

Abstract
Impaired behavioral flexibility and repetitive behavior is a common phenotype in autism and other neuropsychiatric disorders, but the underlying synaptic mechanisms are poorly understood. The trans-synaptic glutamate delta (GluD)-Cerebellin 1-Neurexin complex, critical for synapse formation/maintenance, represents a vulnerable axis for neuropsychiatric diseases. We have previously found that GluD1 deletion results in reversal learning deficit and repetitive behavior. In this study, we show that selective ablation of GluD1 from the dorsal striatum impairs behavioral flexibility in a water T-maze task. We further found that striatal GluD1 is preferentially found in dendritic shafts, and more frequently associated with thalamic than cortical glutamatergic terminals suggesting localization to projections from the thalamic parafascicular nucleus (Pf). Conditional deletion of GluD1 from the striatum led to a selective loss of thalamic, but not cortical, terminals, and reduced glutamatergic neurotransmission. Optogenetic studies demonstrated functional changes at thalamostriatal synapses from the Pf, but no effect on the corticostriatal system, upon ablation of GluD1 in the dorsal striatum. These studies suggest a novel molecular mechanism by which genetic variations associated with neuropsychiatric disorders may impair behavioral flexibility, and reveal a unique principle by which GluD1 subunit regulates forebrain circuits.
Keywords: Cbln1, GluD1, glutamate, striatum, Parafascicular thalamus
Graphical Abstract
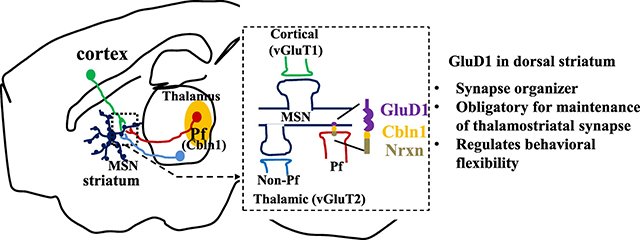
1. Introduction
Impaired behavioral flexibility is a phenotype observed in several neuropsychiatric disorders and most commonly in autism, Tourette syndrome and ADHD. Although previous studies have found that cortical and striatal circuits support cognitive flexibility, the knowledge of synaptic changes that underlie deficits in this behavior, and its relationship to neuropsychiatric disorders remains incomplete. Neurexins are strongly associated with neuropsychiatric disorders and interact with a repertoire of post-synaptic proteins of which neuroligins are most studied (reviewed (Sudhof, 2017, Yuzaki, 2018)). Recent studies have demonstrated that GluDs interact with presynaptic Neurexin via the complement pathway family protein cerebellin 1 (Cbln1) released from presynaptic terminals. Together, the GluD-Cbln1-Neurexin forms a trans-synaptic complex that mediates the development and maintenance of specific synapses in young and adult mice (Uemura et al., 2010; Matsuda et al., 2010). This function has been well established at parallel fiber-Purkinje cell (PF-PC) glutamatergic synapses, where loss of GluD2 or Cbln1 reduces number of PF-PC synapses and impairs normal LTD and motor learning (Yuzaki and Aricescu, 2017).
In contrast to GluD2, the function of GluD1, which is enriched in corticolimbic regions and striatum (Konno et al., 2014; Hepp et al., 2015), is less understood. Nonetheless, copy number variations (CNVs) and single nucleotide polymorphism studies show a strong association of GRID1 gene, that codes for GluD1, with autism (Griswold et al., 2012; Nord et al., 2011; Smith et al., 2009; Glessner et al., 2009) and schizoaffective disorders (Edwards et al., 2012; Fallin et al., 2005; Guo et al., 2007; Greenwood et al., 2011; Treutlein et al., 2009). In addition, missense mutations in GRID1 underlie developmental delay and intellectual disability ((Turner et al., 2017); FENS Neuroscience 2016, C.16.B, Laumonnier and Toutain). Altered GluD1 expression is found in Rett syndrome-patient derived neurons and in a mouse model of Rett syndrome (Livide et al., 2015; Patriarchi et al., 2016). These findings, together with the strong association of neurexin with Tourette syndrome, autism and schizoaffective disorders (Sudhof, 2017), suggest that GluD1-Cbln1-Neurexin is a highly vulnerable node for neuropsychiatric disorders. Thus, a better understanding of the organizing principle of GluD1 in the forebrain may provide mechanisms underlying behavioral phenotypes in neuropsychiatric disorders.
We have previously shown that GluD1 knockout mice exhibit impaired reversal learning and repetitive behaviors (Yadav et al., 2013). In the present study, we probed the potential mechanism of impaired behavioral flexibility due to GluD1 deletion using a conditional knockout strategy. Our findings demonstrate that GluD1 in the dorsal striatum regulates behavioral flexibility in water T-maze test, and contributes to the anatomical and functional integrity of the thalamostriatal over the corticostriatal system, providing a mechanism by which genetic variations associated with neuropsychiatric disorders may result in impaired behavioral flexibility.
2. Materials and methods
Detailed materials and methods are provided as Supplementary Information.
2.1. Animals
Male and female mice were used in this study. GluD1 KO mice (Gao et al., 2007) were on 95% C57BL/6 and remaining 129SvEv background. GluD1flox/flox mice (generous gift from Dr. Pei Lung-Chen, on congenic C75BL/6 background) were crossed with Emx1-Cre (congenic C57BL/6, 005628 Jackson Labs) or Rgs9-cre (mixed C57BL/6 and 129SvEv background (Dang et al., 2006)) driver mice to selectively ablate GluD1 from the corticolimbic region or striatum, as previously described (Liu et al., 2018). In animals without any surgical manipulation, electrophysiology and immunohistochemical studies were carried out at 4–6 weeks of age and 3–4 months of age for behavioral studies. For mice with surgical manipulation all studies were carried out at 3–4 months of age. All experimental protocols were approved by the Creighton University Institutional Animal Care and Use Committee Policies and Procedures.
2.2. Stereotaxic surgery and viral delivery
Mice were anesthetized with isoflurane and placed in a stereotaxic frame. Skull was exposed, and a small hole was drilled through the skull at the coordinates for the dorsal striatum, ventral striatum or parafascicular nucleus (Pf). The injection needle was lowered, and virus particles were delivered at a rate of 1 nl/s (total volume 150 nl) using a UMP3 micro-syringe pump (World Precision Instruments).
3. Behavior
3.1. Water T-maze
On day 1, the acquisition training was started and for every animal the platform was kept on opposite side to the preferred side assessed during habituation on day 0. Ten trials were carried out per day with an inter-trial interval of ~10 min. Following scores were given; 0 for correct choice, 1 for non-platform arm entry (incorrect choice), and 2 if after successfully finding the platform mouse left the platform arm. Once the daily criterion of 80% correct choices for four consecutive days was reached, reversal learning was started in which platform location was switched to the opposite side arm. Total errors (consistent entries between the incorrect arm and start arm), preservative errors (incorrect arm entry) and regressive errors (any entry made after successfully finding the platform) were analyzed.
4. Electrophysiology
Whole-cell electrophysiology voltage-clamp and current-clamp recordings were performed as previously described (Gupta et al., 2015; Gupta et al., 2016; Liu et al., 2018). 300-μm thick parasagittal or approximately 30° oblique horizontal sections were prepared using vibrating microtome. Electrical-evoked corticostriatal EPSCs were induced by a bipolar tungsten electrode placed in the layer 6 of the cortex. Thalamostriatal EPSCs were evoked by 5 ms 470 nM LED light delivered by a fiber optic cannula.
5. Immunohistochemistry
5.1. Confocal microscopy
Immunohistochemistry and confocal imaging was conducted, as previously described (Gupta et al., 2015; Gupta et al., 2016). For vGluT1 and vGluT2 puncta analysis, the central dorsal striatum region was analyzed. Co-localization and puncta number were analyzed using Volocity software (PerkinElmer Inc. Coventry, United Kingdom). The number of puncta per image was filtered based on intensity and size range (≥ 0.1–0.3μm3) and 3–8 images were analyzed per animal.
5.2. Immuno-electron microscopy
Brains were EM immunostained with a highly specific GluD1 antibody (Konno et al., 2014) using the immunoperoxidase method, as described in our previous studies (Raju et al., 2006). Random series of 50–55 micrographs of striatal tissue from the surface of the blocks that contained immunoreactive profiles were taken at 25000×. GluD1-immunoreactive elements that could be categorized as neuronal or glial profiles were counted and expressed as relative percentage of total labeled elements.
6. Results
6.1. Striatal, but not corticolimbic, GluD1 regulates behavioral flexibility
We first evaluated the effect of ablation of GluD1 from the corticolimbic region and striatum, which are the two most enriched regions of GluD1 expression (Konno et al., 2014; Hepp et al., 2015), on behavioral flexibility in a water T-maze task. Selective deletion of GluD1 from corticolimbic region and striatum was achieved by crossing GluD1flox/flox with Emx-1-Cre as previously described (Liu et al., 2018). In water T-maze task, no significant difference was observed in mice with corticolimbic deletion of GluD1 (corticolimbic-GluD1 KO) in habit acquisition learning or in the reversal learning (Fig. 1B–B”‘; Supplementary Fig. 1A). Corticolimbic-GluD1 KO were also similar to wildtype in rotarod test and open field test in total distance traveled (Fig. 1C, D). We then conducted water T-maze test in mice with striatal ablation of GluD1 (St-GluD1 KO), in which most GluD1 protein was abolished from the dorsal striatum (Supplementary Fig. 1D, E). In St-GluD1 KO mice, no significant difference was observed in percent errorless trials across days (Fig. 1F), but they took a significantly shorter time to find the platform (Fig. 1F’). In addition, significantly fewer days were taken to reach the criteria for habit acquisition learning (WT 4.250 ± 0.6756 vs. St-GluD1 KO 1.714 ± 0.4118, p = .0030, unpaired t-test). In reversal learning, St-GluD1 KO mice committed significantly more total errors than WT in the first trial (WT 2.167 ± 0.5198 vs. St-GluD1 KO 4.500 ± 0.9002, p = .0421, unpaired t-test). The number of total errors on day 1 (including trial 1 to trial 10) was significantly higher in St-GluD1 KO mice than WT animals (Fig. 1F”). When the total errors on Day 1 were divided into perseverative or regressive errors, St-GluD1 KO mice made significantly more preservative errors in trial 1 on day 1 (WT 1.583 ± 0.2289 vs. St-GluD1 KO 2.929 ± 0.4625, p = .0174, unpaired t-test) and in total preservative errors on day 1 and day 2 than wildtype mice (Fig. 1F”‘). No difference was observed in the percent trials completed without error during reversal training (Supplementary Fig. 1E). Overall, these results suggest that striatal GluD1, but not corticolimbic GluD1, regulates behavioral flexibility during the water T-maze task. In rotarod test, which is used to assess motor coordination and repetitive behaviors (Rothwell et al., 2014), St-GluD1 KO mice showed greater latency to fall compared to wildtype, with no change in open field test (Fig. 1G, H). We also tested corticlimbic-GluD1 KO and St-GluD1 KO in other behavioral and cognitive tasks. No differences were observed in other motor, sensorimotor and cognitive tests in either genotype (Supplementary Fig. 1B–I). These results suggest that deletion of striatal GluD1 selectively impairs behavioral flexibility, but improves habit learning behaviors.
Fig. 1.

Deletion of GluD1 from the striatum but not corticolimbic region impairs behavioral flexibility. A. Strategy for deletion of GluD1 from corticolimbic region. BB’. No significant difference was observed in the number of percent trials without errors across days during habit acquisition (genotype [F (1, 11) = 0.7664, p = .4000)], Two-way repeated measures ANOVA) or latency to find the platform (genotype [F (1, 11) = 0.1618, p = .6952)]) (N = 6 for WT and N = 8 for Corticolimbic (CL)-GluD1 KO). B″-B”‘. No significant differences were observed in total errors (genotype [F (1, 11) = 0.9646, p = .3471]) or preservative errors (genotype [F (1, 11) = 1.35, p = .2699]) during the reversal learning in corticolimbic-GluD1 KO. C. In the rotarod test, during test phase no significant difference in the latency to fall was observed in WT and CL-GluD1 KO mice (N = 11 for WT and N = 10 for CL-GluD1 KO; Two-way repeated measures ANOVA genotype [F (1, 19) = 1.402, p = .2511)], trials [F (5, 95) = 10.3, p < .0001)] and interaction [F (5, 95) = 0.2416, p = .9430]). D. In the open field test, no significant difference was found between WT and CL-GluD1 KO mice in total distance traveled (N = 10 for WT and N = 14 for CL-GluD1 KO; WT 39.44 ± 2.71, vs CL-GluD1 KO 43.96 ± 1.538, p = .1683, unpaired t-test). E. Strategy to deletion of GluD1 from striatal neurons. F. Mice were tested in water T-maze test. No significant difference was observed in the percent trials without errors on training days between the two groups (N = 12 for WT and N = 14 for St-GluD1 KO). F′. The latency to reach the platform during the training phase was significantly shorter in St-GluD1 KO. Two-way repeated measures ANOVA revealed a significant effect of genotype [F (1, 24) = 6.236, p = .0198)] and time [F (7, 168) = 15.35, p < .0001)] on latency to find platform. Bonferroni’s post-hoc test revealed a significant reduction in latency to find platform in the St-GluD1 KO mice (day 2: p = .0107). F″. In the reversal learning test St-GluD1 KO mice made significantly more total errors across days. Comparison of errors in all ten trials for each day was conducted by two-way repeated measures ANOVA which revealed a significant effect of genotype [F (1, 24) = 31.21, p < .0001)], time [F (5, 120) = 151.9, p < .0001)] and interaction [F (5,120) = 18.19, p < .0001 on total errors. Bonferroni’s post-hoc test revealed a significantly higher number of total errors in the St-GluD1 KO mice (day 1: p ≤ .0001). F″’. St-GluD1 KO mice made more perseverative errors. Comparison of total preservative errors per day across days by two-way repeated measures ANOVA revealed a significant effect of genotype [F (1, 24) = 16.93, p = .0004)], time [F (5, 120) = 167.5, p < .0001)] and interaction [F (5,120) = 12.59, p < .0001] on total preservative errors. Bonferroni’s post-hoc test revealed a significantly higher number of total errors in the St-GluD1 KO mice (day 1: p < .0001; day 2: p = .0105). All data are presented as mean ± SEM. G. In the rotarod test, the latency to fall during test phases was longer in St-GluD1 KO mice than WT (N = 13 for WT and N = 17 for St-GluD1 KO). Two-way repeated measures ANOVA revealed a significant effect of genotype [F (1, 28) = 9.190, p = .0052)], trials [F (5, 140) = 13.55, p < .0001)] and interaction [F (5, 140) = 0.4604, p = .8052] on falling latency. Bonferroni’s post-hoc test revealed a significant difference in falling latency of St-GluD1 KO mice (trial 2 WT 68.36 ± 6.722 vs St-GluD1 KO 101.6 ± 8.405: p = .0270; trial 4 WT 74.47 ± 7.464 vs St-GluD1 KO 105.7 ± 9.552: p = .0458; trial 6 WT 74.85 ± 8.605 vs St-GluD1 KO 108.0 ± 8.649: p = .0275). H. In the open field test, no significant difference was found between WT and St-GluD1 KO mice in total distance traveled (N = 13 for WT and N = 17 for St-GluD1 KO; WT 30.65 ± 2.405 vs St-GluD1 KO 37.39 ± 2.442, p = .0641, unpaired t-test).
6.2. Reduced behavioral flexibility maps to GluD1 in the dorsal striatum
In order to better understand the origin of impaired behavioral flexibility upon GluD1 ablation from the striatum, we tested the effect of local ablation of GluD1 by cre recombinase expression using viral vectors in GluD1flox/flox mice. AAV-eGFP or AAV-eGFP-Cre were injected into the dorsal striatum (centromedial region) of GluD1flox/flox mice (Supplementary Fig. 2A–B), and after sufficient survival time for optimal expression water T-maze test was conducted. Similar to St-GluD1 KO in the water T-maze task, the AAV-eGFP-Cre mice took a significantly shorter time to find the platform than AAV-eGFP animals on testing days 2, 5 and 6 (Fig. 2B’). However, the percentage of trials without errors was not significantly different (Fig. 2B). In the reversal learning task, the deficits in the AAV-eGFP-Cre mice were consistent with those described in St-GluD1 KO mice with more total perseverative errors on day 1 and day 2 than in control animals (Fig. 2B”, B″’). In contrast, mice with ventral striatum GluD1 deletion did not exhibit differences in the number of total or preservative errors in the water T-maze test (Fig. 2F”), indicating that GluD1 function in the dorsal striatum is critical for the regulation of behavioral flexibility. Deletion of GluD1 from the dorsal striatum, but not the ventral striatum, also led to an increase in latency to fall in rotarod test with no change in the total distance traveled in the open field test (Fig. 2C, D, G, H).
Fig. 2.

GluD1 in the dorsal striatum controls behavioral flexibility. A. Strategy for local ablation of GluD1 from dorsal striatum. AAV-eGFP or AAV-eGFP-Cre were stereotaxically injected into the dorsal striatum of GluD1flox/flox mice. B. Striatal AAV-eGFP and AAV-eGFP-Cre mice did not show any significant change in the percent trials without errors on training days (N = 7 for AAV-eGFP and 7 for AAV-eGFP-Cre). B′. The latency to reach the platform during the training (habit acquisition) phase was shorter in AAV-eGFP-Cre compared to the AAV-eGFP group. Two-way repeated measures ANOVA treatment [F (1, 12) = 13.95, p = .0029)], days [F (6, 72) = 5.064, p < .0002)] and interaction [F (6, 72) = 0.5384, p = .7773. Post-hoc Bonferroni’s test revealed significance at day 2: AAV-eGFP 32.24 ± 4.718 vs. AAV-eGFP-Cre 14.53 ± 2.878, p = .0343; day 5: AAV-eGFP 24.16 ± 2.635, vs. AAV-eGFP-Cre 12.16 ± 1.96, p = .0279; day 6: AAV-eGFP 19.64 ± 2.532, vs AAV-eGFP-Cre 7.814 ± 1.729, p = .0316. B″. During reversal learning test, AAV-eGFP-Cre mice made significantly more total errors compared to AAV-eGFP group. Two-way repeated measures ANOVA treatment [F (1, 12) = 19.05, p = .0009)], days [F (5, 60) = 18.49, p < .0001)] and interaction [F (5, 60) = 3.057, p = .0160]. Bonferroni’s post-hoc test revealed significance on day 1 (AAV-eGFP 7.571 ± 0.4809 vs. AAV-eGFP-Cre 17.29 ± 4.932, p < .0002); day 2 (AAV-eGFP 0.8571 ± 0.3401 vs. AAV-eGFP-Cre 7.429 ± 1.11, p < .0220). B″’. Similarly, AAV-eGFP-Cre committed more number of perseverative errors compared to AAV-eGFP group. Two-way repeated measures ANOVA treatment [F (1, 12) = 31.69, p = .0001)], days [F (5, 60) = 28.2, p < .0001)] and interaction [F (5, 60) = 5.064, p = .0006]. Bonferroni’s post-hoc test revealed significance on day 1 (AAV-eGFP 6.429 ± 0.8411 vs AAV-eGFP-Cre 14.14 ± 2.874, p < .0001); day 2 (AAV-eGFP 0.5714 ± 0.2974 vs AAV-eGFP-Cre 7.429 ± 1.11, p < .0001). C. In the rotarod test, the latency to fall during test phase was longer in AAV-eGFP-Cre than AAV-eGFP injected mice (N = 14 for AAV-eGFP and N = 13 for AAV-eGFP-Cre). Two-way repeated measures ANOVA revealed a significant effect of genotype [F (1, 25) = 37.4, p < .0001], trials [F (5, 125) = 5.19, p = .0002] and interaction [F (5, 125) = 1.38, p = .2360] on falling latency. Bonferroni’s post-hoc test revealed a significant difference in falling latency of AAV-eGFP-Cre mice (trial 1 AAV-eGFP 61.52 ± 5.466 vs AAV-eGFP-Cre 104.3 ± 6.22: p < .0001; trial 2 AAV-eGFP 49.94 ± 5.615 vs AAV-eGFP-Cre 84.82 ± 5.487: p < .0002; trial 3 AAV-eGFP 67.91 ± 4.34 vs AAV-eGFP-Cre 98.85 ± 4.789: p < .0014; trial 4 AAV-eGFP 64.35 ± 4.988 vs AAV-eGFP-Cre 94.72 ± 5.995: p < .0018; trial 6 AAV-eGFP 72.28 ± 4.11 vs AAV-eGFP-Cre 101.3 ± 9.748: p < .0032). D. In the open field test, no significant difference was found between AAV-eGFP and AAV-eGFP-Cre injected mice in total distance traveled (N = 16 for AAV-eGFP and N = 17 AAV-eGFP-Cre; AAV-eGFP 47.88 ± 4.587 vs AAV-eGFP-Cre 52.78 ± 3.614, p = .4080, unpaired t-test). E. Strategy of deletion of GluD1 from ventral striatum. AAV-eGFP or AAV-eGFP-Cre vectors were setereotaxically injected into the ventral striatum of GluD1flox/flox mice. F. During the habit acquisition no significant difference was observed between AAV-eGFP and AAV-eGFP-Cre mice, (treatment [F (1, 17) = 2.466, p = .1347, Two-way repeated measures ANOVA) (N = 9 for AAV-eGFP and 10 for AAV-eGFP-Cre). F′. No significant differences were observed in the latency to find the platform, although a trend for higher latency was observed on day 2 (treatment [F (1, 17) = 0.9066, p = .03544)]). F″-F″’. No significant differences were observed in total errors (treatment [F (1, 17) = 4.523, p = .0484]) or preservative errors (treatment [F (1, 17) = 4.055, p = .0601]) during the reversal learning in corticolimbic-GluD1 KO. G. In the rotarod test, during test phase no significant difference in the latency to fall was observed in AAV-eGFP and AAV-eGFP-Cre injected mice (N = 9 for AAV-eGFP and N = 9 for AAV-eGFP-Cre; Two way repeated measures ANOVA genotype [F (1, 16) = 0.3285, p = .5745], trials [F (5, 80) = 4.826, p = .0007] and interaction [F (5, 80) = 0.3245, p = .8968]). H. In the open field test, no significant difference was found between AAV-eGFP and AAV-eGFP-Cre injected mice in total distance traveled (N = 9 for AAV-eGFP and N = 10 AAV-eGFP-Cre; AAV-eGFP 54.76 ± 6.085 vs AAV-eGFP-Cre 48.2 ± 4.254, p = .3917, unpaired t-test). All data are presented as mean ± SEM.
6.3. Cellular and subcellular localization of GluD1 immunoreactivity in the striatum
To determine the potential sites where GluD1 may mediate its functional effects in the striatum, we assessed the localization of GluD1 using light, electron and confocal microscopy. At the light microscopic level, GluD1 immunoreactivity is heavily expressed in the striatum of WT mice, but the staining is completely abolished in GluD1-KO mice, demonstrating the specificity of the GluD1 antibody used in this study (Fig. 3A–B). Although intense across the whole striatum, patches of striatal neuropil display a lower intensity of immunostaining than neighboring regions, a pattern reminiscent of the patch/matrix striatal compartmentation (Herkenham and Pert, 1981). In the electron microscope, striatal GluD1 immunoreactivity was predominantly found in dendritic shafts (47.9 ± 1.2% of total GluD1-labeled elements) and glial processes (37.7 ± 2.5%) over dendritic spines (14.3 ± 2.6%) (Fig. 3B–F). Relevant to these results, GluD1 mRNA has been reported in oligodendrocyte precursor cells (OPCs) (Larson et al., 2016; Saunders et al., 2018), a cell-type known to interact with neurons via axo-glial synapses (Bergles et al., 2000).
Fig. 3.

GluD1 is preferentially localized in striatal dendrites and co-localized with vGluT2 puncta. A-B. Light micrographs showing lack of striatal GluD1 immunostaining in GluD1 KO mice, confirming the specificity of the GluD1 antibody used in our study. In A and A’, asterisks indicate areas of striatum, reminiscent of the striatal patch compartment, with low expression of GluD1 immunoreactivity. C-E. Examples of GluD1-immunoreactive dendritic shafts (Den), spines (sp) and glial processes (Gl) in the mouse dorsal striatum. Unlabeled terminals (u.Te) and spines (u.Sp) are also indicated. Scale bars in A: 1 mm (valid for B); C: 1 μm (valid for DF). F. Histogram showing the relative proportion of the different GluD1- positive elements in the mouse dorsal striatum (N = 3 mice; total surface area of tissue analyzed: 5987.35 μm2; total number of labeled elements sampled: 808). Note the predominance of labeling associated with dendritic shafts and glial processes compared with spines. G. Confocal images of dorsal striatum tissue co-labeled with GluD1 and vGluT1 or vGluT2. Arrows indicate co-localized puncta. Total number of GluD1-labeled punctate structures co-localized with vGluT1 or vGluT2 puncta per 2000 μm2 striatal surface area. A higher percent of GluD1 elements were found in apposition with vGluT2 than vGluT1 puncta; vGluT1 1.12 ± 1.02 vs. vGluT2 10.31 ± 1.46, p = .005, Unpaired t-test (N = 3 animals). All data are presented as mean ± SEM.
The striatum receives major excitatory inputs from the thalamus and cortex that can be distinguished by vGluT2- and vGluT1-positive terminals, respectively (Fremeau Jr et al., 2001). In confocal microscopy, our findings indicate that GluD1-immunoreactive elements were far more frequently co-localized with vGluT2 (thalamic)- than vGluT1 (cortical)-immunoreactive puncta (vGluT1 1.12 ± 1.02 vs. vGluT2 10.31 ± 1.46 GluD1+ elements/2000 μm2 of striatal tissue) (Fig. 3G), thereby suggesting a preferential association of GluD1 with thalamostriatal over corticostriatal terminals.
6.4. Deletion of striatal GluD1 reduces excitatory neurotransmission in MSNs and thalamostriatal vGluT2 expression
To understand the potential synaptic functions of striatal GluD1, we first recorded mEPSC and mIPSC from MSNs in both the constitutive and St-GluD1 KO models. A significant reduction in the frequency, but not the amplitude, of mEPSC was observed in MSNs of St-GluD1 mice (Fig. 4A). No significant change in mIPSC frequency and amplitude was observed (Fig. 4B). In current-clamp recordings, a significant reduction in spike frequency of MSNs at higher current injections was observed in St-GluD1 KO further suggesting reduced excitability of MSNs in these animals (Fig. 4C). No change in the resting membrane potential, input resistance or rheobase was observed (Fig. 4D). When other features of excitability were analyzed a greater accommodation was seen in St-GluD1 KO mice at 300 pA current injection but not 200 pA current injection (Supplementary Fig. 3A, B). No changes were observed in the first spike latency or last inter-spike interval in St-GluD1 KO (Supplementary Fig. 3C, D). Reduction in mEPSC frequency, with no change in mIPSC properties, was also observed in constitutive GluD1 KO (Supplementary Fig. 4A, B).
Fig. 4.

Striatum specific ablation of GluD1 leads to reduced excitatory neurotransmission in MSNs. A. A significant reduction in the frequency of mEPSC was observed in St-GluD1 KO (WT: 6.57 ± 0.7 Hz vs. St-GluD1 KO: 2.99 ± 0.46 Hz, p = .0004, Unpaired t-test). Inter-event interval (IEI) was also significantly reduced (p = .0021, KeS test) (N = 11–12 from 3 to 4 animals per genotype). No change in amplitude was observed (WT: 12.73 ± 0.27 pA vs. St-GluD1 KO: 12.65 ± 0.55 pA, p = .894, Unpaired t-test). B. No significant change in mIPSC frequency or amplitude was observed (frequency: WT: 1 ± 0.08 Hz vs. St-GluD1 KO: 0.86 ± 0.14 Hz, p = .409; amplitude: WT: 11.92 ± 0.69 pA vs. St-GluD1 KO: 12.92 ± 0.8 pA, p = .366; Unpaired t-test) (N = 10–12 from 3 to 4 animals per genotype). All data are presented as mean ± SEM. C. Reduced excitability of MSNs in St-GluD1 KO (N = 7–8/genotype from 3 animals; 280 pA: WT 16.86 ± 2.32 Hz vs. St-GluD1 KO 8 ± 2.6 Hz, p = .016; 300 pA: WT 18.29 ± 2.27 Hz vs. St-GluD1 KO 9.25 ± 2.58 pA, p = .013; Two-way ANOVA with Bonferroni’s post-hoc test). D. No change in resting membrane potential (p = .9645, unpaired t-test), input resistance (p = .1220, unpaired t-test) or rheobase (p = .4617, unpaired t-test) of MSNs in St-GluD1 KO. All data are presented as mean ± SEM. E. A significant reduction in the number of striatal vGluT2, but not vGluT1, puncta was found in St-GluD1 KO (vGluT1: WT 100 ± 7.703% vs. St-GluD1 KO 112.932 ± 15.469% of WT, p = .5649; vGluT2: WT 100 ± 12.482% vs. St-GluD1 KO 32.482 ± 4.341% of WT, p = .0003; unpaired t-test, N = 5 mice/genotype for vGluT1 and 9 mice/genotype for vGluT2). All data are presented as mean ± SEM.
We next assessed the changes in number of vGluT1- and vGluT2-immunoreactive puncta in the dorsal striatum of St-GluD1 KO mice to determine whether the reduced mEPSC frequency seen in MSNs of these animals was due to a specific reduction in the prevalence of cortical or thalamic terminals. This analysis revealed a significant reduction in the prevalence of vGluT2-positive puncta, with no change in vGluT1 puncta counts, in the dorsal striatum of St-GluD1 KO mice (Fig. 4E). A specific reduction in vGluT2 puncta counts in the dorsal striatum was also observed in constitutive GluD1 KO mice (Supplementary Fig. 5A). We also assessed the changes in thalamic and cortical terminals in mice with selective ablation of GluD1 in the dorsal striatum using AAVs. A significant reduction in the number of striatal vGluT2, but not vGluT1, puncta was seen in the GluD1-ablated striatal region (Supplementary Fig. 5B, C).
6.5. Pf-MSN synapses show higher AMPA/NMDA ratio upon deletion of striatal GluD1
The enrichment of GluD1 in dendritic shafts suggests that GluD1 may localize preferentially to projections from parafascicular nuclei of the thalamus (Pf) since it is the major source of asymmetric axo-dendritic synapses in the striatum (Dube et al., 1988; Sadikot et al., 1992). Additionally, Pf shows strong expression of Cbln1 (Miura et al., 2006; Otsuka et al., 2016) which the major synaptogenic partner of GluDs. This hypothesis is further supported by immunohistochemical results demonstrating that GluD1 is preferentially co-localized with thalamic terminals and that loss of GluD1 specifically reduces the number of thalamostriatal terminals. Thus, we determined if St-GluD1 deletion affects glutamatergic transmission and other neuroplastic properties of Pf-MSN synapses. To do so, we used optogenetics to compare the effects of light activation of Pf-striatal axons on MSNs activity between WT and St-GluD1-KO mice. AAV-ChR2 was injected into the Pf, and after sufficient expression, ex vivo optogenetic experiments were conducted.
Mice in which the viral vector injection sites spread outside the nuclear confines of Pf were not used. No change in paired-pulse ratio was observed at Pf-MSN synapses in St-GluD1 KO mice (Fig. 5C). As previously reported (Ellender et al., 2013), the ratio of AMPA/NMDA receptor currents at Pf-MSN synapses was smaller than at corticostriatal synapses in WT mice. However, a significant increase in the AMPA/NMDA receptor current ratio was found in St-GluD1 KO mice (Fig. 5D). Deletion of striatal GluD1 did not significantly affect the rectification index of AMPA currents suggesting potentially no change in composition (Fig. 5E). We also conducted ex vivo optogenetic experiments to determine input-output ratio at Pf-MSN in wildtype and St-GluD1 KO, however these were not informative due to the variability in infection efficiency across animals. As an alternate strategy to evaluate whether the shift in AMPA/NMDA ratio may be due to change in NMDA receptor currents, we measured NMDA receptor sEPSCs. A significant reduction in the frequency of NMDA receptor sEPSC, but no change in amplitude, was observed in GluD1 KO (Supplementary Fig. 6A), suggesting minimal postsynaptic effect. It is also possible that sEPSC recordings failed to detect specific changes of Pf-MSN synapses. We further analyzed synaptoneurosomal preparations from the dorsal striatum of wildtype and St-GluD1 KO mice to examine if there were any changes in the expression of AMPA and NMDA receptor subunits. No change was observed in the expression of total AMPA receptor subunits detected using pan-AMPA antibody in St-GluD1 KO. However, a significant reduction in GluN1 subunit expression was observed in St-GluD1 KO (Supplementary Fig. 6B). Thus, it is possible that reduced NMDA receptor content is responsible for the increase in AMPA/NMDA ratio at Pf-MSN synapses.
Fig. 5.

Specific increase in the AMPA/NMDA ratio at Pf-MSN synapses in St-GluD1 KO. A. Viral vector injections in the Pf (top row) and resulting anterograde labeling of the thalamic projections in the dorsal striatum (bottom row) in WT and St-GluD1 KO mice. B. Light-evoked EPSCs (in the presence of picrotoxin (PCTX)), sensitive to CNQX and AP5, were recorded in striatal MSNs (** p < .01). C. No significant change in paired-pulse ratio across different inter-stimulus interval was observed between WT and KO animals [p > .876 for all intervals, Two-way ANOVA with Bonferroni’s post-hoc test (N = 7–10 from 3 to 4 animals per genotype)]. D. A significant increase in the AMPA/NMDA ratio was observed at Pf-MSN synapses in St-GluD1 KO [WT 0.35 ± 0.05 vs. St-GluD1 KO 0.98 ± 0.24, p = .033; Unpaired t-test (N = 8–10 from 3 to 4 animals per genotype)]. E. No change in the rectification of AMPA receptor responses in St-GluD1 KO; p > .999 for all voltages; Two-way ANOVA with Bonferroni’s post-hoc test (N = 6 from 3 animals per genotype). All data are presented as mean ± SEM.
Because plasticity mechanisms such as long-term depression may underlie reversal learning, we further tested whether the loss of GluD1 affected LTD at Pf-MSN synapses. LTD was induced by activation of mGluR1/5 receptors using DHPG. DHPG produced a similar reduction in amplitude of light-evoked AMPA EPSCs at Pf-MSN synapses in WT vs St-GluD1 KO mice (Fig. 5F). Along the same line, the synaptic depression effects of D-serine on the amplitude of AMPA-mediated EPSCs at Pf-MSN synapses were not significantly different between WT and St-GluD1 KO mice (Fig. 5G). These data suggest that GluD1 is not necessary for these forms of striatal synaptic plasticity.
6.6. No functional changes at corticostriatal synapses upon striatal deletion of GluD1
We next examined whether GluD1 affected properties of corticostriatal synapses in St-GluD1 KO animals. We found no change in paired-pulse facilitation, AMPA/NMDA ratio and spike threshold at cortico-striatal synapses in MSNs of these mice compared to WT (Fig. 6A–C). Cortico-striatal synapses also appeared to be functionally intact in constitutive GluD1 KO with no change in AMPA/NMDA ratio and paired-pulse ratio or DHPG-induced LTD (Supplementary Fig. 7A to C). Additionally, D-serine did not affect DHPG-induced LTD in WT mice (Supplementary Fig. 7D). No change in DHPG-induced LTD was noticed when analyzing mEPSC in MSNs or CA1 neurons which are also enriched in GluD1 (Supplementary Fig. 8A, B). Moreover, unlike dorsal striatum MSNs, CA1 neuron mEPSC characteristics were unchanged in GluD1 KO (Supplementary Fig. 8C), suggesting region-specific effects of GluD1 on excitatory neurotransmission.
Fig. 6.

No change in the function of corticostriatal synapses in St-GluD1 KO. A. No change was observed in the paired-pulse ratio in St-GluD1 KO; p > .999 for all intervals, Two-way ANOVA with Bonferroni’s post-hoc test. (N = 10 from 4 mice per genotype). B. No change in AMPA/NMDA ratio at corticostriatal synapses; WT 2.23 ± 0.19 vs. St-GluD1 KO 2.33 ± 0.35, p = .807, Unpaired t-test (N = 9 from 3 mice per genotype). C. No change in the evoked spike threshold; WT 3.5 ± 0.68 mA vs. St-GluD1 KO 3.13 ± 0.39 mA, p = .629, Unpaired t-test (N = 7–8 from 3 mice per genotype). All data are presented as mean ± SEM.
Recent studies have suggested that activation of metabotropic glutamate receptors 1/5 (mGluR1/5) may induce ion channel currents via the GluD receptors (Ady et al., 2014; Benamer et al., 2018). We examined whether this property may be altered in GluD1 KO which could affect behavior. Thus, we evaluated whether DHPG induces GluD1-dependent ion channel currents in striatal MSNs and CA1 pyramidal neurons. In MSNs, no significant change in baseline was observed with DHPG in both WT and St-GluD1 KO mice (Supplementary Fig. 9A). Similarly, although DHPG application led to a slow current in CA1 hippocampal neurons, its amplitude was not significantly different between WT and GluD1 KO animals (Supplementary Fig. 9B). Together, the behavioral changes induced by GluD1 depletion in the dorsal striatum cannot be explained by dysregulation of mGluR1/5 function.
7. Discussion
7.1. GluD1 functions as a synaptic organizer in the striatum
In addition to the canonical neurexin-neuroligin adhesion molecules, recent studies have identified several proteins that may serve as synaptic organizers by inducing formation and/or maintenance of specific synapses. Among these complement family proteins, C1ql1 and Cblns as well as glia-derived molecules such as hevin, play a critical role in formation/maintenance of cerebellar and cortical synapses (Sudhof, 2017; Yuzaki, 2018). The GluDs, which bind to Cbln1 with their aminoterminal domain, have long been considered orphan receptors due to lack of ligand-gated ion channel activity, are now known as synaptic organizers in the cerebellar cortex. However, their role outside the cerebellar cortex remains largely unknown. Our findings identify a unique synaptic organizer property of GluD1 in the dorsal striatum whereby it plays a role in the maintenance of thalamostriatal, but not corticostriatal, glutamatergic synapses. We found that GluD1 was preferentially localized to dendritic shafts, the main synaptic target of thalamic terminals from the Pf (Dube et al., 1988; Sadikot et al., 1992; Raju et al., 2006), and that ablation of GluD1 led to reduced excitatory neurotransmission in MSNs and decreased prevalence of vGuT2 (putatively thalamic) terminals in the dorsal striatum. Further evidence for GluD1 association with Pf-striatal projections come from our light microscopic data (Fig. 3A) showing a lower level of GluD1 expression in presumed striatal patches, a striatal compartment known to be devoid of Pf inputs (Herkenham and Pert, 1981). Since no change in paired pulse ratio was observed, these results suggest that reduced mEPSC was not due to a change in release probability, but most likely a reduced number of functional glutamatergic synapses. Because of the specific enrichment of Cbln1, the synaptogenic partner of GluD1, in Pf, and the known role of Cbln/GluD complex in synapse formation/maintenance in other brain regions, we suggest that the lower excitatory neurotransmission recorded in striatal neurons may result from a specific loss of Pf-MSN terminals. Since the optogenetic stimulation of Pf-MSN synapses was still intact in St-GluD1 KO, it suggests that loss of GluD1 does not completely eliminate Pf-MSN synapses. Instead, the remaining Pf-MSN synapses exhibit a shift in AMPA/NMDA ratio, but no change in chemical LTD mechanisms. Additional electrophysiology experiments are needed to determine whether deletion of GluD1 affects other forms of plasticity at Pf-MSN synapses. We have previously found a reduction in AMPA receptor subunit expression and an increase in NMDA receptor subunit expression in prefrontal cortex and hippocampus of GluD1 KO, and a contrasting increase in AMPA and kainate subunit expression in the amygdala (Yadav et al., 2012; Yadav et al., 2013). Thus, the increase in AMPA/NMDA ratio at Pf-MSN synapses in St-GluD1 KO is reminiscent of changes observed upon GluD1 deletion in amygdala. In contrast to the thalamostriatal system, the corticostriatal synapses were intact after GluD1 deletion.
The preferential role of GluD1 in the regulation of Pf-MSN synapses is likely due to the strong and specific expression of Cbln1 in the Pf, known as the main source of thalamostriatal projections in rodents (Miura et al., 2006; Otsuka et al., 2016). Although Cbln2 is also expressed in cortical and Pf neurons, its affinity for GluD1 is significantly lower than that of Cbln1 (Wei et al., 2012). Thus, the interactions between GluD1 and Cbln2 may play a relatively minor role in regulating corticostriatal and thalamostriatal synapses, compared to the regulatory functions of GluD1-Cbln1 interactions upon thalamostriatal synapses from Pf. We have previously found that ablation of GluD1 does not affect the excitatory neurotransmission in the prefrontal cortex, hippocampus or nucleus accumbens suggesting that there may be other compensatory mechanisms that normalize the excitatory neurotransmission in these regions ((Gupta et al., 2015, Liu et al., 2018), current study). However, in both complete and St-GluD1 KO mice used in this study, we observed a reduction in the frequency of mEPSC and vGluT2 puncta suggesting a specific reduction in thalamostriatal glutamatergic innervation. Thus, our findings suggest that GluD1 is obligatory for the maintenance of excitatory thalamostriatal synapses on MSNs.
In the cerebellar cortex, a role of GluD2 in mGluR1-induced LTD is well established (Kashiwabuchi et al., 1995) and GluDs are known to interact via other signaling mechanisms with mGluRs (Uemura et al., 2004; Kato et al., 2012; Suryavanshi et al., 2016). Based on these observations and the potential role of this form of plasticity in reversal learning, we evaluated the role of GluD1 in corticostriatal and thalamostriatal synaptic plasticity. Consistent with previous reports (Wu et al., 2015), we found an initial greater depression of corticostriatal than thalamostriatal synapses upon application of DHPG, but GluD1 loss did not affect DHPG-induced LTD at either synapses. Although delta receptors do not function as typical iGluRs, they have a ligandbinding domain similar to the GluN1 subunit where D-serine, glycine and other smaller amino acids can bind (Naur et al., 2007; Yadav et al., 2011; Kristensen et al., 2016). Importantly, this interaction has been found to induce conformational changes in delta receptors and produce GluD2-dependent LTD at immature PF-PC synapses (Kakegawa et al., 2011). We found that D-serine application led to a reduction in the amplitude of EPSCs at Pf-MSN synapses, but this was not significantly affected by ablation of GluD1 subunit, suggesting that GluD1 is not necessary for this form of plasticity. Because this neuroplastic effect was observed in the presence of NMDAR blockers AP5 and MK-801, it suggests that either GluD2 or GluN3-containing NMDA receptors or some unknown mechanisms may be responsible for this form of plasticity. Future studies are needed to clarify this issue. Although we did not test D-serine effect alone at corticostriatal synapses, we did not find a significant effect of D-serine on DHPG-induced LTD, which may be interpreted as saturation of LTD response or that D-serine does not induce LTD at corticostriatal synapses. Thus, although GluD1 is obligatory for maintenance of synapses and potentially synaptic composition, synaptic depression is not affected by loss of GluD1. Thus, GluD1 and GluD2 are similar is some function, but diverge in other. Difference in the C-terminal interaction of the two receptors may be responsible for some of the divergences.
7.2. Thalamostriatal synaptic dysfunction in neuropsychiatric and neurological disorders
As discussed earlier, genetic studies demonstrate strong association of GRID1 with autism and schizoaffective disorders. In addition, Neurexin mutations are associated with Tourette syndrome, autism and szhizoaffective disorders (Sudhof, 2017). Thus, GluD1-Cbln1-Neurexin is a highly vulnerable node for neuropsychiatric disorders. One of the core symptoms in autism, Tourette syndrome and ADHD is increase in repetitive behaviors and impaired behavioral flexibility. Previous studies have used a wide variety of manipulations, primarily lesion and deactivation of Pf, to demonstrate a role of the Pf-striatal system in behavioral flexibility (Bradfield and Balleine, 2017). The thalamostriatal pathway is relevant to Tourette syndrome, ADHD and OCD since DBS of CM/Pf relieve symptoms in some patients (Smith et al., 2014). Despite this information, our knowledge of synaptic mechanisms that underlie behavioral inflexiblity remains incomplete. Using region-specific ablation of GluD1, we found a strong phenotype of impaired behavioral flexibility in water T-maze test in mice with specific GluD1 ablation in the dorsal striatum. Interestingly, these animals also demonstrated faster habit acquisition and time to task completion. In addition, deletion of GluD1 from the dorsal striatum led to better performance in rotarod test with higher latency to fall which is also relevant to higher repetitive behavior. These behavioral findings are consistent with our previous observations that constitutive GluD1 KO mice exhibit greater task completion and lower working memory error in an 8-arm radial maze task, but display slower learning in a water maze reversal task (Yadav et al., 2013). In optogenetic studies, we identified a specific increase in AMPA/NMDA ratio at Pf-MSN synapses upon GluD1 ablation. We hypothesize that this change in synaptic function may enhance plasticity mechanisms which facilitate learning during acquisition phase and impair behavioral flexibility during reversal learning. Future studies using genetic manipulations to control Pf-MSN synapses will be necessary to identify the precise molecular switch that underlies impaired behavioral flexibility.
In addition to cognitive function in neuropsychiatric disorders, the thalamostriatal system is also important in neurological disorders. Pf undergoes severe neuronal loss in Parkinson’s and Huntington diseases (PD and HD) (Henderson et al., 2000; Heinsen et al., 1996) and it has been suggested that degeneration of the Pf-MSNs thalamostriatal system may contribute to cognitive impairments associated with these disorders (Smith et al., 2014). The reports of reduced Pf activity and Pf-MSN neurotransmission in PD models (Parker et al., 2016; Parr-Brownlie et al., 2009), and evidence that deep brain stimulation (DBS) of CM/Pf provides relief of symptoms in PD patients (Krauss et al., 2002; DeLong and Wichmann, 2015; Sharma et al., 2017) further support the functional importance of this system in PD. Our results of a critical role of GluD1 in regulating thalamostriatal synapses together with the evidence that both Cbln1 and Neurexin S4+ variant, which interact with Cbln1-GluD1, are controlled in an activity-dependent manner (Iijima et al., 2009; Iijima et al., 2011) suggest that changes in expression in response to pathological insults can precipitate synaptic dysfunction in neurological disorders such as PD and HD.
8. Conclusion
In conclusion, we have identified a unique role of GluD1 in the organization of neural circuits in the dorsal striatum which provides a mechanism by which genes associated with neuropsychiatric disorders may lead to precipitation of impaired behavioral flexibility. Because both Cbln1 and splicing of neurexin are controlled in an activity-dependent manner (Iijima et al., 2009, Iijima et al., 2011), it is an intriguing possibility that the GluD1-Cbln1-Neurexin complexes at thalamostriatal synapses may serve as a physiological switch to govern behavioral flexibility both in normal and disease states.
Supplementary Material
Acknowledgements
The authors are thankful to Anna Ayala, Lorenzo Rivera, Susan Jenkins and Jean-Francois Pare for excellent technical help.
Funding
This work was supported by grants from the National Science Foundation #1456818 (SMD), National Institutes of Health NS104705 (SMD), National Institutes of Health MH116003 (SMD) and Yerkes National Primate Center National Institutes of Health/ORIP base grant P51OD11132 (YS). The authors note no conflict of interest.
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2020.104746.
References
- Ady V, Perroy J, Tricoire L, Piochon C, Dadak S, Chen X, Dusart I, Fagni L, Lambolez B, Levenes C, 2014. Type 1 metabotropic glutamate receptors (mGlu1) trigger the gating of GluD2 delta glutamate receptors. EMBO Rep. 15, 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benamer N, Marti F, Lujan R, Hepp R, Aubier TG, Dupin AAM, Frebourg G, Pons S, Maskos U, Faure P, Hay YA, Lambolez B, Tricoire L, 2018. GluD1, linked to schizophrenia, controls the burst firing of dopamine neurons. Mol. Psychiatry 23 (3), 691–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Roberts JD, Somogyi P, Jahr CE, 2000. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature 405, 187–191. [DOI] [PubMed] [Google Scholar]
- Bradfield LA, Balleine BW, 2017. Thalamic control of dorsomedial striatum regulates internal state to guide goal-directed action selection. J. Neurosci 37, 3721–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang MT, Yokoi F, Yin HH, Lovinger DM, Wang Y, Li Y, 2006. Disrupted motor learning and long-term synaptic plasticity in mice lacking NMDAR1 in the striatum. Proc. Natl. Acad. Sci. U. S. A 103, 15254–15259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong MR, Wichmann T, 2015. Basal ganglia circuits as targets for neuromodulation in Parkinson disease. JAMA Neurol 72, 1354–1360. [DOI] [PubMed] [Google Scholar]
- Dube L, Smith AD, Bolam JP, 1988. Identification of synaptic terminals of thalamic or cortical origin in contact with distinct medium-size spiny neurons in the rat neostriatum. J. Comp. Neurol 267, 455–471. [DOI] [PubMed] [Google Scholar]
- Edwards AC, Aliev F, Bierut LJ, Bucholz KK, Edenberg H, Hesselbrock V, Kramer J, Kuperman S, Nurnberger JI Jr., Schuckit MA, Porjesz B, Dick DM, 2012. Genome-wide association study of comorbid depressive syndrome and alcohol dependence. Psychiatr. Genet 22, 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellender TJ, Harwood J, Kosillo P, Capogna M, Bolam JP, 2013. Heterogeneous properties of central lateral and parafascicular thalamic synapses in the striatum. J. Physiol 591, 257–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallin MD, Lasseter VK, Avramopoulos D, Nicodemus KK, Wolyniec PS, McGrath JA, Steel G, Nestadt G, Liang KY, Huganir RL, Valle D, Pulver AE, 2005. Bipolar I disorder and schizophrenia: a 440-single-nucleotide polymorphism screen of 64candidate genes among Ashkenazi Jewish case-parent trios. Am. J. Hum. Genet 77,918–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremeau RT Jr., Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, Bellocchio EE, Fortin D, Storm-Mathisen J, Edwards RH, 2001. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron 31,247–260. [DOI] [PubMed] [Google Scholar]
- Gao J, Maison SF, Wu X, Hirose K, Jones SM, Bayazitov I, Tian Y, Mittleman G, Matthews DB, Zakharenko SS, Liberman MC, Zuo J, 2007. Orphan glutamate receptor delta1 subunit required for high-frequency hearing. Mol. Cell. Biol 27, 4500–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, Imielinski M, Frackelton EC, Reichert J, Crawford EL, Munson J, Sleiman PM, Chiavacci R, Annaiah K, Thomas K, Hou C, Glaberson W, Flory J, Otieno F, Garris M, Soorya L, Klei L, Piven J, Meyer KJ, Anagnostou E, Sakurai T, Game RM, Rudd DS, Zurawiecki D, McDougle CJ, Davis LK, Miller J, Posey DJ, Michaels S, Kolevzon A, Silverman JM, Bernier R, Levy SE, Schultz RT, Dawson G, Owley T, McMahon WM, Wassink TH, Sweeney JA, Nurnberger JI, Coon H, Sutcliffe JS, Minshew NJ, Grant SF, Bucan M, Cook EH, Buxbaum JD, Devlin B, Schellenberg GD, Hakonarson H, 2009. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood TA, Lazzeroni LC, Murray SS, Cadenhead KS, Calkins ME, Dobie DJ, Green MF, Gur RE, Gur RC, Hardiman G, Kelsoe JR, Leonard S, Light GA, Nuechterlein KH, Olincy A, Radant AD, Schork NJ, Seidman LJ, Siever LJ, Silverman JM, Stone WS, Swerdlow NR, Tsuang DW, Tsuang MT, Turetsky BI, Freedman R, Braff DL, 2011. Analysis of 94 candidate genes and 12 endophenotypes for schizophrenia from the consortium on the genetics of schizophrenia. Am. J. Psychiatry 168, 930–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griswold AJ, Ma D, Cukier HN, Nations LD, Schmidt MA, Chung RH, Jaworski JM, Salyakina D, Konidari I, Whitehead PL, Wright HH, Abramson RK,Williams SM, Menon R, Martin ER, Haines JL, Gilbert JR, Cuccaro ML, Pericak-Vance MA, 2012. Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum. Mol. Genet 21, 3513–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo SZ, Huang K, Shi YY, Tang W, Zhou J, Feng GY, Zhu SM, Liu HJ, Chen Y, Sun XD, He L, 2007. A case-control association study between the GRID1 gene and schizophrenia in the Chinese northern Han population. Schizophr. Res 93, 385–390. [DOI] [PubMed] [Google Scholar]
- Gupta SC, Yadav R, Pavuluri R, Morley BJ, Stairs DJ, Dravid SM, 2015. Essential role of GluD1 in dendritic spine development and GluN2B to GluN2A NMDAR subunit switch in the cortex and hippocampus reveals ability of GluN2B inhibition in correcting hyperconnectivity. Neuropharmacology 93, 274–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta SC, Ravikrishnan A, Liu J, Mao Z, Pavuluri R, Hillman BG, Gandhi PJ, Stairs DJ, Li M, Ugale RR, Monaghan DT, Dravid SM, 2016. The NMDA receptor GluN2C subunit controls cortical excitatory-inhibitory balance, neuronal oscillations and cognitive function. Sci. Rep 6, 38321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinsen H, Rub U, Gangnus D, Jungkunz G, Bauer M, Ulmar G, Bethke B, Schuler M, Bocker F, Eisenmenger W, Gotz M, Strik M, 1996. Nerve cell loss in the thalamic centromedian-parafascicular complex in patients with Huntington’s disease. Acta Neuropathol. 91, 161–168. [DOI] [PubMed] [Google Scholar]
- Henderson JM, Carpenter K, Cartwright H, Halliday GM, 2000. Degeneration of the Centre median-parafascicular complex in Parkinson’s disease. Ann. Neurol 47, 345–352. [PubMed] [Google Scholar]
- Hepp R, Audrey Hay Y, Aguado C, Lujan R, Dauphinot L, Potier MC, Nomura S, Poirel O, El Mestikawy S, Lambolez B, Tricoire L, 2015. Glutamate receptors of the delta family are widely expressed in the adult brain. Brain Struct. Funct 220 (5), 2797–2815. [DOI] [PubMed] [Google Scholar]
- Herkenham M, Pert CB, 1981. Mosaic distribution of opiate receptors, parafascicular projections and acetylcholinesterase in rat striatum. Nature 291, 415–418. [DOI] [PubMed] [Google Scholar]
- Iijima T, Emi K, Yuzaki M, 2009. Activity-dependent repression of Cbln1 expression: mechanism for developmental and homeostatic regulation of synapses in the cerebellum. J. Neurosci 29, 5425–5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima T, Wu K, Witte H, Hanno-Iijima Y, Glatter T, Richard S, Scheiffele P, 2011. SAM68 regulates neuronal activity-dependent alternative splicing of neurexin-1. Cell 147, 1601–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakegawa W, Miyoshi Y, Hamase K, Matsuda S, Matsuda K, Kohda K, Emi K, Motohashi J, Konno R, Zaitsu K, Yuzaki M, 2011. D-serine regulates cerebellar LTD and motor coordination through the delta2 glutamate receptor. Nat. Neurosci 14,603–611. [DOI] [PubMed] [Google Scholar]
- Kashiwabuchi N, Ikeda K, Araki K, Hirano T, Shibuki K, Takayama C, Inoue Y, Kutsuwada T, Yagi T, Kang Y, 1995. Impairment of motor coordination, Purkinje cell synapse formation, and cerebellar long-term depression in GluR delta 2 mutant mice. Cell 81, 245–252. [DOI] [PubMed] [Google Scholar]
- Kato AS, Knierman MD, Siuda ER, Isaac JT, Nisenbaum ES, Bredt DS, 2012. Glutamatereceptor delta2 associates with metabotropic glutamate receptor1 (mGluR1), protein kinase Cgamma, and canonical transient receptor potential 3 and regulates mGluR1-mediated synaptic transmission in cerebellar Purkinje neurons. J. Neurosci 32, 15296–15308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno K, Matsuda K, Nakamoto C, Uchigashima M, Miyazaki T, Yamasaki M, Sakimura K, Yuzaki M, Watanabe M, 2014. Enriched expression of GluD1 in higher brain regions and its involvement in parallelfiber-interneuron synapse formation in the cerebellum. J. Neurosci 34, 7412–7424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauss JK, Pohle T, Weigel R, Burgunder JM, 2002. Deep brain stimulation of the Centre median-parafascicular complex in patients with movement disorders. J. Neurol. Neurosurg. Psychiatry 72, 546–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen AS, Hansen KB, Naur P, Olsen L, Kurtkaya NL, Dravid SM, Kvist T, Yi F, Pohlsgaard J, Clausen RP, Gajhede M, Kastrup JS, Traynelis SF, 2016. Pharmacology and structural analysis of ligand binding to the Orthosteric site of glutamate-like GluD2 receptors. Mol. Pharmacol 89, 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson VA, Zhang Y, Bergles DE, 2016. Electrophysiological properties of NG2(+) cells: matching physiological studies with gene expression profiles. Brain Res. 1638, 138–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Gandhi PJ, Pavuluri R, Shelkar GP, Dravid SM, 2018. Glutamate delta-1 receptor regulates cocaine-induced plasticity in the nucleus accumbens. Transl. Psychiatry 8, 219 018–0273–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livide G, Patriarchi T, Amenduni M, Amabile S, Yasui D, Calcagno E, Lo Rizzo C, De Falco G, Ulivieri C, Ariani F, Mari F, Mencarelli MA, Hell JW, Renieri A, Meloni I, 2015. GluD1 is a common altered player in neuronal differentiation from both MECP2-mutated and CDKL5-mutated iPS cells. Eur. J. Hum. Genet 23, 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda K, Miura E, Miyazaki T, Kakegawa W, Emi K, Narumi S, Fukazawa Y, Ito-Ishida A, Kondo T, Shigemoto R, Watanabe M, Yuzaki M, 2010. Cbln1 isa ligand for an orphan glutamate receptor delta2, a bidirectional synapse organizer. Science 328, 363–368. [DOI] [PubMed] [Google Scholar]
- Miura E, Iijima T, Yuzaki M, Watanabe M, 2006. Distinct expression of Cbln family mRNAs in developing and adult mouse brains. Eur. J. Neurosci 24, 750–760. [DOI] [PubMed] [Google Scholar]
- Naur P, Hansen KB, Kristensen AS, Dravid SM, Pickering DS, Olsen L, Vestergaard B, Egebjerg J, Gajhede M, Traynelis SF, Kastrup JS, 2007. Ionotropic glutamate-like receptor delta2 binds D-serine and glycine. Proc. Natl. Acad. Sci. U. S. A 104, 14116–14121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nord AS, Roeb W, Dickel DE, Walsh T, Kusenda M,O’Connor KL, Malhotra D, McCarthy SE, Stray SM, Taylor SM, Sebat J, STAART Psychopharmacology Network, King B, King MC, McClellan JM, 2011. Reduced transcript expression of genesaffected by inherited and de novo CNVs in autism. Eur. J. Hum. Genet 19, 727–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka S, Konno K, Abe M, Motohashi J, Kohda K, Sakimura K, Watanabe M, Yuzaki M, 2016. Roles of Cbln1 in non-motor functions of mice. J. Neurosci 36, 11801–11816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker PR, Lalive AL, Kreitzer AC, 2016. Pathway-specific remodeling of thalamostriatal synapses in Parkinsonian mice. Neuron 89, 734–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parr-Brownlie LC, Poloskey SL, Bergstrom DA, Walters JR, 2009. Parafascicular thalamic nucleus activity in a rat model of Parkinson’s disease. Exp. Neurol 217, 269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patriarchi T, Amabile S, Frullanti E, Landucci E, Lo Rizzo C, Ariani F, Costa M, Olimpico F, W Hell J, M Vaccarino F, Renieri A, Meloni I, 2016. Imbalance of excitatory/inhibitory synaptic protein expression in iPSC-derived neurons from FOXG1(+/−)patients and in foxg1(+/−)mice. Eur. J. Hum. Genet 24,871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju DV, Shah DJ, Wright TM, Hall RA, Smith Y, 2006. Differential synaptology of vGluT2-containing thalamostriatalafferents between the patch and matrix compartments in rats. J. Comp. Neurol 499, 231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell PE, Fuccillo MV, Maxeiner S, Hayton SJ, Gokce O, Lim BK, Fowler SC, Malenka RC, Sudhof TC, 2014. Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell 158, 198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadikot AF, Parent A, Smith Y, Bolam JP, 1992.Efferent connections of the centromedian and parafascicular thalamic nuclei in the squirrel monkey: a light and electron microscopic study of the thalamostriatal projection in relation to striatal heterogeneity. J. Comp. Neurol 320, 228–242. [DOI] [PubMed] [Google Scholar]
- Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, Goeva A, Nemesh J, Kamitaki N, Brumbaugh S, Kulp D, McCarroll SA, 2018. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma VD, Mewes K, Wichmann T, Buetefisch C, Willie JT, DeLong M, 2017. Deep brain stimulation of the centromedian thalamic nucleus for essential tremor:a case report. Acta Neurochir. 159, 789–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M, Spence MA, Flodman P, 2009. Nuclear and mitochondrial genome defects in autisms. Ann. N. Y. Acad. Sci 1151, 102–132. [DOI] [PubMed] [Google Scholar]
- Smith Y, Galvan A, Ellender TJ, Doig N, Villalba RM, Huerta-Ocampo I, Wichmann T, Bolam JP, 2014. The thalamostriatal system in normal and diseased states. Front. Syst. Neurosci 8,5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, 2017. Synaptic Neurexin complexes: a molecular code for the logic of neural circuits. Cell 171, 745–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suryavanshi PS, Gupta SC, Yadav R, Kesherwani V, Liu J, Dravid SM, 2016. Glutamate Delta-1 receptor regulates metabotropic glutamate receptor 5 Signaling in the Hippocampus. Mol. Pharmacol 90, 96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treutlein J, Muhleisen TW, Frank J, Mattheisen M, Herms S, Ludwig KU, Treutlein T, Schmael C, Strohmaier J, Bosshenz KV, Breuer R, Paul T, Witt SH, Schulze TG, Schlosser RG, Nenadic I, Sauer H, Becker T, Maier W, Cichon S, Nothen MM, Rietschel M, 2009. Dissection of phenotype reveals possible association between schizophrenia and glutamate Receptor Delta 1 (GRID1) gene promoter. Schizophr. Res 111, 123–130. [DOI] [PubMed] [Google Scholar]
- Turner TN, Yi Q, Krumm N, Huddleston J, Hoekzema K, F Stessman HA, Doebley AL, Bernier RA, Nickerson DA, Eichler EE, 2017. Denovo-Db:a compendium of human De novo variants. Nucleic Acids Res. 45, D804–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura T, Mori H, Mishina M, 2004. Direct interaction of GluRdelta2 with shank scaffold proteins in cerebellar Purkinje cells. Mol. Cell. Neurosci 26, 330–341. [DOI] [PubMed] [Google Scholar]
- Uemura T, Lee SJ, Yasumura M, Takeuchi T, Yoshida T, Ra M, Taguchi R, Sakimura K, Mishina M, 2010. Trans-synaptic interaction of GluRdelta2 and Neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell 141, 1068–1079. [DOI] [PubMed] [Google Scholar]
- Wei P, Pattarini R, Rong Y, Guo H, Bansal PK, Kusnoor SV, Deutch AY, Parris J, Morgan JI, 2012. The Cbln family of proteins interact with multiple signaling pathways. J. Neurochem 121, 717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YW, Kim JI, Tawfik VL, Lalchandani RR, Scherrer G, Ding JB, 2015. Inputand cell-type-specific endocannabinoid-dependent LTD in the striatum. Cell Rep. 10, 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav R, Rimerman R, Scofield MA, Dravid SM, 2011. Mutations in the transmembrane domain M3 generate spontaneously open orphan glutamate delta1 receptor. Brain Res. 1382,1–8. [DOI] [PubMed] [Google Scholar]
- Yadav R, Gupta SC, Hillman BG, Bhatt JM, Stairs DJ, Dravid SM, 2012. Deletion of glutamate delta-1 receptor in mouse leads to aberrant emotional and social behaviors. PLoS One 7, e32969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav R, Hillman BG, Gupta SC, Suryavanshi P, Bhatt JM, Pavuluri R, Stairs DJ, Dravid SM, 2013. Deletion of glutamate delta-1 receptor in mouse leads to enhanced working memory and deficit in fear conditioning. PLoS One 8, e60785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzaki M, 2018. Two classes of secreted synaptic organizers in the central nervous system. Annu. Rev. Physiol 80, 243–262. [DOI] [PubMed] [Google Scholar]
- Yuzaki M, Aricescu AR, 2017. A GluD coming-of-age story. Trends Neurosci. 40, 138–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.