

Abstract
Herein we report the first highly enantioselective allenoate-Claisen rearrangement using doubly axially chiral phosphate sodium salts as catalysts. This synthetic method provides access to β-amino acid derivatives with vicinal stereocenters in up to 95% ee. We also investigated the mechanism of enantioinduction by transition state (TS) computations with DFT as well as statistical modeling of the relationship between selectivity and the molecular features of both the catalyst and substrate. The mutual interactions of charge-separated regions in both the zwitterionic intermediate generated by reaction of an amine to the allenoate and the Na+-salt of the chiral phosphate leads to an orientation of the TS in the catalytic pocket that maximizes favorable noncovalent interactions. Crucial arene–arene interactions at the periphery of the catalyst lead to a differentiation of the TS diastereomers. These interactions were interrogated using DFT calculations and validated through statistical modeling of parameters describing noncovalent interactions.
Graphical Abstract
INTRODUCTION
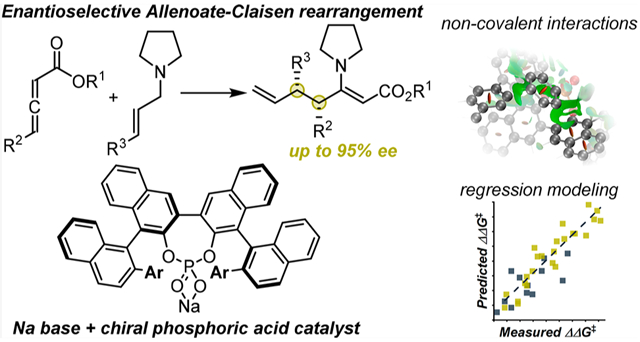
Sigmatropic rearrangements, such as the Claisen rearrangement, are an exceptional synthetic platform for the rapid construction of structural complexity from relatively simple starting materials.1 Despite a century-long history of research on this class of transformations, the development of asymmetric examples remains a significant challenge with success only achieved on a case-by-case basis. As an example, the most traditional strategies to imparting stereocontrol are substrate-derived or auxiliary-controlled methods.2 In contrast, truly catalytic enantioselective Claisen rearrangements have only been reported with a limited number of systems, mostly based on chiral Lewis acids.3 In this context, we were intrigued by the so-called allenoate-Claisen rearrangement,4 an elegant, Lewis acid-catalyzed variant developed by Lambert and MacMillan in 2002, for which a highly enantioselective version remains elusive. In this transformation, an allenoate ester reacts with a tertiary allylamine to form a zwitterionic allyl-vinylammonium intermediate (Figure 1A and B), containing charge separation to facilitate a [3,3]-sigmatropic rearrangement. A Lewis acid catalyst is required to activate the allenoate for nucleophilic addition by coordination to the carbonyl oxygen. This mechanism determines that the prostereogenic domain of the intermediate is separated from the Lewis acid catalyst by >5 Å, which potentially explains the difficulty of rendering this rearrangement enantioselective.
Figure 1.

Hypothesis for an enantioselective catalytic allenoate-Claisen rearrangement.
We envisioned that chiral Lewis acid catalysis could be achieved for this reaction with phosphate counterions featuring an extensive chiral pocket (Figure 1C).4 The weak coordination of phosphates to cations would create an adaptable environment and potentially allow additional stabilizing interactions between the phosphate and the ammonium moiety of the intermediate. Such a mode of interaction would place the chiral “ligand” in closer proximity to the prostereogenic domain of the intermediate, resulting in an environment more likely to render the process stereoselective.
RESULTS AND DISCUSSION
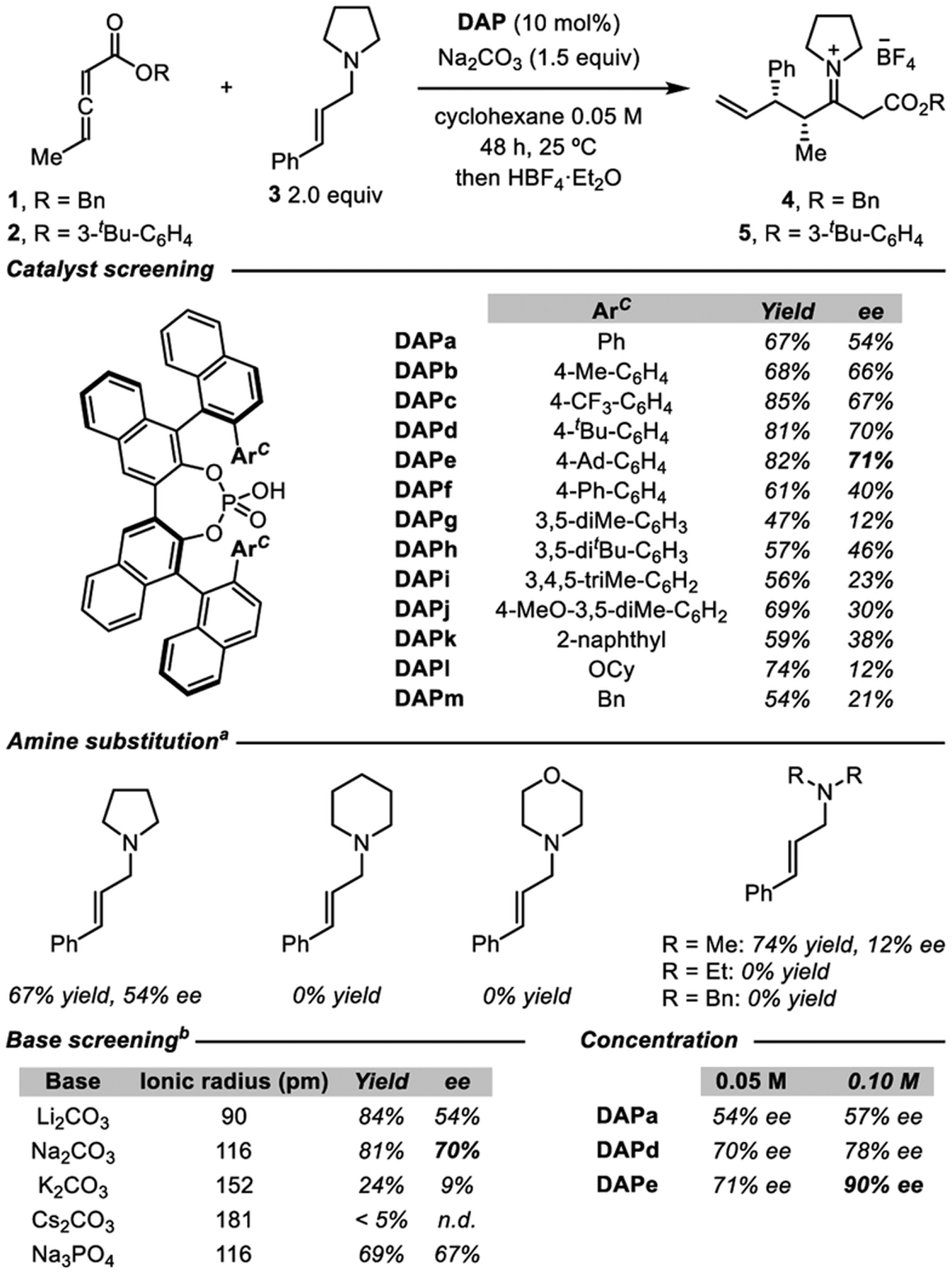
An initial evaluation of typical chiral phosphoric acid derivatives using racemic benzyl 2,3-pentadienoate (1) and (E)-cinnamyl pyrrolidine (3) did not meet with promising results (see the Supporting Information (SI) for details). Therefore, we focused our attention on doubly axially chiral phosphoric acids (DAPs, Figure 1C).5 This unique catalyst architecture can be constructed from the homocoupling of two BINOL scaffolds resulting in a structure containing two chiral axes, while retaining a nondefined central axis. As compared to most common phosphoric acid catalysts, these scaffolds also have a larger chiral pocket, with numerous opportunities for noncovalent interactions (NCIs) to drive asymmetric catalysis (Figure 1D).6 Under this scenario, we found that DAPa provided β-enamino ester 4 in 50% ee with sodium carbonate (Table 1). Further investigations into the ester substituent afforded a moderate increase in selectivity (54% ee) when using 3-tert-butyl-phenol ester derivative 2. Intriguingly, any modification of the amine from pyrrolidine resulted in a dramatic loss of efficiency. Notably, the use of different bases failed to improve results although enantioselectivity was sensitive to the nature of the Lewis acidic cation.
Table 1.
Optimization of the Reaction Using Allenoate 2
|
Using DAPa.
Using DAPd.
As part of the reaction optimization, we developed a new route for the synthesis of DAP catalysts (Scheme 1). The previously reported method to establish the 2,2′-binaphthyl connectivity employed a Suzuki coupling, which required the preparation of two separate coupling partners starting from BINOL. The synthesis was shortened substantially using a Fe-mediated homocoupling for this connection, precluding any prefunctionalization at the 3-position of the precursor. This new synthesis expedited the preparation of a library of DAPs and their subsequent evaluation as catalysts for the allenoate-Claisen rearrangement (Table 1). The most effective derivative was DAPe that incorporates a remote adamantyl group and promotes the formation of 5 in 82% yield and 71% ee. Conducting the reaction at a higher overall concentration afforded significantly improved levels of enantioselectivity (90% ee at 0.1 M) by minimizing the impact of the background reaction in favor of the DAP-catalyzed process.8 In the crystallization of the standard product 5, decarboxylation was observed to form 6, which allowed determination of the absolute configuration using crystal structure analysis (Table 2).
Scheme 1.

Revised Synthesis of DAP Catalysts
Table 2.
Scope of the DAP-Catalyzed Enantioselective Allenoate-Claisen Rearrangement

|
Crystal structure after decarboxylation of 5. BF4− counterion omitted for clarity.
Substrate Scope.
Using the optimized reaction conditions, we explored the scope of the process (Table 2). Significant structural variation in the allenoate component (R2) was possible, affording a diverse range of β-amino-γ,δ-disubstituted esters including functional groups such as esters (11), a halide (13), an ether (15), and unsaturated moieties (17, 19) with excellent levels of stereocontrol. However, a longer alkyl chain had a negative effect on conversion (7, 9). Hypothesizing that a more electrophilic allenoate would increase the reaction rate, we found that trifluoroethanol-derived allenoates led to full conversion while retaining high levels of stereocontrol (products B). For some substrates, the use of trifluoroethanol derivatives increased the ee by up to 20% over the tert-butyl phenol derivatives.9
Aryl allyl amines bearing both electron-rich and electron-poor groups at the para position underwent the reaction, resulting in moderate to good enantiomeric excess (20–24). While good selectivity was observed with a naphthyl allyl amine (27), a meta-(25) and ortho-substituted (26) aryl allyl amine resulted in modest and more noticeable decreases in enantioselectivity, respectively. Moreover, we were pleased to observe that this catalytic system was capable of setting fully substituted carbons, including both a quaternary carbon (28) and a tertiary fluoride center (29), in promising levels of enantioinduction.
Computational Studies.
To gain insight into the mechanism of enantioinduction, we turned to computational chemistry.10–12 The weak, nondirectional interactions between the catalyst and reactants result in a flat potential energy surface (PES) for this system. The size and flexibility of the molecules involved further exacerbated the exploration of the PES. Thus, we aimed to restrict the computational analysis of the full catalyst system to the relevant enantiodetermining step. To identify that step, we began by computing the energy profile of the reaction mechanism in two limiting cases: (a) uncatalyzed and (b) purely Lewis-acid catalyzed using Na+ as a model Lewis acid (Figure 2). The mechanism consists of two steps: (i) nucleophilic addition of the amine to the allenoate and (ii) the [3,3]-sigmatropic rearrangement to the product. We found that, in either case, the addition is reversible and the rearrangement is the rate-limiting transition state (TS). The Lewis acid catalyst greatly facilitates the nucleophilic addition by activating the electrophile, lowering the free energy of activation from 19.1 to 9.5 kcal/mol, and stabilizing the zwitterionic intermediate by up to 13.5 kcal/mol. This intermediate can adopt numerous conformations, and those with a geometry that can undergo the requisite [3,3]-sigmatropic rearrangement are 1.3 and 7.6 kcal/mol higher in free energy than the most stable conformation in the uncatalyzed and Na+-catalyzed reaction, respectively. In turn, the barrier for the rearrangement is not strongly impacted by the Lewis acid catalyst (ΔG‡ = 12.3 and 14.1 kcal/mol without and with Na+-catalyst, respectively). This analysis suggests that, during catalysis with the sodium salt of DAP catalysts, the enantiodetermining step is the rearrangement and it is sufficient to study this step computationally with the full DAP catalyst.
Figure 2.
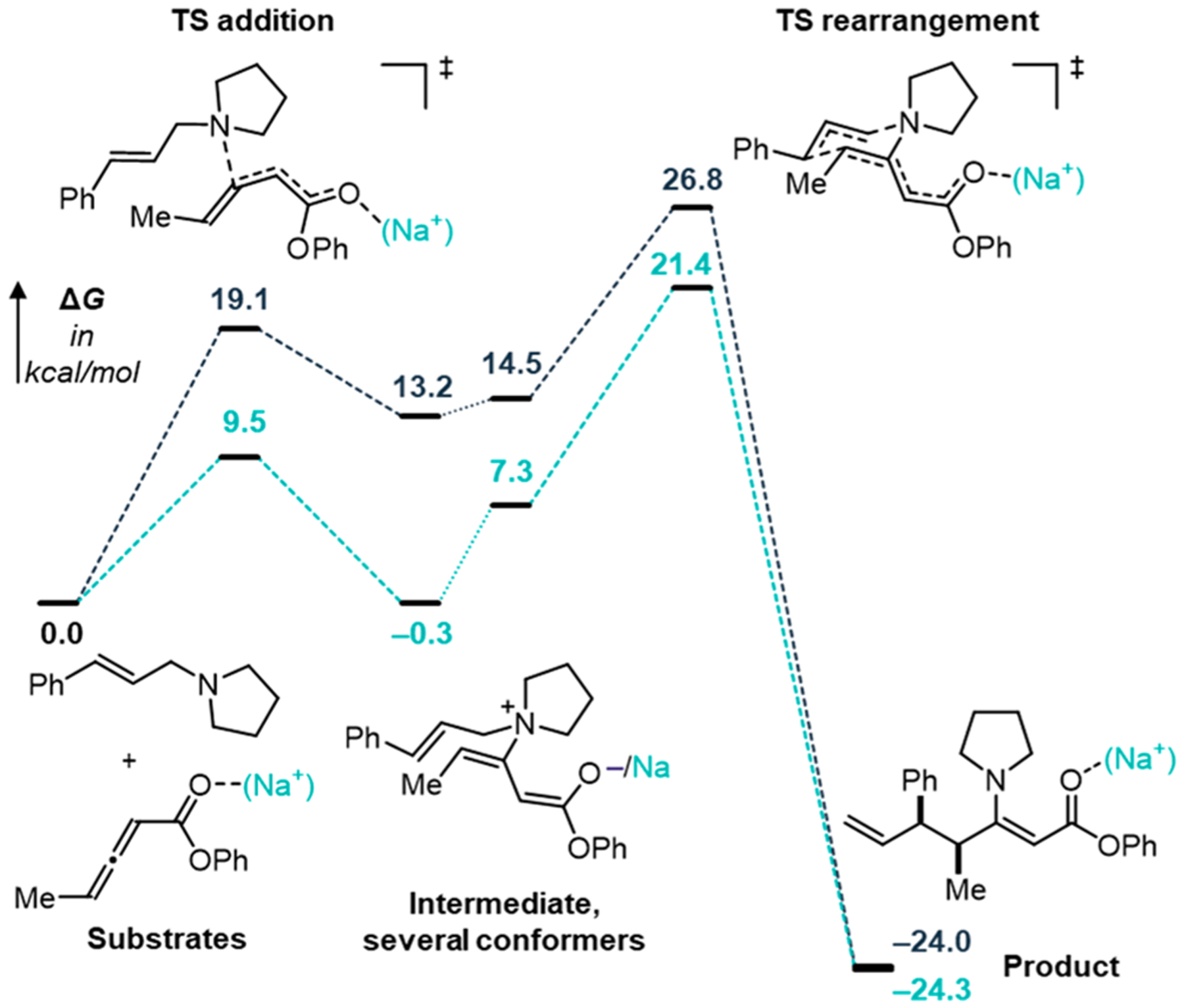
Free energy profile of the mechanism of the allenoate-Claisen reaction between allyl amine 3 and a model allenoate with R = Ph. Black, uncatalyzed; light blue, Na+-catalyzed.
According to computations using DAPb (ArC = 4-Me-C6H4), allyl amine 3 (ArA = Ph) and a model allenoate with R = Ph, the lowest free energies of activation are 13.9 kcal/mol to the major enantiomer and 15.6 kcal/mol to the minor enantiomer, with ΔΔG‡ = 1.7 kcal/mol. The Boltzmann-weighted average of 14 transition states for the rearrangement results in a computed ee of 77%. In comparison, the experimentally observed selectivity with this catalyst and the standard substrate (R = 4-tBu-C6H4) was 66% ee or ~0.9 kcal/mol. The early transition state of the rearrangement still reflects the charge separation that is present in the zwitterionic intermediate. Thus, the interactions between TS and catalyst are dominated by strong electrostatic interactions (Figure 3).13 In both pathways, the sodium cation assembles the transition state structure by several key contacts: an ionic interaction with a phosphate oxygen, a cation–π interaction with one of the outer naphthyl “side-walls” of the catalytic pocket as well as an ionic interaction with the allenoate oxygen and another cation–π interaction with the phenolate moiety at the allenoate. The partially positively charged ammonium moiety interacts via several C–H-anion interactions with the other phosphate oxygen.
Figure 3.

Lowest transition state structures for the major (ΔG‡ = 13.9 kcal/mol) and minor (ΔG‡ = 15.6 kcal/mol, ΔΔG‡ = 1.7 kcal/mol) enantiomer pathways. The insets illustrate the most important NCIs between catalyst and transition state with representative interaction distances in Å (top) and NCI plot visualizations (bottom).13
The transition state is further oriented by NCIs with the same naphthyl side-wall that coordinates Na+. There are two striking differences between the transition states leading to either enantiomer. First, in the major-enantiomer TS, the phenyl group of the allyl amine substrate (ArA) fits into a space at the catalyst that is confined by ArC and one of the “inner” naphthyl units, effectively stacking the three arenes with two edge-to-face arene–arene interactions. Conversely, in the minor-enantiomer TS, that same phenyl group ArA engages in a staggered sandwich-type arene–arene interaction with ArC. The second major difference is the orientation of the phenyl ester group relative to the sodium cation, which results in a weaker cation–π interaction in the minor enantiomer TS. According to distortion–interaction analysis,14 the major enantiomer TS is stabilized by 4.1 kcal/mol greater interaction energy over the minor enantiomer TS, while the distortion energy for both TS is nearly identical.
This analysis suggests why DAP catalysts generally display higher enantioselectivity in this transformation than the more commonly employed chiral phosphoric acids: The extended π-system of the “outer” naphthyl moieties serves to fix the TS in place via mutual interactions with Na+, while at the same time offering a large area for NCIs with the TS and a pocket that is large enough to accommodate the extended TS of the allenoate-Claisen reaction.15
As part of the exploration of the catalyst conformations, we noted that the DAP catalyst can adopt two diastereomeric conformations. In the synthesis, the biaryl axes of the 1,1′-binaphthyl units are introduced as single (Ra)-atropisomers. In the coupling that sets the 2,2′-binaphthyl axis, only the (Ra,Ra,Ra)-diastereomer was obtained, as elucidated by crystal structure analysis of DAPa.6b,16, Our computational study suggests that the central biaryl axis of the catalyst is configurationally flexible at room temperature with ΔG‡ = 12.4 (phosphoric acid, R = H), or 7.8 (sodium phosphate, R = Na) kcal/mol for the inversion (Figure 4A). The relative free energies of the (Ra,Sa,Ra)-diastereomers are +2.8 and +0.3 kcal/mol for R = H and Na, respectively. In agreement with the crystal structure, this relates to a preference for the (Ra,Ra,Ra)-diastereomer in the phosphoric acid. Nevertheless, the low inversion barrier and close relative free energies suggest that both configurations might be in equilibrium under the reaction conditions where the catalyst presumably is present as the sodium salt.
Figure 4.

Influence of the catalyst 1,1′-binaphthyl axis configuration on the selectivity in the allenoate-Claisen rearrangement. (A) Computed free energy profile for the axis inversion of DAPa as acid and as sodium salt. (B) Synthesis of the triply axially chiral phosphoric acid TAPa. (a) Propyne (1 atm), AlCl3 (1.5 equiv), DCM 0.1M, 4 h, rt. (b) FeCl3 (10 mol %), (tBuO)2 (1.5 equiv), DCE 0.2M, 72 h, rt. (c) (1R)-(+)-menthyl chloroformate (1.2 equiv), Et3N (1.2 equiv), DCM 0.18M, 30 min. (d) Separation of diastereoisomers in automated column chromatography system, then: pyrrolidine (12 equiv), THF 0.05M, 2 h, rt. (e) Six steps; see the SI for details. (f) (i) POCl3 (2.0 equiv), pyridine 0.19 M, 95 °C, 2 h; (ii) H2O 0.19 M, 95 °C, 24 h. (C) Crystal structures of DAPa and 34. (g) From ref 6b. (D) Enantioselectivity of DAPd, ent-DAPd, and TAPa in the allenoate-Claisen rearrangement of 2 and 3 under standard conditions.
We computed transition states for the rearrangement with DAPb for both catalyst diastereomers and found the lowest pathways to either enantiomer to proceed via the (Ra,Ra,Ra)-conformation diastereomer of the catalyst interacting with the reaction intermediate in a conformation that ultimately corresponds to the minor enantiomer. Thus, both catalyst diastereomers might contribute to the observed product distribution. Nevertheless, the lowest TS at the (Ra,Sa,Ra)-diastereomer of the catalyst leads to the major enantiomer with ΔΔG‡ = 1.6 kcal/mol, which is nearly the same relative free energy of the lowest minor enantiomer TS. Also, the most stable catalyst–intermediate geometry features the (Ra,Sa,Ra)-configuration.
Synthesis of a Triply Axially Chiral Phosphate Probe.
Attempts to characterize this equilibrium experimentally by circular dichroism and NMR were unsuccessful due to the low solubility of the sodium phosphates. Therefore, we decided to synthesize a triply axially chiral phosphoric acid (TAP) TAPa with ArC = 4-tBu-C6H4 (Figure 4B) on the basis of the hypothesis that it would not be able to undergo axis inversion under the reaction conditions. The 4-substituted binaphthol scaffold rac-32 was obtained in synthetically useful yields by oxidative Fe-catalyzed homocoupling of 4-methyl-2-naphthol 31 which, in turn, was prepared according to a reported procedure via sequential intermolecular acylation–intramolecular Friedel–Crafts reaction from benzoyl chloride and propyne gas. Enantiomerically pure binaphthol 32 was obtained by chromatographic separation of its diastereomeric menthyl carbonate derivatives. After neat deprotection, a synthetic route slightly modified from the one to access the DAP catalyst (see the SI for further details) yielded the triply axially chiral catalyst TAPa. Crystal structure analysis of intermediate 34 revealed that the relative configuration of TAPa is the same as that of DAPa in the crystal structure, but the absolute configuration of all three chiral axes is the opposite (Figure 4C). Due to a change in naming priority imposed by the methyl groups, the axis of the 1,1′-binaphthyl moieties is denoted (Ra) in this opposite configuration. The reaction employing TAPa under the standard conditions provided the product in −95% ee (Figure 4D), providing the same enantiomer of the product as the opposite enantiomer of DAPd (ent-DAPd). Thus, TAPa provides the same product enantiomer as the DAP catalyst of the same absolute configuration, but with improved enantioselectivity. Introduction of four methyl groups into the catalyst could change several features of the catalytic pocket, such as the dihedral angle of the central chiral axis, and these structural changes might contribute to the increased enantioselectivity.
Still, the observation is consistent with the hypothesis that the central “flexible” axis of the DAP catalyst in the transition state is in the configuration that observed in the ground state (crystal structure). Thus, the TAP catalysts, which are less accessible, provide a valuable platform for examining the stereochemical impact of the more synthetically tractable DAP catalysts.
Statistical Modeling.
While the transition state computations provided insight into the mode of enantioinduction, the effect of structural changes of either the catalyst or the allyl amine on the enantioselectivity of the reaction was not apparent. We investigated these structural dependencies using statistical analysis tools.17 By developing multivariable linear regression (MLR) models that relate the properties of the reaction components to the enantioselectivity of the corresponding reaction, we can learn about the factors that determine selectivity using all the available data. Based on the transition state structures, we paid special attention to molecular features that describe the noncovalent interactions between catalyst and allyl amine, in particular these three critical NCIs: (1) ArC(side)–ArA(π) edge-to-face complex and (2) ArA(side)–naphthyl(π) edge-to-face complex in the major TS, and (3) ArC(π)–ArA(π) sandwich complex in the minor TS (Figure 5, also cf. Figure 3). According to the general approach we have previously published,18 each interaction component was represented by simple probes: ArC and ArA were represented as the corresponding H-capped arene, and the naphthyl section of the catalyst was represented by a benzene probe. Then, the NCIs between these probes were quantified by determining the equilibrium interaction distance and energies in idealized interaction geometries resembling the TS geometries. The equilibrium distances were found by interpolation from single point distance scans using symmetry-adapted perturbation theory (SAPT) at the sSAPT0/jun-cc-pVDZ level.19–21 For each interaction geometry, several conformers may exist depending on the symmetry of the probes. This is taken into account by using as separate modeling features both the lowest and highest interaction distance and energy found in any of the interaction conformers as well as their Boltzmann-weighted average. Using the linear catalyst and substrate variation data as the training set (all catalysts with ArA = Ph, all substrates with ArC = 4-Ad-Ph; 24 samples) and all simultaneous variations of both components as the validation set (13 samples), we found that the enantioselectivity of the training set can be estimated reasonably well by a multivariable linear regression model using only these NCI descriptors, with a coefficient of determination (R2) of 0.77 for the training set (Figure 5, top). This adds further credibility to the importance of all three NCIs in the transition states, as well as indicating that structural changes of catalyst or allyl amine impact the enantioselectivity through modulation of these NCIs. In line with this hypothesis, the descriptors for NCIs in the major TS both contribute with a negative coefficient and the NCI descriptor for the minor TS contributes with a positive coefficient (lower energies as well as shorter distances indicate stronger NCIs; thus, a negative coefficient indicates that stronger interactions result in higher enantioselectivity).
Figure 5.
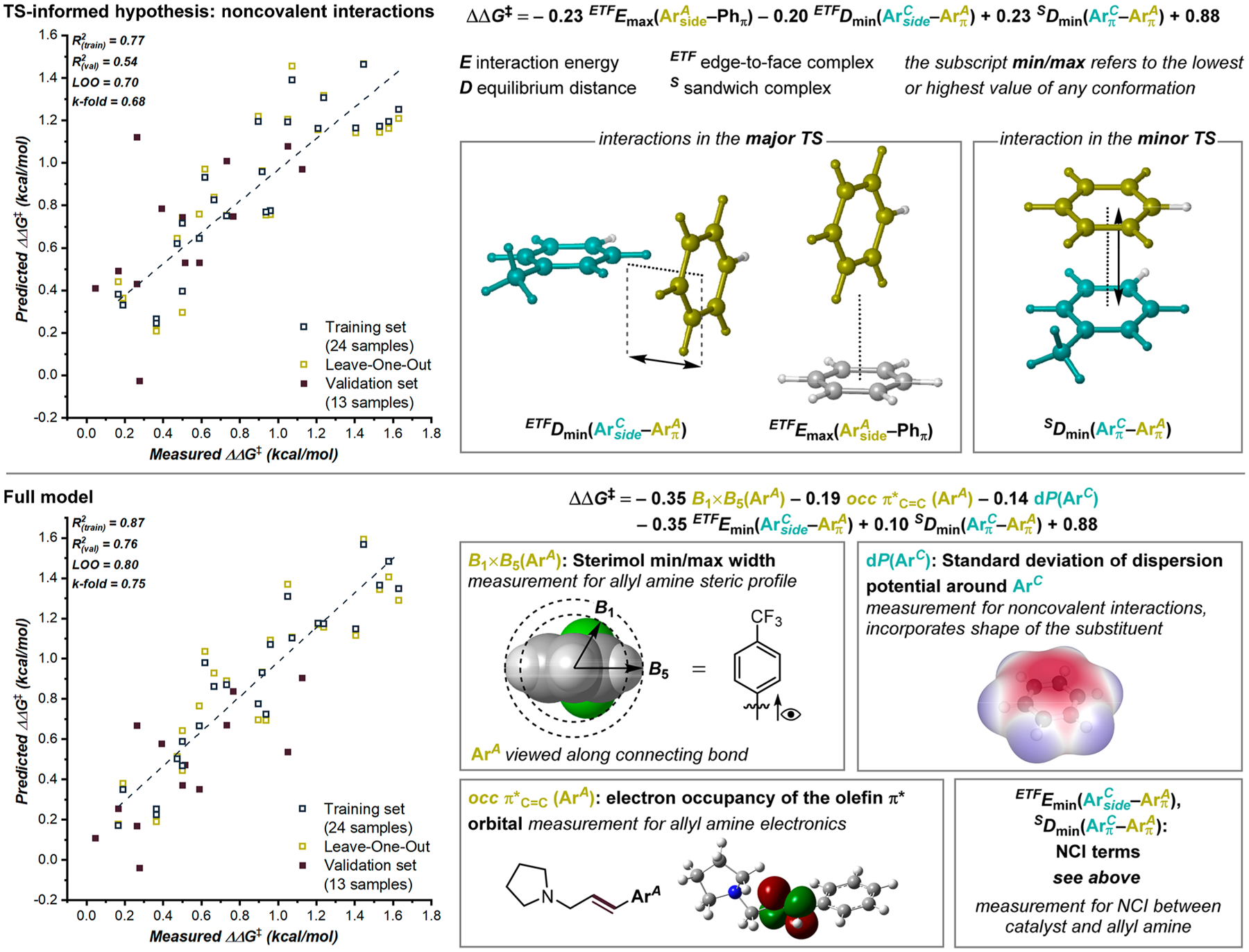
Multivariable regression model relating structural variations of allyl amines and catalysts to the observed enantioselectivity (expressed as ΔΔG‡) of the respective combination. ArC refers to the variable arene residue of the catalysts, and ArA refers to arene substituents at the allyl amine.
Nevertheless, while this model may be an underfit for the linear catalyst and substrate variation data, it cannot predict the simultaneous variations equally well, as indicated by the lower R2 = 0.56 of the validation set. In other words, not all structural effects on the enantioselectivity are represented by the model and by the NCI. Thus, we obtained electronic and steric properties of the catalysts and allyl amines from DFT computations and sought to identify an MLR model combining such features with the NCI descriptors (Figure 5, bottom). This model includes descriptors for the steric profile and the electronic properties of the allyl amine, the dispersion potential of the catalyst substituent, and two of the aforementioned descriptors for NCIs between catalyst and allyl amine. The standard deviation dP of the dispersion potential parameter recently introduced by Pollice and Chen22 relates to the distribution of sites of stronger and weaker dispersion around the substituent and as such is also influenced by the substituent shape. This shape-dependence of two terms in the regression model perhaps represents the complexity of the catalyst–TS interactions. In the DFT computations discussed above, we identified 14 different TS structures for the rearrangement at the catalyst. Thus, it is feasible that the presence of ortho or meta substituents on either component alters the TS geometries to an extent that another TS structure becomes favored over those shown in Figure 3. Thus, the regression model might reflect such changes via shape-sensitive descriptors B1×B5(ArA), dP(ArC), and, to some degree, both NCI terms.
CONCLUSION
In conclusion, we have achieved an enantioselective allenoate-Claisen rearrangement using DAP catalysts under basic conditions and investigated the origins of the unique ability of this catalyst architecture to impart enantiocontrol over this reaction. DFT studies demonstrate the stabilization of charge-separated regions of the transition state via interactions with the sodium cation and the phosphate anion. The enantiomeric transition states are differentiated by numerous noncovalent interactions between the extended catalytic pocket of the DAP catalyst architecture. The impact of structural changes to either the catalyst or the allyl amine on the enantioselectivity can partially be attributed to modulation of arene–arene interactions at the periphery of the catalyst, as suggested by statistical parametrization modeling. Furthermore, the mechanistic investigations revealed the configurational flexibility of DAP molecules. Both the computational analysis and the synthesis of triply axially chiral phosphoric acid catalysts revealed that the more stable conformation of the catalysts is likely responsible for the observed product. It is likely that there will be reactions in which the opposite will be true, and these studies provide a platform for these future examinations. More broadly, these findings provide the impetus for further explorations on the role of flexibility in catalyst-derived selective reactions.
Supplementary Material
ACKNOWLEDGMENTS
This paper is dedicated to Prof. Santos Fustero (University of Valencia) on the occasion of his retirement. We gratefully acknowledge the National Institutes of Health (Grant 1 R01 GM121383 to M.S.S. and R35 GM118190 to F.D.T.) and the European Commission (RG87181 Marie Skłodowska-Curie International Outgoing Fellowship to J.M.) for financial support. T.G. thanks the Leopoldina Fellowship Programme of the German National Academy of Sciences Leopoldina (LPDS 2017-18), M.E. thanks the Deutsche Forschungsgemeinschaft (DFG) for postdoctoral fellowships, and H.-H.L. thanks the Ministry of Science and Technology of the Republic of China (MOST 106-2917-I-003-0040). The support and resources from the Center for High Performance Computing at the University of Utah are gratefully acknowledged. Further computational resources were provided by the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF (ACI-1548562) and provided through allocation TG-CHE180003.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c01637.
Synthetic procedures, characterization data for all new compounds, additional optimization data, computational details, parametrization data, coordinates for all computed structures (PDF)
X-ray crystallographic data for 6 (CCDC #1969562) (CIF)
X-ray crystallographic data for 34 (CCDC 1969563) (CIF)
Coordinates of all computed structures (XYZ)
Descriptors used for statistical modeling (XLSX)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.0c01637
The authors declare no competing financial interest.
Contributor Information
Javier Miró, Department of Chemistry, University of California, Berkeley, California 94720, United States.
Tobias Gensch, Department of Chemistry, University of Utah, Salt Lake City, Utah 84112, United States.
Mario Ellwart, Department of Chemistry, University of California, Berkeley, California 94720, United States.
Seo-Jung Han, Department of Chemistry, University of California, Berkeley, California 94720, United States; Chemical Kinomics Research Center and Division of Bio-Medical Science & Technology, Korea Institute of Science and Technology (KIST), Seoul 02792, Republic of Korea.
Hsin-Hui Lin, Department of Chemistry, University of California, Berkeley, California 94720, United States.
Matthew S. Sigman, Department of Chemistry, University of Utah, Salt Lake City, Utah 84112, United States.
F. Dean Toste, Department of Chemistry, University of California, Berkeley, California 94720, United States.
REFERENCES
- (1).(a) Hiersemann M; Nubbemeyer U The Claisen Rearrangement: Methods and Applications, 1st ed.; Wiley-VCH, 2007. [Google Scholar]; (b) Jones AC; May JA; Sarpong R; Stoltz BM Toward a Symphony of Reactivity: Cascades Involving Catalysis and Sigmatropic Rearrangements. Angew. Chem., Int. Ed 2014, 53, 2556–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Majumdar KC; Alam S; Chattopadhyay B Catalysis of the Claisen Rearrangement. Tetrahedron 2008, 64, 597–643. [Google Scholar]; (d) Martin Castro AM Claisen Rearrangement over the Past Nine Decades. Chem. Rev 2004, 104, 2939–3002. [DOI] [PubMed] [Google Scholar]; (e) Gonda J The Belluš-Claisen rearrangement. Angew. Chem., Int. Ed 2004, 43, 3516–3524. [DOI] [PubMed] [Google Scholar]
- (2).(a) Kadam VD; Rao BSS; Mahesh SK; Chakraborty M; Vemulapalli SPB; Dayaka SN; Sudhakar G Stereoselective Access to the Core Structure of Macroline-Type Indole Alkaloids: Total Synthesis of Macroline and Alstomicine. Org. Lett 2018, 20, 4782–4786. [DOI] [PubMed] [Google Scholar]; (b) Crimmins MT; Knight JD; Williams PS; Zhang Y Stereoselective Synthesis of Quaternary Carbons via the Dianionic Ireland-Claisen Rearrangement. Org. Lett 2014, 16, 2458–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Peng B; Geerdink D; Maulide N Electrophilic Rearrangements of Chiral Amides: A Traceless Asymmetric α-Allylation. J. Am. Chem. Soc 2013, 135, 14968–14971. [DOI] [PubMed] [Google Scholar]; (d) Takao K; Sakamoto S; Touati MA; Kusakawa Y; Tadano K Asymmetric Construction of All-Carbon Quaternary Stereocenters by Chiral-Auxiliary-Mediated Claisen Rearrangement and Total Synthesis of (+)-Bakuchiol. Molecules 2012, 17, 13330–13344. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Liu Z; Mehta SJ; Lee K-S; Grossman B; Qu H; Qu X; Nichol GS; Hruby VJ Thio-Claisen Rearrangement Used in Preparing Anti-β-Functionalized γ,δ-Unsaturated Amino Acids: Scope and Limitations. J. Org. Chem 2012, 77, 1289–1300. [DOI] [PubMed] [Google Scholar]; (f) Qu H; Gu X; Liu Z; Min BJ; Hruby VJ Asymmetric Eschenmoser-Claisen Rearrangement for Anti-β-Substituted γ,δ-Unsaturated Amino Acids. Org. Lett 2007, 9, 3997–4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Yoon TP; MacMillan DWC Enantioselective claisen rearrangements: Development of a first-generation asymmetric acyl-Claisen reaction. J. Am. Chem. Soc 2001, 123, 2911–2912. [DOI] [PubMed] [Google Scholar]; (b) Abraham L; Czerwonka R; Hiersemann M The Catalytic Enantioselective Claisen Rearrangement of an Allyl Vinyl Ether. Angew. Chem., Int. Ed 2001, 40, 4700–4703. [DOI] [PubMed] [Google Scholar]; (c) Linton EC; Kozlowski MC Catalytic Enantioselective Meerwein-Eschenmoser Claisen Rearrangement: Asymmetric Synthesis of Allyl Oxindoles. J. Am. Chem. Soc 2008, 130, 16162–16163. [DOI] [PubMed] [Google Scholar]; (d) Geherty ME; Dura RD; Nelson SG Catalytic Asymmetric Claisen Rearrangement of Unactivated Allyl Vinyl Ethers. J. Am. Chem. Soc 2010, 132, 11875–11877. [DOI] [PubMed] [Google Scholar]; (e) Tan J; Cheon C-H; Yamamoto H Catalytic Asymmetric Claisen Rearrangement of Enolphosphonates: Construction of Vicinal Tertiary and All-Carbon Quaternary Centers. Angew. Chem., Int. Ed 2012, 51, 8264–8267. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Becker J; Butt L; von Kiedrowski V; Mischler E; Quentin F; Hiersemann M Catalytic Asymmetric Claisen Rearrangement of Gosteli-Type Allyl Vinyl Ethers: Total Synthesis of (–)-9,10-Dihydroecklonialactone B. J. Org. Chem 2014, 79, 3040–3051. [DOI] [PubMed] [Google Scholar]; (g) Fontoura Rodrigues TCA; Alves Silva W; Lira Machado AH Recent Advances in the Asymmetric Claisen Rearrangement Promoted by Chiral Organometallic Lewis Acids or Organic Brønsted-Lowry Acids. Curr. Org. Synth 2015, 12, 795–805. [Google Scholar]; (h) Zheng H; Wang Y; Xu C; Xu X; Lin L; Liu X; Feng X Stereodivergent Synthesis of Vicinal Quaternary-Quaternary Stereocenters and Bioactive Hyperolactones. Nat. Commun 2018, 9, 1968. [DOI] [PMC free article] [PubMed] [Google Scholar]; Alternatively, guanidinium salts, N-heterocyclic carbenes and phosphoric acids can also induce excellent levels of enantioselectivity in a variety of Claisen rearrangements:; (i) Uyeda C; Jacobsen EN Enantioselective Claisen Rearrangemnts with a Hydrogen-Bond Donor Catalyst. J. Am. Chem. Soc 2008, 130, 9228–9229. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Kaeobamrung J; Mahatthananchai J; Zheng P; Bode JW An Enantioselective Claisen Rearrangement Catalyzed by N-Heterocyclic Carbenes. J. Am. Chem. Soc 2010, 132, 8810–8812. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Rueping M; Antonchick AP Catalytic Asymmetric Aminoallylation of Aldehydes: A Catalytic Enantioselective Aza-Cope Rearrangement. Angew. Chem., Int. Ed 2008, 47, 10090–10093. [DOI] [PubMed] [Google Scholar]; (l) Maity P; Pemberton RP; Tantillo DJ; Tambar UK Brønsted Acid Catalyzed Enantioselective Indole Aza-Claisen Rearrangement Mediated by an Arene CH–O Interaction. J. Am. Chem. Soc 2013, 135, 16380–16383. [DOI] [PubMed] [Google Scholar]; (m) Rehbein J; Hiersemann M Gosteli-Claisen Rearrangement: DFT Study of Substituent-Rate Effects. J. Org. Chem 2009, 74, 4336–4342. [DOI] [PubMed] [Google Scholar]; (n) Rehbein J; Leick S; Hiersemann M Gosteli-Claisen Rearrangement: Substrate Synthesis, Simple Diastereoselectivity, and Kinetic Studies. J. Org. Chem 2009, 74, 1531–1540. [DOI] [PubMed] [Google Scholar]; (o) Troendlin J; Rehbein J; Hiersemann M; Trapp O Integration of Catalysis and Analysis is the Key - Rapid and Precise Investigation of the Catalytic Asymmetric Gosteli-Claisen Rearrangement. J. Am. Chem. Soc 2011, 133, 16444–16450. [DOI] [PubMed] [Google Scholar]
- (4).(a) Lambert TH; MacMillan DWC Development of a New Lewis Acid-Catalyzed [3,3]-Sigmatropic Rearrangement: The Allenoate-Claisen Rearrangement. J. Am. Chem. Soc 2002, 124, 13646–13647. [DOI] [PubMed] [Google Scholar]; (b) Lambert TH Development of the Lewis Acid Catalyzed Allenoate-Claisen Rearrangement Investigations of Enantioselective Catalysis of the Allenoate-Claisen Rearrangement. Studies Towards the Total Synthesis of Erythrolide E. Ph.D. Dissertation, California Institute of Technology, Pasadena, CA, 2004. [Google Scholar]; (c) Lopes SMM; Santos BS; Palacios F; Pinho E Melo TMVD Microwave-assisted Reactions of Allenic esters: [3 + 2] Anellations and Allenoate-Claisen Rearrangement. Arkivoc 2010, 2010, 70–81. [Google Scholar]
- (5).(a) Phipps RJ; Hamilton GL; Toste FD The progression of chiral anions from concepts to applications in asymmetric catalysis. Nat. Chem 2012, 4, 603–614. [DOI] [PubMed] [Google Scholar]; (b) Parmar D; Sugiono E; Raja S; Rueping M Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates. Chem. Rev 2014, 114, 9047–9153. [DOI] [PubMed] [Google Scholar]
- (6).(a) Guo Q-S; Du D-M; Xu J The Development of Double Axially Chiral Phosphoric Acids and Their Catalytic Transfer Hydrogenation of Quinolines. Angew. Chem., Int. Ed 2008, 47, 759–762. [DOI] [PubMed] [Google Scholar]; (b) Honjo T; Phipps RJ; Rauniyar V; Toste FD A Doubly Axially Chiral Phosphoric Acid Catalyst for the Asymmetric Tandem Oxyfluorination of Enamides. Angew. Chem., Int. Ed 2012, 51, 9684–9688. [DOI] [PubMed] [Google Scholar]; (c) Hiramatsu K; Honjo T; Rauniyar V; Toste FD Enantioselective Synthesis of Fluoro-Dihydroquinazolones and – Benzooxazinones by Fluorination-Initiated Asymmetric Cyclization Reactions. ACS Catal. 2016, 6, 151–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Reviews on the role of NCIs in asymmetric catalysis:; (a) Knowles RR; Jacobsen EN Attractive noncovalent interactions in asymmetric catalysis: Links between enzymes and small molecule catalysts. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 20678–20685. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Toste FD; Sigman MS; Miller SJ Pursuit of Noncovalent Interactions for Strategic Site-Selective Catalysis. Acc. Chem. Res 2017, 50, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Neel AJ; Hilton MJ; Sigman MS; Toste FD Exploiting non-covalent π interactions for catalyst design. Nature 2017, 543, 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8). Control experiments revealed that the desired product is formed in the absence of the chiral phosphoric acid in moderate yields.
- (9). γ-Aryl-substituted allenic aryl- and trifluoroethanol-derived esters appeared to be highly unstable. Attempts to react them immediately after isolation provided intractable mixtures of products. Further limitations inherent to this reaction are discussed in ref 4b.
- (10).Selected examples for the use of computational chemistry to gain insight on reaction mechanisms of chiral phosphoric acid catalysts:; (a) Maji R; Mallojjala SC; Wheeler SE Chiral phosphoric acid catalysis: from numbers to insights. Chem. Soc. Rev 2018, 47, 1142–1158 and references therein.. [DOI] [PubMed] [Google Scholar]; (b) Milo A; Neel AJ; Toste FD; Sigman MS A data-intensive approach to mechanistic elucidation applied to chiral anion catalysis. Science 2015, 347, 737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Reid JP; Goodman JM Goldilocks Catalysts: Computational Insights into the Role of the 3,3′ Substituents on the Selectivity of BINOL-Derived Phosphoric Acid Catalysts. J. Am. Chem. Soc 2016, 138, 7910–7917. [DOI] [PubMed] [Google Scholar]; (d) Maji R; Champagne PA; Houk KN; Wheeler SE Activation mode and origin of selectivity in chiral phosphoric acid-catalyzed oxacycle formation by intramolecular oxetane desymmetrizations. ACS Catal. 2017, 7, 7332–7339. [Google Scholar]; (e) Simón L; Paton RS The True Catalyst Revealed: The Intervention of Chiral Ca and Mg Phosphates in Brønsted Acid Promoted Asymmetric Mannich Reactions. J. Am. Chem. Soc 2018, 140, 5412–5420. [DOI] [PubMed] [Google Scholar]; (f) Zhang J; Yu P; Li S-Y; Sun H; Xiang S-H; Wang J; Houk KN; Tan B Asymmetric phosphoric acid–catalyzed four-component Ugi reaction. Science 2018, 361, No. eaas8707. [DOI] [PubMed] [Google Scholar]
- (11). ωB97X-D/spAug-cc-pVTZ//B97-D3/jun-cc-pVDZ, SMD: cyclohexane in Gaussian 16, rev. A.03.
- (12).(a) Chai J-D; Head-Gordon M Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys 2008, 10, 6615–6620. [DOI] [PubMed] [Google Scholar]; (b) Grimme S; Ehrlich S; Goerigk L Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem 2011, 32, 1456–1465. [DOI] [PubMed] [Google Scholar]; (c) Dunning TH Gaussian basis sets for use in correlated molecular calculations I The atoms boron through neon and hydrogen. J. Chem. Phys 1989, 90, 1007. [Google Scholar]; (d) Kendall RA; Dunning TH; Harrison RJ Electron affinities of the first-row atoms revisited Systematic basis sets and wave functions. J. Chem. Phys 1992, 96, 6796–6806. [Google Scholar]; (e) Marenich AV; Cramer CJ; Truhlar DG Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]; (f) Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams-Young D; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JAJ; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas Ö; Foresman JB; Fox DJ Gaussian 16; Gaussian Inc.: Wallingford CT, 2016. [Google Scholar]
- (13).(a) Johnson ER; Keinan S; Mori-Sánchez P; Contreras-García J; Cohen AJ; Yang W Revealing Noncovalent Interactions. J. Am. Chem. Soc 2010, 132, 6498–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Contreras-García J; Johnson ER; Keinan S; Chaudret R; Piquemal J-P; Beratan DN; Yang W NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput 2011, 7, 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bickelhaupt FM; Houk KN Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem., Int. Ed 2017, 56, 10070–10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Crawford J; Sigman M Conformational Dynamics in Asymmetric Catalysis: Is Catalyst Flexibility a Design Element? Synthesis 2019, 51, 1021–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16). Some catalysts were prepared using the previously reported route that sets the 2,2′-binaphthyl connection via Suzuki coupling, cf. ref 6.
- (17).(a) Sigman MS; Harper KC; Bess EN; Milo A The Development of Multidimensional Analysis Tools for Asymmetric Catalysis and Beyond. Acc. Chem. Res 2016, 49, 1292–1301. [DOI] [PubMed] [Google Scholar]; (b) Santiago CB; Guo J-Y; Sigman MS Predictive and mechanistic multivariate linear regression models for reaction development. Chem. Sci 2018, 9, 2398–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Reid JP; Sigman MS Comparing quantitative prediction methods for the discovery of small-molecule chiral catalysts. Nat. Rev. Chem 2018, 2, 290–305. [Google Scholar]
- (18).Orlandi M; Coelho JAS; Hilton MJ; Toste FD; Sigman MS Parametrization of Non-covalent Interactions for Transition State Interrogation Applied to Asymmetric Catalysis. J. Am. Chem. Soc 2017, 139, 6803–6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19). Interaction energies were obtained with the sSAPT0/jun-cc-pVDZ method implemented in PSI4 1.1.
- (20).(a) Jeziorski B; Moszynski R; Szalewicz K Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev 1994, 94, 1887–1930. [Google Scholar]; (b) Hohenstein EG; Sherrill CD Wavefunction methods for noncovalent interactions. WIREs. Comput. Mol. Sci 2012, 2, 304–326. [Google Scholar]; (c) Sherrill CD Energy Component Analysis of π Interactions. Acc. Chem. Res 2013, 46, 1020–1028. [DOI] [PubMed] [Google Scholar]
- (21).(a) Hohenstein EG; Sherrill CD Density fitting and Cholesky decomposition approximations in symmetry-adapted perturbation theory: Implementation and application to probe the nature of π-π interactions in linear acenes. J. Chem. Phys 2010, 132, 184111. [Google Scholar]; (b) Parker TM; Burns LA; Parrish RM; Ryno AG; Sherrill CD Levels of symmetry adapted perturbation theory (SAPT) I Efficiency and performance for interaction energies. J. Chem. Phys 2014, 140, 094106. [DOI] [PubMed] [Google Scholar]; (c) Gonthier JF; Sherrill CD Density-fitted open-shell symmetry-adapted perturbation theory and application to π -stacking in benzene dimer cation and ionized DNA base pair steps. J. Chem. Phys 2016, 145, 134106. [DOI] [PubMed] [Google Scholar]; (d) Parrish RM; Burns LA; Smith DGA; Simmonett AC; DePrince AE; Hohenstein EG; Bozkaya U; Sokolov AY; Di Remigio R; Richard RM; Gonthier JF; James AM; McAlexander HR; Kumar A; Saitow M; Wang X; Pritchard BP; Verma P; Schaefer HF; Patkowski K; King RA; Valeev EF; Evangelista FA; Turney JM; Crawford TD; Sherrill CD PSI4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability. J. Chem. Theory Comput 2017, 13, 3185–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Pollice R; Chen P A Universal Quantitative Descriptor of the Dispersion Interaction Potential. Angew. Chem., Int. Ed 2019, 58, 9758–9769. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.