

Abstract
Hypoxia-induced neuroinflammation typically causes neurological damage and can occur during stroke, neonatal hypoxic-ischemic encephalopathy, and other diseases. Propofol is widely used as an intravenous anesthetic. Studies have shown that propofol has antineuroinflammatory effect. However, the underlying mechanism remains to be fully elucidated. Thus, we aimed to investigate the beneficial effects of propofol against hypoxia-induced neuroinflammation and elucidated its potential cellular and biochemical mechanisms of action. In this study, we chose cobalt chloride (CoCl2) to establish a hypoxic model. We found that propofol decreased hypoxia-induced proinflammatory cytokines (TNFα, IL-1β, and IL-6) in BV2 microglia, significantly suppressed the excessive production of reactive oxygen species, and increased the total antioxidant capacity and superoxide dismutase activity. Furthermore, propofol attenuated the hypoxia-induced decrease in mitochondrial membrane potential andy 2 strongly inhibited protein expression of nuclear factor-kappa B (NF-κB) subunit p65 and hypoxia inducible factor-1α (Hif-1α) in hypoxic BV2 cells. To investigate the role of NF-κB p65, specific small interfering RNA (siRNA) against NF-κB p65 were transfected into BV2 cells, followed by exposure to hypoxia for 24 h. Hypoxia-induced Hif-1α production was downregulated after NF-κB p65 silencing. Further, propofol suppressed Hif-1α expression by inhibiting the upregulation of NF-κB p65 after exposure to hypoxia in BV2 microglia. In summary, propofol attenuates hypoxia-induced neuroinflammation, at least in part by inhibiting oxidative stress and NF-κB/Hif-1α signaling.
1. Introduction
Microglia are the primary neuroimmune cells important for surveillance and defense, in addition to maintaining homeostasis, being the first sensors of pathophysiological changes, and triggering subsequent cascade reactions [1–3]. Microglia undergo a remarkable transformation into “activated microglia” during brain injury and disease, when numerous genes are switched on. M1-activated microglia mainly secrete proinflammatory cytokines (TNF-α, IL-1β, and IL-6), which consequently promote the development of several central nervous system (CNS) disorders. However, M2-activated microglia primarily secrete anti-inflammatory cytokines, which can facilitate tissue reconstruction [4, 5]. In a hypoxic condition, Hif-1α is stabilized and activates the transcription of genes associated with proinflammation [6, 7]. Moreover, hypoxia impairs mitochondrial function resulting in increased production of reactive oxygen species (ROS), decreased mitochondrial membrane potential, and decreased ATP production [8, 9]. Considering the key role of neuroinflammation in hypoxia-induced nerve damage, neuroinflammation induced by activated microglia (especially M1 polarization) is an important regulator of hypoxia-induced CNS damage.
As a commonly used intravenous anesthetic, propofol has many other functions [10, 11]. In our previous study, we found that propofol can prevent oxidative stress and attenuate mitochondrial dysfunction during focal cerebral ischemia-reperfusion injury [12, 13]. Further, propofol has an anti-inflammatory effect as it inhibits NF-κB activation in mice with allergic asthma [14]. NF-κB subunit p65 has been reported to induce basal level expression of Hif-1α mRNA and protein [15–17]. However, the mechanism by which propofol attenuates hypoxia-induced neuroinflammation in activated microglia is relatively unclear. Therefore, we designed this study to investigate the protective effect of propofol on hypoxia-induced neuroinflammation associated with microglia and whether this occurs through the inhibition of oxidative stress and NF-κB/Hif-1α signaling.
2. Materials and Methods
2.1. Microglial Cell Culture
Since BV2 microglia originate from mouse brains, they share several phenotypic characteristics with primary microglia. Thus, we chose these cells as the model for the current study. The cells were obtained from the National Infrastructure of Cell Line Resource (China). Microglia were grown according to the recommended conditions described by the cell bank. The medium contained high-glucose Dulbecco's modified Eagle's medium (DMEM) (Corning, USA) with 10% fetal bovine serum (FBS) (Corning, USA) and 1% penicillin-streptomycin. The cells were incubated at 37°C in a humidified atmosphere of 95% air and 5% CO2. After growing them to 80% confluence, cells were subcultured two or three times every week. Microglia were seeded in a 6-well plate for 24 h for subsequent experiments, and the inoculation density was 0.5 × 106 cells/ml.
2.2. Cell Treatment
In this study, 300 μM CoCl2 was used to establish hypoxia. According to the preliminary results of cell viability, the 50% inhibitive concentration (IC50) of CoCl2 was 300 μM at 24 h. Moreover, Hif-1α protein was expressed stably. CoCl2 was dissolved in distilled water to 300 mM. CoCl2 solution was filtered with a 0.22 mm Millipore membrane and stored at -20°C. Serum-free medium was used to dilute CoCl2 for cell treatment. Propofol was used to pretreat cells for 3 h before CoCl2 treatment. Following propofol treatment, the cells were washed twice with PBS.
2.3. Real-Time Quantitative PCR (qPCR)
Microglia were treated as indicated, and total RNA was collected with a TRIzol reagent (Sigma, America). cDNA was prepared using 1 μg of total RNA as the template with a reverse transcription kit (Takara, Japan). The reverse transcription apparatus was set up according to the kit instructions. Real-time PCR analysis was performed using an ABI Prism 7500 fast real-time PCR System (Applied Biosystems, America) with SYBR green. ΔΔCT values were used for analysis. qPCR was set up with a SYBR green PCR master mix (Roche, Switzerland), specific primers, cDNA, and double-distilled water. The conditions for PCR cycles were as follows: predenaturation (94°C for 2 min), denaturation (94°C for 15 s), annealing, and extension (60°C for 30 s). β-Actin was used as a housekeeper gene control, and untreated cells were used as a control to normalize the relative amounts of target gene expression. qPCR primer sequences are shown in Table 1.
Table 1.
Primer sequence.
| Primer | Sequence (from 5′ to 3′) |
|---|---|
| IL-6-F | CTCTGCAAGAGACTTCCATCC |
| IL-6-R | GAATTGCCATTGCACAACTC |
| iNOS-F | TTCACAGCTCATCCGGTACG |
| iNOS-R | CCATCAGCTTGCAAGACCAG |
| IL-1β-F | TAACCTGCTGGTGTGTGACG |
| IL-1β-R | TGTCGTTGCTTGGTTCTCCT |
| TNF-α-F | GTCCGGGCAGGTCTACTTTG |
| TNF-α-R | GGGGCTCTGAGGAGTAGACA |
| β-Actin-F | GGAGATTACTGCCCTGGCTCCTA |
| β-Actin-R | GACTCATCGTACTCCTGCTTGCTG |
2.4. Enzyme-Linked Immunosorbent Assay (ELISA)
Microglia were treated as indicated, and cell supernatants were used to measure the concentration of TNF-α, IL-1β, and IL-6 with ELISA kits (BOSTER, China), according to the manufacturer's instructions. The absorbance, using an iMark microplate reader (SpectraMax M3, Molecular Devices, America), was measured at a wavelength of 450 nm. The cytokine concentrations were determined based on reference standard curves.
2.5. Western Blotting
After treating microglia as indicated, cells were lysed with RIPA buffer containing a protease inhibitor and phosphatase inhibitor (BOSTER, China) at a ratio of 100 : 1 : 1. After the cells were completely lysed, the supernatant was taken, and the protein concentration was measured using a BCA protein quantitative kit (BOSTER, China). The same amount of protein was separated by sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene fluoride (PVDF) membrane (Roche, Switzerland). Tween 20 (BOSTER, China) in Tris-buffered saline (BOSTER, China), containing 5% skim milk powder, was used to block the membrane for 2 h. Primary antibodies against NF-κB p65 (1 : 1400, ab16502, Abcam), Hif-1α (1 : 1000, 14179, Cell Signaling Technology), and GAPDH (1 : 1000, TA-08, ZSGB-Bio) were then incubated with PVDF membranes at 4°C overnight. Next, secondary antibodies (peroxidase-labeled) were incubated with the blots at 25°C for 1 h. The protein bands were observed using the enhanced chemiluminescence (ECL) system (GE Healthcare, UK), and relative protein expression was quantified using ImageJ software.
2.6. Reactive Oxygen Species Assay Using Flow Cytometry
Drug intervention was completed as expected, and cells were stained with DCFH-DA (10 μM) for 20 min at 37°C in the dark. Following this, the cells were washed twice with PBS. Cells were removed from the plate surface and transferred to polypropylene FACS tubes with 500 μl PBS. Finally, a MoFlo XDP flow cytometer (Beckman, USA) was used for measurements and FlowJo software for data analysis.
2.7. Observation of Mitochondrial Membrane Potential Using Confocal Laser Scanning Microscopy
BV-2 cells were first seeded in confocal dishes. After treatment with different drugs, cells were stained with TMRM (20 nM, T668, Thermo Fisher Scientific) for 30 min at 37°C in the dark. The cells were then washed twice with PBS. Subsequently, the fluorescence intensity was measured by confocal laser scanning microscopy at an excitation wavelength of 561 nm, and data were analyzed using ImageJ software.
2.8. NF-κB p65 siRNA Transfections
siRNA targeted at NF-κB p65 (Sangon Biotech, China) was used to knock down NF-κB p65. The target sequence was 5′-CAACCATGGCTGAAGGAAA-3′. First, 100 pmol NF-κB p65 siRNA duplex was diluted into 250 μl Opti-MEM medium. Second, 5 μl lipofectamine 2000 (Lipo2000) transfection reagent (Thermo Fisher Scientific, America) was diluted with 250 μl Opti-MEM medium and incubated for 5 min at room temperature in a separate tube. Third, the aforementioned solutions were mixed gently and incubated for 30 min at 25°C to form a transfection complex. Next, 70% confluent BV2 cells to be used in the study were washed thrice with PBS. Following this, 1500 μl Opti-MEM medium and 500 μl transfection complex were added. The cells were incubated for 24 h at 37°C. Western blotting was performed to detect NF-κB p65 protein levels.
2.9. Statistical Analysis
Statistical analyses were performed using IBM SPSS Statistics 24 software. All data presented are representative of at least three independent experiments. The data are shown as the mean ± standard errors of the mean (S.E.M.). Independent Student's t-test was used for comparisons between two groups, and one-way analysis of variance (ANOVA) with the SNK post hoc test was used to compare multiple groups. The result was considered to be statistically significant when the P value was less than 0.05.
3. Results
3.1. CoCl2 Exposure Induces the Expression of Proinflammatory Cytokines in BV2 Cells
To observe the effect of CoCl2 treatment time on inflammatory responses in microglia, BV2 cells were treated with CoCl2 (300 μM) for 3, 6, 12, and 24 h. TNF-α, IL-1β, IL-6, and iNOS mRNA expression was detected by qPCR. As shown in Figure 1, TNF-α, IL-1β, IL-6, and iNOS mRNA expression was significantly increased in CoCl2-treated cells at 12 and 24 h compared to that in control cells (∗∗P < 0.01). Further, compared to that in control cells, IL-1β and iNOS expression was significantly increased in cells treated with CoCl2 at 6 h (∗P < 0.05). Based on the results, we infer that the expression of cytokines in BV2 cells was remarkably increased with prolonged CoCl2 incubation times.
Figure 1.
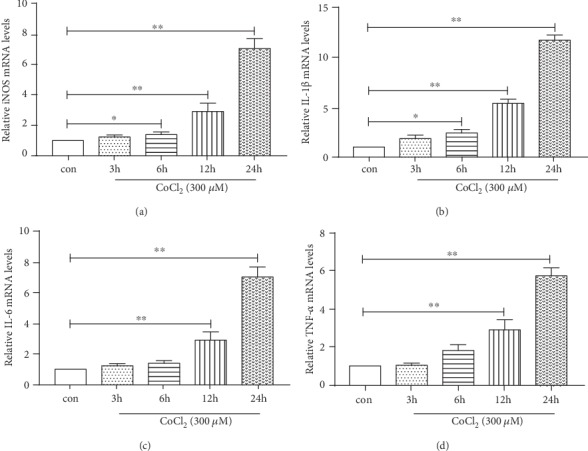
CoCl2 induces iNOS, IL-1β, IL-6, and TNF-α mRNA expression in microglia. BV2 cells were treated with 300 μM of CoCl2 for the indicated times (3, 6, 12, and 24 h). iNOS, IL-1β, IL-6, and TNF-α mRNA expression was detected by qPCR. The relative amounts of transcripts were calculated using the 2−ΔΔCT formula. β-Actin mRNA was used as the internal control. (a) iNOS mRNA expression at different times. (b) IL-1β mRNA expression at different times. (c) IL-6 mRNA expression at different times. (d) TNF-α mRNA expression at different times. Data from at least three independent experiments are expressed as the mean ± S.E.M. ANOVA with the SNK post hoc test was used to assess differences between groups. ∗P < 0.05, ∗∗P < 0.01 versus the control group.
3.2. Propofol Inhibits CoCl2-Induced Production of Proinflammatory Cytokines
To determine whether propofol has an effect on proinflammatory cytokines, we pretreated cells with different concentrations of propofol (25, 50, and 100 μM) for 3 h before CoCl2 stimulation for 24 h and then measured TNF-α, IL-1β, and IL-6 protein and mRNA levels. As displayed in Figure 2, CoCl2 increased both protein and mRNA levels of TNF-α, IL-1β, and IL-6 compared to control cells. Propofol inhibited protein secretion of TNF-α and IL-1β at 25, 50, and 100 μM concentrations; however, protein secretion of IL-6 was inhibited at 50 and 100 μM propofol concentrations only. Furthermore, propofol reduced mRNA levels of TNF-α, IL-1β, and IL-6 at 25, 50, and 100 μM concentrations. To summarize, propofol could reduce CoCl2-induced proinflammatory cytokine levels in BV2 cells, but not in a concentration-dependent manner.
Figure 2.
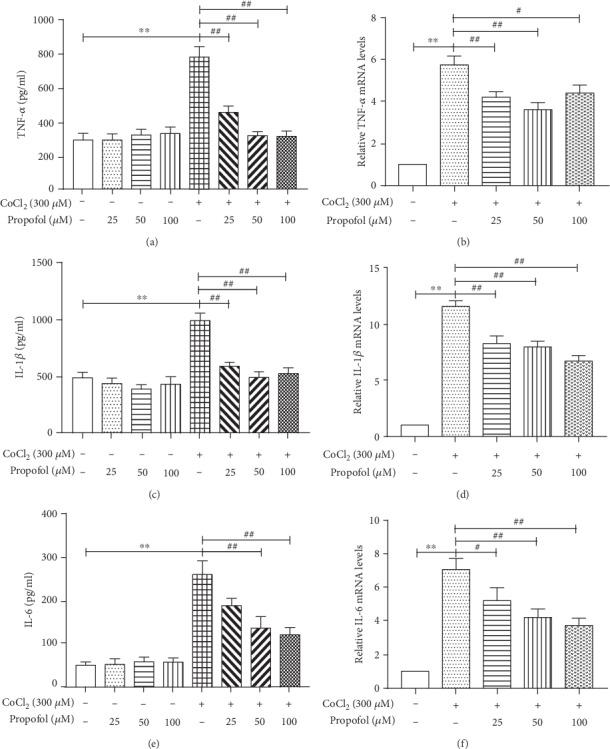
Propofol reduces the secretion of TNF-α, IL-1β, and IL-6 in CoCl2-treated microglia. BV2 cells were pretreated with propofol (25, 50, and 100 μM) for 3 h, which was followed by treatment with CoCl2 (300 μM) for 24 h. IL-1β, TNF-α, and IL-6 were analyzed by ELISA and qPCR. (a) TNF-α concentration after different treatments. (b) Relative TNF-α mRNA levels after different treatments. (c) IL-1β concentration after different treatments. (d) Relative IL-1β mRNA levels after different treatments. (e) IL-6 concentration after different treatments. (f) Relative IL-6 mRNA levels after different treatments. Data from at least three independent experiments are expressed as the mean ± S.E.M. Independent Student's t-test was used for comparisons between two groups, and ANOVA with the SNK post hoc test was used for multiple groups. ∗∗P < 0.01 versus the control group; ##P < 0.01 versus the CoCl2 group.
3.3. Propofol Ameliorates CoCl2-Induced Oxidative Stress
To observe the effect of propofol on CoCl2-induced oxidative stress, cells were pretreated with propofol for 3 h before CoCl2 stimulation for 24 h. Subsequently, ROS, superoxide dismutase (SOD) activity, and total antioxidant capacity (T-AOC) were detected. As shown in Figures 3(a) and 3(b), CoCl2 induced higher ROS production in BV2 cells compared to control cells, whereas propofol decreased ROS levels compared to CoCl2-treated cells. As displayed in Figures 3(c) and 3(d), CoCl2 decreased SOD and T-AOC activity compared to that in control cells, but propofol ameliorated this reduction. In summary, propofol could ameliorate CoCl2-induced oxidative stress in BV2 cells.
Figure 3.
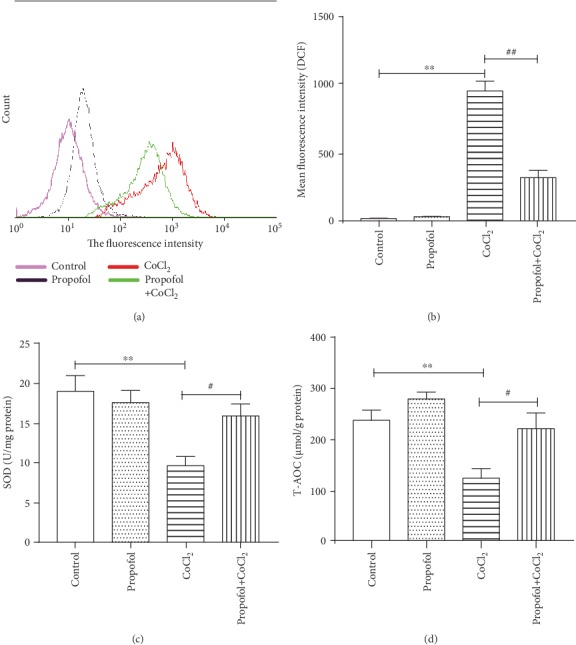
Propofol ameliorates CoCl2-induced oxidative stress in microglia. BV2 cells were pretreated with propofol (50 μM) for 3 h, followed by treatment with CoCl2 (300 μM) for 24 h. ROS were analyzed by flow cytometry. SOD and T-AOC were measured using specific kits. (a) ROS levels detected by flow cytometry. (b) The mean fluorescence intensity of DCF staining reflects ROS levels. (c) SOD levels after different treatments. (d) T-AOC levels after different treatments. Data from at least three independent experiments are expressed as the mean ± S.E.M. Independent Student's t-test was used for comparisons between two groups. ∗∗P < 0.01 versus the control group; #P < 0.05 versus the CoCl2 group; ##P < 0.01 versus the CoCl2 group.
3.4. Propofol Ameliorates the Decrease in Mitochondrial Membrane Potential in CoCl2-Treated Microglia
To determine whether propofol affects the CoCl2-induced decrease in mitochondrial membrane potential in BV2 cells, we pretreated cells with propofol for 3 h before CoCl2 stimulation for 24 h. We then measured mitochondrial membrane potential by confocal laser scanning microscopy. As shown in Figure 4, CoCl2 induced a decrease in mitochondrial membrane potential in BV2 cells compared to control cells, whereas propofol attenuated this decrease.
Figure 4.
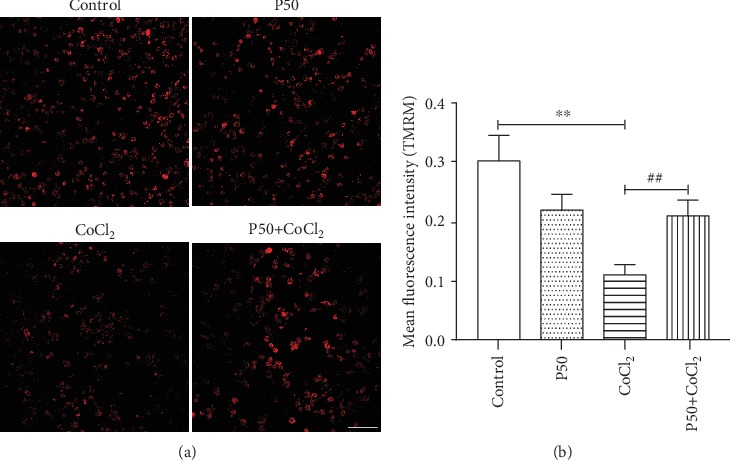
Propofol ameliorates the decrease in mitochondrial membrane potential in CoCl2-treated microglia. BV2 cells were pretreated with propofol (50 μM) for 3 h followed by treatment with CoCl2 (300 μM) for 24 h. Confocal laser scanning microscopy was used to observe mitochondrial membrane potential. Scale bar: 100 μm. (a) TMRM staining observed by confocal laser scanning microscopy. (b) The mean fluorescence intensity of TMRM staining reflects the membrane potential. The average fluorescence value is shown as the mean ± S.E.M. Data are based on at least three independent experiments. Independent Student's t-test was used for comparisons between two groups. ∗∗P < 0.01 versus the control group, #P < 0.05 versus the CoCl2 group. p50: propofol 50 μM.
3.5. Propofol Attenuates Hypoxia-Induced Neuroinflammation via NF-κB/Hif-1α Signaling
To examine the mechanism by which propofol decreased CoCl2-induced secretion of proinflammatory cytokines in BV2 cells, we investigated two proteins that propofol could potentially affect within the cell, namely, NF-κB p65 and Hif-1α. First, we pretreated cells with different concentrations of propofol (25, 50, and 100 μM) for 3 h before CoCl2 stimulation for 24 h and then examined NF-κB p65 and Hif-1α production by western blotting. As illustrated in Figures 5(a)–5(c), compared to the CoCl2-treated group, propofol significantly suppressed NF-κB p65 and Hif-1α production (P < 0.05). To further investigate the intrinsic mechanism with respect to NF-κB p65, siRNA against NF-κB p65 were transfected into BV2 cells and incubated for 24 h, followed by exposure to hypoxia for 24 h. As shown in Figures 5(d), 5(f), and 5(g), Hif-1α and IL-1β were downregulated in NF-κB p65-silenced and CoCl2-treated cells compared to that in only CoCl2-treated cells (P < 0.05). To summarize, propofol suppressed Hif-1α production by inhibiting the upregulation of NF-κB p65 in CoCl2-treated BV2 microglia. Propofol attenuated hypoxia-induced inflammation in BV2 cells, at least in part by NF-κB/Hif-1α signaling.
Figure 5.
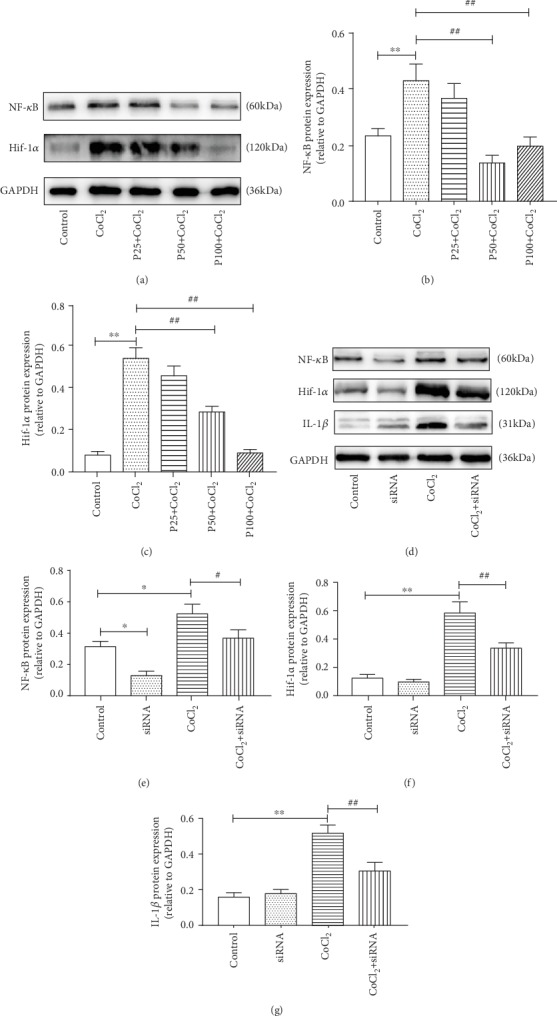
Propofol suppresses NF-κB/Hif-1α production in CoCl2-treated BV2 cells. BV2 cells were pretreated with propofol (25, 50, and 100 μM) for 3 h followed by treatment with CoCl2 for 24 h, and cells were examined for NF-κB p65, Hif-1α, and IL-1β production by western blotting (a–c). (a) The protein levels of NF-κB and Hif-1α. (b) The gray value for NF-κB is displayed as a percentage of GAPDH protein levels. (c) The gray value for Hif-1α is displayed as a percentage of GAPDH protein levels. We first pretreated cells with siRNA specific for NF-κB p65 for 24 h, followed by CoCl2 (300 μM) treatment for 24 h, and then examined NF-κB p65, Hif-1α, and IL-1β production by western blotting (d–g). (d) The protein levels of NF-κB p65, Hif-1α, and IL-1β. (e) The gray value for NF-κB is displayed as a percentage of GAPDH protein levels. (f) The gray value for Hif-1α is displayed as a percentage of GAPDH protein levels. (g) The gray value for IL-1β is displayed as a percentage of GAPDH protein levels. Gray values are shown as the mean ± S.E.M. Data are based on at least three independent experiments. Independent Student's t-test was used for comparisons between two groups, and ANOVA with the SNK post hoc test was used for multiple groups. ∗P < 0.05 versus the control group; ∗∗P < 0.01 versus the control group; #P < 0.05 versus the CoCl2 group; ##P < 0.01 versus the CoCl2 group.
4. Discussion
Propofol, widely used as a short-acting intravenous anesthetic, has chemical properties similar to those of tocopherol, a phenolic free radical scavenger. Moreover, it is lipophilic and can quickly enter cells and subcellular membrane compartments. Thus, propofol has many other functions besides its anesthetic effect [18, 19]. In our previous study, we found that propofol can prevent oxidative stress and attenuate mitochondrial dysfunction upon focal cerebral ischemia-reperfusion injury [12, 13]. Furthermore, it has an antineuroinflammatory effect and can also inhibit NF-κB activation [20]. NF-κB subunit p65 was reported to contribute to basal levels of Hif-1α mRNA and protein expression [15–17, 21]. During hypoxia, Hif-1α is stabilized and activates the transcription of genes associated with proinflammation [6, 7]. However, the protective effect of propofol against hypoxia-induced neuroinflammation has rarely been reported. In this study, we found that propofol has a protective effect against hypoxia-induced neuroinflammation, at least in part by inhibiting oxidative stress and NF-κB/Hif-1α signaling.
Microglia are the primary neuroimmune cells participating in surveillance and defense, first sensing pathophysiological changes in the brain, and triggering subsequent cascade reaction [1–3, 22]. Hypoxia contributes to many central systemic diseases, such as stroke, neonatal hypoxic-ischemic encephalopathy, Alzheimer's disease, Parkinson's disease, vascular dementia, and epilepsy [23–26]. Hypoxia leads to activation of M1 microglia, which mainly secrete neurotoxic substances such as proinflammatory cytokines (TNF-α, IL-1β, and IL-6) and ROS [27, 28]. Proinflammatory cytokines and ROS can directly damage neurons and trigger subsequent cascade reactions. Therefore, inhibiting hypoxia-induced proinflammatory cytokine production is the key to regulating the neuroinflammatory response after hypoxia.
During hypoxia, Hif-1α is stabilized and binds to Hif-1β and subsequently migrates to the nucleus where it activates transcription of genes associated with inflammation and oxidative stress, causing tissue damage [6, 7, 29, 30]. In this study, we chose CoCl2 treatment to mimic hypoxia. CoCl2 is known as a hypoxia-mimetic agent in cell line models, as it induces biochemical and molecular reactions similar to those observed under hypoxic conditions [31–34]. In our study, we observed that with prolonged incubation with CoCl2, the expression of TNF-α, IL-1β, IL-6, and iNOS significantly increased in BV2 cells (Figure 1). As reported [35], Hif-1α was also markedly upregulated in hypoxia-induced BV2 cells compared to normoxic cells (Figures 5(a) and 5(d)). It turns out that CoCl2-induced inflammation in BV2 cells is associated with Hif-1α. Moreover, proinflammatory cytokines (TNFα, IL-1β, and IL-6) were decreased in hypoxic BV2 cells pretreating with propofol (Figure 2). Propofol also inhibited Hif-1α protein expression in hypoxic BV2 cells (Figures 5(a) and 5(c)).
It is known that hypoxia impairs mitochondrial function resulting in ROS production, decreased mitochondrial membrane potential, and metabolic changes [8, 9]. ROS are proinflammatory factors which initiate inflammatory cascade reactions and are the main signaling molecules that regulate macrophage phagocytosis and killing [8, 36]. ROS oxidizes Fe2+ to Fe3+, an important cofactor that inhibits prolyl hydroxylase activity, and indirectly stabilizes Hif-1α protein [37]. However, overproduction of ROS can lead to oxidative stress, resulting in decreased SOD and T-AOC activity. SOD can also inhibit the activation of NF-κB to limit the inflammatory response [38]. Further, oxidative stress amplifies microglia inflammatory responses, resulting in neuron injury [39–41]. In the present study, we found that ROS production was increased in hypoxia-treated microglia (Figures 3(a) and 3(b)), whereas SOD and T-AOC activities were decreased (Figures 3(c) and 3(d)). In contrast, propofol was able to restore antioxidant activity (both SOD and T-AOC) in hypoxia-treated microglia (Figures 3(c) and 3(d)), thus inhibiting the production of ROS and inflammatory responses. It should be noted that the concentration of propofol was a key factor in this process and that high concentrations (100 μM) were found to reduce antioxidant activity. This might be due to cytotoxicity at high concentrations [42, 43]. This decrease in antioxidant activity (both SOD and T-AOC) in hypoxia-treated microglia is consistent with previous studies [9, 44].
In addition, overproduction of ROS also results in the loss of mitochondrial membrane potential and aggravates mitochondrial dysfunction [8, 45, 46]. Furthermore, the effect of propofol on mitochondrial membrane potential and mitochondrial function is related to dose, cell type, and administration route [12, 47]. Studies have shown that propofol can prevent the collapse in membrane potential in liver and brain mitochondria during ischemia [11, 12]. In this study, we found that propofol could attenuate hypoxia-induced decreases in mitochondrial membrane potential (Figure 4) but failed to do so at 50 μM concentration.
To explore the mechanism by which propofol affects hypoxia-induced inflammation in microglia, further experiments were carried out. NF-κB is a key transcription factor that is associated with hypoxia-induced inflammation [29, 48–50]. Propofol can inhibit NF-κB activation, thereby playing an anti-inflammatory role. In vivo, NF-κB is a prerequisite for constitutive Hif-1α expression [17, 21]. At the same time, activated NF-κB regulates Hif-1α expression and protects against thrombin, hydrogen peroxide, and even short-term hypoxia in vitro [15, 51]. To investigate the role of NF-κB p65 in propofol-mediated protective effects, we knocked down NF-κB p65 via siRNA transfection, followed by exposure to hypoxia for 24 h. We observed that hypoxia-induced Hif-1α and IL-1β production was downregulated with NF-κB p65 silencing (Figures 5(d), 5(f), and 5(g)). In addition, pretreatment with propofol significantly inhibited the upregulation of NF-κB p65 and Hif-1α production in BV2 microglia exposed to CoCl2 (Figures 5(a)–5(c)).
To summarize, our data show that propofol attenuates hypoxia-induced inflammation in BV2 microglia, in addition to reducing the production of ROS and enhancing antioxidant activity, at least in part by regulating the NF-κB/Hif-1α signaling pathway. Further studies have to be performed to explore the effectiveness of propofol in clinical trials and to standardize the mode of administration with an appropriate dosage.
Acknowledgments
This project was supported by a Grant from the Natural Science Foundation of China (No. 81271456).
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
Additional Points
Highlights. (1) Propofol reduces CoCl2-induced proinflammatory cytokine levels in BV2 cells. (2) Propofol alleviates CoCl2-induced oxidative stress and decrease in mitochondrial membrane potential. (3) Propofol attenuates hypoxia-induced neuroinflammation via inhibiting NF-κB/Hif-1α signaling.
Conflicts of Interest
The authors declare no competing interests.
Authors' Contributions
XP and WY worked on study design, CL and SL handled the study conduct, XP and CL participated in data analysis, XP and SQ wrote the paper, and all authors revised the paper.
References
- 1.Wendeln A. C., Degenhardt K., Kaurani L., et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. 2018;556(7701):332–338. doi: 10.1038/s41586-018-0023-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li Q., Barres B. A. Microglia and macrophages in brain homeostasis and disease. Nature Reviews Immunology. 2018;18(4):225–242. doi: 10.1038/nri.2017.125. [DOI] [PubMed] [Google Scholar]
- 3.Rothhammer V., Borucki D. M., Tjon E. C., et al. Microglial control of astrocytes in response to microbial metabolites. Nature. 2018;557(7707):724–728. doi: 10.1038/s41586-018-0119-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fumagalli M., Lombardi M., Gressens P., Verderio C. How to reprogram microglia toward beneficial functions. Glia. 2018;66(12):2531–2549. doi: 10.1002/glia.23484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orihuela R., McPherson C. A., Harry G. J. Microglial M1/M2 polarization and metabolic states. British Journal of Pharmacology. 2016;173(4):649–665. doi: 10.1111/bph.13139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palazon A., Goldrath A. W., Nizet V., Johnson R. S. HIF transcription factors, inflammation, and immunity. Immunity. 2014;41(4):518–528. doi: 10.1016/j.immuni.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walmsley S. R., Chilvers E. R., Thompson A. A., et al. Prolyl hydroxylase 3 (PHD3) is essential for hypoxic regulation of neutrophilic inflammation in humans and mice. The Journal of Clinical Investigation. 2011;121(3):1053–1063. doi: 10.1172/JCI43273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koshikawa N., Hayashi J., Nakagawara A., Takenaga K. Reactive oxygen species-generating mitochondrial DNA mutation up-regulates hypoxia-inducible factor-1alpha gene transcription via phosphatidylinositol 3-kinase-Akt/protein kinase C/histone deacetylase pathway. The Journal of Biological Chemistry. 2009;284(48):33185–33194. doi: 10.1074/jbc.M109.054221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu W., Wei W., Lu M., et al. Neuroprotective effect of chitosan oligosaccharide on hypoxic-ischemic brain damage in neonatal rats. Neurochemical Research. 2017;42(11):3186–3198. doi: 10.1007/s11064-017-2356-z. [DOI] [PubMed] [Google Scholar]
- 10.Adembri C., Venturi L., Tani A., et al. Neuroprotective effects of propofol in models of cerebral ischemia: inhibition of mitochondrial swelling as a possible mechanism. Anesthesiology. 2006;104(1):80–89. doi: 10.1097/00000542-200601000-00014. [DOI] [PubMed] [Google Scholar]
- 11.Bellanti F., Mirabella L., Mitarotonda D., et al. Propofol but not sevoflurane prevents mitochondrial dysfunction and oxidative stress by limiting HIF-1α activation in hepatic ischemia/reperfusion injury. Free Radical Biology & Medicine. 2016;96:323–333. doi: 10.1016/j.freeradbiomed.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 12.Li J., Yu W., Li X. T., Qi S. H., Li B. The effects of propofol on mitochondrial dysfunction following focal cerebral ischemia-reperfusion in rats. Neuropharmacology. 2014;77:358–368. doi: 10.1016/j.neuropharm.2013.08.029. [DOI] [PubMed] [Google Scholar]
- 13.Yu W., Gao D., Jin W., Liu S., Qi S. Propofol prevents oxidative stress by decreasing the ischemic accumulation of succinate in focal cerebral ischemia-reperfusion injury. Neurochemical Research. 2018;43(2):420–429. doi: 10.1007/s11064-017-2437-z. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Q., Wang L., Chen B., Zhuo Q., Bao C., Lin L. Propofol inhibits NF-κB activation to ameliorate airway inflammation in ovalbumin (OVA)-induced allergic asthma mice. International Immunopharmacology. 2017;51:158–164. doi: 10.1016/j.intimp.2017.08.015. [DOI] [PubMed] [Google Scholar]
- 15.Azoitei N., Becher A., Steinestel K., et al. PKM2 promotes tumor angiogenesis by regulating HIF-1α through NF-κB activation. Molecular Cancer. 2016;15:p. 3. doi: 10.1186/s12943-015-0490-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y., Sui H., Jiang C., et al. Dihydroartemisinin increases the sensitivity of photodynamic therapy via NF-κB/HIF-1α/VEGF pathway in esophageal cancer cell in vitro and in vivo. Cellular Physiology and Biochemistry. 2018;48(5):2035–2045. doi: 10.1159/000492541. [DOI] [PubMed] [Google Scholar]
- 17.Liu R., Liao X. Y., Pan M. X., et al. Glycine exhibits neuroprotective effects in ischemic stroke in rats through the inhibition of M1 microglial polarization via the NF-κB p65/Hif-1α signaling pathway. Journal of Immunology. 2019;202(6):1704–1714. doi: 10.4049/jimmunol.1801166. [DOI] [PubMed] [Google Scholar]
- 18.Marik P. E. Propofol: an immunomodulating agent. Pharmacotherapy. 2005;25:28S–33S. doi: 10.1592/phco.2005.25.5_part_2.28s. [DOI] [PubMed] [Google Scholar]
- 19.Vasileiou I., Xanthos T., Koudouna E., et al. Propofol: a review of its non-anaesthetic effects. European Journal of Pharmacology. 2009;605(1-3):1–8. doi: 10.1016/j.ejphar.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 20.Cheng L., Chen Z., Wang L., Lan Y., Zheng L., Wu F. Propofol partially attenuates complete freund's adjuvant-induced neuroinflammation through inhibition of the ERK1/2/NF‐κB pathway. Journal of Cellular Biochemistry. 2019;120(6):9400–9408. doi: 10.1002/jcb.28215. [DOI] [PubMed] [Google Scholar]
- 21.Rius J., Guma M., Schachtrup C., et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453(7196):807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neumann H., Kotter M. R., Franklin R. J. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132:288–295. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merelli A., Rodriguez J. C. G., Folch J., Regueiro M. R., Camins A., Lazarowski A. Understanding the role of hypoxia inducible factor during neurodegeneration for new therapeutics opportunities. Current Neuropharmacology. 2018;16(10):1484–1498. doi: 10.2174/1570159X16666180110130253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serdar M., Kempe K., Rizazad M., et al. Early pro-inflammatory microglia activation after inflammation-sensitized hypoxic-ischemic brain injury in neonatal rats. Frontiers in Cellular Neuroscience. 2019;13:p. 237. doi: 10.3389/fncel.2019.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davies A. L., Desai R. A., Bloomfield P. S., et al. Neurological deficits caused by tissue hypoxia in neuroinflammatory disease. Annals of Neurology. 2013;74(6):815–825. doi: 10.1002/ana.24006. [DOI] [PubMed] [Google Scholar]
- 26.Vezzani A., Balosso S., Ravizza T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nature Reviews. Neurology. 2019;15(8):459–472. doi: 10.1038/s41582-019-0217-x. [DOI] [PubMed] [Google Scholar]
- 27.Butturini E., Boriero D., Carcereri de Prati A., Mariotto S. STAT1 drives M1 microglia activation and neuroinflammation under hypoxia. Archives of Biochemistry and Biophysics. 2019;669:22–30. doi: 10.1016/j.abb.2019.05.011. [DOI] [PubMed] [Google Scholar]
- 28.Zhang F., Zhong R., Li S., et al. Acute hypoxia induced an imbalanced M1/M2 activation of microglia through NF-κB signaling in Alzheimer's disease mice and wild-type littermates. Frontiers in Aging Neuroscience. 2017;9:p. 282. doi: 10.3389/fnagi.2017.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor C. T., Doherty G., Fallon P. G., Cummins E. P. Hypoxia-dependent regulation of inflammatory pathways in immune cells. The Journal of Clinical Investigation. 2016;126(10):3716–3724. doi: 10.1172/JCI84433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y., Murugesan P., Huang K., Cai H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: novel therapeutic targets. Nature Reviews Cardiology. 2020;17(3):170–194. doi: 10.1038/s41569-019-0260-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan X., Liu Y., Xie T., Liu F. α-Tocopherol protected against cobalt nanoparticles and cocl<sub>2</sub> induced cytotoxicity and inflammation in Balb/3T3 cells. Immunopharmacology and Immunotoxicology. 2018;40(2):179–185. doi: 10.1080/08923973.2018.1424901. [DOI] [PubMed] [Google Scholar]
- 32.Kwak J., Choi S. J., Oh W., Yang Y. S., Jeon H. B., Jeon E. S. Cobalt chloride enhances the anti-inflammatory potency of human umbilical cord blood-derived mesenchymal stem cells through the ERK-HIF-1α-microRNA-146a-mediated signaling pathway. Stem Cells International. 2018;2018:4978712. doi: 10.1155/2018/4978763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun Z., Mohamed M. A. A., Park S. Y., Yi T. H. Fucosterol protects cobalt chloride induced inflammation by the inhibition of hypoxia-inducible factor through PI3K/Akt pathway. International Immunopharmacology. 2015;29(2):642–647. doi: 10.1016/j.intimp.2015.09.016. [DOI] [PubMed] [Google Scholar]
- 34.Li J., Wang T., Jiang X. F. Inhibition of miR-337-3p involved in the protection of CoCl2‐induced injury in PC12 cells via activating JAK2/STAT3 signaling pathway. Journal of Cellular Biochemistry. 2019;120(11):19076–19086. doi: 10.1002/jcb.29230. [DOI] [PubMed] [Google Scholar]
- 35.Lu D. Y., Liou H. C., Tang C. H., Fu W. M. Hypoxia-induced iNOS expression in microglia is regulated by the PI3-kinase/Akt/mTOR signaling pathway and activation of hypoxia inducible factor-1α. Biochemical Pharmacology. 2006;72(8):992–1000. doi: 10.1016/j.bcp.2006.06.038. [DOI] [PubMed] [Google Scholar]
- 36.Cui Q., Wang J. Q., Assaraf Y. G., et al. Modulating ROS to overcome multidrug resistance in cancer. Drug Resistance Updates. 2018;41:1–25. doi: 10.1016/j.drup.2018.11.001. [DOI] [PubMed] [Google Scholar]
- 37.Tannahill G. M., Curtis A. M., Adamik J., et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496(7444):238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang J., Park E. J., Jou I., Kim J. H., Joe E. H. Reactive oxygen species mediate a beta(25-35)-induced activation of BV-2 microglia. Neuroreport. 2001;12(7):1449–1452. doi: 10.1097/00001756-200105250-00030. [DOI] [PubMed] [Google Scholar]
- 39.Duarte A. I., Santos P., Oliveira C. R., Santos M. S., Rego A. C. Insulin neuroprotection against oxidative stress is mediated by Akt and GSK-3beta signaling pathways and changes in protein expression. Biochimica et Biophysica Acta. 2008;1783(6):994–1002. doi: 10.1016/j.bbamcr.2008.02.016. [DOI] [PubMed] [Google Scholar]
- 40.Kaur C., Ling E. A. Antioxidants and neuroprotection in the adult and developing central nervous system. Current Medicinal Chemistry. 2008;15(29):3068–3080. doi: 10.2174/092986708786848640. [DOI] [PubMed] [Google Scholar]
- 41.Ransohoff R. M., Schafer D., Vincent A., Blachere N. E., Bar-Or A. Neuroinflammation: ways in which the immune system affects the brain. Neurotherapeutics. 2015;12(4):896–909. doi: 10.1007/s13311-015-0385-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Twaroski D. M., Yan Y., Zaja I., Clark E., Bosnjak Z. J., Bai X. Altered mitochondrial dynamics contributes to propofol-induced cell death in human stem cell-derived neurons. Anesthesiology. 2015;123(5):1067–1083. doi: 10.1097/ALN.0000000000000857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krzisch M., Sultan S., Sandell J., Demeter K., Vutskits L., Toni N. Propofol anesthesia impairs the maturation and survival of adult-born hippocampal neurons. Anesthesiology. 2013;118(3):602–610. doi: 10.1097/ALN.0b013e3182815948. [DOI] [PubMed] [Google Scholar]
- 44.Hou R. C.-W., Wu C.-C., Yang C.-H., Jeng K.-C. G. Protective effects of sesamin and sesamolin on murine BV-2 microglia cell line under hypoxia. Neuroscience Letters. 2004;367(1):10–13. doi: 10.1016/j.neulet.2004.05.073. [DOI] [PubMed] [Google Scholar]
- 45.Song H. P., Chu Z. G., Zhang D. X., Dang Y. M., Zhang Q. PI3K-AKT pathway protects cardiomyocytes against hypoxia-induced apoptosis by mitoKATP-mediated mitochondrial translocation of pAKT. Cellular Physiology and Biochemistry. 2018;49(2):717–727. doi: 10.1159/000493037. [DOI] [PubMed] [Google Scholar]
- 46.Zhang L., Qi X., Zhang G., Zhang Y., Tian J. Saxagliptin protects against hypoxia-induced damage in H9c2 cells. Chemico-Biological Interactions. 2020;315 doi: 10.1016/j.cbi.2019.108864. [DOI] [PubMed] [Google Scholar]
- 47.Yang N., Liang Y., Yang P., Yang T., Jiang L. Propofol inhibits lung cancer cell viability and induces cell apoptosis by upregulating microRNA-486 expression. Brazilian Journal of Medical and Biological Research. 2017;50(1) doi: 10.1590/1414-431X20165794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen X., Li X., Zhang W., et al. Activation of AMPK inhibits inflammatory response during hypoxia and reoxygenation through modulating JNK-mediated NF-κB pathway. Metabolism. 2018;83:256–270. doi: 10.1016/j.metabol.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang T., Sun J., Wei B., Liu S. SENP1-mediated NEMO de-SUMOylation inhibits intermittent hypoxia induced inflammatory response of microglia in vitro. Journal of Cellular Physiology. 2019;235(4):3529–3538. doi: 10.1002/jcp.29241. [DOI] [PubMed] [Google Scholar]
- 50.Barnes P. J., Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. The New England Journal of Medicine. 1997;336(15):1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 51.Walmsley S. R., Print C., Farahi N., et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. The Journal of Experimental Medicine. 2005;201(1):105–115. doi: 10.1084/jem.20040624. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.