

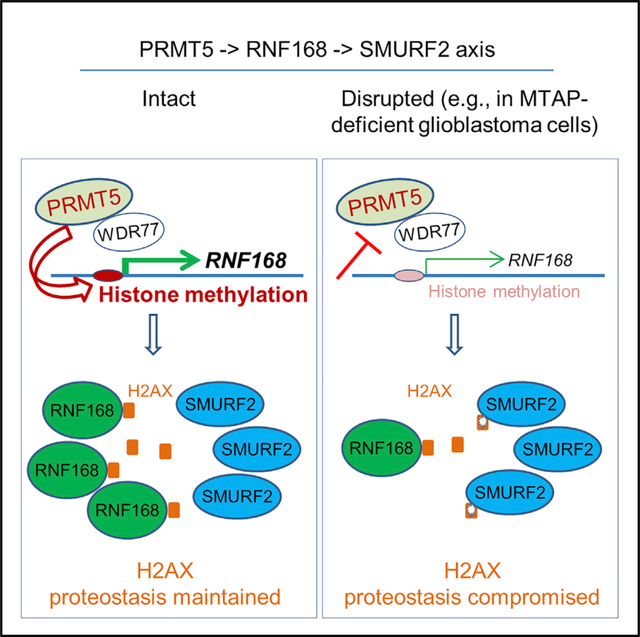
SUMMARY
H2AX safeguards genomic stability in a dose-dependent manner; however, mechanisms governing its proteostasis are poorly understood. Here, we identify a PRMT5-RNF168-SMURF2 cascade that regulates H2AX proteostasis. We show that PRMT5 sustains the expression of RNF168, an E3 ubiquitin ligase essential for DNA damage response (DDR). Suppression of PRMT5 occurs in methylthioadenosine phosphorylase (MTAP)-deficient glioblastoma cells and attenuates the expression of RNF168, leading to destabilization of H2AX by E3 ubiquitin ligase SMURF2. RNF168 and SMURF2 serve as a stabilizer and destabilizer of H2AX, respectively, via their dynamic interactions with H2AX. In supporting an important role of this signaling cascade in regulating H2AX, MTAP-deficient glioblastoma cells display higher levels of DNA damage spontaneously or in response to genotoxic agents. These findings reveal a regulatory mechanism of H2AX proteostasis and define a signaling cascade that is essential to DDR and that is disrupted by the loss of a metabolic enzyme in tumor cells.
In Brief
Du et al. identify a signaling cascade that regulates the abundance of H2AX, an essential protein in mediating the DNA damage response. The study links the effect of MTAP loss, a common genetic alteration in cancers, to cancer cells’ response to DNA damage insults (e.g., genotoxic agents).
Graphical Abstract
INTRODUCTION
Cells respond to DNA double-strand breaks (DSBs) by activating the DNA damage response (DDR) machinery. Central to the proper functioning of the DDR machinery is the phosphorylation of H2AX (γH2AX) and its localization to the DSB sites. These events are accompanied by further functionally critical posttranslational modifications on H2AX, the recruitment of additional DNA repair and checkpoint proteins, and the activation of downstream DNA damage signaling cascade (Yuan et al., 2010). As such, H2AX plays essential roles in maintaining genome stability and suppressing tumorigenesis, and unsurprisingly, it does so in a dose-sensitive manner (Bassing et al., 2003; Celeste et al., 2003). Intriguingly, despite the strong evidence linking H2AX’s function to its protein abundance, the fundamental aspect of its biochemistry, the regulation of its proteostasis, remains mostly unclear.
Human cells employ a sophisticated proteostasis network to maintain proteome homeostasis and integrity in development and diseases (Sala et al., 2017). It has been well established that cancer cells rewire their proteasome system to adapt to cellular stress and meet proliferation needs, such that proteasome targeting has been an actively pursued cancer therapeutic strategy (Liu and Ye, 2011; Manasanch and Orlowski, 2017). The ubiquitin proteasome system, including three major types of E3 ligases grouped by their characteristic domains and mechanisms of ubiquitin transferring, is a major pathway responsible for controlling protein proteostasis (Morreale and Walden, 2016; Schnell and Hicke, 2003). Two previous studies suggest a link between H2AX’s ubiquitination and its destabilization. For example, it was found that upon DSBs, H2AX is stabilized by a signal cascade induced by ATM kinase to allow efficient γH2AX foci formation (Atsumi et al., 2015). In addition, under oxidative stress, H2AX in breast cancer cells was found to undergo degradation associated with an enhanced interaction with RNF168, a RING-type E3 ligase (Gruosso et al., 2016). Paradoxically, most studies point to major roles of ubiquitination in modifying the functionality of H2AX; it has been well established that coordinated mono- and poly-ubiquitination by E3 ubiquitin ligases, notably RING finger protein 8 (RNF8) and RING finger protein 168 (RNF168), are critical in DDR protein assembly and signaling cascade activation (Huen et al., 2007; Mailand et al., 2007; Mattiroli et al., 2012).
Our initial research focuses on the roles of methylthioadenosine phosphorylase (MTAP) loss in tumorigenesis, cancer progression, and shaping cancer cell biology. MTAP loss, via homozygous deletion or epigenetic silencing, is common in numerous cancer types, including sarcomas, and cancers of breast, lung, skin, and blood. In particular, MTAP loss is prevalent in glioblastoma (GBM); it occurs in half of all patients (Behrmann et al., 2003; Gao et al., 2013; Hellerbrand et al., 2006; Li et al., 2014). Normally, MTAP functions by metabolizing the byproduct of the polyamine pathway, methylthioadenosine (MTA), to eventually produce adenine and methionine, resulting in the salvage of these metabolites (Kamatani and Carson, 1980; Kryukov et al., 2016; Mavrakis et al., 2016). Recent studies revealed that the MTAP-loss-induced accumulation of MTA inhibits protein arginine methyltransferase 5 (PRMT5) and by doing so renders tumor cells more sensitive to PRMT5 inhibition (Kryukov et al., 2016; Mavrakis et al., 2016). To gain new insight into the pathogenic consequences underlying MTAP loss in GBMs, we investigated the effect of MTAP loss on gene expression and various cellular processes in GBM cells. These efforts led us to uncover a previously unknown signaling cascade regulating H2AX proteostasis in GBM cells. Specifically, we identify that SMURF2, a homologous to the E6-AP carboxyl terminus (HECT)-type E3 ubiquitin ligase, is a negative regulator of H2AX stability and that RNF168 acts as a H2AX stabilizer by countering the effects of SMURF2. We further demonstrate that the sustained expression of RNF168 requires PRMT5; when PRMT5 activity is suppressed, as is the case in MTAP-deficient GBM cells, RNF168 downregulation results in compromised H2AX proteostasis, which is associated with higher levels of spontaneous or induced DSBs.
RESULTS AND DISCUSSION
H2AX Proteostasis Is Compromised in MTAP-Deficient GBM Cells
MTAP loss occurs in about half of all GBM patients (Behrmann et al., 2003; Gao et al., 2013; Hellerbrand et al., 2006; Li et al., 2014). While two major consequences of MTAP loss in tumor cells—defective purine production (Bertino et al., 2011; Lubin and Lubin, 2009) and attenuation of PRMT5 activity (Kryukov et al., 2016; Marjon et al., 2016; Mavrakis et al., 2016)—have provided potential therapeutic opportunities, the effects of the loss of this metabolic enzyme on key cellular processes remain unknown. We found that ablating MTAP function, via treatment with methylthio-DADMe-Immucillin-A (MTDIA), an inhibitor of MTAP’s enzymatic activity (Basu et al., 2011), or MTAP knockout, sensitized GBM cell lines to radiation or doxorubicin (Figures S1A–S1C). MTAP-deficient cells also demonstrated higher levels of basal and induced DSBs, as measured by alkaline comet assays (Figures 1A–1C, S1D, and S1E). Unexpectedly, this higher level of DSBs was not accompanied by increased gH2AX, an early marker for cellular response to DSBs (Fernandez-Capetillo et al., 2004) (Figures 1D, 1E, and S2A). We monitored the formation of gH2AX foci and the total gH2AX levels at different time points following a DNA damage stimulation. We found that in MTAP-deficient cells, γH2AX induction was delayed and weakened, yet extended, consistent with a compromised DDR and an attenuated capacity to resolve the DNA damage (Figures 1F–1H and S2B–S2E). Indeed, we observed a reduced basal level of total H2AX protein upon MTAP inhibition or MTAP knockout in GBM cell lines and in a transformed human astrocyte model (Hansen et al., 2019; Li et al., 2016) (Figures 1D, 1E, 1G–1I, 2A, and 2B). Chromatin fractionation experiments revealed this effect of MTAP loss in both chromatin-associated and chromatin-free H2AX fractions (Figure 2C). Notably, this MTAP-inhibition-induced reduction in H2AX protein was not associated with a lower level of H2AFX transcript (Figure 2D) and was rescued by the proteasome inhibitor MG132 (Figure 2E). H2AX stability assays revealed an accelerated rate of H2AX degradation in the MTAP-deficient cells (Figure 2F).
Figure 1. Loss of MTAP Compromises GBM Cells’ Response to DNA Damage.

(A) T98G cells, pre-treated with vehicle control (DMSO) or with MTDIA (3 μM, 24 h), were exposed to doxorubicin (0.1 μM) for 24 h (in the presence of DMSO or MTDIA), and an alkaline comet assay was performed. Representative image of alkaline comet assays and quantification of the tail moment are shown.
(B) Quantification of the tail moment for U251MG cells that were pre-treated with DMSO or MTDIA (3 μM) for 24 h before being exposed to radiation (representative images of the alkaline comet assay are shown in Figure S1D).
(C) Quantification of the tail moment for the parental U251MG cell line and its derivative MTAP-ko line exposed to radiation (representative images of the alkaline comet assay are shown in Figure S1E).
(D) U251MG cells, pre-treated with DMSO or MTDIA, were exposed to doxorubicin (0.1 μM) for 24 h (in the presence of DMSO or MTDIA), and immunoblots for γH2AX and H2AX were performed.
(E) The parental U251MG cell line and its derivative MTAP-ko cell lines were treated with doxorubicin (0.1 μM) for 24 h before immunoblots were performed to determine the abundance of γH2AX and H2AX. An anti-MTAP immunoblot confirmed the loss of MTAP in the MTAP-ko cell line.
(F) Schematic of the doxorubicin treatment time course experiments in U251MG cells.
(G) U251MG cell line (CRISPR control) and its derivative, MTAP-ko line, were treated as depicted in (F), and an anti-γH2AX immunoblot was performed.
(H) Quantification of γH2AX immunoblot bands in (G), as assayed.
(I) Primary human astrocytes (passage 5 or 6) and OMRP (transformed cells; CRISPR control and the matched MTAP-ko lines) were used for immunoblots.
Mann-Whitney U test was used in (A)–(C); Student’s t test was used for (H). *p < 0.05; **p < 0.01; n.s., no significance. Error bars represent mean ± SD.
Figure 2. Loss of MTAP Function Compromises H2AX Proteostasis in GBM Cells.

(A) U251MG cells were treated with different doses of MTDIA for 24 h or treated with MTDIA (3 μM) for indicated time points, and immunoblots were performed to determine the abundance of H2AX (H2B protein level remained unchanged, serving as an additional control).
(B) The abundance of H2AX was examined in parental GBM cell lines, a transformed human astrocyte cell line (OMRP), and their matched MTAP-ko derivatives.An anti-MTAP immunoblot confirmed the loss of MTAP in the MTAP-ko cell line, and an anti-H2B immunoblot served as an additional control for loading. The quantification of the relative abundance of H2AX in each lane is shown in the bottom panel.
(C) U251MG cells were used for chromatin fractionation, and both chromatin-associated and chromatin-free fractions were used for immunoblots (note thespecific and clean immunoblot signaling; the slightly different migration of proteins between the two fractions was most likely due to a difference in buffers).
(D) qRT-PCR determined the level of the H2AFX transcript in U251MG cells following MTDIA treatment for 24 h.
(E) U251MG cells were exposed to DMSO or MTDIA (3 μM), without or with MG132 (10 μM), for 24 h, and immunoblotting was performed to determine the abundance of H2AX (an anti-p53 immunoblot served as an additional control confirming the effect of MG132).
(F) Parental U251MG or its MTAP-ko derivative line was treated with cycloheximide for indicated time points, and the abundance of H2AX was determined by immunoblotting (quantification of relative H2AX abundance compared to the starting time point is shown in the bottom panel).
(G) U251MG cell line knockdown control (shCtrl) or H2AFX knockdown (shH2AFX) were used for alkaline comet assays, and quantification of the tail moment is shown (representative images of the comet assay are shown in Figure S3B).
(H) U251MG cell lines (CRISPR control or MTAP-ko), without or with overexpression of exogenous H2AX, were used for alkaline comet assays, and quantification of the tail moment is shown (representative images of the alkaline comet assay are shown in Figure S3D).
Student’s t-test was used for statistical analysis, except for (G) and (H), where Mann-Whitney U test was used. *p < 0.05, **p < 0.01. Error bars represent mean ± SD.
Subsequent experiments validated the contribution of reduced H2AX abundance to the higher level of DSBs in MTAP-deficient GBM cells. First, a loss of H2AX, induced by H2AFX knockdown (Figure S3A), led to a higher level of DSBs than was observed in MTAP-deficient cells (Figures 2G and S3B). Furthermore, overexpression of exogenous H2AX in MTAP null GBM cells partially mitigated MTAP-loss-induced DSBs (Figures 2H, S3C, and S3D). These findings were in agreement with the critical, dose-dependent function of H2AX in the DDR (Bassing et al., 2003; Celeste et al., 2003).
Collectively, these results suggest that MTAP loss-of-function compromises GBM cells’ response to DSBs, and it promotes proteasome-mediated degradation of H2AX. They also support that lower H2AX abundance, potentially together with other factors such as compromised PRMT5 activity (Hamard et al., 2018), contributes to the higher level of DSBs in MTAP null cells.
RNF168 Expression Is Attenuated in MTAP-Deficient GBM Cells
To gain further insight into the effect of MTAP deficiency on the DDR, we analyzed the correlation between the expression of MTAP and DDR genes in GBMs from the The Cancer Genome Atlas (TCGA) via the GlioVis data portal (Bowman et al., 2017). The expression of CDKN2A, a gene that is frequently co-deleted with MTAP (Gao et al., 2013), displayed a positive correlation with that of MTAP, serving as a control (Figure S4A). We then examined the expression of 193 previously curated genes involved in DDR, repair, and genome stability (Chae et al., 2016; Wood et al., 2005). Among the 170 genes for which expression information was available, 64% (108/170) displayed a significant correlation with the expression level of MTAP, within which 73% (79/108) showed a reduced expression in the low MTAP group of GBMs (Table S1). Consistent with the result from the MTDIA treatment, no significant correlation between MTAP and H2AFX expression was found (Table S1). One of the most strongly correlated genes was RNF168, which encodes a RING finger class E3 ubiquitin ligase that regulates ubiquitination of H2AX during DDR and DNA repair (Gatti et al., 2015; Luijsterburg et al., 2017) and is an essential mediator of cellular response to and repairs of DSBs (Doil et al., 2009; Stewart et al., 2009). Six independent datasets were examined (Bao et al., 2014; Bowman et al., 2017; Gravendeel et al., 2009; Madhavan et al., 2009), and in each case, a reduced level of RNF168 transcript was detected in low MTAP-expressing GBMs compared with the high MTAP-expressing group (Figure S4B). Notably, other E3 ubiquitin ligases that have been found to function along with RNF168 in this particular cellular process—RNF2, RNF8, and HUWE1 (Atsumi et al., 2015; Huen et al., 2007; Mailand et al., 2007; Wu et al., 2011)—displayed no such correlation with MTAP expression (Figures S4C–S4E).
Three lines of evidence support a direct regulatory relationship between MTAP deficiency and RNF168 transcripts. First, MTAP knockout (MTAP-ko) GBM cell lines had a reduced level of RNF168 expression (both transcript and protein) when compared to their matched parental lines (Figures 3A and 3B). Second, the treatment of the MTAP-intact parental GBM cell lines with MTDIA (Basu et al., 2011) also led to a downregulated expression of RNF168 (Figure 3C). Finally, restoration of MTAP expression in a GBM-patient-derived MTAP null GBM cell line resulted in upregulated RNF168 transcription (Figure 3D). This regulatory relationship exists for RNF168, but not for its functionally close partner E3 ubiquitin ligases, RNF8 (Huen et al., 2007; Mailand Et al.,2007) and RNF2 (Pan et al., 2011) (Figures S4F–S4I). The expected, important role of RNF168in mediating cellular response to DNA damage in the context of GBM cells was confirmed by an alkaline comet assay (Figures 3E and 3F). Collectively, these results suggest that MTAP loss in GBMs leads to a reduced expression of RNF168, providing a mechanism linking MTAP loss to GBM cells’ compromised response to DNA damage.
Figure 3. RNF168 Expression Is Attenuated in MTAP-Deficient GBM Cells.

(A and B) The expression of RNF168 was determined in the parental U251MG cell line and its derivative MTAP-ko cell line by (A) qRT-PCR and (B) immunoblotting (the quantification of the relative abundance of RNF168 in each lane is shown in the right panel).
(C) GBM cell lines were treated with DMSO or MTDIA (3 μM) for 24 h, and the expression of RNF168 was determined by qRT-PCR (left panel) and by immunoblotting (right panel).
(D) The RNF168 transcript level in a patient-derived, naturally MTAP null GBM cell line (GBM #13–0302), without or with exogenous MTAP restored (via retroviral delivery, denoted MTAP+), was determined by qRT-PCR (note that no MTAP transcript was detected in the “vector” line).
(E) Immunoblot detection of RNF168 in U251MG cells without (shCtrl) or with knockdown of RNF168 (shRNF168).
(F) U251MG cell lines, without or with RNF168 knockdown, were treated with vehicle control (DMSO) or with doxorubicin (0.1 μM, 24 h), and an alkaline comet assay was performed. Quantification of the tail moment is shown in the right panel.
Student’s t-test was used for statistical analysis, except for tail-moment quantification, where Mann-Whitney U test was used. *p < 0.05, **p < 0.01. Error bars represent mean ± SD.
PRMT5 Activates RNF168 Transcription by Maintaining the Activating Histone Marks in the Promoter
A direct downstream molecular target suppressed by MTAP loss in cancer cells is PRMT5 (Kryukov et al., 2016; Marjon et al., 2016; Mavrakis et al., 2016), a bifunctional transcriptional regulator (Chen et al., 2017; Deng et al., 2017; Fabbrizio et al., 2002; LeBlanc et al., 2012; Tarighat et al., 2016). This led us to hypothesize that PRMT5 positively regulates the transcription of RNF168 in GBM cells. We found that shRNA-mediated knockdown of PRMT5 in GBM cells led to a reduction of RNF168 expression (Figures 4A and S5A), and overexpression of PRMT5 resulted in upregulated RNF168 expression (Figures 4B and S5B). In corroborating these findings, a selective small molecular inhibitor of PRMT5, EPZ015666 (Chan-Penebre et al., 2015), similarly caused a reduced level of RNF168, but not of RNF8, in GBM cells (Figure 4C).
Figure 4. PRMT5 Is Essential in Maintaining the Transcription of RNF168 in GBM Cells.

(A) The expression of RNF168 in U251MG cells without (shCtrl) or with PRMT5 knockdown (shPRMT5) was determined by qRT-PCR and by anti-RNF168 immunoblotting (PRMT5 knockdown was confirmed by an anti-PRMT5 immunoblot, as shown in the right panel).
(B) The expression of RNF168 in U251MG cells without (vector) or with exogenous PRMT5 overexpression (PRMT5+) was determined by qRT-PCR and by anti-RNF168 immunoblotting (PRMT5 overexpression was confirmed by an anti-PRMT5 immunoblot, as shown in the right panel).
(C) The expression of RNF168 was determined in GBM cell lines treated with a PRMT5 inhibitor, EPZ015666 (0.3 μM for 24 h), by immunoblotting (an anti-RNF8 immunoblot served as a control to confirm the effect was specific for RNF168). Anti-H3R8me2s (s, symmetrical) was used as the control to confirm the inhibitor’s effect.
(D) Chromatin immunoprecipitation (ChIP)-qPCR was performed to determine the PRMT5-associated gene activation histone mark in the promoter of RNF168 (fragment 4 [F4], as illustrated in Figure S5E) in the parental U251MG cell line and its MTAP-ko derivative line.
(E) ChIP-qPCR was performed to determine the PRMT5-associated gene activation histone mark in the promoter of RNF168 (F4) in U251MG without (shCtrl) or with PRMT5 knockdown (shPRMT5).
(F) ChIP-qPCR was performed to determine the association of PRMT5 with the promoter of RNF168 (F1–F6, as illustrated in Figure S5E) in U251MG cells.
(G) U251MG cell lines, without or with PRMT5 knockdown, were treated with vehicle control (DMSO) or doxorubicin (0.1 μM, 24 h), and an alkaline comet assay was performed. Quantification of the tail moment is shown in the right panel.
Student’s t-test was used, except for quantification of tail moment, where Mann-Whitney U test was used. *p < 0.05, **p < 0.01. Error bars represent mean ± SD.
PRMT5 can activate gene transcription through modulating histone methylation, most notably mono-methylation on arginine 2 and symmetric di-methylation on arginine 8 of histone 3 (H3R2me1 and H3R8me2s, respectively) (Chen et al., 2017; Deng et al., 2017; LeBlanc et al., 2012; Tarighat et al., 2016). MTDIA or EPZ015666 treatment led to a global reduction in H3R2me1, H3R8me2s, and to a lesser extent, symmetric methylation of histone H4 arginine 3 (H4R3me2s, a PRMT5-mediated gene suppression marker) (Zhao et al., 2009) in the U251MG cell line (Figures S5C and S5D). We then examined these histone marks in the ∼1.5-kilobase (kb) region spanning the promoter of RNF168 and revealed positive enrichment for H3R2me1 and H3R8me2s, but not for H4R3me2s, in the ∼200 base pair (bp) upstream of the RNF168 transcriptional start site (Figure S5E). Knockout of MTAP or knockdown of PRMT5 led to reduced H3R2me1 in the same ∼200-bp locus (Figures 4D and 4E). Finally, we confirmed the presence of PRMT5 in the promoter region of RNF168, particularly in the ∼200 bp surrounding the transcriptional start site (Figure 4F). Thus, these results uncovered an essential role of PRMT5 in sustaining RNF168 expression: PRMT5 accomplishes this by adding activating histone methylation marks, including H3R2me1 and potentially H3R2me2s, likely in concert with another epigenetic regulator, PRMT7 (Migliori et al., 2012). PRMT5 has been shown to function in the cellular response to DNA damage via post-translationally modifying RUVBL1 and consequentially potentiating TIP60’s enzymatic activity (Clarke et al., 2017). This prompted us to test whether PRMT5-dependent expression of RNF168 was affected by DNA damage. We found that PRMT5-mediated RNF168 expression was not further potentiated by DNA damage, suggesting that PRMT5 regulates steady-state RNF168 expression (Figure S5F). As RNF168 is a critical protein in driving DDR signaling (Mattiroli et al., 2012), these results suggest that PRMT5 also drives the cellular response to DNA damage via a transcriptional regulation mechanism and provides a mechanism explaining the reduced expression level of RNF168 in MTAP-deficient GBM cells. Consistent with such a function and the DSB phenotypes in MTAP-deficient cells, knockdown of PRMT5 resulted in a higher DSB level (Figure 4G).
The PRMT5-RNF168 Axis Maintains the Proteostasis of H2AX
The stepwise, causative events of MTAP deficiency, attenuated PRMT5 activity, downregulated RNF168 expression, and lower abundance of the H2AX protein in MTAP-deficient cells led us to hypothesize that the PRMT5-RNF168 axis regulates H2AX proteostasis. Several complementary results support this hypothesis. First, exogenous expression of RNF168 led to a higher basal level of the H2AX protein (Figure 5A) and protected H2AX from MTDIA-induced reduction (Figure 5B). Conversely, knockdown of RNF168 led to a lower level of H2AX (Figure 5C). This effect was reversed by the proteasome inhibitor, MG132 (Figure 5D), reminiscent of what was observed in MTAP-deficient GBM cells. In agreement with the essential role of PRMT5 in sustaining RNF168 expression, overexpression of PRMT5, its essential co-activator WDR77 (Burgos et al., 2015), or both similarly stabilized H2AX (Figures 5E and 5F). Complementarily, knockdown of PRMT5 or WDR77 resulted in an opposite effect on H2AX (Figures 5G and 5H). Finally, an H2AX protein abundance was similarly reduced by the PRMT5 inhibitor, EPZ015666 (Figures 5I–5K), and was rescued by MG132 (Figure 5L), confirming the stabilizing effects of the PRMT5-RNF168 axis on H2AX proteostasis. Notably, MTAP-expressing cells displayed a stronger response to EPZ015666 when compared to their MTAP-deficient counterparts (Figure 5K), in agreement with the previous finding that PRMT5 activity is compromised in MTAP-deficient cells (Kryukov et al., 2016; Marjon et al., 2016; Mavrakis et al., 2016).
Figure 5. The PRMT5-RNF168 Axis Controls the Proteostasis of H2AX.

(A) The abundance of H2AX in U251MG cells, without or with exogenous RNF168 overexpression (RNF168+), was determined by immunoblotting.
(B) The abundance of H2AX in U251MG cell lines (control or exogenous RNF168-overexpressing line) in response to treatment with MTDIA (3 μM, 24 h) was determined by immunoblotting.
(C) The abundance of H2AX in GBM cell lines, without or with RNF168 knockdown (shRNF168), was determined by immunoblotting (an anti-RNF168 immunoblot confirmed the knockdown of RNF168).
(D) U251MG cells, without or with RNF168 knockdown, were treated with vehicle control or with MG132 (10 μM), and the abundance of H2AX was determined by immunoblotting (the quantification of the relative abundance of H2AX in each lane is shown in the bottom panel).
(E) The abundance of H2AX without or with exogenous PRMT5 overexpression in U251MG cells was determined by immunoblotting (the quantification of the relative abundance of H2AX in each lane is shown in the bottom panel).
(F) U251MG cell lines overexpressing exogenous PRMT5, PRMT5 coactivator WDR77, or both were used for immunoblots with indicated antibodies.
(G) The abundance of H2AX in control or in PRMT5 knockdown U251MG cells was determined by immunoblotting (the quantification of H2AX abundance in each lane is shown in the bottom panel).
(H) The abundance of RNF168 and H2AX in U251MG cells, without or with WDR77 knockdown, was determined by immunoblots (WDR77 knockdown was confirmed by anti-WDR77 immunoblot).
(I) Immunoblot detection of H2AX in U251MG cells treated with EPZ015666 for 24 h (anti-H3R8me2s, a histone methylation mark induced by PRMT5, served as acontrol to confirm the blockage of the PRMT5’s function; anti-H3 served as an additional negative/loading control). “s” in H3R8me2s denotes “symmetrical.”
(J) OMRP cells were treated with the PRMT5 inhibitor EPZ015666 (0.3 μM, 24 h), and immunoblots were performed.
(K) GBM cell line U138MG (naturally MTAP null), without or with exogenous MTAP restoration, was treated with EPZ015666 (0.3 μM, 24 h), and immunoblots were performed (note the expected increased RNF168 and H2AX abundance when MTAP was restored).
(L) U251MG cells were treated with vehicle or with EPZ015666 (0.3 μM, 24 h), with or without the presence of MG132 (10 μM), and immunoblotting was performed to determine the abundance of H2AX. Error bars represent mean ± SD.
Several confounding factors that could potentially alter the H2AX abundance have been ruled out in the following experiments. First, another functionally H2AX-related gene, APEX2 (Willis et al., 2013), also displayed a strong correlation with MTAP in GBMs (Table S1) and was found to positively regulate H2AX abundance (Figure S6A). However, APEX2’s expression was not attenuated by the loss of MTAP in several GBM cell line models (Figures S6B and S6C), suggesting the correlation in GBMs was not due to a direct causative effect exerted by MTAP status and that APEX2 was not a contributing factor in inducing H2AX reduction in MTAP-deficient GBM cells. Second, chronic oxidative stress was found to promote H2AX degradation in breast cancer cells (Gruosso et al., 2016), yet we found no difference in the reactive oxygen species (ROS) level between MTAP-deficient and control cells (Figure S6D). Finally, cell proliferation, which was found to affect H2AX stability (Atsumi et al., 2015), was not a contributing factor, as MTAP-deficient cells displayed no difference in proliferation compared to the control MTAP-intact GBM cells (Figure S6E).
Collectively, these results support the notion that the PRMT5-RNF168 axis functions as an important regulator, potentially together with other factors, in maintaining the H2AX homeostasis in GBMs.
SMURF2 Interacts with and Destabilizes H2AX
RNF168 and HUWE1 are two E3 ligases previously found to act as negative regulators of H2AX stability under specific circumstances. The role of RNF168 was described in the context of chronic oxidative stress in breast cancer cells (Gruosso et al., 2016), opposite to the described results here. The function of HUWE1 as a negative regulator of H2AX was implicated in the process of cells’ initial response to DNA damage (Atsumi et al., 2015), a scenario that was ruled out by HUWE1 knockdown (Figure S7A). To identify the E3 ubiquitin ligase responsible for the negative proteostasis of H2AX, we initially tested an inhibitor, MLN4924, that blocks the function of the well-established family of Cullin-Ring E3 ligases (Soucy et al., 2009). Treating GBM cells with this inhibitor led to a reduction in the H2AX protein level (Figure S7B), in agreement with the aforementioned protective effect of RNF168, a RING-type E3 ligase, on H2AX. Next, we tested the effect of Heclin, a specific inhibitor of HECT-type E3 ligases, without affecting RING ligases (Mund et al., 2014). In contrast to MLN4924, Heclin induced a potent upregulation of the H2AX protein in a fashion reminiscent of the effect on TP53, a protein that is susceptible to HECT-type E3 ligase-mediated degradation (Bernassola et al., 2008; Chen et al., 2005) (Figure S7C). Further, it also protected H2AX from MTDIA-induced reduction (Figure S7D), suggesting the activity of HECT-type E3 ligase(s) played a major role in the destabilization of H2AX. The initial study identifying Heclin demonstrated its inhibitory effect on HUWE1 (defined as “other HECTs” based on domain structure) and three NEDD4 family members: SMURF2, NEDD4, and WWP1 (Mund et al., 2014; Scheffner and Kumar, 2014). We therefore examined the role of the latter three E3 ligases and found that only the knockdown of SMURF2 led to a higher level of the H2AX protein (Figures 6A, S7E, and S7F) and protected H2AX from MTDIA-induced degradation (Figure 6B). In complementary experiments, overexpression of exogenous SMURF2 destabilized the H2AX protein (Figure 6C) and concurrently led to a higher level of poly-ubiquitination of H2AX (Figure 6D). Supporting evidence for the role of SMURF2 in facilitating H2AX degradation was obtained via reciprocal co-immunoprecipitation (co-IP) experiments, which revealed the interaction of SMURF2 with H2AX (Figures 6D–6F).
Figure 6. SMURF2 Negatively Regulates H2AX Proteostasis.

(A) Immunoblot detection of H2AX in U251MG and T98G cell lines without or with SMURF2 knockdown (SMURF2 knockdown was confirmed by an anti-SMURF2 immunoblot). The quantification of the relative abundance of H2AX in each lane is shown in the bottom panels.
(B) U251MG cells, without or with SMURF2 knockdown, were treated with vehicle control or with MTDIA (3 μM, 24 h), and immunoblotting was performed to determine the abundance of H2AX.
(C) Immunoblot detection of H2AX in U251MG cells without (vector) or with exogenous Myc-tagged SMURF2 overexpression (SMURF2+), under the treatment of DMSO or MG132 (10 μM, 24 h).
(D) Control U251MG cell line and its derivative lines expressing exogenous FLAG-tagged H2AX, Myc-tagged SMURF2, or both were used for anti-FLAG co-immunoprecipitation and immunoblot analysis to determine the poly-ubiquit ination of H2AX following overexpression of SMURF2. Note the stronger ubiquitination signal (bold arrow) in lane 8 compared to lane 7. The heavy bands (marked by blue arrowhead) were likely due to the antibody used for IP or non-specific immune blot signaling.
(E) U251MG cells expressing exogenous FLAG-tagged H2AX were used for co-immunoprecipitation with control IgG or with an anti-FLAG antibody, and immunoblotting was performed.
(F) U251MG cells expressing exogenous Myc-tagged SMURF2 were used for co-immunoprecipitation with control IgG or with an anti-Myc antibody, and immunoblotting was performed.
(G) A simulation-based model depicts the interaction of SMURF2’s HECT domain with H2AX.
(H) FLAG-tagged H2AX expressing U251MG cell lines, expressing exogenous Myc-tagged SMURF2, or SMURF2ΔHECT mutant were used for anti-FLAG co-immunoprecipitation (co-IP) and immunoblots with indicated antibodies.
(I) The same set of cell lines were used for immunoblots with indicated antibodies. Blue arrowheads denote non-specific bands (likely due to antibodies used for IPs).
Note that for co-immunoprecipitations in (D)–(F) and (H), cells were treated with MG132 (10 μM, to ensure equal H2AX abundance) 1 day before the co-immunoprecipitation experiments. Error bars represent mean ± SD.
To gain further mechanistic insight into the regulation of H2AX by SMURF2, we took advantage of available crystal structures of H2AX (Shao et al., 2012; Singh et al., 2012; Stucki et al., 2005) and of SMURF2’s HECT domain (Chong et al., 2006, 2010; Jäckl et al., 2018; Ogunjimi et al., 2005; Wiesner et al., 2007) to simulate their interaction using a previously described docking tool (Tovchigrechko and Vakser, 2006; Vakser, 1996a, 1996b, 1997; Vakser et al., 1999). Among the resultant docking models (Figures S8A and S8B), the one with the maximum negative free energy change predicts a highly specific interaction, in which the C terminus of H2AX inserts into the α7 helix in the small subdomain of SMURF2’s HECT domain (Figure 6G; Video S1). Consequently, the C terminus of H2AX comes into close proximity with the small subdomain in the HECT’s N-lobe, which is the predicted location of ubiquitin conjugating enzyme (E2) binding (Ogunjimi et al., 2005). In agreement with this model of interaction, the HECT domaintruncated mutant of SMURF2 (SMURF2DHECT), when overexpressed, had a diminished capacity to interact with H2AX (Figure 6H) and failed to reduce H2AX’s abundance (Figure 6I). Collectively, the above experimental results and the structure and interaction modeling suggest that SMURF2 interacts with and destabilizes H2AX.
RNF168 Blocks SMURF2 from Accessing H2AX by Modifying the N Terminus of H2AX
The simulation-based model, together with the opposing effects of SMURF2 and RNF168 on H2AX proteostasis, led us to test the following hypotheses: (1) SMURF2 targets the C terminus of H2AX for ubiquitination and by doing so induces H2AX degradation; and (2) RNF168 protects H2AX from being accessed by SMURF2 by modifying the N terminus of H2AX.
Three lysine (K) residues in H2AX—two in the N-terminal tail (K13 and K15) and one in the C terminus (K119)—are known to be modified by ubiquitination (Gruosso et al., 2016; Mattiroli et al., 2012). Notably, K13 and K15, but not K119, were found to serve as direct substrates for RNF168 in response to DNA damage (Mattiroli et al., 2012). In supporting the ubiquitination of the C-terminal tail K119 by SMURF2, we found that only the mutation of K119 to alanine (A), and not that of K13 or K15, stabilized H2AX upon MTAP inhibition (Figure 7A). In agreement, the mutation of K119 protected H2AX from SMURF2-mediated destabilization (Figure 7B). Furthermore, unlike wild-type (WT) H2AX, the H2AX-K119A mutant displayed no increase in poly-ubiquitination level in response to exogenous SMURF2 expression (Figure 7C). Collectively, these results support that SMURF2 targets the ubiquitination of the C-terminal K119 in mediating H2AX degradation.
Figure 7. The Negative Regulatory Role of SMURF2 in Controlling H2AX Proteostasis Is Opposed by RNF168.

(A) U251MG cell lines without or with the expression of exogenous FLAG-tagged H2AX (wild-type [WT], K13A, K15A, or K119A) were treated with vehicle control or with MTDIA (3 μM) for 24 h, and immunoblotting was performed to detect the abundance of the FLAG-tagged proteins.
(B) U251MG cell lines expressing exogenous FLAG-tagged H2AX (wild-type [WT] or K119A mutant), without or with exogenous Myc-tagged SMURF2 expression, were used for an immunoblot analysis.
(C) Control U251MG cell line and its derivative lines expressing exogenous FLAG-tagged H2AX or H2AX-K119A mutant, each without or with the overexpression of exogenous SMURF2, were used for anti-FLAG co-immunoprecipitation and immunoblot analysis. Note the stronger polyubiquitination signal (bold arrow) in lane 6 when compared to lane 5; heavy bands (marked by blue arrowhead) were non-specific, likely due to the antibody used for IP or non-specific immune blot signaling. The specificity of anti-FLAG IP was confirmed in Figure 6D, lanes 5 and 6.
(D and E) U251MG cells expressing exogenous FLAG-tagged H2AX, (D) without or with RNF168 overexpression or (E) without or with RNF168 knockdown, were used for anti-FLAG co-immunoprecipitation and immunoblot analysis.
(F) U251MG cells expressing exogenous FLAG-tagged H2AX were treated with vehicle control or with MTDIA (3 μM, 24 h) were used for anti-FLAG co-immunoprecipitation, and immunoblotting was performed.
(G) U251MG cells expressing exogenous FLAG-tagged H2AX, treated with vehicle control or with EPZ015666 (0.3 μM, 24 h), were used for anti-FLAG co-immunoprecipitation and immunoblot analysis.
(H) FLAG-tagged H2AX expressing U251MG cell lines, without or with SMURF2 knockdown, were used for anti-FLAG co-immunoprecipitation and immunoblotswith indicated antibodies
(I) FLAG-tagged H2AX expressing U251MG cell lines, without or with SMURF2 overexpression, were used for anti-FLAG co-immunoprecipitation and immunoblots with indicated antibodies. Note that for both (H) and (I), as in all co-immunoprecipitation experiments, cells were treated with MG132 (10 μM, to ensure equal H2AX abundance) 1 day before the co-immunoprecipitation experiments. Blue arrowheads marked non-specific immunoblot bands.
Next, to gain insight into the RNF168-mediated H2AX stabilization, we determined the effect of RNF168’s abundance on the SMURF2-H2AX interaction. We found that overexpression of RNF168 attenuated the interaction between H2AX and SMURF2 (Figure 7D). Complementarily, knockdown of RNF168 led to more a potentiated H2AX and SMURF2 interaction (Figure 7E). As expected, this interaction was also potentiated by MTAP inhibition (Figure 7F) or PRMT5 inhibition (Figure 7G), further supporting reduced RNF168 abundance as an enabling factor in the destabilization of H2AX under each condition. Notably, manipulating the abundance of SMURF2 did not lead to measurable changes in the interaction between H2AX and RNF168 (Figures 7H and 7I), suggesting the dominant role of RNF168 in interacting with H2AX. These results are in agreement with the different nature of the two E3 ligases’ interactions with H2AX (i.e., RNF168 functions as a post-translation modification enzyme protecting H2AX from the degradation, while SMURF2 acts as an E3 ligase inducing H2AX degradation). Finally, while both K13 and K15 were found to be direct substrates for RNF168 in response to DNA damage (Mattiroli et al., 2012), we found only the mutation at K13, not at K15, resulted in an enhanced interaction between H2AX and SMURF2 (Figure S9A). In supporting the expected RNF168-mediated ubiquitination of H2AX, the immunoprecipitation of exogenous FLAG-H2AX and anti-Ub-K63 immunoblot confirmed the increased Ub-K63 ubiquitination of H2AX in response to RNF168 overexpression (Figure S9B). Collectively, these results suggest that the RNF168-modulated H2AX-SMURF2 interaction involves RNF168-mediated poly-ubiquitination of H2AX at K13 in the N-terminal tail. We speculate that such a modification potentially poses a physical hindrance, preventing the C-terminal tail of H2AX from fully inserting into the small subdomain of SMURF2’s HECT domain, as illustrated in the docking simulation (Figure 6G; Video S1). We note that further studies will be needed to illuminate the interplay among the two functionally opposing E3 ubiquitin ligases and H2AX, and quantitative in vivo analysis of the ubiquitination of K resides involved (e.g., via quantitative mass spectrometry) will be necessary to more definitively illuminate the functional outcomes of these interactions. Nevertheless, the previously mentioned results collectively support a model in which RNF168 protects H2AX from SMURF2 by modifying H2AX’s N-terminal tail and attenuating the interaction of SMURF2 with H2AX. Consequently, the depletion of RNF168 leads to H2AX destabilization by allowing its enhanced interaction with SMURF2.
In summary, this study reveals an axis of PRMT5-RNF168-SMURF2 in GBM cells that exerts a delicate control on the proteostasis of H2AX, an essential, dose-sensitive guardian of genome stability (Bassing et al., 2003; Celeste et al., 2003). This axis is disrupted in GBM cells with an MTAP deficiency, whose loss of function is a common event in multiple cancer types and in GBM in particular. The identification of such a regulatory cascade—involving a metabolic enzyme, a protein modification enzyme, and two distinct classes of E3 ubiquitin ligases of opposing effects—reveals the far-reaching consequence of MTAP loss in GBM cells and raises the question of the pathogenic effect of aberrant H2AX levels in various stages of GBM development. It also provides insight into the cellular control of protein homeostasis and how such a process can be affected by cancer-associated alterations.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yiping He (yiping.he@duke.edu). All plasmids used and genetically engineered cell lines generated in the study will be freely available without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
Glioblastoma cell lines including U251MG, T98G, and U138MG (all male cell lines; U251MG was described in Hansen et al., 2019), and T98G and U138MG were gifts from Dr. Darell D. Bigner) were cultured in Dulbecco’s Modified Eagle’s medium (GIBCO # 11995–065), supplemented with 10% (v/v) fetal bovine serum (FBS; Corning cat #35–010-CV). Normal human astrocytes (Lonza, Clonetics NHA, cat# CC-2565) were transformed by following previously described procedure to obtain a transformed cell line (OMRP) (Hansen et al., 2019; Li et al., 2016). GBM patient-derived cell line (#13–0302, described in Hansen et al., 2019) and the OMRP cell line were cultured in Neural Stem Cell medium (STEMCELL, cat# 05751) supplemented with EGF, FGF and heparin (20 ng, 10 ng, and 2 μg per ml, respectively). All cell lines were maintained in a humidified atmosphere at 37°C and with 5% CO2. For CRISPR-mediated gene knockout in U251MG and T98G cell lines, pSpCas9(BB)-2A-GFP (px458, addgene plasmid #48138) was used for delivering the pair of sgRNAs (targeting exon 1 and exon 6 of MTAP, sequences were listed in Table S2) and cell clones with successful gene knockout were confirmed by immunoblot (cell line authentication was performed using short tandem repeat (STR) profiling to match derivative cell lines to confirm the identity of parental and derivative MTAP knockout lines). For MTAP knockout in OMRP, LentiCRISPRv2-blast (Addgene, plasmid #83480) was used to deliver the pair of sgRNAs seven days after primary human astrocytes were transduced with the OMRP virus cocktail, and the successful knockout of MTAP was confirmed by immunoblot. Cell lines with stable knockdown of PRMT5, WDR77, HUWE1, NEDD4, RNF168, and SMURF2 were obtained using lentiviral expression construct pLKO.1 (Addgene, #10878) (shRNA sequences were listed in Table S2) and selected using puromycin (1 μg/ml). Cell lines with exogenous overexpression of H2AX, PRMT5, RNF168 and SMURF2 were obtained using retroviral construct MSCV (Addgene, plasmid #24828). Cells were typically transduced and cultured for seven days before they were used for experiments.
METHOD DETAILS
Plasmid construction and mutagenesis
The Flag-tagged H2AX plasmids and Myc-tagged SMURF2 plasmid were constructed using the In-Fusion Kit (Takara Bio, Cat# 121416). The Flag-H2AX mutants and the SMURF2ΔHECT mutant were constructed using QuikChange Mutagenesis Kit (Agilent Tech, Cat# 200521). Primers used were listed in Table S2. All plasmids were verified by Sanger sequencing. All lentivirus and retrovirus was packaged using 293FT (Invitrogen, #R700–07) cells and cells were transduced with a MOI of 3 – 5 and selected by puromycin (for three passages).
Cell clonogenic formation assay and cell viability measurements
Clonogenic formation assays were performed using the following protocol. Cells (in some cases cells were pre-treated with vehicle control or with MTDIA for 24 h) plated at a cell-density of 70%–80% were exposed to radiation of different doses, seeded on 6-well plates at a density of 500 cells per well with fresh medium, and allowed to grow further for 10–14 days to form colonies. The colonies were fixed with 4% (v/v) paraformaldehyde and stained with 0.1% (w/v) crystal violet. For cell proliferation or viability in response to Doxorubicin, cells were seeded at 1 × 104 cells per well in 96-well microtiter plates. 24 h later, cells were treated with different doses of Doxorubicin for 24 h, and numbers of live cells were determined using Cell Counting kit-8 (CCK8) assay (CCK-8, Dojin Laboratories, Kumamoto, Japan). Briefly, CCK8 solution was added to each well to a final concentration of 10% (v/v), and the plates were incubated at 37°C for 1 h. Absorbance in individual wells was determined at 450 nm using microplate reader infinite M200 PRO (TECAN, Männedorf, Switzerland). Student’s t test was used to calculate p value.
Alkaline comet assay
Alkaline comet assay was performed as previously described (Langie et al., 2015). Briefly, cells (when MTDIA was used, cells were pre-treated with MTDIA for 24 h) were suspended in 0.5% (w/v) low-melting-point agarose following treatment (radiation or Doxorubicin) at a density of 1 × 104 per ml. Comet formation was induced by electrophoresis of cells (constant voltage of 1 V per cm) in alkaline electrophoresis solution (300mM NaOH, 1mM EDTA, pH > 13). For each sample, duplicate slides were processed. The tail moment was defined as the product of the percentage of DNA in the tail and the displacement between the head and the tail of the comet. At least 50 nuclei were evaluated in each slide. All measurements were carried out using the software ImageJ OpenComet (Gyori et al., 2014). Tail-moments between different groups were compared using nonparametric test (Mann–Whitney U test).
Analysis of expression correlation of DNA damage response/repair genes in patients
Gene expression profiles and their correlations in GBM and glioma patients were obtained from the GlioVis data portal (http://gliovis.bioinfo.cnio.es/) (Bowman et al., 2017). Datasets used included those from TCGA (Cancer Genome Atlas Research Network, 2008), the Ivy Glioblastoma Atlas Project (http://glioblastoma.alleninstitute.org/), and three additional published datasets (Gravendeel et al., 2009; Madhavan et al., 2009; Bao et al., 2014). The expression correlation status of DNA damage response/repair genes with the MTAP expression are presented in Table S1.
Protein extraction, immunoblotting and immunoprecipitation
Total cellular protein extracts were obtained by using lysis buffer composed of 1% SDS (w/v), 1 mM DTT and protease inhibitor cocktail (ROCHE, Cat# 04693132001) in PBS buffer. The lysis solution was mixed with the equal volume of 2X loading buffer (BIO-RAD, Cat# 161–0737) and heated at 95C for 10 min before being used for immunoblot analysis. For analyzing H2AX stability, cells were treated with Cycloheximide (20 μM) for selected time points before total protein extracts were similarly prepared.
All the cells used for immunoprecipitation (IP) of E3-ligase or ubiquitination analyses were pre-treated with MG132 overnight to prevent protein degradation. Protein extracts for immunoprecipitation were prepared using p300 lysis buffer as previously reported (Gruosso et al., 2016). Cells were lysed in p300 buffer for 30 min on ice. Cells were then centrifuged at 12,000 rpm, 4°C, for 15 min each. Protein concentration in the supernatant was determined by BCA assay (Thermo, Cat# 23225). The supernatants were used for immunoblotting (input) or for co-immunoprecipitation. For immunoprecipitation, Flag beads (Sigma, #A2220) or c-myc beads (Sigma, #A7470) were added to cell lysates (20 μL of beads per ml of lysates) and incubated at 4°C for 8–12 h. The immunoprecipitates were washed three times with lysis buffer and eluted with SDS loading buffer by boiling for 10 min before being used for immunoblot analysis.
Immunoblotting was performed as previously described (Du et al., 2017). In brief, protein lysates were resolved using Bis-Tris gels (12% or 4%–12% of SDS-PAGE gel, Novex, cat# NP0341BOX and NP0335BOX) in MOPS running buffer (Novex, cat# NP0341BOX). Proteins were transferred onto PVDF membranes (Immobilon, Millipore) and incubated overnight with primary antibodies (see Key Resources Table for detailed information on antibodies) at 4°C. Then blots were incubated with horseradish peroxidase-conjugated secondary antibody (Cell Signaling Tech, Anti-mouse IgG: Cat #7076; Anti-rabbit IgG: Cat #7074) followed by detection with enhanced chemoluminescence by the Gel Doc XR+ System (BIO-RAD, Hercules, USA). Quantification of immunoblot signals was performed by using Image Lab 5.0™ (BIO-RAD, Hercules, USA). All IP and immunoblot experiments shown were repeated in at least two independent experiments, and assays for H2AX protein abundance were repeated in at least three experiments.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| MTAP | Cell Signaling | Cat#4158; RRID: AB_1904054 |
| H2AX | Abcam | Cat#ab10475-100; RRID: AB_297215 |
| H2AX | Cell Signaling | Cat#7631; RRID: AB_10860771 |
| β-Actin | Cell Signaling | Cat#3700S; RRID: AB_2242334 |
| α-Tubulin | Cell Signaling | Cat#3873S; RRID: AB_1904178 |
| γH2AX | Abcam | Cat#ab22551; RRID: AB_447150 |
| Anti-Flag Tag | Sigma | Cat#F1804; RRID: AB_262044 |
| Anti-Myc Tag | Invitrogen | Cat#MA1-21316; RRID: AB_558473 |
| P53 | Cell Signaling | Cat#2524; RRID: AB_331743 |
| Ubiquitin | Abcam | Cat#ab7780; RRID: AB_306069 |
| PRMT5 (for WB) | Invitrogen | Cat#PA5-30383; RRID: AB_2547857 |
| PRMT5 (for ChIP) | Epigentek | Cat#A-3005-050 |
| RNF168 | Sigma | Cat#WH0165918M1; RRID: AB_2180101 |
| β-catenin | Abcam | Cat#ab2982-500 |
| WDR77 | Bio-Rad | Cat#VPA00644 |
| H3R2me1 (for WB and ChIP) | Epigentek | Cat#A-3713 |
| H3R3me2s (for WB and ChIP) | Epigentek | Cat#A-3706 |
| H4R3me2s (for WB and ChIP) | Abcam | Cat#ab5823; RRID: AB_10562795 |
| RNF8 | Abcam | Cat#ab4183-100; RRID: AB_304352 |
| HUWE1 | Cell Signaling | Cat#5695S; RRID: AB_10922588 |
| NEDD4 | Cell Signaling | Cat#3607; RRID: AB_2149311 |
| SMURF2 | Cell Signaling | Cat#12024; RRID: AB_2797800 |
| K63-linkage polyubiquitin | Cell Signaling | Cat#5621; RRID: AB_10827985 |
| WWP1 | ThermoFisher | Cat#A302-949A-M; RRID: AB_2780873 |
| RING1B | Cell Signaling | Cat#5694; RRID: AB_10705604 |
| Myc Tag | Cell Signaling | Cat#2278; RRID: AB_490778 |
| Anti-Mouse IgG HRP-linked secondary | Cell Signaling | Cat#7076; RRID: AB_330924 |
| Anti-Rabbit IgG HRP-linked secondary | Cell Signaling | Cat#7074; RRID: AB_2099233 |
| Bacterial and Virus Strains | ||
| XL10-Gold Ultracompetent cells | Agilent | Cat#200315 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM cell culture medium | GIBCO | Cat#11995-065 |
| Neural Stem Cell culture medium | STEMCELL | Cat#05751 |
| MTDIA (MTAP inhibitor) | MedKoo Biosciences | Cat#407244 |
| Doxorubicin | Sigma | Cat#44583 |
| Cycloheximide | Sigma | Cat#46401 |
| MG132 | Sigma | Cat#M7449 |
| EPZ015666 (PRMT5 inhibitor) | Sigma | Cat#SML1421 |
| MLN4924 | Focus Biomolecules | Cat#10-1311 |
| Heclin | Focus Biomolecules | Cat#10-1534 |
| Protease Inhibitor Cocktail | Roche | Cat#04693132001 |
| 2X Laemmli Sample Buffer | Bio-Rad | Cat#161-0737 |
| Flag beads (for IP) | Sigma | Cat#A2220 |
| c-Myc beads (for IP) | Sigma | Cat#A7470 |
| A-G agarose beads (for ChIP) | ThermoFisher | Cat#20421 |
| NuPAGE MOPS SDS running buffer (20X) | ThermoFisher | Cat#NP0001 |
| RIPA Buffer | Santa Cruz | Cat#sc-24948 |
| Critical Commercial Assays | ||
| In-Fusion Kit | Takara Bio | Cat#121416 |
| QuikChange Mutagenesis Kit 394 | Agilent Tech | Cat#200521 |
| Cell Counting Kit-8 (CCK8) | Dojindo Molecular Technologies | Cat#CK04-20 |
| BCA protein quantification assay | Thermo | Cat#23225 |
| RNA extraction kit | QIAGEN | Cat#80204 |
| EcoDry RNA to cDNA Premix Kit | TaKaRa | Cat#639549 |
| SYBR Green PCR Kit | QIAGEN | Cat#204054 |
| Dihydroethidium (DHE) fluorescent probe (ROS detection) | Cayman | Cat#601290 |
| Deposited Data | ||
| Atomic coordinates for SMURF2’s HECT domain | RCSB Protein Data Bank | 1ZVD |
| Atomic coordinates for H2AX | This paper | Mendeley: cbhjvwt3wb/1 (https://doi.org/10.17632/cbhjvwt3wb.1) |
| Experimental Models: Cell Lines | ||
| U251MG | Sigma | cat #09063001 |
| T98G | From peer lab | Dr. Darell D Bigner |
| U138MG | from peer lab | Dr. Darell D Bigner |
| 293FT | Invitrogen | R700-07 |
| GBM #13-0302 patient-derived cell line | Duke University | Reference cited |
| Clonetics Normal human astrocytes (NHA) | Lonza | Cat#CC-2565 |
| Oligonucleotides | ||
| sgRNA sequences (See Table S2) | This paper | N/A |
| RT-qPCR primers (See Table S2) | This paper | N/A |
| H2AX mutant construction primers (See Table S2) | This paper | N/A |
| SMURF2 mutant construction primers (See Table S2) | This paper | N/A |
| ChIP-qPCR primers (See Table S2) | This paper | N/A |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-GFP (px458) | Feng Zhang Lab; Nat Protoc. 8:2281 | Addgene plasmid #48138 |
| LentiCRISPRv2-Blast | Mohan Babu Lab (unpublished data) | Addgene plasmid #83480 |
| pLKO.1 | David Root Lab; Cell 124: 1283 | Addgene plasmid #10878 |
| MSCV | Lin He Lab; Genes Dev. 23: 2839 | Addgene plasmid #24828 |
| Flag-H2AX-MSCV | This paper | N/A |
| Flag-H2AX-K13A, -K15A, -K119A - MSCV | This paper | N/A |
| Myc-SMURF2-MSCV | This paper | N/A |
| SMURF2ΔHECT-MSCV | This paper | N/A |
| shRNA sequences (See Table S2) | This paper | N/A |
| Software and Algorithms | ||
| Image Lab 5.0 ™ | Bio-Rad | N/A |
| CFX Maestro | Bio-Rad | N/A |
| ImageJ (version 1.8.0) | NIH | https://imagej.nih.gov/ij/ |
| SWISS-MODEL | Benkert et al., 2011; Waterhouse et al., 2018 | https://swissmodel.expasy.org/assess |
| GRAMM (docking simulation) | Tovchigrechko and Vakser, 2006 | http://vakser.compbio.ku.edu/resources/gramm/grammx/ |
| LSM ZEN microscope software | Zeiss | N/A |
| Other | ||
| Immobilon-P PVDF membrane | Millipore | Cat#IPVH00010 |
| Novex Bis-Tris SDS-PAGE 12% gel | ThermoFisher | Cat#NP0341BOX |
| Novex Bis-Tris SDS-PAGE 4–12% gel | ThermoFisher | Cat#NP0335BOX |
| Gel Doc XR+ System | Bio-Rad | N/A |
| Infinite M200 PRO plate reader | Tecan | N/A |
| CFX96TM Real-Time System | Bio-Rad | N/A |
| Bioruptor (sonicator) | Diagenode | N/A |
| Zeiss 880 confocal microscope | Zeiss | N/A |
Whole gel images for all immunoblots, with molecular weight markers marked, are available from the corresponding author upon request.
Reverse transcription and quantitative real-time PCR (RT-qPCR)
Total RNA was extracted using the RNA extraction kit from QIAGEN (Cat# 80204). 1 μg of total RNA was reverse-transcribed using a RNA to cDNA Premix Kit (TaKaRa, Cat# 639549) following the manufacturer’s protocols. The cDNA was amplified by use of the SYBR Green PCR Kit (QIAGEN, Cat# 204054) with gene-specific primers listed in Table S2. RT-qPCR was performed using CFX96TM Real-Time System (BIO-RAD, Hercules, USA), and the results were analyzed using CFX Maestro Software (BIO-RAD, Hercules, USA). Internal control genes used for gene expression normalization included GAPDH and ACTB.
Chromatin Immunoprecipitation (ChIP) and quantitative PCR (ChIP-qPCR)
Formaldehyde cross-linking and chromatin immunoprecipitations (ChIP) was performed as previously described (Chen et al., 2017) with modifications. Briefly, cells were harvested at 70%–80% confluence, washed twice with PBS, and cross-linked with 1% (v/v) formaldehyde for 10 min. Then the cells were lysed on ice using RIPA buffer (Santa Cruz, Cat# sc-24948) for 30 min, followed by sonication using Bioruptor (Diagenode SA, Belgium) for 30min at high power. Ten percent of total lysate was set aside as input for later quantification. Immunoprecipitation was carried out using antibodies specific for H3R2me1, H3R8me2s, H4R3me2s or PRMT5, or using an IgG control antibody, with 1 ug of each antibody in 1 mL of lysate. The immunoprecipitation was done using A/G agarose beads (ThermoFisher, Cat# 20421) (20 μL per tube) overnight in 4°C. The thermocycler used for qPCR was the same as the one used for aforementioned RT-qPCR. Primers for the RNF168 promoter region are listed in the Table S2. The enrichment of each protein in the promoter region was evaluated by the comparison of the percentage of pulled-down DNA to the input (% input) between ChIP group and IgG group. Statistical analysis was performed using Student’s t test.
Immunofluorescent Staining
Immunofluorescence was performed as previously described (Gruosso et al., 2016), with modifications. Briefly, cells were fixed in 4% (v/v) paraformaldehyde for 20 min, permeabilized in 0.25% (v/v) Triton X-100 for 5 min, washed with PBS, rinsed and then blocked with 5% (v/v) bovine serum albumin (BSA) in PBS for 30 min. Cells were stained with DAPI (0.1 μg per ml) for DNA detection, together with specific antibodies followed by fluorescein coupled secondary antibody (antibodies used were listed in Key Resources Table. Slides were scanned using a Zeiss 880 confocal microscope. Images were acquired under identical settings including fluorescence signal intensity and exposure time, saved as czi files, and converted to JPEG using LSM ZEN software. γH2AX foci were analyzed using ImageJ (version 1.8.0, National Insititutes of Health, Bethesda, U.S.). Briefly, foci with a diameter greater than 0.8 μm were detected and used for quantification (Gruosso et al., 2016). The number of foci was counted in 50 nuclei for each experimental group. The mean values of foci between different groups were compared using a t test.
Reactive oxidation species (ROS) assay
ROS was detected using Dihydroethidium (DHE) fluorescent probe assay (DHE Kit from Cayman, Cat# 601290); with the Antimycin treatment as the positive control, and the N-acetyl Cysteine treatment as the negative control. The excitation wavelength was 480 nm, and the emission wavelength was 570 nm.
Simulation of protein-protein interaction
Atomic coordinates for SMURF2’s HECT domain were obtained from RCSB protein Data Bank (reference 1ZVD). Atomic coordinates for H2AX were calculated by SWISS-MODEL (https://swissmodel.expasy.org/) based on its primary sequence (https://www.ncbi.nlm.nih.gov/protein/NP_002096.1), and were validated by comparing to previous studies (Shao et al., 2012; Singh et al., 2012; Stucki et al., 2005). The quality of these models was assessed using SWISS-MODEL (https://swissmodel.expasy.org/assess) (Benkert et al., 2011; Waterhouse et al., 2018). The structure assessment of these models is presented in Figure S10. The docking simulation was performed using GRAMM (accessed via http://vakser.compbio.ku.edu/resources/gramm/grammx/) (Tovchigrechko and Vakser, 2006). Parameters used for simulation included the following: Potential range equal to atoms radii (30.0,−1.0, and 0.0); 6.5 for Grid step; 32 for Grid size; Energy values are divided by 178.5; Angle for rotations by 10 degrees.
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments for Immunoblot, Co-IP assay, CCK8 assay, colony formation assay, loci-specific ChIP and RT-qPCR were repeated in at least three independent experiments. Results obtained are presented as mean ± s.d. Mean values between two groups were compared using Student’s t-test if they follow normal distributions, otherwise they were compared using a nonparametric test. Multiple groups of mean values were compared using ANOVA. All tests were two-sided, deemed statistically significant if p < 0.05.
DATA AND CODE AVAILABILITY
The whole gel images for all immunoblots and all original IF images are available from the corresponding author upon request. The atomic coordinate data for H2AX (used for modeling SMURF2 HECT domain’s interaction with H2AX) has been deposited in Mendeley Data. The identifier for the H2AX atomic coordinate data reported in this paper is Mendeley: cbhjvwt3wb/1 (https://data.mendeley.com/datasets/cbhjvwt3wb/1).
Supplementary Material
Highlights.
H2AX proteostasis is dynamically and conversely regulated by RNF168 and SMURF2
PRMT5 maintains the expression of RNF168, which protects H2AX from SMURF2
A disrupted PRMT5-RNF168-SMURF2 axis causes decreased H2AX levels and defective DDR
ACKNOWLEDGMENTS
We thank Dr. Chuan-Yuan Li (Duke University) for providing the set of OMRP plasmids used for the transformation of human astrocytes and Dr. Darell D. Bigner (Duke University) for providing GBM cell lines (T98G and U138MG). We thank colleagues in the Duke Functional Genomics Shared Resource for generating some of the lentivirus reagents (for gene knockdown) used in the study. This work was supported by the National Institute of Neurological Disorders and Stroke of the NIH, award R01NS101074 (Y.H.), the NIH Duke SPORE in Brain Cancer (P50 CA190991), and the Southeastern Brain Tumor Foundation’s Karen’s Legacy Memorial Award (Y.H.). C.D. was partially supported by the Peking University Cancer Hospital and thanks Drs. Aiwen Wu, Jiafu Ji, and Jin Gu and Mr. Tiefu Kui for their support and encouragement. R.S. was partially supported by a scholarship from the China Scholarship Council (201506175043). L.J.H. was a recipient of a National Cancer Institute National Research Service Award (F30CA206336). C.J.M. was a recipient of an F31 fellowship from the NIH (F31CA203399). Y.H. was a recipient of National Comprehensive Cancer Network (NCCN) Young Investigator Award.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.08.031.
REFERENCES
- Atsumi Y, Minakawa Y, Ono M, Dobashi S, Shinohe K, Shinohara A, Takeda S, Takagi M, Takamatsu N, Nakagama H, et al. (2015). ATM and SIRT6/SNF2H Mediate Transient H2AX Stabilization When DSBs Form by Blocking HUWE1 to Allow Efficient γH2AX Foci Formation. Cell Rep.13, 2728–2740. [DOI] [PubMed] [Google Scholar]
- Bao ZS, Chen HM, Yang MY, Zhang CB, Yu K, Ye WL, Hu BQ, Yan W, Zhang W, Akers J, et al. (2014). RNA-seq of 272 gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in secondary glioblastomas. Genome Res.24, 1765–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassing CH, Suh H, Ferguson DO, Chua KF, Manis J, Eckersdorff M, Gleason M, Bronson R, Lee C, and Alt FW (2003). Histone H2AX:a dosage-dependent suppressor of oncogenic translocations and tumors. Cell 114, 359–370. [DOI] [PubMed] [Google Scholar]
- Basu I, Locker J, Cassera MB, Belbin TJ, Merino EF, Dong X, Hemeon I, Evans GB, Guha C, and Schramm VL (2011). Growth and metastases of human lung cancer are inhibited in mouse xenografts by a transition state analogue of50-methylthioadenosine phosphorylase. J. Biol. Chem 286, 4902–4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrmann I, Wallner S, Komyod W, Heinrich PC, Schuierer M, Buettner R, and Bosserhoff AK (2003). Characterization of methylthioadenosin phosphorylase (MTAP) expression in malignant melanoma. Am. J. Pathol.163, 683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkert P, Biasini M, and Schwede T(2011). Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27, 343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernassola F, Karin M, Ciechanover A, and Melino G (2008). The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer Cell 14, 10–21. [DOI] [PubMed] [Google Scholar]
- Bertino JR, Waud WR, Parker WB, and Lubin M (2011). Targeting tumors that lack methylthioadenosine phosphorylase (MTAP) activity: current strategies. Cancer Biol. Ther 11, 627–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman RL, Wang Q, Carro A, Verhaak RG, and Squatrito M (2017). GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro-oncol.19, 139–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos ES, Wilczek C, Onikubo T, Bonanno JB, Jansong J, Reimer U, and Shechter D (2015). Histone H2A and H4 N-terminal tails are positioned by the MEP50 WD repeat protein for efficient methylation by the PRMT5 arginine methyltransferase. J. Biol. Chem 290, 9674–9689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste A, Difilippantonio S, Difilippantonio MJ, Fernandez-Capetillo O, Pilch DR, Sedelnikova OA, Eckhaus M, Ried T, Bonner WM, and Nussenzweig A (2003). H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell 114, 371–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae YK, Anker JF, Carneiro BA, Chandra S, Kaplan J, Kalyan A, Santa-Maria CA, Platanias LC, and Giles FJ (2016). Genomic landscape of DNA repair genes in cancer. Oncotarget 7, 23312–23321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan-Penebre E, Kuplast KG, Majer CR, Boriack-Sjodin PA, Wigle TJ, Johnston LD, Rioux N, Munchhof MJ, Jin L, Jacques SL, et al. (2015). A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol 11, 432–437. [DOI] [PubMed] [Google Scholar]
- Chen D, Kon N, Li M, Zhang W, Qin J, and Gu W (2005). ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell 121, 1071–1083. [DOI] [PubMed] [Google Scholar]
- Chen H, Lorton B, Gupta V, and Shechter D (2017). A TGFβ-PRMT5-MEP50 axis regulates cancer cell invasion through histone H3 and H4 arginine methylation coupled transcriptional activation and repression. Oncogene 36, 373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong PA, Lin H, Wrana JL, and Forman-Kay JD (2006). An expanded WW domain recognition motif revealed by the interaction between Smad7 and the E3 ubiquitin ligase Smurf2. J. Biol. Chem 281, 17069–17075. [DOI] [PubMed] [Google Scholar]
- Chong PA, Lin H, Wrana JL, and Forman-Kay JD (2010). Coupling of tandem Smad ubiquitination regulatory factor (Smurf) WW domains modulates target specificity. Proc. Natl. Acad. Sci. USA 107, 18404–18409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke TL, Sanchez-Bailon MP, Chiang K, Reynolds JJ, Herrero-Ruiz J, Bandeiras TM, Matias PM, Maslen SL, Skehel JM, Stewart GS, et al. (2017). PRMT5-Dependent Methylation of the TIP60 Coactivator RUVBL1 Is a Key Regulator of Homologous Recombination. Mol. Cell 65, 900–916.e907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Shao G, Zhang HT, Li C, Zhang D, Cheng L, Elzey BD, Pili R, Ratliff TL, Huang J, and Hu CD (2017). Protein arginine methyltransferase 5 functions as an epigenetic activator of the androgen receptor to promote prostate cancer cell growth. Oncogene 36, 1223–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J, et al. (2009). RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 136, 435–446. [DOI] [PubMed] [Google Scholar]
- Du C, Huang D, Peng Y, Yao Y, Zhao Y, Yang Y, Wang H, Cao L, Zhu WG, and Gu J (2017). 5-Fluorouracil targets histone acetyltransferases p300/CBP in the treatment of colorectal cancer. Cancer Lett.400, 183–193. [DOI] [PubMed] [Google Scholar]
- Fabbrizio E, El Messaoudi S, Polanowska J, Paul C, Cook JR, Lee JH, Negre V, Rousset M, Pestka S, Le Cam A, and Sardet C (2002). Negative regulation of transcription by the type II arginine methyltransferase PRMT5. EMBO Rep. 3, 641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Lee A, Nussenzweig M, and Nussenzweig A (2004). H2AX: the histone guardian of the genome. DNA Repair (Amst.) 3, 959–967. [DOI] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatti M, Pinato S, Maiolica A, Rocchio F, Prato MG, Aebersold R, and Penengo L (2015). RNF168 promotes noncanonical K27 ubiquitination to signal DNA damage. Cell Rep. 10, 226–238. [DOI] [PubMed] [Google Scholar]
- Gravendeel LA, Kouwenhoven MC, Gevaert O, de Rooi JJ, Stubbs AP, Duijm JE, Daemen A, Bleeker FE, Bralten LB, Kloosterhof NK, et al. (2009). Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 69, 9065–9072. [DOI] [PubMed] [Google Scholar]
- Gruosso T, Mieulet V, Cardon M, Bourachot B, Kieffer Y, Devun F, Dubois T, Dutreix M, Vincent-Salomon A, Miller KM, and Mechta-Grigoriou F (2016). Chronic oxidative stress promotes H2AX protein degradation and enhances chemosensitivity in breast cancer patients. EMBO Mol. Med 8, 527–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D, and Clement MV (2014). OpenComet: an automated tool for comet assay image analysis. Redox Biol. 2, 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamard PJ, Santiago GE, Liu F, Karl DL, Martinez C, Man N, Mookhtiar AK, Duffort S, Greenblatt S, Verdun RE, and Nimer SD (2018). PRMT5 Regulates DNA Repair by Controlling the Alternative Splicing of Histone-Modifying Enzymes. Cell Rep. 24, 2643–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen LJ, Sun R, Yang R, Singh SX, Chen LH, Pirozzi CJ, Moure CJ, Hemphill C, Carpenter AB, Healy P, et al. (2019). MTAP Loss Promotes Stemness in Glioblastoma and Confers Unique Susceptibility to Purine Starvation. Cancer Res. 79, 3383–3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellerbrand C, Mühllbauer M, Wallner S, Schuierer M, Behrmann I, Bataille F, Weiss T, Schölmerich J, and Bosserhoff AK (2006). Promoter-hypermethylation is causing functional relevant downregulation of methylthioadenosine phosphorylase (MTAP) expression in hepatocellular carcinoma. Carcinogenesis 27, 64–72. [DOI] [PubMed] [Google Scholar]
- Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, and Chen J (2007). RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 131, 901–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäckl M, Stollmaier C, Strohäker T, Hyz K, Maspero E, Polo S, and Wiesner S (2018). β-Sheet Augmentation Is a Conserved Mechanism of Priming HECT E3 Ligases for Ubiquitin Ligation. J. Mol. Biol 430(18 Pt B), 3218–3233. [DOI] [PubMed] [Google Scholar]
- Kamatani N, and Carson DA (1980). Abnormal regulation of methylthioadenosine and polyamine metabolism in methylthioadenosine phosphorylasedeficient human leukemic cell lines. Cancer Res. 40, 4178–4182. [PubMed] [Google Scholar]
- Kryukov GV, Wilson FH, Ruth JR, Paulk J, Tsherniak A, Marlow SE, Vazquez F, Weir BA, Fitzgerald ME, Tanaka M, et al. (2016). MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 351, 1214–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langie SA, Azqueta A, and Collins AR (2015). The comet assay: past, present, and future. Front. Genet 6, 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc SE, Konda S, Wu Q, Hu YJ, Oslowski CM, Sif S, and Imbalzano AN (2012). Protein arginine methyltransferase 5 (Prmt5) promotes gene expression of peroxisome proliferator-activated receptor γ2 (PPAR γ 2) and its target genes during adipogenesis. Mol. Endocrinol 26, 583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CF, Fang FM, Kung HJ, Chen LT, Wang JW, Tsai JW, Yu SC, Wang YH, Li SH, and Huang HY (2014). Downregulated MTAP expression in myxofibrosarcoma: A characterization of inactivating mechanisms, tumor suppressive function, and therapeutic relevance. Oncotarget 5, 11428–11441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Liu X, Sampson JH, Bigner DD, and Li CY (2016). Rapid Reprogramming of Primary Human Astrocytes into Potent Tumor-Initiating Cells with Defined Genetic Factors. Cancer Res.76, 5143–5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, and Ye Y (2011). Proteostasis regulation at the endoplasmic reticulum: a new perturbation site for targeted cancer therapy. Cell Res.21, 867–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubin M, and Lubin A (2009). Selective killing of tumors deficient in methylthioadenosine phosphorylase: a novel strategy. PLoS ONE 4, e5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg MS, Typas D, Caron MC, Wiegant WW, van den Heuvel D, Boonen RA, Couturier AM, Mullenders LH, Masson JY, and van Attikum H (2017). A PALB2-interacting domain in RNF168 couples homologous recombination to DNA break-induced chromatin ubiquitylation. eLife 6, e20922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavan S, Zenklusen JC, Kotliarov Y, Sahni H, Fine HA, and Buetow K (2009). Rembrandt: helping personalized medicine become a reality through integrative translational research. Mol. Cancer Res 7, 157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, and Lukas J (2007). RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 131, 887–900. [DOI] [PubMed] [Google Scholar]
- Manasanch EE, and Orlowski RZ (2017). Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol 14, 417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marjon K, Cameron MJ, Quang P, Clasquin MF, Mandley E, Kunii K, McVay M, Choe S, Kernytsky A, Gross S, et al. (2016). MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis.Cell Rep. 15, 574–587. [DOI] [PubMed] [Google Scholar]
- Mattiroli F, Vissers JH, van Dijk WJ, Ikpa P, Citterio E, Vermeulen W, Marteijn JA, and Sixma TK (2012). RNF168 ubiquitinates K13–15 on H2A/ H2AX to drive DNA damage signaling. Cell 150, 1182–1195. [DOI] [PubMed] [Google Scholar]
- Mavrakis KJ, McDonald ER 3rd, Schlabach MR, Billy E, Hoffman GR, deWeck A, Ruddy DA, Venkatesan K, Yu J, McAllister G, et al. (2016). Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 351, 1208–1213. [DOI] [PubMed] [Google Scholar]
- Migliori V,Müller J, Phalke S, Low D, Bezzi M, Mok WC, Sahu SK, Gunaratne J, Capasso P, Bassi C, et al. (2012). Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat. Struct. Mol. Biol 19, 136–144. [DOI] [PubMed] [Google Scholar]
- Morreale FE, and Walden H (2016). Types of Ubiquitin Ligases. Cell 165, 248.e241. [DOI] [PubMed] [Google Scholar]
- Mund T, Lewis MJ, Maslen S, and Pelham HR (2014). Peptide and small molecule inhibitors of HECT-type ubiquitin ligases. Proc. Natl. Acad. Sci. USA 111, 16736–16741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogunjimi AA, Briant DJ, Pece-Barbara N, Le Roy C, Di Guglielmo GM, Kavsak P, Rasmussen RK, Seet BT, Sicheri F, and Wrana JL (2005). Regulation of Smurf2 ubiquitin ligase activity by anchoring the E2 to the HECT domain. Mol. Cell 19, 297–308. [DOI] [PubMed] [Google Scholar]
- Pan MR, Peng G, Hung WC, and Lin SY (2011). Monoubiquitination of H2AX protein regulates DNA damage response signaling. J. Biol. Chem 286, 28599–28607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala AJ, Bott LC, and Morimoto RI (2017). Shaping proteostasis at the cellular, tissue, and organismal level. J. Cell Biol. 216, 1231–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffner M, and Kumar S (2014). Mammalian HECT ubiquitin-protein ligases: biological and pathophysiological aspects. Biochim. Biophys. Acta 1843, 61–74. [DOI] [PubMed] [Google Scholar]
- Schnell JD, and Hicke L (2003). Non-traditional functions of ubiquitin and ubiquitin-binding proteins. J. Biol. Chem 278, 35857–35860. [DOI] [PubMed] [Google Scholar]
- Shao Z, Li F, Sy SM, Yan W, Zhang Z, Gong D, Wen B, Huen MS, Gong Q, Wu J, and Shi Y (2012). Specific recognition of phosphorylated tail of H2AX by the tandem BRCT domains of MCPH1 revealed by complex structure. J. Struct. Biol 177, 459–468. [DOI] [PubMed] [Google Scholar]
- Singh N, Basnet H, Wiltshire TD, Mohammad DH, Thompson JR, Héroux A, Botuyan MV, Yaffe MB, Couch FJ, Rosenfeld MG, and Mer G (2012). Dual recognition of phosphoserine and phosphotyrosine in histone variant H2A.X by DNA damage response protein MCPH1. Proc. Natl. Acad. Sci. USA 109, 14381–14386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, et al. (2009). An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736. [DOI] [PubMed] [Google Scholar]
- Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, Nakada S, Ylanko J, Olivarius S, Mendez M, et al. (2009). The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 136, 420–434. [DOI] [PubMed] [Google Scholar]
- Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, and Jackson SP (2005). MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123, 1213–1226. [DOI] [PubMed] [Google Scholar]
- Tarighat SS, Santhanam R, Frankhouser D, Radomska HS, Lai H, Anghelina M, Wang H, Huang X, Alinari L, Walker A, et al. (2016). The dual epigenetic role of PRMT5 in acute myeloid leukemia: gene activation and repression via histone arginine methylation. Leukemia 30, 789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovchigrechko A, and Vakser IA (2006). GRAMM-X public web server for protein-protein docking. Nucleic Acids Res. 34, W310–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakser IA (1996a). Low-resolution docking: prediction of complexes for underdetermined structures. Biopolymers 39, 455–464. [DOI] [PubMed] [Google Scholar]
- Vakser IA (1996b). Main-chain complementarity in protein-protein recognition. Protein Eng. 9, 741–744. [DOI] [PubMed] [Google Scholar]
- Vakser IA (1997). Evaluation of GRAMM low-resolution docking methodology on the hemagglutinin-antibody complex. Proteins (Suppl 1), 226–230. [PubMed] [Google Scholar]
- Vakser IA, Matar OG, and Lam CF (1999). A systematic study of low-resolution recognition in protein–protein complexes. Proc. Natl. Acad. Sci. USA 96, 8477–8482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. (2018). SWISSMODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46 (W1), W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesner S, Ogunjimi AA, Wang HR, Rotin D, Sicheri F, Wrana JL, and Forman-Kay JD (2007). Autoinhibition of the HECT-type ubiquitin ligase Smurf2 through its C2 domain. Cell 130, 651–662. [DOI] [PubMed] [Google Scholar]
- Willis J, Patel Y, Lentz BL, and Yan S (2013). APE2 is required for ATR-Chk1 checkpoint activation in response to oxidative stress. Proc. Natl. Acad. Sci. USA 110, 10592–10597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood RD, Mitchell M, and Lindahl T (2005). Human DNA repair genes, 2005. Mutat. Res 577, 275–283. [DOI] [PubMed] [Google Scholar]
- Wu CY, Kang HY, Yang WL, Wu J, Jeong YS, Wang J, Chan CH, Lee SW, Zhang X, Lamothe B, et al. (2011). Critical role of monoubiquitination of histone H2AX protein in histone H2AX phosphorylation and DNA damage response. J. Biol. Chem 286, 30806–30815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Adamski R, and Chen J (2010). Focus on histone variant H2AX: to be or not to be. FEBS Lett. 584, 3717–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Rank G, Tan YT, Li H, Moritz RL, Simpson RJ, Cerruti L, Curtis DJ, Patel DJ, Allis CD, et al. (2009). PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat. Struct. Mol. Biol 16, 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The whole gel images for all immunoblots and all original IF images are available from the corresponding author upon request. The atomic coordinate data for H2AX (used for modeling SMURF2 HECT domain’s interaction with H2AX) has been deposited in Mendeley Data. The identifier for the H2AX atomic coordinate data reported in this paper is Mendeley: cbhjvwt3wb/1 (https://data.mendeley.com/datasets/cbhjvwt3wb/1).