended polymeric nanoparticles in high-resistivity water was placed over a carbon copper grid with a contrast agent, the excess sample was removed, and the sample was dried for 10 min before placing the grid on the TEM chamber. Particle size analysis of TEM data was done using ImageJ software.44 Multiple images of the six different ALGN-graft-PLGA and SLGN-graft-PLGA preparations used in particle size analysis and the results are listed in the SI. Each particle was measured approximately three times across the center in different angles to account for the shape polydispersity in core and shell measurements.
Nanoparticle Characterization by Small-Angle Neutron Scattering (SANS)
Freeze-dried ALGN-graft-PLGA 1:2 nanoparticles with cryoprotectant trehalose were resuspended in D2O and prefiltered using 0.45 μm poly(vinylidene difluoride) (PVDF) syringe filters to obtain nanoparticles in solution. Small-angle neutron scattering (SANS) data were obtained by the NG7 SANS instrument at the NIST Center for Neutron Research (NCNR) at the National Institute of Standards and Technology (NIST) and the EQ-SANS instrument at the Spallation Neutron Source (SNS) of Oak Ridge National Laboratory (ORNL).46,47 All measurements were conducted at 20 °C in standard 2 mm cells. Measurements were statistically averaged. Data were reduced using Igor Pro and Mantid.48 D2O and the empty cell were measured separately and subtracted. Absolute intensities were accomplished using water as the calibration standard. The momentum transfer, Q = 4π/λ sin(ϑ), with the scattering angle, ϑ, and λ, the wavelength. For NG7 SANS, the sample-to-detector distance was fixed to 1, 4 and 13 m, and the neutron wavelength was λ = 6 Å. This covers a Q range from ∼0.002 to ∼0.6 Å–1. For EQ-SANS, the sample-to-detector distance was fixed to 2.5, 4, and 8 m, for a minimum wavelength λmin = 2.5 Å, covering a Q range from 0.002 to ∼0.6 Å–1.
Nanoparticle Characterization by Small-Angle X-ray Scattering (SAXS)
Freeze-dried ALGN-graft-PLGA and SLGN-graft-PLGA particles were resuspended in H2O to obtain 12.5 mg/mL solutions. All small-angle X-ray scattering (SAXS) data were obtained at the beamline 4-2 of Stanford Synchrotron Radiation Lightsource (SSRL).49 An automated sampler system was used to load 40 μL aliquots of samples to the capillary cell. Each sample was exposed to the X-ray beam 24 times in 1 s exposures as the flow cell gently moves the sample back and forth to minimize radiation damage by continuous exposure. There was no visible difference. Therefore, the 24 scans were statistically averaged, and radial averaging over the same data yields the intensity as a function of the momentum transfer, similar to the procedure followed in SANS measurements. The comparison of the 24 scans also excludes data from samples with visible radiation damage. The data were obtained at a beam energy of 11 keV, at two detector distances (3.4 and 1.1 m), to explore a Q range of 0.003–1 A°–1 using a Pilatus 3 X 1M detector. Data reduction was done using standard SSRL protocols implemented in the software Blu-Ice.50
Acknowledgments
This work was supported by the National Science Foundation under NSF EPSCoR Track 2 RII, OIA 1632854, the USDA National Institute of Food and Agriculture, AFRI project #2017-07878, the USDA National Institute of Food and Agriculture, AFRI project #2018-07406, and the USDA-NIFA Hatch Project #1008750. The neutron scattering work is supported by the U.S. Department of Energy (DOE) under EPSCoR Grant no. DE-SC0012432 with additional support from the Louisiana Board of Regents. A portion of this research used resources at the EQ-SANS beamline of Spallation Neutron Source, a DOE Office of Science User Facility operated by the Oak Ridge National Laboratory. Access to NG7 SANS was provided by the Center for High Resolution Neutron Scattering, a partnership between the National Institute of Standards and Technology and the National Science Foundation under Agreement no. DMR-1508249. We acknowledge the support of the National Institute of Standards and Technology, U.S. Department of Commerce, in providing the neutron research facilities used in this work. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract no. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health and the National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH. We would like to thank Thomas Weiss for his guidance during SAXS experiments.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c00168.
TEM Images of ALGN-graft-PLGA and SLGN-graft-PLGA nanoparticles used for particle size analysis; distribution of ALGN-graft-PLGA and SLGN-graft-PLGA particle size and core size; ALGN-graft-PLGA and SLGN-graft-PLGA polymeric nanoparticle characterization based on TEM particle analysis (PDF)
The authors declare the following competing financial interest(s): C.E.A. and C.M.S. have intellectual property including one approved patent related to the described research.
Supplementary Material
References
- Ulery B. D.; Nair L. S.; Laurencin C. T. Biomedical Applications of Biodegradable Polymers. J. Polym. Sci., Part B: Polym. Phys. 2011, 49, 832–864. 10.1002/polb.22259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitriu S.; Popa V.. Polymeric Biomaterials; Dumitriu S.; Popa V., Eds.; CRC Press, 2013. [Google Scholar]
- Lü J.-M.; Wang X.; Marin-Muller C.; Wang H.; Lin P. H.; Yao Q.; Chen C. Current Advances in Research and Clinical Applications of PLGA-Based Nanotechnology. Expert Rev. Mol. Diagn. 2009, 9, 325–341. 10.1586/erm.09.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor D. N.; Bhatia A.; Kaur R.; Sharma R.; Kaur G.; Dhawan S. PLGA: A Unique Polymer for Drug Delivery. Ther. Delivery 2015, 6, 41–58. 10.4155/tde.14.91. [DOI] [PubMed] [Google Scholar]
- Lewandowski I.Lignocellulosic Energy Crops, Production, and Provision. In Encyclopedia of Sustainability Science and Technology; Meyer R. A., Ed.; Springer: New York, NY, 2013; pp 1–14. [Google Scholar]
- Agrawal A.; Kaushik N.; Biswas S. Derivatives and Applications of Lignin: An Insight. Scitech J. 2014, 1, 30–36. [Google Scholar]
- Nilsson T. Y.; Wagner M.; Inganäs O. Lignin Modification for Biopolymer/Conjugated Polymer Hybrids as Renewable Energy Storage Materials. ChemSusChem 2015, 8, 4081–4085. 10.1002/cssc.201500570. [DOI] [PubMed] [Google Scholar]
- Upton B. M.; Kasko A. M. Strategies for the Conversion of Lignin to High-Value Polymeric Materials: Review and Perspective. Chem. Rev. 2016, 116, 2275–2306. 10.1021/acs.chemrev.5b00345. [DOI] [PubMed] [Google Scholar]
- Garrison T.; Murawski A.; Quirino R. Bio-Based Polymers with Potential for Biodegradability. Polymers 2016, 8, 262 10.3390/polym8070262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duval A.; Lawoko M. A Review on Lignin-Based Polymeric, Micro- and Nano-Structured Materials. React. Funct. Polym. 2014, 85, 78–96. 10.1016/j.reactfunctpolym.2014.09.017. [DOI] [Google Scholar]
- Kamimura N.; Takahashi K.; Mori K.; Araki T.; Fujita M.; Higuchi Y.; Masai E. Bacterial Catabolism of Lignin-Derived Aromatics: New Findings in a Recent Decade: Update on Bacterial Lignin Catabolism. Environ. Microbiol. Rep. 2017, 9, 679–705. 10.1111/1758-2229.12597. [DOI] [PubMed] [Google Scholar]
- Ge Y.; Li Z. Application of Lignin and Its Derivatives in Adsorption of Heavy Metal Ions in Water: A Review. ACS Sustainable Chem. Eng. 2018, 6, 7181–7192. 10.1021/acssuschemeng.8b01345. [DOI] [Google Scholar]
- Mandlekar N.; Cayla A.; Rault F.; Giraud S.; Salaün F.; Malucelli G.; Guan J.-P.. An Overview on the Use of Lignin and Its Derivatives in Fire Retardant Polymer Systems. Lignin - Trends and Applications; InTech, 2018. [Google Scholar]
- Glasser W. G.Lignin-Based Polymers. Reference Module in Materials Science and Materials Engineering; Elsevier, 2016. [Google Scholar]
- Laurichesse S.; Avérous L. Synthesis, Thermal Properties, Rheological and Mechanical Behaviors of Lignins-Grafted-Poly(ε-Caprolactone). Polymer 2013, 54, 3882–3890. 10.1016/j.polymer.2013.05.054. [DOI] [Google Scholar]
- Park I.-K.; Sun H.; Kim S.-H.; Kim Y.; Kim G. E.; Lee Y.; Kim T.; Choi H.-R.; Suhr J.; Nam J.-D. Solvent-Free Bulk Polymerization of Lignin-Polycaprolactone (PCL) Copolymer and Its Thermoplastic Characteristics. Sci. Rep. 2019, 9, 7033 10.1038/s41598-019-43296-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Wu H.; Kessler M. R. K. High Bio-Content Polyurethane Composites with Urethane Modified Lignin as Filler. Polymer 2015, 69, 52–57. 10.1016/j.polymer.2015.05.046. [DOI] [Google Scholar]
- Jiang S.; Kai D.; Dou Q. Q.; Loh X. J. Multi-Arm Carriers Composed of an Antioxidant Lignin Core and Poly(Glycidyl Methacrylate-Co-Poly(Ethylene Glycol)Methacrylate) Derivative Arms for Highly Efficient Gene Delivery. J. Mater. Chem. B 2015, 3, 6897–6904. 10.1039/C5TB01202C. [DOI] [PubMed] [Google Scholar]
- Buono P.; Duval A.; Verge P.; Averous L.; Habibi Y. New Insights on the Chemical Modification of Lignin: Acetylation versus Silylation. ACS Sustainable Chem. Eng. 2016, 4, 5212–5222. 10.1021/acssuschemeng.6b00903. [DOI] [Google Scholar]
- Buono P.; Duval A.; Averous L.; Habibi Y. Thermally Healable and Remendable Lignin-Based Materials through Diels – Alder Click Polymerization. Polymer 2017, 133, 78–88. 10.1016/j.polymer.2017.11.022. [DOI] [Google Scholar]
- Salanti A.; Zoia L.; Simonutti R.; Orlandi M. Epoxidized Lignin Derivatives as Bio-Based Cross-Linkers Used in the Preparation of Epoxy Resins. BioResources 2018, 13, 2374–2396. 10.15376/biores.13.2.2374-2396. [DOI] [Google Scholar]
- Delgado N.; Ysambertt F.; Chávez G.; Bravo B.; García D. E.; Santos J. Valorization of Kraft Lignin of Different Molecular Weights as Surfactant Agent for the Oil Industry. Waste Biomass Valorization 2019, 10, 3383–3395. 10.1007/s12649-018-0352-4. [DOI] [Google Scholar]
- Duval A.; Avérous L. Cyclic Carbonates as Safe and Versatile Etherifying Reagents for the Functionalization of Lignins and Tannins. ACS Sustainable Chem. Eng. 2017, 5, 7334–7343. 10.1021/acssuschemeng.7b01502. [DOI] [Google Scholar]
- Liu L.-Y.; Cho M.; Sathitsuksanoh N.; Chowdhury S.; Renneckar S. Uniform Chemical Functionality of Technical Lignin Using Ethylene Carbonate for Hydroxyethylation and Subsequent Greener Esterification. ACS Sustainable Chem. Eng. 2018, 6, 12251–12260. 10.1021/acssuschemeng.8b02649. [DOI] [Google Scholar]
- Sun Y.; Ma Z.; Xu X.; Liu X.; Liu L.; Huang G.; Liu L.; Wang H.; Song P. Grafting Lignin with Bioderived Polyacrylates for Low-Cost, Ductile, and Fully Biobased Poly(Lactic Acid) Composites. ACS Sustainable Chem. Eng. 2020, 8, 2267–2276. 10.1021/acssuschemeng.9b06593. [DOI] [Google Scholar]
- Kajihara M.; Aoki D.; Matsushita Y.; Fukushima K. Synthesis and Characterization of Lignin-Based Cationic Dye-Flocculant. J. Appl. Polym. Sci. 2018, 135, 46611 10.1002/app.46611. [DOI] [Google Scholar]
- Setälä H.; Alakomi H.-L.; Paananen A.; Szilvay G. R.; Kellock M.; Lievonen M.; Liljeström V.; Hult E.-L.; Lintinen K.; Österberg M.; et al. Lignin Nanoparticles Modified with Tall Oil Fatty Acid for Cellulose Functionalization. Cellulose 2020, 27, 273–284. 10.1007/s10570-019-02771-9. [DOI] [Google Scholar]
- Zhao S.; Abu-Omar M. M. Synthesis of Renewable Thermoset Polymers through Successive Lignin Modification Using Lignin-Derived Phenols. ACS Sustainable Chem. Eng. 2017, 5, 5059–5066. 10.1021/acssuschemeng.7b00440. [DOI] [Google Scholar]
- Grossman A.; Vermerris W. Lignin-Based Polymers and Nanomaterials. Curr. Opin. Biotechnol. 2019, 56, 112–120. 10.1016/j.copbio.2018.10.009. [DOI] [PubMed] [Google Scholar]
- Ganewatta M. S.; Lokupitiya H. N.; Tang C. Lignin Biopolymers in the Age of Controlled Polymerization. Polymers 2019, 11, 1176 10.3390/polym11071176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan A.; Fatehi P. Self-Assembly of Kraft Lignin-Acrylamide Polymers. Colloids Surf., A 2019, 572, 230–236. 10.1016/j.colsurfa.2019.04.002. [DOI] [Google Scholar]
- Hong N.; Li Y.; Zeng W.; Zhang M.; Peng X.; Qiu X. Ultrahigh Molecular Weight, Lignosulfonate-Based Polymers: Preparation, Self-Assembly Behaviours and Dispersion Property in Coal–Water Slurry. RSC Adv. 2015, 5, 21588–21595. 10.1039/C5RA02157J. [DOI] [Google Scholar]
- Liu Z.; Qie R.; Li W.; Hong N.; Li Y.; Li C.; Wang R.; Shi Y.; Guo X.; Jia X. Preparation of Avermectin Microcapsules with Anti-Photodegradation and Slow-Release by the Assembly of Lignin Derivatives. New J. Chem. 2017, 41, 3190–3195. 10.1039/C6NJ03795J. [DOI] [Google Scholar]
- Yiamsawas D.; Baier G.; Thines E.; Landfester K.; Wurm F. R. Biodegradable Lignin Nanocontainers. RSC Adv. 2014, 4, 11661–11663. 10.1039/C3RA47971D. [DOI] [Google Scholar]
- Liu K.; Zheng D.; Lei H.; Liu J.; Lei J.; Wang L.; Ma X. Development of Novel Lignin-Based Targeted Polymeric Nanoparticle Platform for Efficient Delivery of Anticancer Drugs. ACS Biomater. Sci. Eng. 2018, 4, 1730–1737. 10.1021/acsbiomaterials.8b00260. [DOI] [PubMed] [Google Scholar]
- Zigoneanu I. G.; Astete C. E.; Sabliov C. M. Nanoparticles with Entrapped α-Tocopherol: Synthesis, Characterization, and Controlled Release. Nanotechnology 2008, 19, 105606 10.1088/0957-4484/19/10/105606. [DOI] [PubMed] [Google Scholar]
- Astete C. E.; Sabliov C. M. Synthesis and Characterization of PLGA Nanoparticles. J. Biomater. Sci., Polym. Ed. 2006, 17, 247–289. 10.1163/156856206775997322. [DOI] [PubMed] [Google Scholar]
- Riley T.; Heald C. R.; Stolnik S.; Garnett M. C.; Illum L.; Davis S. S.; King S. M.; Heenan R. K.; Purkiss S. C.; Barlow R. J.; et al. Core–Shell Structure of PLA–PEG Nanoparticles Used for Drug Delivery. Langmuir 2003, 19, 8428–8435. 10.1021/la020911h. [DOI] [Google Scholar]
- Yang B.; Lowe J. P.; Schweins R.; Edler K. J. Small Angle Neutron Scattering Studies on the Internal Structure of Poly(Lactide-Co-Glycolide)-Block-Poly(Ethylene Glycol) Nanoparticles as Drug Delivery Vehicles. Biomacromolecules 2015, 16, 457–464. 10.1021/bm501519u. [DOI] [PubMed] [Google Scholar]
- Schneider G. J.; Vollnhals V.; Brandt K.; Roth S. V.; Göritz D. Correlation of Mass Fractal Dimension and Cluster Size of Silica in Styrene Butadiene Rubber Composites. J. Chem. Phys. 2010, 133, 094902 10.1063/1.3469827. [DOI] [PubMed] [Google Scholar]
- Rice S. A.Small Angle Scattering of X-Rays; Guinier A.; Fournet G., Eds.; Translated by C. B. Wilson and with a Bibliographical Appendix by K. L. Yudowitch. Wiley: New York, 1955. [Google Scholar]
- Gupta S.; Camargo M.; Stellbrink J.; Allgaier J.; Radulescu A.; Lindner P.; Zaccarelli E.; Likos C. N.; Richter D. Dynamic Phase Diagram of Soft Nanocolloids. Nanoscale 2015, 7, 13924–13934. 10.1039/C5NR03702F. [DOI] [PubMed] [Google Scholar]
- Gosecka M.; Gosecki M. Characterization Methods of Polymer Core–Shell Particles. Colloid Polym. Sci. 2015, 293, 2719–2740. 10.1007/s00396-015-3728-z. [DOI] [Google Scholar]
- Rueden C. T.; Schindelin J.; Hiner M. C.; DeZonia B. E.; Walter A. E.; Arena E. T.; Eliceiri K. W. ImageJ2: ImageJ for the next Generation of Scientific Image Data. BMC Bioinformatics 2017, 18, 671 10.1186/s12859-017-1934-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolnik B. S.; Burgess D. J. Effect of Acidic PH on PLGA Microsphere Degradation and Release. J. Controlled Release 2007, 122, 338–344. 10.1016/j.jconrel.2007.05.034. [DOI] [PubMed] [Google Scholar]
- Rosov N.; Rathgeber S.; Monkenbusch M. Neutron Spin Echo Spectroscopy at the NIST Center for Neutron Research. ACS Symp. Ser. 1999, 103–116. 10.1021/bk-2000-0739.ch007. [DOI] [Google Scholar]
- Heller W. T.; Cuneo M.; Debeer-Schmitt L.; Do C.; He L.; Heroux L.; Littrell K.; Pingali S. V.; Qian S.; Stanley C.; et al. The Suite of Small-Angle Neutron Scattering Instruments at Oak Ridge National Laboratory. J. Appl. Crystallogr. 2018, 51, 242–248. 10.1107/S1600576718001231. [DOI] [Google Scholar]
- Kline S. R. Reduction and Analysis of SANS and USANS Data Using IGOR Pro. J. Appl. Crystallogr. 2006, 39, 895–900. 10.1107/S0021889806035059. [DOI] [Google Scholar]
- Smolsky I. L.; Liu P.; Niebuhr M.; Ito K.; Weiss T. M.; Tsuruta H. Biological Small-Angle X-Ray Scattering Facility at the Stanford Synchrotron Radiation Laboratory. J. Appl. Crystallogr. 2007, 40, s453–s458. 10.1107/S0021889807009624. [DOI] [Google Scholar]
- McPhillips T. M.; McPhillips S. E.; Chiu H.-J.; Cohen A. E.; Deacon A. M.; Ellis P. J.; Garman E.; Gonzalez A.; Sauter N. K.; Phizackerley R. P.; et al. Blu-Ice and the Distributed Control System: Software for Data Acquisition and Instrument Control at Macromolecular Crystallography Beamlines. J. Synchrotron Radiat. 2002, 9, 401–406. 10.1107/S0909049502015170. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.