

SUMMARY
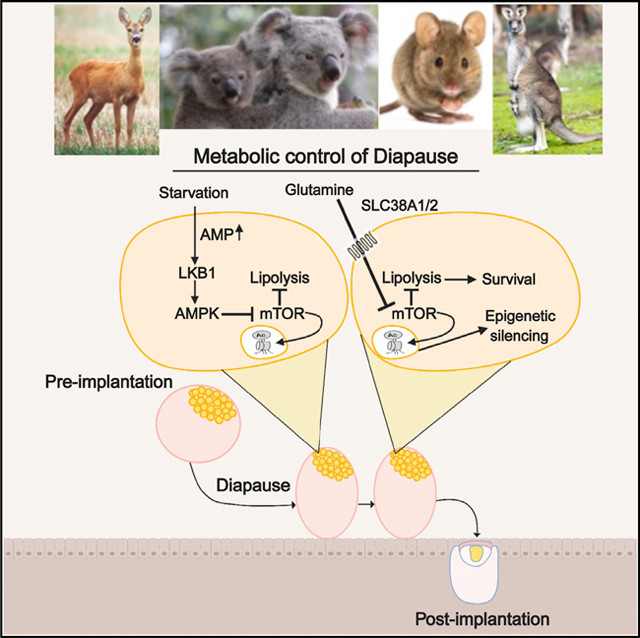
Regulation of embryonic diapause, dormancy that interrupts the tight connection between developmental stage and time, is still poorly understood. Here, we characterize the transcriptional and metabolite profiles of mouse diapause embryos and identify unique gene expression and metabolic signatures with activated lipolysis, glycolysis, and metabolic pathways regulated by AMPK. Lipolysis is increased due to mTORC2 repression, increasing fatty acids to support cell survival. We further show that starvation in pre-implantation ICM-derived mouse ESCs induces a reversible dormant state, transcriptionally mimicking the in vivo diapause stage. During starvation, Lkb1, an upstream kinase of AMPK, represses mTOR, which induces a reversible glycolytic and epigenetically H4K16Ac-negative, diapause-like state. Diapause furthermore activates expression of glutamine transporters SLC38A1/2. We show by genetic and small molecule inhibitors that glutamine transporters are essential for the H4K16Ac-negative, diapause state. These data suggest that mTORC1/2 inhibition, regulated by amino acid levels, is causal for diapause metabolism and epigenetic state.
In Brief
Hussein et al. report that, during starvation, mTOR is repressed through LKB1-AMPK, inducing a reversible metabolically active but epigenetically silenced embryonic diapause-like state that upregulates expression of the glutamine transporters SLC38A1/2. These transporters are required for the H4K16ac-negative, diapause state.
Graphical Abstract
INTRODUCTION
The term diapause describes a reversible, environmentally inducible state of suspended embryonic development that is associated with delayed blastocyst implantation. Diapause has been described in more than 130 mammalian species, indicating an exceptional degree of evolutionary conservation in the coordination between birth and favorable environmental conditions (Fenelon et al., 2014). However, the molecular controls of the entry and exit of this reversible dormant stage are poorly understood.
Diapause can be triggered experimentally in mice through ovariectomy (Yoshinaga and Adams, 1966) or estrogen deprivation (MacLean Hunter and Evans, 1999; Paria et al., 1993) and occurs at day 3.5 (E3.5) of embryonic development. Pre-implantation, diapause blastocyst, comprised of inner cell mass (ICM) surrounded by trophoblasts remains developmentally arrested until implantation is triggered (Fenelon et al., 2014; McLaren, 1968). While some studies have been performed to understand the cellular and molecular changes that occur when embryos enter diapause, a comprehensive analysis of metabolites is required to dissect the metabolic regulation of diapause state. It has been shown that some metabolic activities including protein and DNA synthesis as well as carbohydrate metabolism are reduced in diapause (Fenelon et al., 2014; Menke and McLaren, 1970; Pike, 1981; Van Blerkom et al., 1978; Hamatani et al., 2004; Liu et al., 2012; Martin and Sutherland, 2001; Fu et al., 2014). Furthermore, amino acids in the uterine fluids have been shown to affect diapause (Renfree and Fenelon, 2017; Gardner and Lane, 1993; Van Winkle et al., 2006), although the downstream targets for this regulation have not been dissected.
Autophagy, a metabolic pathway that generates nutrients required for cellular survival during starvation, has shown to be activated during diapause (Lee et al., 2011). Furthermore, autophagy can be regulated by mTOR (Kim and Guan, 2015; Nicklin et al., 2009). Accordingly, recent work has revealed that inhibition of the mTOR pathway or depletion of transcription factor, Myc can induce a diapause-like state (Bulut-Karslioglu et al., 2016; Scognamiglio et al., 2016). However, how mTOR is downregulated in diapause and the signals that reactivate mTOR in blastocyst development are not well understood in any mammal (Fenelon et al., 2014; Van Blerkom et al., 1978; Scognamiglio et al., 2016; Shaw and Renfree, 1986; Renfree and Shaw, 2014; Murphy, 2012; He et al., 2019).
Our study shows a key molecular mechanism and metabolic and epigenetic regulation for entry and exit of diapause. We found that both diapause and diapause-like cells have highly reduced H4K16Ac epigenetic marks, upregulated glycolytic signature, reduced mitochondrial activity and reduced fatty acid b-oxidation. We further show that mTOR-dependent H4K16Ac epigenetic marks are inhibited by nutrient-starvation-dependent LKB1-induced AMPK activation, as well as diapause-enriched glutamine transporter activity (Slc38A1/2).
RESULTS
Diapause Is Associated with a Unique Transcriptional State
To understand the processes underlying embryonic diapause, we characterized the transcriptional profiles of the cells contributing to the future embryo in pre-implantation (ICM, day 3.5 postfertilization), post-implantation (Epi, day 6.5), and diapause (dICM, day 8.5—induced on day 2.5 and harvested on day 8.5) mouse embryos using RNA sequencing (RNA-seq) (Figures 1A–1C; Table S1A). We identified 12 hallmark pathways significantly enriched in genes upregulated in the diapause state (Figure S1A; Tables S2A–S2L), including: TNFa signaling via NF-kB, the p53 pathway, hypoxia, and cholesterol homeostasis (FDR < 0.1). Significantly downregulated pathways in diapause included the late estrogen response pathway (reflecting the experimentally induced diapause by estrogen depletion). We further analyzed the transcriptomes by using principal component analysis (PCA) and found three distinct clusters: pre-implantation, post-implantation, and diapause. This defines a robust diapause gene expression signature (Boroviak et al., 2015) (Figures 1B and 1C; Table S1A).
Figure 1. Gene Expression and Splice Variants Separate Diapause Stage from ICM and Post-implantation Stages.

(A) Schematic diagram depicting outline of the experiment.
(B) PCA of our RNA-seq data showed that diapause is in a distinct transcriptional state compared to pre- and post-implantation epiblasts. (* = Boroviak et al., 2015)
(C) Scatter plot of gene contributions to PC1 and PC2 in the PCA plot in (B) showing genes that are specifically up- or down-regulated in diapause compared topre-/post-implantation.
(D) Metabolic pathway enrichment of genes differentially expressed between diapause and ICM.
(E) PCA plot using transcript splicing rate of our RNA-sequence samples clearly separates diapause and pre-/post-implantation samples.
(F) Sashimi plot of Lkb1 exons 8–10. Bar height represents expression level (FPKM). Arcs connect two exons that are spliced together.
(G) qPCR analysis of Lkb1 splice variant expression in ICM, dICM, and epiblast. n = 2 for each group, p = 0.001965 for ICM versus dICM, and p = 0.000102 for dICM versus Epi.
(H) Immunostaining of pre-implantation and diapause embryos with antibodies against both Lkb1 isoforms (red), OCT4 (magenta), or stained with DAPI (blue). Scale: 22 μm.
Metabolic genes contributed significantly to the 2nd principal component (Figure 1B; Table S1A), corresponding to differences between diapause and pre- and post-implantation (Figure 1C). The gluconeogenesis gene, fructose bisphosphatase 2 (Fbp2) was significantly downregulated in diapause, and its downregulation is known to promote glycolysis (Li et al., 2013). In contrast, phosphofructo-2-kinase/fructose-2,6-biphosphatase 2 (Pfkfb2), which catalyzes the formation of fructose 2,6-bisphosphate (F2,6P2), is upregulated in diapause (Figure 1C). F2,6P2 strongly activates glycolysis by activating the key glycolytic enzyme phosphofructokinase-1 (PFK-1) (Hue and Rider, 1987). The significant changes observed in these rate-limiting enzymes suggest a major change in the regulation of glycolysis as a blastocyst transitions from the pre-implantation to the diapause stage. Furthermore, genes associated with lipid transfer (e.g., cholesterol transfer protein Stard4) are also upregulated in diapause. Interestingly, it was recently shown that cholesterol biosynthesis affects intestinal stem cell proliferation (Wang et al., 2018). However, it is not yet understood how cholesterol affects cellular quiescence versus cell division. We further used a hypergeometric test to identify the enrichment of up- and downregulated genes in 64 metabolic pathways of the human metabolic network and found that diapause is characterized by its unique metabolism, including upregulation of glycolysis, upregulation of cholesterol synthesis, and uptake (Figure 1D; Table S1B), as discussed above.
Another metabolic pathway regulator, solute carrier family 38, member 1 (Slc38a1), a sodium-dependent glutamine transporter that affects mTORC1 pathway (Liu et al., 2017), was significantly upregulated during diapause (Figure 1C).
Diapause Is Associated with a Unique Transcript Splice Variant State
In addition to its specific gene expression, we found that diapause has distinct transcript splicing variants (Figure 1E). PCA based on transcript splicing rate (Table S3) across RNAseq samples separated diapause stage from pre- and postimplantation stages. The fact that both differential splicing and differential gene expression analysis cluster diapause samples separate from pre- and post-implantation samples clearly strengthens the finding that diapause is characterized by a unique transcriptomic state.
Specific examples of alternative splicing events that may be beneficial for diapause can be observed with many transcripts, including Pitx1, Mbd2, Tfe3, and Lkb1 (Figures 1F and S1B–S1D; Table S2M). In particular, the short isoform of Pitx1 (644 bp, 123 amino acids) is expressed in diapause (dICM), while the longer isoform (2,451 bp, 315 amino acids) is only expressed in pre-implantation ICM. Pitx1 is a homeobox transcription factor that regulates multiple genes involved in early development and prolactin expression (Quentien et al., 2002). Interestingly, the diapause-specific splice variant is missing the DNA-binding domain possibly rendering the transcription factor unable to activate development. It is therefore plausible that the specific, dominant isoforms play a role in pluripotency during diapause.
Diapause Is Associated with a LKB1 Isoform Switch
RNA-seq results identified a dramatic difference in splice variants for the key metabolic kinase, Lkb1, between the three developmental stages. A sashimi plot, which visualizes raw RNA-seq reads along splice junctions, was used to examine Lkb1-alternative splicing in more detail (Figure 1F). We found that exon 9a is expressed in ICM and Epi, but not in dICM. Multiple reads span exon 8 and 9a in pre-implantation ICM supporting the expression of Lkb1 short (Lkb1s) isoform in that stage. On the other hand, in ICM, dICM, and Epi, multiple reads span the junctions between exon 8 to 9b and 9b to 10, supporting the expression of Lkb1 long (Lkb1L) isoform in all these stages (Figure 1F). Therefore, the Lkb1 short isoform is expressed in pre-implantation ICM and post-implantation epiblast (Epi) but not in diapause (dICM). We validated the RNA-seq data with quantitative PCR (qPCR) and found that Lkb1L isoform was present in all stages whereas Lkb1s isoform was present in pre-implantation ICM and post-implantation Epi, but not in diapause (dICM) (Figure 1G). We show that Lkb1 protein can be detected in mouse blastocyst ICM, both in pre-implantation and diapause stage (Figure 1H).
Starvation Induces a Diapause-like Transcriptional Program in Pre-implantation ICM-Derived mESCs
Since nutritional deprivation is one of the ways embryonic facultative diapause is induced in some animals, we tested the outcome of starvation in mouse pre-implantation ICM-derived embryonic stem cells (mESCs). We performed RNA-seq on mESCs that were cultured under pre-implantation naive conditions, with and without overnight starvation (using DMEM that lacks glucose and FBS). Results were analyzed in the context of the transcriptional profiles of ICM, dICM, and Epiblast. We calculated eigen vectors from the PCA in Figure 1B and projected the in vitro starvation data into the resulting space (Figure 2A; Table S4A). Importantly, the RNA-seq analysis revealed that our in vitro starvation condition of mESCs produced a gene expression signature, which highly mimics the diapause state and thus can be regarded as an in vitro model of diapause-like state.
Figure 2. Lkb1 Activation by Starvation Induces Diapause-like State.

(A) Starvation moved mESC samples (R1/Lkb1) toward a diapause state. Cell line samples are projected in the PCA of in vivo samples using a previously described method (Kho et al., 2004).* = Boroviak et al., 2015.
(B) Model of Lkb1 and starvation. AMPK is activated by starvation and is a target of Lkb1. Activated AMPK inhibits mTOR and induces diapause.
(C) Immunostaining of day 3.5 and diapause embryos with antibody against pAMPKa2(Thr172) (green) and OCT4 (blue). Scale: 20 μm.
(D) 1-h exposure to AICAR, starvation, or 2-DG (10 min) increased the activity of AMPK compared to untreated cells. beta-actin 1: pmTOR, pAMPKand pS6; betaactin 2: mTOR, AMPK and S6.
(E) Phosphorylation of mTOR, Akt(S473), S6, ULK1(S757), and acetylation of H4K16 in R1(LL) cells decreased after 24 h in starvation, whereas p4EBP1 did notchange. Beta-actin 1, pmTOR and pS6; beta-actin 2, mTOR and S6; beta-actin 3, H4K16ac; beta-actin 4, pAkt(S473) and p4EBP1; beta-actin 5, Akt.
(F) Immunostaining of mouse ESCs with or without starvation. Staining was performed to detect pluripotent marker, OCT4 (green). The nuclei of all cells were stained blue with DAPI. Scale: 21 μm.
(G) Lkb1 siRNAs effectively knock down expression of Lkb1 at the protein level compared to the control luciferase siRNA. The efficiency of the transient siRNA approach was assessed at the protein level by western blotting coupled with densitometry for Lkb1, mTOR, and S6 in transiently transfected R1(LL) cells.
(H) mTOR, and S6 phosphorylation were significantly reduced in luciferase siRNA control under starvation compared to Lkb1 siRNA under starvation. Beta-actin 1: Lkb1 and pmTOR; beta-actin 2: pS6; beta-actin 3: mTOR and S6. n = 3 for pmTOR, p = 0.010 for control versus KD; n = 3 for pS6, p = 0.0039 for control versus KD; n = 3 for Lkb1, p = 0.035 for control versus KD, p = 0.0068 for control+starv versus KD+starv, p = 0.0081 for control versus KD+starv, p = 0.014 for control+starv versus KD.
(I) A simplified diagram of the mTOR signaling pathway and mTOR Complexes 1 and 2 inhibitor, INK-128. See text for details.
(J) Phosphorylation of mTOR, Akt(S473), S6, 4EBP1, ULK1(S757), and acetylation of H4K16 in R1(LL) cells decreased after INK-128 treatment for 24h. Beta-actin1: pmTOR, pAkt(S473), pS6, and p4EBP1, beta-actin 2: mTOR, S6, and 4EBP1, beta-actin 3: Akt, beta-actin 4: pULK1 and H4K16ac.
To dissect the similarity between diapause and starvation models, we identified all downregulated and upregulated overlapping genes between starved versus non-starved ESCs and diapause versus ICM (Table S4B) and analyzed their biological connectivity by STRING- and GO-analysis (Figures S2A–S2D). This analysis revealed similar changes in cell-cycle controllers, epigenetic modifiers, metabolic remodeling (e.g., phosphocholine lipases and amino acid transport), and stress response genes both in in vivo diapause and starvation models. One of the major differences between the two models, however, is that the in vivo diapause ensures efficient free fatty acid accumulation in cells, not only with transporters but also triglyceride lysis process, while the starvation model expresses less genes in these pathways, presumably affecting fatty acid (FA) levels with fewer parallel mechanisms. In summary, using PCA, our transcriptomic data of both diapause and starved pre-implantation mESCs suggest that starved mouse ESCs mimic diapause.
We hypothesized that nutrient starvation might induce this diapause-like state through the LKB1 pathway (Figure 2B). We therefore tested whether any known components in the LKB1 pathway affect this diapause-like state. LKB1 is an upstream activating kinase for AMP-activated protein kinase (AMPK) (Figure S3). In response to energetic stress, LKB1 can negatively regulate mTOR signaling through LKB1-dependent phosphorylation of AMPK (Shaw et al., 2004). By immunofluorescence we confirmed that pAMPK was observed in diapause ICM (Figure 2C). To test the action of pAMPK, pmTOR, and pS6 in starved or stressed pre-implantation ICM-derived ESCs, we cultured mESCs in starvation media (1h), in 2-Deoxy-D-glucose (2-DG) (10 min), or 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR) (1h), an analog of adenosine monophosphate (AMP) that stimulates AMPK activity (Figure 2D). 2-DG is a synthetic glucose analog that inhibits glycolysis. Upon entering the cell, 2-DG is phosphorylated by hexokinase (HK) and 2-DG-6-P cannot be further metabolized through glycolysis. It accumulates and noncompetitively inhibits HK and competitively inhibits phosphoglucose isomerase (PGI) (Ralser et al., 2008; Kurtoglu et al., 2007; Urakami et al., 2013). We identified significant AMPK activation upon starvation and exposure to AICAR or 2-DG, as indicated by increased AMPK phosphorylation compared to untreated cells (Figure 2D). Total AMPK levels were the same across all samples, regardless of culture conditions. The presence of 2-DG resulted in the complete loss of mTOR (Ser2448) phosphorylation but not phosphorylation of S6. Total mTOR and S6 levels remained the same. However, 24 h of starvation resulted in the complete absence of both mTOR and S6 phosphorylation, and lack of epigenetic mark of transcriptionally active cells, H4K16Ac, as seen previously in diapause state (Shogren-Knaak et al., 2006; Deng et al., 2013; Bulut-Karslioglu et al., 2016) (Figure 2E). Phosphorylation of Akt(S473) and ULK1(S757) also decreased in starvation compared to control, whereas phosphorylation of 4EBP1 did not change (Figure 2E). Immunostaining analysis revealed that starved mESCs maintain the expression of the pluripotency factor OCT4 (Figure 2F). These results suggest that under extended nutrient starvation, AMPK is activated, and mTOR and its targets are inactivated in mESCs.
Next, we wanted to address the role of LKB1 in starvationdependent mTOR inactivation through AMPK. To do this, we determined whether the transient depletion of Lkb1 in wildtype mESCs would attenuate mTOR dephosphorylation in response to AMPK activation through starvation. Lkb1-siRNA was used to transiently knockdown (KD) the expression of Lkb1 (Figures 2G, 2H, and S2E–S2H). When the Lkb1 KD and control cells, transfected with a siRNA targeting luciferase, were starved, control cells showed a significantly larger reduction of mTOR phosphorylation compared to Lkb1 KD samples (Figure 2H). These data suggest that Lkb1 is required for starvation-induced mTOR inhibition.
The Dormant ESC Diapause-like State In Vitro Is Reversible
mTOR is a key nutrient sensor and regulator of cell growth and proliferation (Zoncu et al., 2011). Inhibition of mTOR using INK128, an inhibitor of both mTOR complexes (Hsieh et al., 2012), induces a diapause-like pluripotent state (Bulut-Karslioglu et al., 2016). Since diapause is a natural process that can be reversed (implantation takes place after exit from diapause), we wanted to determine if the dormancy was also reversible in our in vitro models. Using naive mESCs, we showed that INK-128 inhibits phosphorylation of the downstream substrates of mTORC1/2 (S6 and 4EBP1, Akt Ser473) after 24 h in culture, and it also decreased the epigenetic mark of transcriptionally active cells, H4K16Ac (Figures 2I and 2J). We further showed INK-128 and the starvation-induced diapause-like state are both reversible (Figures 3B–3D). These two reversible treatments were used as surrogate in vitro models to induce the diapause-like state by reversible inactivation of mTOR in naive mESCs.
Figure 3. Reversible Diapause-like State Can Be Induced In Vitro by Starvation or Inhibition of mTOR.

(A) Generation of Lkb1 short only isoform. A schematic of Talen-mediated gene editing of the endogenous Lkb1 locus using donor construct that expresses Lkb1-short form-specific exon 9A (Figure 1F) fused directly in frame with exon 8 followed by 2A peptide. This modified Lkb1 locus expresses only short form of Lkb1.
(B and C) Pharmacological inhibition of mTOR and starvation can induce a reversible diapause-like state. (B) mTOR inhibition by INK-128, is reversible as indicated by the rephosphorylation of mTOR, S6 and 4EBP1 24 h after removal of INK-128 in R1(LL) and R1(SS) cells. Beta-actin 1: pmTOR, pS6 and p4EBP1, beta-actin 2: mTOR, S6 and 4EBP1. (C). Starvation abolishes phosphorylation of mTOR and its substrates, S6 kinase, S6, ULK1, and 4EBP1 in 24 h. This inhibition is reversible as indicated by the rephosphorylation of those proteins 24 h after removal of starvation media from culture both in R1(LL) and R1(SS) cells. Intriguingly, the mTOR signal in the untreated and the reversed R1(SS) samples is reduced compared to their respective R1(LL) samples. Beta-actin 1: pmTOR, pS6 and p4EBP1, beta-actin 2: mTOR, S6 kinase, S6 and 4EBP1, beta-actin 3: pS6 kinase, beta-actin 4: pULK1, beta-actin 5: H4K16ac.
(D) Quantification of pmTOR protein bands from C using ImageJ software. n = 3 for each sample, p = 0.0027 for R1(LL) versus R1(SS) 24h after starvation removal. (E) Western blot of pAMPK in mESCs expressing different forms of Lkb1 splice variants. The phosphorylation of AMPK is increased in response to 2DG both in R1(LL) and R1(SS). However, higher levels of AMPK phosphorylation are observed in R1(SS) even in the absence of 2-DG.
(F) A schematic representation of the two splice forms of Lkb1.
LKB1 Lacking Regulatory C-Terminal Domain Is Constitutively Active
We hypothesized that the Lkb1 isoform switch that we observed in diapause embryos might contribute to the diapause-like metabolic state. We therefore created two mESC lines using talenbased gene editing: 1. A heterozygous line in which one allele was modified to only express the short isoform of Lkb1 (SL) and 2. A homozygous line in which both alleles were modified to express only the short isoform of Lkb1 (SS) (Figures 3A and S3). Like R1(LL), R1(SS) can be used to induce reversible diapause-like state in vitro using both starvation and INK-128 (Figures 3B–3D).
Interestingly, the phospho-mTOR protein levels in the untreated and the reversed samples of R1(SS) were reduced 35% and 40%, respectively, when compared to their respective control samples of R1(LL) (Figures 3C and 3D). To test this further, we investigated the phosphorylation state of the LKB1 downstream target, AMPK, using cell lines R1(LL) and R1(SS). We found AMPK was phosphorylated in the presence of 2-DG with either splice variant (Figure 3E). However, R1(SS) phosphorylates AMPK even in the absence of 2-DG. This suggests that R1(SS) constitutively activates AMPK. The two corresponding LKB1 proteins have different C-terminal sequences generating a 50 kDa form (Lkb1L) and a 48 kDa form (Lkb1s) (Denison et al., 2009) (Figure 3F). The C-terminal region is proposed to be a site of regulation of this protein (Towler et al., 2008). A previous study suggested that Lkb1 short has an essential role in spermiogenesis and fertility and that males lacking Lkb1 short were sterile (Towler et al., 2008). We concluded that in ESCs the differences in mTOR and AMPK activities between Lkb1 splice variants are due to the fact that the Lkb1 kinase without the C-terminal regulatory domain (R1(SS)) was partially constitutively active, even without external cues such as starvation.
Lkb1 Regulatory C-Terminal Domain Is Required for Dynamic Changes in Mitochondrial Respiratory Activity during Diapause-like State
As described above, our gene expression analysis identified a diapause associated upregulation of glycolytic, pyruvate, and cholesterol metabolic pathways in vivo and in vitro (Figure 1D). Thus, we sought to assess the functional metabolic profile of mESCs cultured in a diapause-like state.
Measurement of oxygen consumption rate (OCR) determines the level of mitochondrial respiration. No differences in OCR in response to FCCP, an uncoupling agent that disrupts mitochondrial membrane potential, were detected in the cell lines with different Lkb1 splice variants after a 3-h starvation (Figures 4A and 4B). However, after overnight starvation, there was a reduced OCR after FCCP injection in the starved mESC lines with short Lkb1 R1(SS), compared to R1(LL) (Figures 4C and 4D). These data indicate that mouse ESCs expressing the Lkb1 long isoform, which is the only isoform that is present in the diapause state, can withstand stress better than mouse ESCs bearing either one or two copies of the Lkb1 short form and that the stress response is biologically linked to Lkb1 isoform composition of different cell types.
Figure 4. Lkb1 Short Does Not Respond Dynamically to Starvation, and Diapause-like State Cells Have Higher Glycolytic Activity Compared to Control Metabolic Flux in Mouse ESCs with Different Lkb1 Splice Variants.

(A–F) Short Lkb1 does not respond dynamically to starvation. Metabolic flux of mouse ESCs with different Lkb1 splice variants using Seahorse analyzer.
(A) Representative trace of OCR changes is shown under a MitoStress protocol.
(B) Quantification of A (4 independent experiments).
(C) Representative trace of OCR changes of mouse ESCs with R1(SS) or R1(SL) lines have reduced OCR changes in response to FCCP compared to R1(LL) splicevariant of Lkb1.
(D) Quantification of C (4 independent experiments). n = 28 per group, p = 0.0076 R1(LL) versus R1(SL), p < 0.0001 R1(LL) versus R1(SS).
Mouse ESC lines with short Lkb1 splice variant cannot respond dynamically to stress.
(E) mESCs with Lkb1 short splice variant show high mitochondrial beta-oxidation when substrate, fatty acid palmitate (Palm) is offered for oxidation in normalconditions. n = 7 per group, p = 0.0088 R1(LL) versus R1(SL), p = 0.062 R1(LL) versus R1(SS).
(F) The mESC lines with Lkb1 SL and SS splice variants show lower fatty acid beta-oxidation levels than R1(LL, blue). n = 7 per group, p = 0.073 R1(LL) versus R1(SL).
(G) MitoTracker Green staining of R1(LL) and R1(SS). No change in mitochondrial mass was detected between R1(LL) and R1(SS) in normal or starvationconditions.
(H) Representative trace of ECAR changes in response to glucose, oligomycin, and 2-DG is shown under a glucose stress protocol.
(I and J) Using starvation and INK-128, naive mESCs, R1(LL), and R1(SS), under starvation or INK-128, have higher glycolytic capacity (I) and glycolytic reserve (J) compared to naive mESCs under normal conditions.
(I) n = 12 for R1(LL) and R1(SS), n = 9 for R1(LL)+INK-128, n = 15 for R1(LL)+starv, n = 22 for R1(SS)+starv, n = 8 for R1(SS)+INK-128. p < 0.0001 for R1(LL) versusR1(LL)+INK-128, p < 0.0001 for R(LL) versus R1(LL)+starv, p < 0.0007 for R1(LL)+starv versus R1(LL)+INK-128, p < 0.0026 for R1(SS) versus R1(SS)+INK-128, p < 0.0001 for R1(SS) versus R1(SS)+starv, p = 0.0088 for R1(LL) versus R1(SS), p = 0.0109 for R1(LL)+starv versus R1(SS)+starv.
(J) n values are the same as Figure 4I. p = 0.0063 for R1(LL) versus R1(LL)+INK-128, p < 0.0001 for R(LL) versus R1(LL)+starv, p < 0.0001 for R1(LL)+starv versus R1(LL)+INK-128, p = 0.0022 for R1(SS) versus R1(SS)+starv, p = 0.0030 for R1(SS)+starv versus R1(SS)+INK-128, p = 0.0176 for R1(LL) versus R1(SS).
(K) No glycolytic capacity differences were observed between R1(LL) and R1(SS) under starvation when AICAR is added to R1(LL). n = 11 for R1(LL)+starv, n = 12 for the rest, p = 0.537 for R1(LL)+starv+AICAR versus R1(SS)+starv, p = 0.0171 for R1(LL)+starv versus R1(LL)+starv+AICAR, p = 0.5267 for R1(SS)+starv versus R1(SS)+AICAR, p = 0.0350 for R1(LL)+starv versus R1(SS)+starv.
(L) Compound C, an inhibitor of AMPK, reduces the glycolytic capacity observed under starvation for both Lkb1 splice variants. n = 4 for R1(SS), n = 5 for R1(LL) and R1(LL)+10 mM compound C, rest, n = 6. p = 0.0027 for R1(LL)+starv versus R1(LL)+starv+10 mM compound C, p < 0.0001 for R1(SS)+starv versus R1(SS)+starv+10 mM compound C, p = 0.5978 for R1(LL) versus R1(LL)+10 mM compound C, p = 0.0056 for R1(SS) versus R1(SS)+10 mM compound C.
(M) Model of AMPK pathway. AMPK, once phosphorylated by Lkb1, can stimulate glucose uptake and inhibit mTOR while INK-128 inhibits mTOR.
Using palmitate as a substrate, the level of fatty acid b-oxidation in Lkb1 splice variants R1(LL), R1(SS), and R1(SL) were tested by performing a palmitate-oxidation assay with the Seahorse metabolic flux analyzer. Without starvation, the R1(SS) and R1(SL) mESC lines showed higher FA oxidation compared to the wild-type line (Figure 4E). However, after overnight starvation, both SS and SL lines exhibited decreased OCR compared to the wild-type R1(LL) (Figure 4F). These results indicate that after starvation, mouse ESC lines, R1(SS) and R1(SL), have a lower mitochondrial respiration capacity than R1(LL) and suggests that mouse ESCs with the Lkb1 short isoform have a compromised stress response.
To better understand the reduced mitochondrial response to starvation (diapause-like state) in the ESC line that was lacking the long form of Lkb1, normally seen in diapause, we tested if Lkb1 short isoform alters mitochondrial mass, with or without starvation. Using FACS analysis after Mitotracker Green stains and qPCR analysis to determine the amount of mitochondrial DNA we observed no significant changes in mitochondrial mass and amount due to forced expression of Lkb1 short isoform (Figures 4G and S4A). These experiments show that the changes we see in Figures 4C–4F are due to functional and not structural defects. We therefore analyzed the gene expression data under different conditions, and we observed dramatic defects in nutrient-transporters in samples with short form Lkb1 (SS and SL) compared to controls (R1(LL)) (Figures S4B–S4D). The long isoform of Lkb1 elicited a dynamic increase in nutrient-transporter expression after challenge (starvation), whereas Lkb1 short isoform lacked this response (Figure S4D). These data support and give a mechanistic explanation as to why mESCs with the Lkb1 short form, R1(SS) do not have the same dynamic mitochondrial activity as control ESCs with a long Lkb1 form, R1(LL), that can be regulated by multiple signaling pathways.
Diapause-like State In Vitro Has High Glycolytic Activity, as Seen In Vivo
Based on our RNA-seq results, glycolysis was the most significantly upregulated metabolic pathway in diapause and diapause-like state (Figures 1D and 2A). We therefore analyzed glycolytic capacity of the cells in the induced diapause-like state. A glycolytic stress test assay was performed to measure the glycolytic function using the Seahorse flux analyzer. We measured extracellular acidification rate (ECAR), which is predominately the result of the excretion of lactic acid derived from pyruvate that was generated through glycolysis (Wu et al., 2007).
mESCs under starvation or in the presence of INK-128 had a higher glycolytic capacity compared to untreated mESCs (Figures 4H and 4I). The result agreed with the diapause and diapause-like state RNA-seq data that showed upregulation of glycolysis (Figures 1D and 2A). Using starvation and INK-128, mESCs in a diapause-like state showed higher glycolytic reserve compared to control mESCs (Figures 4H and 4J)
Interestingly, the cells with only Lkb1 short form without regulatory C terminus (R1(SS)) had significantly higher glycolytic capacity both in the presence and absence of starvation compared to R1(LL) cells (Figures 4H and 4I). R1(SS) cells also had higher glycolytic reserve compared to R1(LL) cells under no starvation (Figures 4H–4J). No glycolytic activity changes were detected with INK-128 for both splice variants (Figures 4H–4J).
To further investigate and find an explanation for the variations between INK-128 and starvation treatments with respect to the glycolytic capacity of Lkb1 splice variants, we postulated that this was due to the observed differential activation of the Lkb1 target, AMPK (Figure 3E). We tested this hypothesis by activating AMPK by AICAR in R1(LL) conditions and analyzing the glycolytic capacity of these cells. Interestingly, there was no difference in the glycolytic capacity between the Lkb1 splice variants under starvation when AICAR was added to the long form of Lkb1 (Figure 4K). Furthermore, we tested if Lkb1SS effect in glycolysis requires AMPK, and show that AMPK inhibition using compound C reduces the starvation-induced glycolysis seen in SS. The results unequivocally show that the induction of glycolysis is AMPK dependent (Figure 4L). These data support the finding that Lkb1 short form is constitutively active and thereby activates the AMPK and glucose uptake more than the regulatable Lkb1 form.
When AMPK is phosphorylated, it inhibits the mTOR pathway (Inoki et al., 2003; Gwinn et al., 2008) (Figure 2). In addition, AMPK activation causes GLUT4 expression and translocation in skeletal muscles (Holmes et al., 1999; Kurth-Kraczek et al., 1999; Zheng et al., 2001). We observed increased glucose transporter levels in R1(SS) (Figure S4E), in agreement with constitutive activation of AMPK in the presence of the Lkb1 short isoform. INK-128 only inhibits mTOR, whereas activated AMPK can stimulate glucose uptake in addition to inhibiting mTOR. These data suggest that the glycolytic activity differences between the experimental ICM and diapause-like states are due to preferential AMPK activation in the diapause-like state (Figures 2B and 4M). Similarly, higher glycolytic activity observed in the cell line expressing only the short Lkb1 isoform may be due to constitutive activation of AMPK observed in this line.
Metabolite Analysis of In Vivo Diapause State
We analyzed the metabolome of diapause in vivo embryos to better understand this unique metabolic stage. Three and a half days after mating, the pre-implantation blastocysts were collected or diapause was induced by Tamoxifen and Depo provera as previously described (Yoshinaga and Adams, 1966). Diapause embryos were collected five days after diapause induction and both diapause and pre-implantation embryos were flash frozen for metabolite analysis (Figure 5A).
Figure 5. Overall Changes of Lipids and Polar Metabolites between Diapause and Pre-implantation Blastocyst.

(A) Schematic diagram depicting outline of the metabolomics experiment for pre-implantation or diapause embryos.
(B) Chemical enrichment analysis of diapause blastocyst compared to pre-implantation blastocyst show dramatic differences in lipid composition. Node size reflects the number of metabolites and color indicates direction of change, blue is decreased and red increased. The y axis indicates enrichment p values calculated by Kolmogorov-Smirnov-test and x axis show the polarity of chemical clusters.
(C) Relative lipid classes between diapause and pre-implantation blastocyst.
(D) Log2 fold change of a total of 243 polar metabolites that were identified between diapause and pre-implantation blastocyst.
(E) Schematic diagram of sphingolipid metabolism showing metabolites and genes that are upregulated in diapause compared to pre-implantation. D, diapause;PI, pre-implantation; TAG, Triacylglycerol; DAG, Diacylglycerol; ASAH1, N-acylsphingosine amidohydrolase 1; ACSS1, acyl-CoA synthetase short-chain family member 1; FASN, fatty acid synthase.
(F) One-carbon metabolism pathway indicating metabolites that are increased or decreased and their connections with other pathways.
We first performed lipidomics to identify the lipid constituents of the diapause and pre-implantation blastocysts. We used chemical similarity enrichment analysis (ChemRICH) (Barupal and Fiehn, 2017), a statistical enrichment approach to analyze lipidomic data (Figure 5B; Table S5A). In diapause, the major enriched lipid class compared to pre-implantation blastocysts was free fatty acids (FA), which made up about 54% of total lipid content, followed by the main phospholipid phosphatidylcholine (PC, 26%). FA and PC contents of diapause were much higher than in pre-implantation blastocyst (16% and 11%, respectively) (Figures 5B, 5C, S5A, and S5B). In diapause, triacylglycerol (TAG) and acylcarnitine (AC) constituted 14% and 1% of total lipid contents, respectively, significantly lower than those of pre-implantation blastocysts (38% and 29%, respectively) (Figures 5B and 5C). Phosphatidylethanolamine (PE), sphingomyelin (SM), ceramide (Cer), lysophosphatidylcholine (LPC), and diacylglycerol (DAG) constituted about 0.4%–3% of total lipids in both diapause and pre-implantation stages (Figures S5C–S5G). The high level of acylcarnitines in pre-implantation blastocyst suggests increased fatty acid transport into the mitochondria for beta-oxidation. In contrast, the high abundance of fatty acids in diapause suggest an increased synthesis and/or decreased beta-oxidation in diapause blastocyst.
We observed multiple categories of fatty acids in diapause and pre-implantation blastocysts: saturated fatty acids (SFAs), mono-unsaturated fatty acids (MUFAs), and poly-unsaturated fatty acids (PUFAs) (Figures S5H and S5I). MUFAs and PUFAs were enriched in diapause in free fatty acid pool. Interestingly, high levels of PUFAs are shown to be important for the prolonged survival of Caenorhabditis elegans dauer larva (Lam et al., 2017). This suggests that PUFAs might also be important for the survival of mouse embryos in diapause. SFAs were also highly enriched in diapause (Figures S5H and S5I). Interestingly, it has been reported that in cancer, when the cells are under hypoxia, there is a build-up of saturated fatty acid, similar to diapause (Ackerman et al., 2018). Furthermore, since long chain SFAs are known to induce inflammation (Yang et al., 2015; Lancaster et al., 2018) and might activate NF-kB (Yang et al., 2015), it is possible that SFAs that accumulate in diapause are stimulating NF-kB, and thereby survival of the diapause embryo. Interestingly, a recent work has shown that inflammatory NF-kB can induce stem cell quiescence (Chen et al., 2019).
In addition to lipidomic analysis, we measured polar metabolites in diapause and pre-implantation blastocysts using hydrophilic interaction chromatography (HILIC) and gas chromatography (GC) with accurate mass tandem mass spectrometry. These analyses identified 234 polar metabolites in the early embryos (Figures 5E and 5F; Table S5B). Using statistical analysis, we found multiple highly differential metabolites in these two stages, including AMP, serine, indoxyl sulfate, fructose-1-phosphate, and ergothioneine (ET; a scavenger of hydroxyl radicals [Franzoni et al., 2006]), in addition to leucine degradation intermediates such as 3-hydroxyisovaleroly carnitine (Figures 5E and 5F; Table S5C). AMP among metabolites that were up in diapause corroborates the gene expression data, and further supports AMPK activation during diapause (Figure S4F). Since serine can be generated by serine synthesis pathway from glycolysis (de Koning et al., 2003), serine upregulation in diapause may in part be due to high glycolysis at this stage. Furthermore, since indoxyl sulfate has shown to activate NF-kB through reactive oxygen species (ROS), followed by expression of p53 that suppresses cell proliferation (Shimizu et al., 2011), its upregulation in diapause may be involved in halting the cell cycle observed in diapause.
The dramatic upregulation of fatty acids seen in diapause may be partly due to serine-derived lipid metabolism (Gao et al., 2018) (Figures 5C–5E). A recent study found that serine levels were critical to avoid mitochondrial fragmentation by regulating ceramide and sphingomyelin metabolism and sphingolipid synthesis (Gao et al., 2018). We found intermediate lipid metabolites derived from serine were enriched in diapause and the enzyme that generates free fatty acids from ceramide, N-acylsphingosine amidohydrolase 1 (ASAH1), was significantly upregulated (Figure 5E).
Fatty acid synthesis may also be upregulated in diapause since the enzymes that generate free fatty acids from acetate and acetyl-CoA, acyl-CoA synthetase short (ACSS1), and fatty acid synthase (FASN), respectively, were significantly upregulated in diapause state. A previous study suggested that acetyl-CoA can enhance acetylation levels of histones at FASN promoters, a positive feed-forward regulation to upregulate FASN expression (Gao et al., 2016). Furthermore, a dramatic downregulation of TAG and DAG were observed in diapause, suggesting a release of free fatty acids from their breakdown (Figures 5C and 5E).
One of the ways free fatty acids can be utilized in diapause is to incorporate them into highly enriched phosphatidylcholines (PCs). Accumulation of PCs can stimulate phosphatidylcholine phospholipase C (PC-PLC), which is significantly upregulated in diapause (Figure 5E). Previous studies suggest that PCs play a critical role in NF-kB activation (Kouba et al., 2001). We show that TNFa signaling via NF-kB pathway is upregulated in diapause (Figures 5E and S1A; Table S2A). These data suggest that PCs in diapause may aid in cell survival through NF-kB pathway (Vuong et al., 2015; Schutze et al., 1992; Zamorano€ et al., 2003).
We previously found that the metabolic enzyme NNMT and its substrate, S-Adenosyl methionine (SAM), can regulate human pre- and post-implantation pluripotency in vitro by regulating histone methylation (Sperber et al., 2015). Interestingly, compounds that indicate SAM utilization beyond histone methylation (e.g., 1-MNA) are upregulated in diapause suggesting that epigenetic modifications may be differentially regulated in diapause.
The polar metabolite analysis revealed differential regulation of one-carbon metabolism between pre-implantation and diapause embryos (Figure 5F). In diapause state, high serine may be used to generate SAM since serine can donate one-carbon units to support the synthesis of methionine (Maddocks et al., 2016). SAM furthermore acts as a vivid methyl donor since accumulation of SAH and 1MNA are observed (Figure 5F). SAH interestingly was not used downstream the pathway, indicated by low levels of the downstream metabolites and the enzymes involved in the process (Figure 5E). Since previous studies suggest that upregulation of SAH activates NF-kB pathway, it is plausible that the NFkB activation in diapause is partly effected by high SAH levels (Barroso et al., 2016). Cystine levels were moderately elevated possibly due to autophagy (Lee et al., 2011) (diapause upregulation of genes involved in autophagy, Gpnmb, Ulk2, Uvrag, Gabarapl1,and Wipi1) (Li et al., 2010; Tsuyuki et al., 2014) (Table S1C) and serine increase in diapause. High hypotaurine and taurine levels suggest that cysteine is utilized to generate metabolites with antioxidant activities in diapause. The enrichment of antioxidants indicates the importance of preventing these dormant cells from potential oxidation (Figure S5J).
Lipolysis in Diapause-like State and in Rictor KO
We observed a significant decrease of TAGs and DAGs and increase in fatty acid levels in diapause compared to pre-implantation state, consistent with increased lipolysis in diapause (Figure 6A). Interestingly, lipolysis has previously been connected with mTOR inhibition and starvation adaptation (Cai et al., 2016; Tao et al., 2016). We therefore proceeded to analyze the lipidomics profile in Lkb1 splice variants in starved versus nonstarved mESCs (Figures 6B and S6). Our data show that starved mESCs have an increase of lipolysis as seen by TAG and DAG depletion (Figure 6B). Interestingly, PUFAs are highly enriched in R1(SS) splice variant in free fatty acid pool (Figure S6H). To test if mTORC2 inhibition in mESCs increases lipolysis, we generated mESC Rictor KO clones and analyzed their lipidomic profile (Figures 6D–6F and S6). Rictor KO mESCs were generated using CRISPR-Cas9 system (Figures 6C and 6D). As expected, Rictor KO clones do not express Rictor protein and do not phosphorylate AKT(S473). In addition, mTORC2 also affects mTORC1 activity in mESCs, as seen by decrease of mTOR, S6, and S6K phosphorylation (Figure 6E). Rictor KO mutants show upregulation of lipolysis as seen by the reduced levels of TAGs (Figure 6F). Upregulation of lipolysis was therefore observed in diapause, diapause-like state, and Rictor KO mESCs, suggesting that mTOR inhibition is causal for lipolysis in diapause (Figure 6).
Figure 6. Lipolysis Is Upregulated in Embryonic Diapause, in Diapause-like State, and in Rictor Knockout mESCs.

(A) Abundance of TAG, DAG, FA, and AC in pre-implantation and diapause blastocyst.
(B) Schematic diagram depicting outline of the Lkb1 splice variant metabolomics experiment and abundance of TAG, DAG, FA, and AC in Lkb1 splice variants,R1(LL) and R1(SS), in control and starvation conditions.
(C) A simplified diagram of the mTOR signaling pathway, showing components, and downstream targets of mTORC1/2.
(D) Schematic diagram of CRISPR-Cas9 system–mediated homozygous Rictor (mTORC2 specific component) KO in mESCs. Sequence and chromatogram(homozygote) of Rictor KO clone 14 mESCs.
(E) Screening of Rictor KO clones (top panel) by western blotting and analysis of Rictor knockout effect on mTOR and its downstream targets (bottom panel). (Beta-actin-1: pAkt and pmTOR, Beta-actin-2: pS6 Kinase and pS6, Beta-actin-3: Akt, S6 and 4EBP1, Beta-actin-4: mTOR, Beta-actin-5: pULK1 and p4EBP1, Beta-actin-6: S6 Kinase).
(F) Schematic diagram depicting outline of the Rictor KO metabolomics experiment and Rictor KO effect on lipolysis. TAGs are downregulated in Rictor knockout clones compared to wildtype.
InhibitionoftheGlutamineTransporterSLC38A1Blocks the mTOR-Dependent Diapause State
Amino acid transporters are essential for a variety of cellular processes such as nutrient uptake and energy metabolism (McGivan and Pastor-Anglada, 1994). They are also required for activation of mTORC1. SLC38A1 mediates the saturable, pH-sensitive, and electrogenic cotransport of glutamine and sodium ions. Previous studies have shown that glutamine is an essential and rate-limiting sensitizing factor that enables essential amino acids (EAAs) and growth factors to regulate mTOR in different tissues (Jewell et al., 2015; Altman et al., 2016).
Slc38a1 was highly upregulated in our diapause and diapauselike state gene expression data (Figures 1C, 7A, S7A, and S7B; Tables S6A and S6B). We hypothesized that upregulation of the glutamine transporter SLC38A1 is required for the diapause state. We tested this hypothesis by inducing diapause and treating the diapause embryos with a glutamine transporter inhibitor, L-g-glutamyl-p-nitroanilide (GPNA) (Figures 7B and 7C). The drug is not amenable to in vivo studies of diapause. Hence, to address this question, we utilized ex vivo assays for blastocysts. We induced diapause in vivo and tested the blastocyst diapause with and without the glutamine transporter inhibitor using epigenetic state as an endpoint assay (H4K16Ac). We show with GPNA treatments that mouse embryos require glutamine transporters for diapause (Figures 7B and 7C).
Figure 7. Inhibition of the Glutamine Transporters Leads to Exit from Diapause.

(A) Glutamine transporters, SLC38A1, SLC38A2, and SLC1A5 are upregulated in diapause and starvation-induced diapause-like state.
(B) Schematic diagram depicting outline of the glutamine transporter inhibition in mouse diapause embryos.
(C) Immunostaining of diapause embryos with or without GPNA treatment (1h) for epigenetic, H4K16ac mark (magenta), and DAPI (blue). n = 4 diapaused embryos per treatment, p < 0.0001 for diapaused embryos versus diapaused embryo+10 mM GPNA. Scale: 20 μm.
(D) Hypothetical model of mouse embryos in diapause state. Pre-implantation embryos enter the diapause state through Lkb1-AMPK dependent down regulation of mTOR. Diapause is associated with upregulation of SLC38A1/2 and glycolysis, low mitochondrial activity, low fatty acid beta-oxidation (FAO), increased NF-κB activity, and downregulation of mTOR. Downregulation of mTOR leads to upregulation of lipolysis, resulting in breakdown of TAGs. Glutamine transporters (SLC38A1/2) are required for epigenetic diapause state, plausibly through prolonged mTOR inhibition. PM, plasma membrane.
We also designed gRNAs for mouse SLC38A1 and A2 to test their function in diapause stage (Table S7A). We electroporated mouse zygotes with SLC38A1/2 guides using CRISPR/Cas9 system and tested for potential mutations and function. Both SLC38A1 and A2 are upregulated in diapause state. These proteins (and other SNAT glutamine transporters) most likely act in a redundant manner, making KO experiments complicated. However, we made a small number of mutations with both SLC38A1 and A2 and cultured them to blastocyst (about 4 days), and induced diapause-like state using INK-128. We showed that mutations in these glutamine transporters are detrimental for functional diapause (Figures S7C–S7F).
DISCUSSION
Future treatments manipulatingquiescence in aging stem cells,or cancer stem cells should significantly improve modern medicine. However, the regulation of quiescence in any stem cell stage or type is poorly understood. We have identified regulators of an in vivo quiescent stage, embryonic diapause, the environmentally inducible suspended stage in development and have generated an in vitro diapause-like model for further dissecting the process. With these new tools, we have shown that downregulation of mTOR by starvation-induced Lkb1-AMPK activation is necessary for the induction of a diapause-like state and that glutamine transporter SLC38A1 primes for mTOR-dependent exit from the quiescence. Forced expression of a non-diapause Lkb1 splice variant results in a constitutive diapause-like state due to a phospho-AMPK dependent increase in glucose transporters and decrease in mTOR activation. These data also explain why amino acid composition in uterine fluid is critical for diapause regulation (RenfreeandFenelon,2017).TheglutaminetransporterSlc38a1is essential for diapause state. These data show that amino acid composition and Lkb1 splice variant regulate mTOR-dependent diapause metabolic and epigenetic state (Figure 7D). Since metabolism has recently been shown to be a critical determinant of cellular fate, it is likely to play a role in quiescence and may also contribute to the epigenetic state of diapause stage (Sperber et al., 2015; Mathieu and Ruohola-Baker, 2017). These studies set the stage to better understand how cells communicate to synchronize this enigmatic, reversibly paused embryonic diapause stage (Figure 7D).
Diapause ICM, in contrast to ICM, has upregulated PFKPB2 levels catalyzing the formation of F2,6P2 that activates the key glycolytic enzyme PFK-1. Similarly, diapause-like state has high glycolytic activity and Lkb1 splice variants affect the glycolytic activity. These data have revealed a metabolic state for dormant ESCs. While previous studies showed transcriptome profile of diapause that is associated with downregulation of metabolism (Renfree and Fenelon, 2017), the present study reveals a significant increase in a critical metabolic pathway, glycolysis. Since diapause is a cell cycle and growth dormant stage, an unanswered question is why diapause requires such a high glycolytic rate.
The present study suggests that the short Lkb1 splice variant is eliminated in normal diapause since the produced protein cannot be regulated as the wild-type Lkb1. Lkb1 short codes for a protein that has a C-terminal sequence lacking potentially critical regulation sites, a phosphorylation site (serine 431 in mouse) and a farnesylation site (cysteine 433 in mouse) (Denison et al., 2009). It is possible that these sites are involved in differential regulation and/or localization of the two forms of Lkb1 protein (Towler et al., 2008). Lkb1-based activation of AMPK under energetic stress has multiple beneficial metabolic effects. However, having a constitutive activation of AMPK by Lkb1 short splice variant leads to a chronic, low-level activation of AMPK, and this can have an adverse metabolic consequence in the long-term. Constant activation of AMPK was found to cause obesity and diminishes b-cell function in mice while similar phenotype and increased adiposity were observed in humans bearing a mutation that leads to chronic activation of AMPK (Yavari et al., 2016).
The molecular mechanisms for regulation of the signaling pathways that control diapause are not well understood. In our studies, we observed high expression levels of genes involved with leucine degradation, high levels of leucine degradation metabolites and high levels of glutamine transporters in diapause. Pharmacological inhibition of the glutamine transporter SLC38A1 blocked diapause-like state, consistent with the hypothesis that amino acid glutamine levels are critical regulators of mTOR activity and thereby the diapause state (Gonzá lez et al., 2012; Jewell et al., 2015). We propose that the regulated high glutamine levels inhibit mTOR and thereby allow the blastocyst diapause state.
Taken together, this study has revealed that diapause and starvation-induced diapause-like cells have highly upregulated glycolytic activity, similar to that observed in cancer cells. We have also shown critical regulators of the mTOR pathway as the gateway to entry and exit from diapause. Starvation induces Lkb1/AMP/AMPK-dependent repression of mTOR pathway and thereby entry to diapause, while glutamine transporters are required for the diapause state. Stem cells and cancer stem cells share the capacity of controlled entry to and exit from a dormant state. Examples of abnormal control of these quiescent states include loss of regeneration due to aging and recurrence of cancer after remission. With the onset of aging, stem cells lose the ability to re-enter the cell cycle and regenerate after injury (Artoni et al., 2017), while aggressive cancer cells with stem cell properties can enter quiescence and become resistant to conventional chemotherapy by withdrawing from the cell cycle. In our study, we identified candidate regulators of diapause. Future studies will shed light on the functional roles of these regulators in stem cell quiescence, normal, and pathological conditions.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
This study did not generate new unique reagents. Further information and request for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hannele Ruohola-Baker (hannele@uw.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Isolation of Mouse Embryos
All embryos were recovered from superovulated (Day −2 injection with Pregnant Mare Serum Gonadotropin (PMS), Day 0 injection with human Chorionic Gonadotropin (hCG)) C57BL/6 females (Charles River). The day of the vaginal plug was considered day 0.5. The third day following detection of a vaginal plug was referred to as day 3.5. Embryonic diapause was induced by intraperitoneal injection of tamoxifen and subcutaneous administration of 0.5 mg of Depo Provera on day 2.5 (MacLean Hunter and Evans, 1999). Diapause blastocysts were harvested 6 days later. Day 3.5 and diapause blastocysts were flushed from the uterus of superovulated pregnant females. For the isolation of ICM, the zona pellucida was first removed from day 3.5 embryos by incubation in acid Tyrode’s solution. To remove the trophoectoderm through immune-surgery, blastocysts were placed in a rabbit anti-mouse polyclonal antibody (Rockland Immunochemicals) for 20 min at 37C, followed by guinea pig serum complement for 20–30 min at 37C. The lysed trophectoderm cells were removed and the isolated ICM was placed in lysis buffer to isolate RNA. The derivation of epiblast (Epi) from day 6.5 post-implantation embryos has been described previously (Brons et al., 2007). Each biological triplicate for ICM and dICM and duplicate for Epi samples contained lysates from approximately 50 embryos. Our work has been performed in compliance with ethical regulations, IACUC protocol # 4152–02.
Generation of Lkb1 Isoforms in Mouse ESC Lines Using TALEN-Based Gene Editing
Two mESC lines were created using Talen-based gene editing: 1. A heterozygous line in which one allele was modified to only express the short isoform of Lkb1 (SL) and 2. A homozygous line in which both alleles were modified to express only the short isoform of Lkb1 (SS). A schematic diagram illustrating the donor construct, the wild-type (wt) Lkb1 locus and the targeted allele is shown in Figure S3A. Exon 8 is the last common exon of the long and short forms of Lkb1. The short form of Lkb1 has exon 9A while the long form of Lkb1 has exons 9B/10. The modified Lkb1 locus will express only the short form of Lkb1 since the short form-specific exon 9A is fused directly in frame with exon 8 followed by the 2A peptide and the neomycin/puromycin resistance cassette followed by a poly A site.
mESCs were transfected with the TALEN pairs and donor plasmid. Neo resistant clones were analyzed for homologous recombination using PCR (Figure S3B). We have also analyzed the expression of both Lkb1 long and Lkb1 short using primers that are specific to each form using RT-PCR (Figure S3C). Lkb1 long is about 55-kDa on western blot and Lkb1 short form is about 48-kDa (Figure S3D). The control mESC line, R1(LL; wildtype for both alleles) expresses predominantly the Lkb1 long form while short form is expressed at a very low level. In the m/m mESC line, both alleles are modified to express Lkb1 short form only (SS). Since Lkb1 short form is expressed with an additional 22 amino acids (2A peptide), its size on the western blot is the same as the Lkb1 long form. Since the modified alleles express Lkb1 short form with 2A peptide, the wild-type short form disappeared in m/m lane of the western blot (Figure S3D).
Generation of mESCs Expressing Lkb1 Short Isoform Only
TALEN-mediated gene editing of the endogenous Lkb1 locus was employed. Exon 8 is the last common exon of the long and short forms of Lkb1. Exon 9A and 9B are short form- and long form-specific exons, respectively. The wild-type Lkb1 locus was modified using donor construct, that expresses exon 9 followed by 2A peptide by homologous recombination. The modified Lkb1 locus expressed only the short form of Lkb1 since the short form-specific exon 9A is fused directly in-frame with exon 8 followed by 2A peptide and Neo followed by a poly A site. mESCs were transfected with the TALEN pairs and donor plasmid. Neo resistant clones were analyzed for homologous recombination using PCR. Primer pairs were used to amplify specifically either the wild-type or the modified locus.
mESC Culture
R1 mESCs from A. Nagy (Samuel Lunenfeld Research Institute, Toronto, Canada) were cultured on feeder free plates coated with 0.1% (w/v) gelatin (Sigma) 37C for 10 min. Medium for mESCs contained DMEM (Invitrogen, Paisley, UK), supplemented with 20% ESC-qualified fetal bovine serum, 1mM sodium pyruvate, 1mM non-essential amino acids (NEAA), 1mM penicillin/streptomycin (all from Invitrogen, Carlsbad), 0.1mM 2- b-mercaptoethanol (Sigma Aldrich, St. Louis, MO), with addition of 10ng/mL mouse LIF (EMD Millipore), 1 mM GSK inhibitor (CHIR99021, Selleckchem) and 1 mM MEK inhibitor (PD0325901, Selleckchem). mESCs were passaged every 2–3 days as a single-cell suspension using 0.05% trypsin/EDTA (Life Technologies).
METHOD DETAILS
RNA-Seq
Total RNA was extracted using Qiagen Micro RNeasy kit and amplified following protocols of WT-Ovation Pico kit (Nugen). The quality and quantity of RNAs were determined on an Agilent 2100 Bioanalyzer (Agilent Technologies). Cluster generation of the cDNA library was performed on a cBot Cluster Station (Illumina) and the samples were sequenced on Genome Analyzer IIx (Illumina). Transcripts expressed in ICM, dICM or Epi with >1.4-fold change and a p value < 0.05 (adjusted for multiple testing with the Benjamini-Hochberg procedure) were considered to be differentially expressed. The UCSC genome browser tracks of RNA-Seq reads are available at: http://www.cs.washington.edu/homes/ruzzo/papers/Margaretha/.
Data Analysis
RNA-seq samples were aligned to NCBIM37 using Tophat (Trapnell et al., 2009) (version 2.0.13). Gene-level read counts were quantified using htseq-count (Anders et al., 2015) using Ensembl GRCh37 gene annotations. Genes with total expression above 2 normalized read counts in at least 2 RNA-seq samples were kept for further analysis. prcomp function from R was used to for principal component analysis. DeSeq (Anders and Huber, 2010) was used for differential gene expression analysis. Genes with fold change >1.5 and FDR <0.1 were considered differentially expressed. topGO R package (Alexa et al., 2006) was used for Gene Ontology enrichment analysis. Hallmark gene sets were used for pathway enrichment analysis (Liberzon et al., 2015). Combat (Johnson et al., 2007) was used to correct batch effects between RNA-seq samples of this study and Boroviak et al. (2015). In vitro starvation RNA-seq samples were projected onto the PCA using eigen vectors calculated from the combined in vivo RNA-seq samples of this study and Boroviak et al.
Metabolomics
Mouse Embryos
Mouse embryos were isolated as mentioned above. Briefly, embryonic diapause was induced by the injection of tamoxifen with Depo Provera on day 2.5 and diapause blastocyst was harvested 5 days later. Triplicate samples for diapause (n= 134) and duplicate for pre-implantation blastocyst (114) were collected and washed with B2 media at room temperature. Embryos were flash frozen in liquid nitrogen before performing metabolite analysis (HILIC, GC-MS and lipidomics).
Mouse ESCs
Mouse ESCs (R1(LL) and R1(SS) and rictor knockout mESCs were plated (13105 cells/35 mm plate, minimum triplicate plates for each group) and starved overnight on day 2. Cells were harvested on day 3 and flash frozen in liquid nitrogen before performing metabolite analysis (HILIC, GC-MS and lipidomics).
Sample Preparation for LC-QTOFMS Analysis
A total of 134 and 114 pre-implantation and diapause mouse embryos respectively were extracted with 225 ml of methanol at 20C containing an internal standard mixture of PE(17:0/17:0), PG(17:0/17:0), PC(17:0/0:0), C17 sphingosine, ceramide (d18:1/17:0), SM (d18:0/17:0), palmitic acid-d3, PC (12:0/13:0), cholesterol-d7, TG (17:0/17:1/17:0)-d5, DG (12:0/12:0/0:0), DG (18:1/2:0/0:0), MG (17:0/0:0/0:0), PE (17:1/0:0), LPC (17:0), LPE (17:1), and 750 mL of MTBE (methyl tertiary butyl ether) (Sigma Aldrich) at 20C containing the internal standard cholesteryl ester 22:1. Samples were shaken for 6 min at 4C with an Orbital Mixing Chilling/Heating Plate (Torrey Pines Scientific Instruments). After shaking, 188 ml of LC-MS grade water (Fisher) was added. Samples were vortexed, centrifuged and the upper (non-polar) and bottom (polar) layers were collected (350 mL and 125 mL, respectively in two aliquots) and evaporated to dryness.
One dried 350-mL aliquot of the non-polar layer was re-suspended in 100 mL of methanol:toluene (9:1, v/v) mixture containing 50 ng/ml Cuda ((12- [[(cyclohexylamino)carbonyl]amino]-dodecanoic acid) (Cayman Chemical). One dried 125-mL aliquot of the polar layer was resuspended in an acetonitrile:water (4:1, v/v) mixture with 5 mg/ml Val-Try-Val (Sigma). Samples were then vortexed then centrifuged and prepared for lipidomic or polar metabolite analysis. Method blanks and pooled human plasma (BioreclamationIVT) were included as quality control samples.
Chromatographic and Mass Spectrometric Conditions for Lipidomic LC-QTOF Analysis
For analysis of the non-polar phase, re-suspended samples were injected at 3 mL and 5 mL for ESI positive and negative modes respectively, onto a Waters Acquity UPLC CSH C18 (100-mm length 3 2.1 mm id; 1.7-mm particle size) with an additional Waters Acquity VanGuard CSH C18 pre-column (5 mm 3 2.1 mm id; 1.7-mm particle size) maintained at 65C was coupled to a Vanquish Horizon UHPLC (Thermo Fisher Scientific). To improve lipid coverage, different mobile phase modifiers were used for positive and negative mode analysis (Cajka, 2016). For positive mode 10-mM ammonium formate and 0.1% formic acid were used and 10-mM ammonium acetate (Sigma–Aldrich) was used for negative mode. Both positive and negative modes used the same mobile phase composition of (A) 60:40 v/v acetonitrile:water (LC-MS grade) and (B) 90:10 v/v isopropanol:acetonitrile. The gradient started at 0 min with 15% (B), 0–2 min 30% (B), 2–2.5 min 48% (B), 2.5–11 min 82% (B), 11–11.5 min 99% (B), 11.5–12 min 99% (B), 12– 12.1 min 15% (B), and 12.1–15 min 15% (B). A flow rate of 0.6 mL/min was used. For data acquisition a quadrupole/orbital ion trap mass spectrometer Q Exactive HF with a HESI-II ion source (Thermo Fisher Scientific). Simultaneous MS1 and MS/MS (datadependent MS/MS) acquisition was used. The parameters were ESI polarity, positive or negative; sheath gas pressure, 60 psi; aux gas flow, 25 arbitrary units; sweep gas flow, 2 arbitrary units; spray voltage, ±3.6 kV; capillary temperature, 300C; aux gas heater temperature, 370C; MS1 mass range m/z 60–900; MS1 resolving power, 30,000 FWHM (m/z 200); number of data-dependent scans per cycle: 3; MS2 resolving power, 15,000 FWHM; acquisition speed: 2 MS1 spectra/s; normalized collision energy, 20, 30, 40%.
Chromatographic and Mass Spectrometric Conditions for Polar Metabolite HILIC-QTOFMS Analysis
Hydrophilic interaction liquid chromatography (HILIC) method was used for analysis of the polar phase. Five microliters of re-suspended sample were injected onto a Waters Acquity UPLC BEH amide column (150-mm length 3 2.1 mm id; 1.7-mm particle size) with an additional Waters Acquity VanGuard BEH amide pre-column (5 mm 3 2.1 mm id; 1.7-mm particle size) maintained at 45C coupled to a Vanquish Horizon UHPLC (Thermo Fisher Scientific). The mobile phases were prepared with 10-mM ammonium formate and 0.125% formic acid (Sigma–Aldrich) in either 100% LC-MS grade water for mobile phase (A) or 95:5 v/v acetonitrile:water for mobile phase (B). Gradient elution was performed from 100% (B) at 0–2 min to 70% (B) at 7.7 min, 40% (B) at 9.5 min, 30% (B) at 10.25 min, 100% (B) at 12.75 min, isocratic until 16.75 min with a column flow of 0.4 mL/min. Spectra were collected using a quadrupole/orbital ion trap mass spectrometer Q Exactive HF with a HESI-II ion source (Thermo Fisher Scientific). Simultaneous MS1 and MS/MS (data-dependent MS/MS) acquisition was used. The parameters were ESI polarity, positive or negative; sheath gas pressure, 60 psi; aux gas flow, 25 arbitrary units; sweep gas flow, 2 arbitrary units; spray voltage, ±3.6 kV; capillary temperature, 300C; aux gas heater temperature, 370C; MS1 mass range m/z 120–1200; MS1 resolving power, 30,000 FWHM (m/z 200); number of data-dependent scans per cycle: 3; MS2 resolving power, 15,000 FWHM; acquisition speed: 2 MS1 spectra/s; normalized collision energy, 20, 30, 40%.
LC-MS Data Processing Using MS-DIAL
Both lipidomic and HILIC data processing was performed using MS-DIAL (Tsugawa et al., 2015) for peak picking, alignment, and identification. For both lipidomic and polar metabolite analysis, in house m/z and retention time libraries were used in addition to MS/MS spectra databases in msp format (Kind et al., 2013).
Lkb1 Splice Variant Analysis
We used Isolator Software to identify alternative splicing events. Isolator uses a Bayesian hierarchical model to boost signal in small sample number settings (Jones, 2016). Isolator has been previously used to find differential alternative splicing in cardiomyocyte maturation (Kuppusamy et al., 2015). Reads were aligned to mouse GRCm38 using HISAT2 (Kim et al., 2015). Ensembl gene 38, release 92 were used for annotation.
Starvation Studies
mESCs were serum (fetal bovine serum) and glucose starved for 24 h. Cells were cultured only with DMEM (0 g/L D-glucose, Invitrogen) with addition of 10ng/mL mouse LIF (EMD Millipore), 1 mM GSK inhibitor (CHIR99021, Selleckchem) and 1 mM MEK inhibitor (PD0325901, Selleckchem). To reverse starvation-induced effects, starvation media was removed from the culture after 24 h and mESCs were cultured with DMEM (4.5 g/L D-glucose, Invitrogen), supplemented with 20% ESC-qualified fetal bovine serum, 1mM sodium pyruvate, 1mM non-essential amino acids (NEAA), 1mM penicillin/streptomycin (all from Invitrogen), 0.1mM 2- b-mercaptoethanol (Sigma Aldrich), with addition of 10ng/mL mouse LIF (EMD Millipore), 1 mM GSK inhibitor (CHIR99021, Selleckchem) and 1 μM MEK inhibitor (PD0325901, Selleckchem).
mTORC1/2 Inhibition
mTORC1/2 activity was inhibited in mESCs (Lkb1 long and Lkb1 short) by treatment with INK-128 for 24 h (200nM, Medchem).
Generation of Rictor KO mESCs
One million of R1 mESCs were electroporated with SpCas9 (0.3mM, Sigma) and gRNA (1.5mM, Synthego) as RNP complex (assembled 15min at room temperature) using Amaxa Nucleofector (mouse ESC kit, Lonza) in presence of ROCK inhibitor. Individual colonies were hand-picked and plated into 96-well plates. After passaging onto 24-well plates, proteins were extracted from the clones and RICTOR protein expression was assessed by Western blot analysis. DNA of the clones was extracted using Quick Extract DNA extraction solution (Epicentre#QE09050) and nested PCR was performed around RICTOR gRNA targeting site. The PCR products were purified using EXO-SAP enzyme (ThermoFisher) and sent for Sanger sequencing analysis (through Genewiz). The gRNA sequences and nested PCR primers are listed in Table S7.
OCR and ECAR Measurement Using Seahorse Cellular Flux Assays
Mouse ESCs were seeded onto 96-well Seahorse plates at 23104 cells/well. Cells were cultured in DMEM with or without glucose, depending on starvation criteria, overnight. For mTORC1/2 inhibition, cells were treated with INK-128 overnight (200 nM, Medchem). Culture media were exchanged for base media (unbuffered DMEM (Sigma D5030) supplemented with sodium pyruvate (Gibco, 1mM) and with 25-mM glucose (for Mitostress assay), or 25-mM glucose and 50mM carnitine (for palmitate assay), 1 h prior to the assay. Substrates and selective inhibitors were injected during the measurements to achieve final concentrations of glucose (2.5mM), 4-(trifluoromethoxy) phenylhydrazone (FCCP, 300nM–500nM), oligomycin (2.5 mM), antimycin (2.5mM), rotenone (2.5mM), palmitate (50mM in BSA), BSA and ETO (50mM). The OCR and ECAR values were normalized to the number of cells present in each well, quantified by the hoechst staining (HO33342; Sigma-Aldrich). Changes in OCR and ECAR in response to substrates and inhibitors addition were defined as the maximal change after the chemical injection compared to the last OCR or ECAR value before the injection. Glycolytic capacity is a measure of the maximum rate of conversion of glucose to lactate after using oligomycin to inhibit ATP synthase in the electron transport chain (ETC) and thus block generation of ATP. Measurements of ECAR before and after 2-DG addition is a readout of the maximum glycolytic capacity of the cell. Glycolytic reserve is a measure of ECAR differences before and after oligomycin and indicates the ability of a cell to respond to an energetic demand.
Glutamine Transporter Inhibition in Diapause Embryos
Embryonic diapause was induced by intraperitoneal injection of tamoxifen and subcutaneous administration of 0.5 mg of Depo Provera on day 2.5 of embryonic development in 4 to 7-week-old mice for various strains (C57BL/6, B6C3 and B6D2). The diapause blastocysts were harvested 6 days later in M2 solution supplemented with INK128 (200nM). The embryos were cultured in KSOM media with or without 10 mM of the glutamine transporter inhibitor L-g-Glutamyl-p-nitroanilide (GPNA, MP Biomedicals) for 1h. Embryo were then stained with H4K16Ac.
CRISPR KO of Glutamine Transporters in Mouse Zygotes
Female mice from various strains were superovulated by intraperitoneal injection of 5 IU (for 4 weeks old C57BL/6 mice) or 7.5IU (for 7 weeks old B6C3 mice) of PMSG, followed by intraperitoneal injection of 5 IU hCG (for 4 weeks old C57BL/6 mice) or 7.5IU (for 7 weeks old B6C3 mice) 48 h later. Superovulated females were mated with adult males and euthanatized the next morning. Zygotes were harvested from the oviducts in M2 solution. RNP complexes were prepared with 4mM SpCas9 (Sigma) and 4mM of gRNA against Slc38a1 (Synthego) and gRNA against Slc38a2 (Synthego), and incubated for 15min at room temperature. 2 batches of 40 embryos were electroporated with RNP complex in electroporation buffer (Chen et al, 2016) using BioRad electroporator (GenePulser Xcell: 30V, 10ms, 4pulses) and cultured in KSOM media for 4 days and supplemented with INK128 (200nM) for an additional 24h before imaging and DNA extraction. DNA was extracted using Quick Extract DNA extraction solution (Epicentre#QE09050) and nested PCR were performed for around Slc38a1 and Slc38a2 gRNA target sites. The PCR products were purified using EXO-SAP enzyme (ThermoFisher) or gel extraction (Qiagen) and sent for Sanger sequencing analysis (through Genewiz) to check the genotyping of the embryos. The gRNA sequences and nested PCR primers are listed in Table S7.
Design of Guide RNA (gRNA) Targeting Exons 3 and 4 of Slc38a1/2
We designed gRNAs that target exons 3 and 4 of Slc38a1and Slc38a2, respectively. Non-Homology End Joining (NHEJ) repair created indels at the gRNA cutting site. In Slc38a1, the deletions generated a frameshift that led to the creation of a premature stop codon in exon 4, resulting in a truncated protein in the first transmembrane domain which renders the transporter non-functional. In Slc38a2, there was a deletion of 2 amino acids (alanine and isoleucine) at residues 83 and 84. Previous studies have shown that mutations in the residue 82 of SLC38A2 reduce the affinity of the transporter for sodium (Na+) and therefore the transport of glutamine, suggesting that the first transmembrane domain is crucial for the function of SLC38 transporters (Zhang et al., 2008, 2009).
Measurement of Mitochondrial Mass and Mitochondrial DNA Amount
Mitochondrial mass was measured using flow cytometry after MitoTracker Green staining of R1(LL) and R1(SS) mESCs before or after overnight starvation. Briefly, cell were washed with PBS and incubated 50 nM of MitoTracker green for 30 min before analysis on a Canto I flow cytometer (BD Biosciences). Data analysis was performed using the FlowJo software (Tree Star, Ashland, OR, USA).
Mitochondrial DNA amount was evaluated by measuring the ratio of mitochondrial DNA (mtDNA) versus nuclear DNA (nDNA) by qPCR analysis. Briefly, the DNA of R1(LL) and R1(SS) mESCs after overnight starvation was isolated using DNAzol (Invitrogen) following manufacturer’s protocol. mtDNA was measured using mt-Co1 primers mt-Co1-F 50-CAGTCTAATGCTTACTCAGC-30 and mt-Co1-R 50-GGGCAGTTACGATAACATTG-30, and nDNA was measured using Gapdh primers Gapdh-F 50GGGAA GCCCATCACCATCTTC-30 and Gapdh-R 50AGAGGGGCCATCCACAGTCT-30. Each reaction contained 10ng of DNA extract, 13 SYBR Green Master Mix, and 300nM of each primer. The qPCR was performed using a 7300 real-time PCR system (Applied Biosystems) and the ratios mtDNA/nDNA were measured.
Protein Extraction and Western Blot Analysis
For protein analysis, 13105 cells were plated on 35 mm plates. Cells were lysed directly on the plate with lysis buffer containing 20mM Tris-HCl pH 7.5, 150mM NaCl, 15% glycerol, 1% Triton X-100, 1M b-glycerolphosphate, 0.5M NaF, 0.1M sodium pyrophosphate, orthovanadate, PMSF and 2% SDS. 25 U of Benzonase nuclease (EMD Chemicals, Gibbstown, NJ) was added to the lysis buffer right before use. Proteins were quantified by Bradford assay (Bio-Rad), using BSA (bovine serum albumin) as Standard using the EnWallac Vision. The protein samples were combined with the 43 Laemli sample buffer with 10% b-mercaptoethanol (Bio-Rad #1610747), heated (95 C, 5mins) and run on SDS-PAGE (protean TGX pre-casted gradient gel, 4%–20%, Bio-Rad) and transferred to the nitro-cellulose membrane (Bio-Rad) by semi-dry transfer (Bio-Rad). Membrane were blocked for 1 h with 5% milk or 5% BSA (for antibodies detecting for phosphorylated proteins), and incubated in the primary antibodies overnight in 4C. The antibodies used for western blot were b-actin (Cell Signaling 4970 (1:10000), pAkt (Ser 473) (Cell Signaling 9271, 1:1000), Akt (Cell Signaling 9272, 1:1000), phospho-mTOR (Ser 2448) (Cell Signaling 5536, 1:1000), mTOR (Cell Signaling 2972, 1:1000), pS6 (Cell Signaling 2215, 1:1000), S6 (Cell Signaling 2117, 1:1000), p4EBP1 (Cell Signaling 236B4, 1:1000), 4EBP1 (Santa Cruz, sc-9977, 1:1000), p-p70 S6 kinase (T389 Cell Signaling 108D2, 1:1000), p70 S6 kinase (Cell Signaling 49D7, 1:1000), pULK1 (S757 Cell Signaling D7O6U, 1:1000), Rictor (cell Signaling 53A2, 1:1000), histone 4 (Lys16) acetylation (H4K16Ac) (Millipore Sigma 07–329, 1:1000), AMPKa (Cell Signaling 2532, 1:1000), and pAMPKa (Thr172 Cell Signaling 40H9, 1:1000). The membranes were then incubated with secondary antibodies (1:10000, goat anti-rabbit or goat anti-mouse IgG HRP conjugate (Bio-Rad) for 1 h and detection was performed using the Immobilon-luminol reagent assay (EMP Millipore).
Immunofluorescence Staining
Mouse ESC cells were fixed in 4% paraformaldehyde in PBS for 5 min, permeabilized for 10 min in 0.1% Triton X-100 and blocked for 1 h in 5% serum in PBS. The cells were then incubated in primary antibody overnight at 4 C, washed with PBS (2310 min), incubated with the secondary antibody and stained with 1mg/ml DAPI in 1% serum in PBS for 1 h at 37 C, washed (335min). Mounting media was composed of 2% of n-propyl gallate in 90% glycerol and 10% PBS. Analysis was done on a Leica TCS-SPE Confocal microscope using a 403 objective and Leica Software. The antibodies used for immunostaining were anti-Oct-4 (Santa Cruz, 1:100), anti-pAMPKa (Thr172 Cell Signaling 40H9, 1:100), anti-ATP synthase beta subunit (Abcam, Ab14730, 1:100) and Alexa 488- or 647-conjugated secondary antibody (Molecular Probes).
Whole-Mount Immunofluorescence
The zona pellucida was removed from day 3.5 embryos by incubation in acid Tyrode’s solution. Embryos were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15–20 min at room temperature, rinsed in PBS+0.1% Triton X-100, permeabilized in 0.25% Triton X-100 for 15–20 min, rinsed in PBS+0.1% Triton X-100 and blocked in blocking buffer for at least 1 h at room temperature or overnight at 4C. Blocking buffer was made of PBS supplemented with 10% FBS and 0.1% Triton X-100. The primary antibodies used for immunostaining were anti-RUNX1 (OriGene, TA307515), anti-Oct-4 (Santa Cruz, sc-5279), anti-pAMPKa (Thr172 Cell Signaling 40H9) and anti-Lkb1 (Santa Cruz, sc-5638). Primary antibodies were diluted 1:100 in blocking buffer, and embryos were incubated with the appropriate antibodies at 4 C overnight. They were rinsed (3310 min) in blocking buffer, and incubated with secondary antibodies for 1 h at room temperature. Alexa Fluor secondary antibodies (Invitrogen) were used at 1:500 dilution in blocking buffer. Embryos were then incubated in hoechst 33342 (Invitrogen, 1 mg/mL) for 10 min at room temperature, rinsed (3310 min) in blocking buffer, and mounted on a 35-mm glass-bottom dish (Mat Tek Corporation, P35G-1.5–14-C) in a PBS droplet overlaid with mineral oil (Sigma). Images were taken with a Nikon A1R confocal microscope (603 water immersion objective) or with a Zeiss LSM 510 Meta confocal microscope (403 water immersion objective). ImageJ 1.44 and NIS-Elements Viewer 3.20 software was used to visualize the data. All embryo images are individual laser confocal sections.
QUANTIFICATION AND STATISTICAL ANALYSIS
R was used to perform statistical analysis between groups. Transcripts expressed in ICM, dICM or Epi with >1.4-fold change and a p value < 0.05 (adjusted for multiple testing with the Benjamini-Hochberg procedure) were considered to be differentially expressed. Genes with fold change >1.5 and FDR<0.1 were considered differentially expressed. p values % 0.05 were considered significant. PCA plots were generated using the prcomp function from R with a log10 transformation of the data and 95% confidence intervals were generated using the Euclidian method. Graphical abstract was created with Biorender.com. ns: p >0.05; *: = p % 0.05; **= p % 0.01; ***: p % 0.001; ****: p % 0.0001.
All the statistical details of the experiments and the figures they correspond to can be found in this section. Data are represented as mean + SEM, and Student’s t test was used unless specified otherwise. n represents number of replicate experiments used in the experiment unless otherwise specified as number of animals in the experiment.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Monoclonal anti- β-actin | Cell Signaling | Cat#4970; RRID: AB_2223172 |
| Rabbit polyclonal anti-phospho-Akt (Ser473) | Cell Signaling | Cat#9271; RRID: AB_329825 |
| Rabbit polyclonal anti-Akt | Cell Signaling | Cat#9272; RRID: AB_329827 |
| Rabbit Monoclonal anti-phospho-mTOR (Ser2448) | Cell Signaling | Cat#5536; RRID: AB_10691552 |
| Rabbit Polyclonal anti-mTOR | Cell Signaling | Cat#2972; RRID: AB_330978 |
| Rabbit Polyclonal anti-Phospho-S6 (S240/244) | Cell Signaling | Cat#2215; RRID: AB_331682 |
| Rabbit Monoclonal anti-S6 Ribosomal Protein | Cell Signaling | Cat#2217; RRID: AB_331355 |
| Rabbit Monoclonal anti-Phospho-4EBP1 | Cell Signaling | Cat#;2855 RRID: AB_560835 |
| Mouse Monoclonal anti-4EBP1 | Santa Cruz | Cat#sc-9977; RRID: AB_626621 |
| Rabbit Monoclonal anti-Phospho-p70 S6 Kinase (Thr389) |
Cell Signaling | Cat#9234; RRID: AB_2269803 |
| Rabbit Monoclonal anti-p70 S6 Kinase | Cell Signaling | Cat#2708; RRID: AB_390722 |
| Rabbit Monoclonal anti-Phospho-ULK1 (S757) | Cell Signaling | Cat#14202; RRID: AB_2665508 |
| Rabbit Monoclonal anti-Rictor | Cell Signaling | Cat#2114; RRID: AB_2179963 |
| Rabbit Polyclonal Anti-Acetyl-Histone H4 (Lys16) | Millipore Sigma | Cat#07-329; RRID: AB_310525 |
| Rabbit Polyclonal anti-AMPKα | Cell Signaling | Cat#2532; RRID: AB_330331 |
| Rabbit Monoclonal anti-Phospho-AMPKα (Thr172) | Cell Signaling | Cat#2535; RRID: AB_331250 |
| Goat Anti-Mouse IgG (H+L)HRP Conjugate | Bio-Rad | Cat#1721011; RRID: AB_11125936 |
| Goat Anti-Rabbit IgG (H+L)HRP Conjugate | Bio-Rad | Cat#1706515; RRID: AB_11125142 |
| Mouse Monoclonal anti-ATP synthase beta subunit | Abcam | Cat# Ab14730; RRID: AB_301438 |
| Mouse Monoclonal anti-Oct3/4 | Santa Cruz | Cat#sc-5279; RRID: AB_628051 |
| Mouse Monoclonal anti-Lkb1 | Santa Cruz | Cat#sc-32245; RRID: AB_627890 |
| Alexa Fluor 488 Goat-Anti-Mouse | Thermo Fisher Scientific | Cat#A11029; RRID: AB_138404 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Mouse LIF Protein | Millipore Sigma | Cat#ESG1107 |
| Gelatin from Porcine Skin | Sigma-Aldrich | Cat#G1890 |
| Matrigel | Corning | Cat#356231 |
| Agarose Low Melting Point | RPI | Cat#9012-36-6 |
| Fetal Bovine Serum | BioWest | Cat#S1620 |
| PD0325901 (MEK inhibitor) | Selleckchem | Cat#S1036 |
| Y-27632 2HCl (ROCK inhibitor) | Selleckchem | Cat#S1049 |
| CHIR-99021 (GSK3 inhibitor) | Selleckchem | Cat#S1263 |
| Sapanisertib (INK-128) (mTORC1/2 inhibitor) | Medchem | Cat#HY-13328 |
| Hoechst 33342 Solution | Sigma-Aldrich | Cat#62249 |
| 2-Deoxy-D-glucose | Sigma-Aldrich | Cat#D8375 |
| L-γ-Glutamyl-p-nitroanilide (GPNA) (Glutamine Transporter inhibitor) | MP Biomedicals | Cat#67953-08-6 |
| AICAR (AMPK activator) | Sigma-Aldrich | Cat#A9978 |
| Compound C (AMPK inhibitor) | Sigma-Aldrich | Cat#171260 |
| Lipofectamine RNAiMAX | Thermo Fisher Scientific | Cat#13778075 |
| OPTIMEM Reduced Serum Medium | Thermo Fisher Scientific | Cat#31985 |
| DMEM, high glucose, GlutaMAX Supplement | Thermo Fisher Scientific | Cat#10566 |
| DMEM, no glucose | Thermo Fisher Scientific | Cat#11966 |
| MEM Non-Essential Amino Acids (NEAA) | Thermo Fisher Scientific | Cat#11140 |
| Sodium Pyruvate | Thermo Fisher Scientific | Cat#11360 |
| Trypsin-EDTA (0.05%), phenol red | Thermo Fisher Scientific | Cat#25300 |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat#M7522 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | Cat#15070 |
| Experimental Models: Cell Lines | ||
| Mouse ESC R1 (LL) | Nagy lab | Nagy et al., 1993 |
| Mouse ESC R1 (SL) | this manuscript | N/A |
| Mouse ESC R1 (SS) | this manuscript | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 | Jackson Laboratories | Cat#000664 |
| C57BL/6 | Charles River Laboratories | Cat#027 |
| B6C3 | Jackson Laboratories | Cat#100010 |
| B6D2 | Jackson Laboratories | Cat#100006 |
| Software and Algorithms | ||
| R | The R Foundation | https://www.r-project.org/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Microsoft Excel | Microsoft | https://products.office.com/en-us/excel |
| Microsoft PowerPoint | Microsoft | https://products.office.com/en-us/powerpoint |
| Prism GraphPad Software | Prism | https://www.graphpad.com/scientificsoftware/prism/ |
| Biorender App | Biorender | https://app.biorender.com |
Highlights.
Diapause is associated with increased lipolysis and glutamine transporter expression
Upregulation of lipolysis in diapause is associated with downregulation of mTORC2
Starvation results in a diapause-like state, enriched in glutamine transporters
Inhibition of glutamine transporters leads to exit from the diapause epigenetic state
ACKNOWLEDGMENTS
We thank members of the Ruohola-Baker laboratory for helpful discussions throughout this work. We thank Logeshwaran Somasundaram, Ammar Alghadeer, and Alex Karin Fischer for technical help. We thank the Transgenic Resource Program and Lynn & Mike Garvey Imaging Core at the University of Washington. This work is supported by the ISCRM Fellows Program Award for A.M.H., ISCRM Innovation Pilot Award for J.M., and support from Hahn family and grants from the National Institute of Health R01GM097372, R01GM97372-03S1, and R01GM083867 for H.R.-B. and 1P01GM081619 for C.B.W. and H.R.-B.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
DATA AND CODE AVAILABILITY
The accession number for the sequencing data reported in this paper is NCBI GEO: GSE143494.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.devcel.2019.12.018.
REFERENCES
- Ackerman D, Tumanov S, Qiu B, Michalopoulou E, Spata M, Azzam A, Xie H, Simon MC, and Kamphorst JJ (2018). Triglycerides promote lipid homeostasis during hypoxic stress by balancing fatty acid saturation. Cell Rep 24, 2596–2605.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexa A, Rahnenfuhrer J, and Lengauer T. (2006). Improved scoring of func-€ tional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607. [DOI] [PubMed] [Google Scholar]
- Altman BJ, Stine ZE, and Dang CV (2016). From Krebs to clinic: glutamine metabolism to cancer therapy. Nat. Rev. Cancer 16, 749. [DOI] [PubMed] [Google Scholar]
- Anders S, and Huber W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W. (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artoni F, Kreipke RE, Palmeira O, Dixon C, Goldberg Z, and RuoholaBaker H. (2017). Loss of foxo rescues stem cell aging in Drosophila germ line. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barroso M, Kao D, Blom HJ, Tavares de Almeida I, Castro R, Loscalzo J, and Handy DE (2016). S-adenosylhomocysteine induces inflammation through NFkB: a possible role for EZH2 in endothelial cell activation. Biochim. Biophys. Acta 1862, 82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barupal DK, and Fiehn O. (2017). Chemical similarity enrichment analysis (ChemRICH) as alternative to biochemical pathway mapping for metabolomic datasets. Sci. Rep 7, 14567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroviak T, Loos R, Lombard P, Okahara J, Behr R, Sasaki E, Nichols J, Smith A, and Bertone P. (2015). Lineage-specific profiling delineates the emergence and progression of naive pluripotency in mammalian embryogenesis. Dev. Cell 35, 366–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brons IG, Smithers LE, Trotter MW, Rugg-Gunn P, Sun B, Chuva de Sousa Lopes SM, Howlett SK, Clarkson A, Ahrlund-Richter L, Pedersen RA, et al. (2007). Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 448, 191–195. [DOI] [PubMed] [Google Scholar]
- Bulut-Karslioglu A, Biechele S, Jin H, Macrae TA, Hejna M, Gertsenstein M, Song JS, and Ramalho-Santos M. (2016). Inhibition of mTOR induces a paused pluripotent state. Nature 540, 119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Dong LQ, and Liu F. (2016). Recent advances in adipose mTOR signaling and function: therapeutic prospects. Trends Pharmacol. Sci 37, 303–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cajka T. (2016). Increasing lipidomic coverage by selecting optimal mobilephase modifiers in LC–MS of blood plasma. Metabolomics 12, 1–11. [Google Scholar]
- Chen M, Reed RR, and Lane AP (2019). Chronic inflammation directs an olfactory stem cell functional switch from neuroregeneration to immune defense. Cell Stem Cell 25, 501–513.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Lee B, Lee AY, Modzelewski AJ, and He L. (2016). Highly efficient mouse genome editing by CRISPR ribonucleoprotein electroporation of zygotes. J. Biol. Chem 291, 14457–14467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Koning TJ, Snell K, Duran M, Berger R, Poll-The BT, and Surtees R. (2003). L-serine in disease and development. Biochem. J 371, 653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Berletch JB, Ma W, Nguyen DK, Noble WS, and Shendure J. (2013). Upregulation of the mammalian X chromosome is associated with enhanced transcription initiation, MOF-mediated H4K16 acetylation, and longer RNA half-life. Dev. Cell 25, 55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denison FC, Hiscock NJ, Carling D, and Woods A. (2009). Characterization of an alternative splice variant of Lkb1. J. Biol. Chem 284, 67–76. [DOI] [PubMed] [Google Scholar]
- Fenelon JC, Banerjee A, and Murphy BD (2014). Embryonic diapause: development on hold. Int. J. Dev. Biol 58, 163–174. [DOI] [PubMed] [Google Scholar]
- Franzoni F, Colognato R, Galetta F, Laurenza I, Barsotti M, Di Stefano R, Bocchetti R, Regoli F, Carpi A, Balbarini A, et al. (2006). An in vitro study on the free radical scavenging capacity of ergothioneine: comparison with reduced glutathione, uric acid and trolox. Biomed. Pharmacother 60, 453–457. [DOI] [PubMed] [Google Scholar]
- Fu Z, Wang B, Wang S, Wu W, Wang Q, Chen Y, Kong S, Lu J, Tang Z, Ran H, et al. (2014). Integral proteomic analysis of blastocysts reveals key molecular machinery governing embryonic diapause and reactivation for implantation in mice. Biol. Reprod 90, 52. [DOI] [PubMed] [Google Scholar]
- Gao X, Lee K, Reid MA, Sanderson SM, Qiu C, Li S, Liu J, and Locasale JW (2018). Serine availability influences mitochondrial dynamics and function through lipid metabolism. Cell Rep 22, 3507–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Lin SH, Ren F, Li JT, Chen JJ, Yao CB, Yang HB, Jiang SX, Yan GQ, Wang D, et al. (2016). Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat. Commun 7, 11960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner DK, and Lane M. (1993). Amino acids and ammonium regulate mouse embryo development in culture. Biol. Reprod 48, 377–385. [DOI] [PubMed] [Google Scholar]
- Gonzá lez IM, Martin PM, Burdsal C, Sloan JL, Mager S, Harris T, and Sutherland AE (2012). Leucine and arginine regulate trophoblast motility through mTOR-dependent and independent pathways in the preimplantation mouse embryo. Dev. Biol 361, 286–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, and Shaw RJ (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamatani T, Daikoku T, Wang H, Matsumoto H, Carter MG, Ko MS, and Dey SK (2004). Global gene expression analysis identifies molecular pathways distinguishing blastocyst dormancy and activation. Proc. Natl. Acad. Sci. USA 101, 10326–10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Zhang H, Wang J, Liu M, Sun Y, Guo C, Lu J, Wang H, and Kong S. (2019). Blastocyst activation engenders transcriptome reprogram affecting X-chromosome reactivation and inflammatory trigger of implantation. Proc. Natl. Acad. Sci. USA 116, 16621–16630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes BF, Kurth-Kraczek EJ, and Winder WW (1999). Chronic activation of 50-AMP-activated protein kinase increases GLUT-4, hexokinase, and glycogen in muscle. J. Appl. Physiol 87, 1990–1995. [DOI] [PubMed] [Google Scholar]
- Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, et al. (2012). The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hue L, and Rider MH (1987). Role of fructose 2,6-bisphosphate in the control of glycolysis in mammalian tissues. Biochem. J 245, 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Zhu T, and Guan KL (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. [DOI] [PubMed] [Google Scholar]
- Jewell JL, Kim YC, Russell RC, Yu FX, Park HW, Plouffe SW, Tagliabracci VS, and Guan KL (2015). Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 347, 194–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, and Rabinovic A. (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Jones DC (2016). Isolator: accurate and stable analysis of isoform-level expression in RNA-seq experiments. BioRxiv. [Google Scholar]
- Kho AT, Zhao Q, Cai Z, Butte AJ, Kim JY, Pomeroy SL, Rowitch DH, and Kohane IS (2004). Conserved mechanisms across development and tumorigenesis revealed by a mouse development perspective of human cancers. Genes Dev. 18, 629–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YC, and Guan KL (2015). mTOR: a pharmacologic target for autophagy regulation. J. Clin. Invest 125, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind T, Liu KH, Lee DY, DeFelice B, Meissen JK, and Fiehn O. (2013). LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat. Methods 10, 755–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouba DJ, Nakano H, Nishiyama T, Kang J, Uitto J, and Mauviel A. (2001). Tumor necrosis factor-alpha induces distinctive NF-kappa B signaling within human dermal fibroblasts. J. Biol. Chem 276, 6214–6224. [DOI] [PubMed] [Google Scholar]
- Kuppusamy KT, Jones DC, Sperber H, Madan A, Fischer KA, Rodriguez ML, Pabon L, Zhu WZ, Tulloch NL, Yang X, et al. (2015). Let-7 family of microRNA is required for maturation and adult-like metabolism in stem cell-derived cardiomyocytes. Proc. Natl. Acad. Sci. USA 112, E2785–E2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurth-Kraczek EJ, Hirshman MF, Goodyear LJ, and Winder WW (1999). 50 AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes 48, 1667–1671. [DOI] [PubMed] [Google Scholar]
- Kurtoglu M, Gao N, Shang J, Maher JC, Lehrman MA, Wangpaichitr M, Savaraj N, Lane AN, and Lampidis TJ (2007). Under normoxia, 2deoxy-D-glucose elicits cell death in select tumor types not by inhibition of glycolysis but by interfering with N-linked glycosylation. Mol. Cancer Ther 6, 3049–3058. [DOI] [PubMed] [Google Scholar]
- Lam SM, Wang Z, Li J, Huang X, and Shui G. (2017). Sequestration of polyunsaturated fatty acids in membrane phospholipids of Caenorhabditis elegans dauer larva attenuates eicosanoid biosynthesis for prolonged survival. Redox Biol. 12, 967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster GI, Langley KG, Berglund NA, Kammoun HL, Reibe S, Estevez E, Weir J, Mellett NA, Pernes G, Conway JRW, et al. (2018). Evidence that TLR4 is not a receptor for saturated fatty acids but mediates lipid-induced inflammation by reprogramming macrophage metabolism. Cell Metab 27, 1096–1110.e5. [DOI] [PubMed] [Google Scholar]
- Lee JE, Oh HA, Song H, Jun JH, Roh CR, Xie H, Dey SK, and Lim HJ (2011). Autophagy regulates embryonic survival during delayed implantation. Endocrinology 152, 2067–2075. [DOI] [PubMed] [Google Scholar]
- Li B, Castano AP, Hudson TE, Nowlin BT, Lin SL, Bonventre JV, Swanson KD, and Duffield JS (2010). The melanoma-associated transmembrane glycoprotein Gpnmb controls trafficking of cellular debris for degradation and is essential for tissue repair. FASEB J 24, 4767–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wang J, Xu H, Xing R, Pan Y, Li W, Cui J, Zhang H, and Lu Y. (2013). Decreased fructose-1,6-bisphosphatase-2 expression promotes glycolysis and growth in gastric cancer cells. Mol. Cancer 12, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdó ttir H, Ghandi M, Mesirov JP, and Tamayo P. (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Calvisi DF, Kiss A, Cigliano A, Schaff Z, Che L, Ribback S, Dombrowski F, Zhao D, and Chen X. (2017). Central role of mTORC1 downstream of YAP/TAZ in hepatoblastoma development. Oncotarget 8, 73433–73447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu WM, Pang RT, Cheong AW, Ng EH, Lao K, Lee KF, and Yeung WS (2012). Involvement of microRNA lethal-7a in the regulation of embryo implantation in mice. PLoS One 7, e37039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean Hunter SM, and Evans M. (1999). Non-surgical method for the induction of delayed implantation and recovery of viable blastocysts in rats and mice by the use of tamoxifen and Depo-Provera. Mol. Reprod. Dev 52, 29–32. [Google Scholar]
- Maddocks OD, Labuschagne CF, Adams PD, and Vousden KH (2016). Serine metabolism supports the methionine cycle and DNA/RNA methylation through de novo ATP synthesis in cancer cells. Mol. Cell 61, 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PM, and Sutherland AE (2001). Exogenous amino acids regulate trophectoderm differentiation in the mouse blastocyst through an mTORdependent pathway. Dev. Biol 240, 182–193. [DOI] [PubMed] [Google Scholar]
- Mathieu J, and Ruohola-Baker H. (2017). Metabolic remodeling during the loss and acquisition of pluripotency. Development 144, 541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGivan JD, and Pastor-Anglada M. (1994). Regulatory and molecular aspects of mammalian amino acid transport. Biochem. J 299, 321–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren A. (1968). A study of blastocysts during delay and subsequent implantation in lactating mice. J. Endocrinol 42, 453–463. [DOI] [PubMed] [Google Scholar]
- Menke TM, and McLaren A. (1970). Carbon dioxide production by mouse blastocysts during lactational delay of implantation or after ovariectomy. J. Endocrinol 47, 287–294. [DOI] [PubMed] [Google Scholar]
- Murphy BD (2012). Embryonic diapause: advances in understanding the enigma of seasonal delayed implantation. Reprod. Domest. Anim 47, 121–124. [DOI] [PubMed] [Google Scholar]
- Nagy A, Rossant J, Nagy R, Abramow-Newerly W, and Roder JC (1993). Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc. Natl. Acad. Sci. USA 90, 8424–8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, et al. (2009). Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136, 521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paria BC, Huet-Hudson YM, and Dey SK (1993). Blastocyst’s state of activity determines the “window” of implantation in the receptive mouse uterus. Proc. Natl. Acad. Sci. USA 90, 10159–10162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike IL (1981). Comparative studies of embryo metabolism in early pregnancy. J. Reprod. Fertil Suppl. 29, 203–213. [PubMed] [Google Scholar]
- Quentien MH, Manfroid I, Moncet D, Gunz G, Muller M, Grino M, Enjalbert A, and Pellegrini I. (2002). Pitx factors are involved in basal and hormone-regulated activity of the human prolactin promoter. J. Biol. Chem 277, 44408–44416. [DOI] [PubMed] [Google Scholar]
- Ralser M, Wamelink MM, Struys EA, Joppich C, Krobitsch S, Jakobs C, and Lehrach H. (2008). A catabolic block does not sufficiently explain how 2-deoxy-D-glucose inhibits cell growth. Proc. Natl. Acad. Sci. USA 105, 17807–17811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renfree MB, and Fenelon JC (2017). The enigma of embryonic diapause. Development 144, 3199–3210. [DOI] [PubMed] [Google Scholar]
- Renfree MB, and Shaw G. (2014). Embryo-endometrial interactions during early development after embryonic diapause in the marsupial tammar wallaby. Int. J. Dev. Biol 58, 175–181. [DOI] [PubMed] [Google Scholar]
- Schutze S, Potthoff K, Machleidt T, Berkovic D, Wiegmann K, and € Krö nke M. (1992). TNF activates NF-kappa B by phosphatidylcholine-specific phospholipase C-induced “acidic” sphingomyelin breakdown. Cell 71, 765–776. [DOI] [PubMed] [Google Scholar]
- Scognamiglio R, Cabezas-Wallscheid N, Thier MC, Altamura S, Reyes A, Prendergast ÁM, Baumgartner D, Carnevalli LS, Atzberger A, Haas S, et al. (2016). Myc depletion induces a pluripotent dormant state mimicking diapause. Cell 164, 668–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw G, and Renfree MB (1986). Uterine and embryonic metabolism after diapause in the tammar wallaby, Macropus eugenii. J. Reprod. Fertil 76, 339–347. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, and Cantley LC (2004). The Lkb1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 6, 91–99. [DOI] [PubMed] [Google Scholar]
- Shimizu H, Bolati D, Adijiang A, Muteliefu G, Enomoto A, Nishijima F, Dateki M, and Niwa T. (2011). NF-kB plays an important role in indoxyl sulfate-induced cellular senescence, fibrotic gene expression, and inhibition of proliferation in proximal tubular cells. Am. J. Physiol. Cell Physiol 301, C1201–C1212. [DOI] [PubMed] [Google Scholar]
- Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, and Peterson CL (2006). Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311, 844–847. [DOI] [PubMed] [Google Scholar]
- Sperber H, Mathieu J, Wang Y, Ferreccio A, Hesson J, Xu Z, Fischer KA, Devi A, Detraux D, Gu H, et al. (2015). The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat. Cell Biol 17, 1523–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Ma YC, Yang ZS, Zou CG, and Zhang KQ (2016). Octopamine connects nutrient cues to lipid metabolism upon nutrient deprivation. Sci. Adv 2, e1501372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towler MC, Fogarty S, Hawley SA, Pan DA, Martin DM, Morrice NA, McCarthy A, Galardo MN, Meroni SB, Cigorraga SB, et al. (2008). A novel short splice variant of the tumour suppressor Lkb1 is required for spermiogenesis. Biochem. J 416, 1–14. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsugawa H, Cajka T, Kind T, Ma Y, Higgins B, Ikeda K, Kanazawa M, VanderGheynst J, Fiehn O, and Arita M. (2015). MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 12, 523–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuyuki S, Takabayashi M, Kawazu M, Kudo K, Watanabe A, Nagata Y, Kusama Y, and Yoshida K. (2014). Detection of WIPI1 mRNA as an indicator of autophagosome formation. Autophagy 10, 497–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urakami K, Zangiacomi V, Yamaguchi K, and Kusuhara M. (2013). Impact of 2-deoxy-D-glucose on the target metabolome profile of a human endometrial cancer cell line. Biomed. Res 34, 221–229. [DOI] [PubMed] [Google Scholar]
- Van Blerkom J, Chavez DJ, and Bell H. (1978). Molecular and cellular aspects of facultative delayed implantation in the mouse. Ciba Found. Symp, 141–172. [DOI] [PubMed] [Google Scholar]
- Van Winkle LJ, Tesch JK, Shah A, and Campione AL (2006). System B0,+ amino acid transport regulates the penetration stage of blastocyst implantation with possible long-term developmental consequences through adulthood. Hum. Reprod. Update 12, 145–157. [DOI] [PubMed] [Google Scholar]
- Vuong B, Hogan-Cann AD, Alano CC, Stevenson M, Chan WY, Anderson CM, Swanson RA, and Kauppinen TM (2015). NF-kB transcriptional activation by TNFa requires phospholipase C, extracellular signal-regulated kinase 2 and poly(ADP-ribose) polymerase-1. J. Neuroinflamm 12, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Rong X, Palladino END, Wang J, Fogelman AM, Martıń MG, Alrefai WA, Ford DA, and Tontonoz P. (2018). Phospholipid remodeling and cholesterol availability regulate intestinal stemness and tumorigenesis. Cell Stem Cell 22, 206–220.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, et al. (2007). Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. Cell Physiol 292, C125–C136. [DOI] [PubMed] [Google Scholar]
- Yang X, Haghiac M, Glazebrook P, Minium J, Catalano PM, and Hauguel-de Mouzon S. (2015). Saturated fatty acids enhance TLR4 immune pathways in human trophoblasts. Hum. Reprod 30, 2152–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavari A, Stocker CJ, Ghaffari S, Wargent ET, Steeples V, Czibik G, Pinter K, Bellahcene M, Woods A, Martıńez de Morentin PB, et al. (2016). Chronic activation of g2 AMPK induces obesity and reduces b cell function. Cell Metab 23, 821–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshinaga K, and Adams CE (1966). Delayed implantation in the spayed, progesterone treated adult mouse. J. Reprod. Fertil 12, 593–595. [DOI] [PubMed] [Google Scholar]
- Zamorano J, Rivas MD, Garcia-Trinidad A, Qu CK, and Keegan AD (2003). Phosphatidylcholine-specific phospholipase C activity is necessary for the activation of STAT6. J. Immunol 171, 4203–4209. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Albers T, Fiumera HL, Gameiro A, and Grewer C. (2009). A conserved Na(+) binding site of the sodium-coupled neutral amino acid transporter 2 (SNAT2). J. Biol. Chem 284, 25314–25323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Gameiro A, and Grewer C. (2008). Highly conserved asparagine 82 controls the interaction of Na+ with the sodium-coupled neutral amino acid transporter SNAT2. J. Biol. Chem 283, 12284–12292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D, MacLean PS, Pohnert SC, Knight JB, Olson AL, Winder WW, and Dohm GL (2001). Regulation of muscle GLUT-4 transcription by AMP-activated protein kinase. J. Appl. Physiol 91, 1073–1083. [DOI] [PubMed] [Google Scholar]
- Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, and Sabatini DM (2011). mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334, 678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.