

Abstract
Objective:
Vascular endothelial cells (ECs) normally maintain vascular homeostasis and are regulated by proinflammatory cytokines and reactive oxygen species (ROS). A human genome-wide association study identified that ASK1-interacting protein-1 ([AIP1], also identified as DAB2IP) gene variants confer susceptibility to cardiovascular disease (CVD), but the underlying mechanism is unknown.
Approach and Results:
We detected a normal AIP1 form (named AIP1A) in the healthy aorta, but a shorter form of AIP1 (named AIP1B) was found in diseased aortae that contained atherosclerotic plaques and graft arteriosclerosis. AIP1B transcription in resting ECs was suppressed through epigenetic inhibition by RIF1/H3K9 methyltransferase-mediated H3K9me3 and this inhibition was released by proinflammatory cytokines. AIP1A, but not AIP1B, was downregulated by proteolytic degradation through a Smurf1-dependent pathway in ECs under inflammation. Therefore, AIP1B was the major form present during inflammatory conditions. AIP1B, which lacks the N-terminal pleckstrin homology domain of AIP1A, localized to the mitochondria and augmented TNF-induced mitochondrial ROS generation and EC activation. EC-specific AIP1B transgenic mice (AIP1B-ECTG) exhibited augmented ROS production, EC activation, and neointima formation in vascular remodeling models.
Conclusions:
Our current study suggests that a shift from anti-inflammatory AIP1A to pro-inflammatory AIP1B during chronic inflammation plays a key role in inflammatory vascular diseases.
Keywords: AIP1, isoform, epigenetics, RIF1, H3K9 methylation, atherosclerosis
Subject codes: atherosclerosis, cell signaling, epigenetics
Graphical Abstract
INTRODUCTION
Cardiovascular disease (CVD) is the leading cause of death in the United States. It is clear that inflammatory mediators and oxidative stress are pathogenic mechanisms for CVD1, 2. The vascular cell type that normally limits inflammation and prevents CVD is the endothelial cell (EC). Inflammation and oxidative stress activate intracellular signaling cascades that induce EC activation. EC activation is characterized by proinflammatory molecule expression, a decrease in vascular nitric oxide (NO) bioavailability, and apoptosis. Identification of critical mediators that regulate EC homeostasis and promote EC phenotypic changes would provide novel strategies to prevent and treat CVD. We previously identified that ASK1-interacting protein-1 (AIP1) is highly expressed in the vascular endothelium and functions as a signaling adaptor to mediate phenotypic changes in ECs that contribute to CVD3–14. A human genome-wide association study (GWAS) also identified that AIP1 (also identified as DAB2IP) gene variants confer susceptibility to CVD including abdominal aortic aneurysm, peripheral vascular disease, the early onset of myocardial infarction, and pulmonary embolism15, 16. However, the underlying mechanism by which AIP1 contributes to CVD is unknown.
AIP1 was initially identified as an ASK1- or DAB2-interacting protein, and it contains multiple functional domains including a pleckstrin homology (PH) domain, a protein kinase C conserved domain (C2), and a Ras GTPase-activating protein (GAP) domain within the N-terminal half. A period-like domain, a proline-rich region, and a coiled-coil and leucine-zipper (CC/LZ) domain are within the C-terminal half of the protein3. AIP1 suppresses tumor necrosis factor-α (TNF)- and toll-like receptor (TLR)-induced NF-κB signaling. Additionally, AIP1 inhibits interferon-gamma (IFN-γ)-induced JAK2 pathways3–13. Moreover, mice with a global AIP1 deletion (AIP1-knockout [KO]) exhibit enhanced inflammatory responses in ischemic hindlimb, inflammatory sponge, carotid ligation, atherosclerosis, and graft arteriosclerosis models10, 12, 14, 17. Additional studies suggest that an EC-specific deletion (AIP1-ECKO) also augments inflammation in various animal models14, 18. In the process of studying how AIP1 regulates atherosclerosis and graft arteriosclerosis, we detected a novel isoform of AIP1; therefore, we named the previous isoform AIP1A and the newly discovered isoform AIP1B. This unexpected discovery prompted us to examine the function and regulation of AIP1B.
Previous studies have shown that expression of AIP1A is primarily regulated by epigenetic DNA and histone methylation. DNA methylation in the promoter of a number of genes occurs frequently in prostate tumors, but is rarely found in normal prostate tissues. CpG islands are CpG-rich areas that range from 200 base-pairs to several kilo-bases in length and are usually located near the promoters of highly expressed genes. Notably, these regions are the sites of almost all hypermethylation in human tumors19. DNA methylation triggers a series of downstream events that eventually lead to the formation of compact chromatin structures and reduced accessibility for RNA Polymerase II binding20. AIP1A, which acts as a tumor suppressor, is silenced by DNA methylation in cancers21. AIP1A is also regulated by histone modifications22. Histone methylation can trigger gene activation or inactivation, depending on the target amino acid residues and the extent of the methylation (me1, me2, or me3)23. Among the histone methylation marks, the active tri-methylated lysine 4 in histone H3 (H3K4me3) is enriched at promoter sites and creates an open chromatin structure; therefore, H3K4me3 activates transcription. The tri-methylated lysines, 9 or 27, in histone H3 (H3K9me3, H3K27me3) act as docking sites for transcription repressors and create a compact chromatin structure (heterochromatin); therefore, H3K9me3 and H3K27me3 repress transcription24, 25. H3K27 methylation is primarily methylated by enhancer of zeste homolog 2 (EZH2), which is the catalytic subunit of polycomb repressive complex 2 (PRC2). H3K9 methylation primarily occurs through methyltransferases G9a and GLP24–27. It is known that H3K9 methyltransferases can be recruited to histones by RIF1 and that RIF1 interacts and stabilizes the H3K9 methylation complex28. It is also reported that elevated EZH2 levels are associated with cancer progression. Importantly, EZH2 silences AIP1A expression in human cancers. Furthermore, an association between an EZH2 complex, which contains EED and SUZ12, and the AIP1A gene promoter is found in cancer cells, including prostate cancer, but is absent in normal prostate epithelial cells. However, the mechanism of AIP1A regulation within the vasculature has not been investigated.
Here, we report that in response to inflammation, AIP1A was proteolytically degraded whereas AIP1B was transcriptionally upregulated through increased H3K9 methylation. Moreover, AIP1B was localized to the mitochondria and exhibited opposing vascular functions when compared to AIP1A. AIP1A promoted mitochondrial ROS (mitoROS) production, EC dysfunction, and vascular disease progression.
MATERIALS AND METHODS
The authors declare that all supporting data are available within the article or in the online-only Data Supplement or from the corresponding author on request.
Clinical specimens.
Tissue collection and analysis were approved by Yale University School of Medicine and the New England Organ Bank review boards. Human coronary arteries were obtained from cardiac transplant recipients with chronic rejection that were undergoing re-transplantation, from cardiomyopathy recipients that were undergoing first-time transplantation, and from organ donors without cardiac disease. Our procurement techniques were previously described in detail29. Briefly, the arteries were procured in the operating room. The presence of disease was diagnosed macroscopically by an experienced cardiac surgeon and coded without patient identifiers. The samples were embedded in optimal cutting temperature (OCT) compound and frozen immediately.
Development of the vascular endothelial AIP1B transgenic mouse.
The VE-cadherin promoter in a TA vector was obtained from Dr. Laura Benjamin (Harvard Medical School, Boston, MA)30, 31. The 3′-UTR (untranslated region) from bovine growth hormone (bGH) was cloned downstream of the VE-cadherin promoter to generate a pVE-pA vector. Human AIP1B cDNA with an HA-tag sequence at the 3′-end was inserted into the EcoRI and XbaI restriction enzyme sites between the VE-cadherin promoter and bGH pA to obtain a pVE-AIP1B plasmid30, 31. The plasmid was linearized with XhoI digestion and pronuclear injection was performed at the Yale Transgenic Core. The founder transgenics were identified by polymerase chain reaction (PCR) of tail DNA with a 5′ AIP1B primer and a 3′ HA primer. AIP1B-ECTG transgenic mice were backcrossed with C57BL/6J mice for more than 6 generations before beginning experiments.
Real-time PCR.
Aortae or carotid arteries were cut, immersed in water, briefly centrifuged, and then resuspended in TRIzol reagent (Invitrogen). Total RNA was extracted from tissue sections using the RNeasy Kit (QIAGEN) according to the manufacturer’s instructions. Reverse transcription was performed using random hexamer primers and oligo-dTs according to the Multiscribe RT system protocol (Applied Biosystems). Real-time (RT-PCR) reactions were performed using TaqMan 2× PCR Master Mix, TaqMan PCR reagents, and commercially available TaqMan gene expression probes for mouse cytokines, chemokines, and adhesion molecules or the housekeeping gene, GAPDH. The samples and data were analyzed using an iQ5 machine and its system interface software (Bio-Rad). The expression level of each target gene was normalized to GAPDH, and the results are presented as relative copy numbers.
Cell culture, transfection, cytokines and inhibitors
Human umbilical vein endothelial cells (HUVECs) were obtained from the Yale University Vascular Biology and Therapeutics Tissue Culture Core Facility. Human aortic endothelial cells were isolated from tissue explants. Briefly, the thoracic aorta was gently cleaned of peri-adventitial fat and connective tissue and then cut into five small segments (1.0–1.5 mm thick). The aortic segments were placed on Matrigel (BD Biosciences) and incubated in DMEM media supplemented with 15% fetal bovine serum, 50 μg/ml heparin, 30 μg/ml EC growth factors (ECGS), and penicillin-streptomycin. After sufficient outgrowth of non-fibroblast-like cells was observed, the tissue fragments were removed. At confluence, the cells were passaged using dispase (Worthington) and then cultured for 2 days in culture medium containing D-valine (Sigma-Aldrich) to eliminate fibroblast cells. The subsequent cell passaging was performed with trypsin-EDTA. Immunohistochemical staining of the endothelial monolayer showed strong positive expression of the endothelial marker, von Willebrand factor. For all experiments reported in this study, only passages 3 to 5 of primary cultured cells were used. Commercial siRNA was purchased from Santa Cruz or Ambion, and used at a concentration of 20 μM. The siRNA knockdown was performed as described previously3, 32, and the protocol was modified from the manufacturer’s protocol for Oligofectamine (Invitrogen). For cells in one well of a 6-well plate, 2 μl siRNA and 8 μl Oligofectamine were mixed in OPTIMEM I and incubated at room temperature for 30 minutes. HAECs were cultured to 90% confluence in 6-well plates, followed by transfection with the siRNA-Oligofectamine mixture in OPTIMEM for 12 hours. Regular culture medium was added for the remaining 36 hours of cell culture. Cells were then treated as indicated and harvested. Human recombinant TNF (R&D Systems) was used at 10 ng/ml. mitoTEMPO was purchased from Sigma-Aldrich and ASK1 inhibitor GS444217 was purchased from Gilead Company.
Lipid Binding Assay.
Phosphatidylinositol phosphate (PIP) and membrane lipid strips (Echelon Biosciences) were immersed in blocking buffer (1x PBS, 1% fat free milk, 0.1% Tween-20) for 1 hr. Strips were then probed for 2 hr at 25°C with the indicated GST fusion protein (50 ng/ml) in the presence of an anti-GST antibody (Sigma). Blots were then washed in blocking buffer 3 times for 10 min each and probed with an HRP-conjugated anti-rabbit IgG (GE healthcare) secondary antibody for 30 min, also in blocking buffer. Bound protein was detected using enhanced chemiluminescence (ECL) (GE healthcare).
5’ RACE for the AIP1 transcripts in control and RIF1 knockdown ECs.
HUVECs were transfected with control siRNA or RIF1 siRNA. At 48 h post-transfection, total RNAs were prepared with TRIzol reagent (Ambion, Carlsbad, CA, USA). Next, 5’ rapid amplification of cDNA ends (RACE) was performed according to the manufacturer’s instructions (Invitrogen, AM1700). Briefly, total RNA was treated with calf intestinal phosphatase (CIP) at 37°C for 1 h and extracted with acid-phenol:chloroform (Invitrogen, AM9720), followed by chloroform and precipitation with isopropanol. The purified RNA was treated with Tobacco Acid Pyrophosphatase (TAP) at 37°C for 1 h and ligated with the 5’ adaptor. The ligated RNA was reverse transcribed at 42°C for 1 h. Gene specific nested PCR was performed with nested PCR primers (Outer F:5’-GCTGATGGCGATGAATGAACACTG-3’, Outer R:5’-CTCCTCCTTGGTCTTGGCAC-3’, Inner F:5’-CGCGGATCCGAACACTGCGTTTGCTGGCTTTGATG-3’, Inner R:5’-CGGTACAGGTGGACAGTGAC −3’). The PCR products were analyzed by agarose gel electrophoresis and the bands of interest were extracted for DNA sequencing (Yale DNA Sequencing Core Facility).
ROS detection.
MitoTracker Red CM-H2XROS (MitoSOX, for superoxide) and dihydrorhodamine 123 (DHR123, for H2O2) were used to detect mitoROS. All probes were purchased from Molecular Probes. Cells were treated with TNF (10 ng/ml) for the indicated times. Cells were then loaded with 5 μM of the indicated probe and were incubated at 37°C for 20 min. Next, the cells were immediately detached, and flow cytometry analysis (BD Biosciences) was completed. The fluorescence signal was recorded on the FL1 (green) or FL3 (red) channels and analyzed with BD CellQuest software. ROS in tissue or cell samples were also detected under immunofluorescence microscopy followed by quantifications for mean fluorescence intensity or counting of positive cells.
Immunofluorescence microscopy.
Cells and frozen tissue slides were fixed with 4% paraformaldehyde for 15 min at room temperature, permeabilized with 0.1% Triton X-100 for 5 min, and then blocked in bovine serum albumin for 30 min. Slides were stained for 2 h at room temperature using the appropriate primary antibodies, followed by incubation for 1 h with Alexa Fluor 488- or 594-conjugated secondary antibodies (Molecular Probes). The stained cells were covered with mounting medium containing DAPI for 10 min in a dark environment. Images were captured with a fluorescence microscope (ZEISS).
Preparation of subcellular fractions.
Subcellular HUVEC fractions were prepared as described. To obtain the cytosolic fraction, cells were washed with PBS and resuspended in 50 ⊓l of 250 mM sucrose and 70 mM Tris (pH 7.0) in the presence of a protease inhibitor mixture (Roche Diagnostics Corp., Indianapolis, Indiana, USA). Next, 10 ⊓l of 4 mg/ml digitonin was added, followed by incubation at room temperature for 2 min. Two microliters of cells were stained with trypan blue followed by direct observation with light phase contrast microscopy to verify of 90–95% cell lysis. This approach prepared a cytosolic fraction that was essentially free of mitochondrial contamination33. After centrifugation at 600 × g for 2 min at room temperature, the supernatant was collected as the cytosolic fraction. To prepare the mitochondrial fraction, the cells were washed once with PBS and resuspended in ice-cold hypotonic buffer (10 mM NaCl, 1.5 mM CaCl2, 10 mM Tris-Cl, pH 7.5) in the presence of a protease inhibitor mixture. After a 10 min incubation on ice, MS buffer (210 mM mannitol, 70 mM sucrose, 5 mM EDTA, 5 mM Tris, pH 7.6) was added, and the cells were homogenized with 30 strokes of a Dounce homogenizer. Plasma membrane disruption was monitored by trypan blue staining. After removing the nuclear fraction by two successive centrifugation events at 3000 × g for 10 min, the supernatant was centrifuged at 12,000 × g for 10 min. The pellet was collected as the mitochondrial fraction and resuspended in lysis buffer (50 mM HEPES, pH 7.0, 500 mM NaCl, and 1% Nonidet P-40) that was supplemented with a mixture of protease inhibitors. After measuring protein concentration (Bio-Rad reagents), the fractions were analyzed by western blotting for the indicated proteins.
Western blotting.
For artery and cell culture immunoblots, protein was extracted from homogenized tissues or cells in lysis buffer and boiled in SDS sample buffer for 10 min. Samples with equal protein amounts were separated by SDS-PAGE and transferred to polyvinylidene fluoride membranes (Bio-Rad Laboratories). The membranes were probed with antibodies against AIP1A, as described previously3. Primary antibodies against AIP1A (Min lab), pan-AIP1 (Min lab), FLAG (Sigma), HA (Sigma), and GAPDH (Sigma) were used.
Chromatin immunoprecipitation-qPCR (ChIP-qPCR).
HUVEC cells were washed with PBS and fixed with 1% paraformaldehyde (Electron Microscopy Sciences) for 10 mins while rotating and then stopped with 125 mM Glycine (Sigma) for 5 minutes. After wash with PBS 3 times, cells were scraped and collected. The cell pellet was washed with buffer P1 (0.25% Triton, 10 mM EDTA, 0.5 mM EGTA,10 mM Tris pH7.5), Buffer P2 (200 mM NaCl, 10 mM EDTA, 0.5 mM EGTA, 10 mM Tris pH7.5) and Buffer P3(0.1% SDS, 1 mM EDTA, 1 mM Tris-HCl pH7.6) with 1X complete Protease Inhibitor Cocktail (Roche). Cell pellet then was lysed in 130 μL SDS lysis buffer (50 mM Tris pH 8.0, 10 mM EDTA, 1% SDS, 50 mM PMSF) and was sonicated using an S220 Focused-ultrasonicator (Covaris). The DNA fragment length is between 200 to 600 bp. The fragmented chromatin was diluted in 10x volume ChIP dilution buffer (16.7 mM Tris pH 8.0, 1.1% Triton X-100, 167 mM NaCl) and was centrifuged at 14,000 rpm for 10 min. Aliquot the supernatant into different groups and add beads (Dynabeads Protein G, Invitrogen) pre-bound with antibody. The binding was carried out at 4 °C while rotating overnight. The beads were washed once each with low salt buffer (50 mM Tris pH 8.0, 150 mM NaCl, 0.1% SDS, 0.5% Deoxycholate,1%NP40, 1mM EDTA), high salt buffer (50 mM Tris pH 8.0, 500 mM NaCl, 0.1% SDS, 0.5% Deoxycholate, 1% NP40, 1 mM EDTA), and twice with LiCl wash buffer (50 mM Tris pH 8.0, 250 mM LiCl, 0.5% Deoxycholate, 1% NP40, 1 mM EDTA), Morohashi RIPA buffer (50 mM Tris pH7.5, 150 mM NaCl, 5 mM EDTA, 0.5% NP40, 0.1% SDS), DOC/Triton Buffer (25 mM Tris pH7.5, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 0.5% DOC), and TE buffer (10 mM Tris pH 8.0, 1 mM EDTA). The beads were eluted by 200 ul Direct Elution Buffer (10 mM Tris-HCl pH8, 0.3M NaCl, 5 mM EDTA pH8, 0.5%SDS) and reverse crosslinked overnight at 65C. Then Proteinase K was added and incubated at 55 °C for 3 hr. DNA was extracted by phenol/chloroform and then purified by MinElute PCR Purification kits (QIAGEN). ChIP DNA was analyzed by qPCR and data were normalized to input DNA.
The selected regions were chosen according to the H3K4me3 or Pol II S5 ChIP peaks reported in ENCODE. The qPCR primers were:
Pol II S5: 1A-L (F: GCTCCTTGCAAAGACCTCAG R: AGGAGCAGGACGTCAAACTG)), 1A-S (F: CCTCAAGGGCTCCATCAAG, R: GGGCTCACCTCTCATTGTCC), 1B (F: GGTCAGGAAGGAAACGCATA R: AAGCTCTCCCAGGCTACTCC), GAPDH (F: CTCTGCTCCTCCTGTTCGAC, R: CTAGCCTCCCGGGTTTCTCT).
H3K4me3: 1A-L(F: GATGGAAGAGGCAACAGGAA, R: AGACTCTGCATGGGCTCTGT), 1A-S(F: TCTCCACTCTCAGGGGTGTC, R: AGGTTGAACCACCTCCTCCT), 1B(F: TCTACGCAGACATCCACTGC, R: CAACTCACACTGGGCTTCCT), GAPDH (F: CTCTGCTCCTCCTGTTCGAC, R: CTAGCCTCCCGGGTTTCTCT).
Animal procedures and atherosclerotic lesion analysis.
All of the experiments were approved by the Yale University’s Institutional Animal Care Use Committee. For high fat-diet feeding, at age of 6 weeks, ApoE−/− mice were fed with a Western-type diet containing 1.25% cholesterol (Research Diets, D12108C) for 10 weeks to induce accelerated atherosclerosis. Atherosclerotic lesion analysis was performed as we described previously34. Mice were perfused with phosphate buffered saline (PBS) followed by 4% paraformaldehyde (PFA) and subjected to immunostaining.
Carotid artery ligation.
AIP1B-ECTG mice were bred to a C57BL/6J background for more than 6 generations. Both male and female mice that were 8–10 weeks of age were used to select age- and sex-matched WT and AIP1B-ECTG mice for experiments. After anesthesia, the left external carotid artery was exposed through a midline cervical incision and ligated with a 6–0 silk suture immediately proximal to the bifurcation35–37. For sham operations, a suture was tied around the artery in a non-constricting fashion. The right carotid artery was not ligated and was an internal control. For morphometry and immunohistochemistry experiments, the left and right common carotid arteries were harvested after perfusion fixation with 10% formalin through the left ventricle and immediately embedded in OCT (Tissue-Tek). For medial remodeling studies, the perfusion-fixed vessels were post-fixed overnight in 4% paraformaldehyde and embedded in paraffin for optimal histological analysis of VSMCs and the extracellular matrix. For western blotting, RT-PCR, and enzyme-linked immunosorbent assay (ELISA) studies, the right and left common carotid arteries were perfused with saline, isolated, and snap frozen.
In situ dihydroethidium fluorescence (DHE) was performed as previously described14, 31, 38. Briefly, the unfixed tissues were cut into 10 μM-thick sections, and incubated with 2×10−6 mol/dL DHE at 37°C for 30 min in a light-protected humidified chamber. Pictures were taken from four random areas from five sections per mice using a Kodak digital camera mounted on a light microscope or equipped with fluorescence microscope.
Morphometric analysis.
Morphometric analysis was performed with hematoxylin and eosin (H&E) stained specimen sections that were embedded in OCT using computer-assisted microscopy. The internal elastic lamina perimeter, external elastic lamina (EEL) perimeter, and medial thickness at four quadrants were measured and averaged over 10 separate sections using ImageJ software (National Institutes of Health). The number of nuclei in each medial lamellar unit and the number of medial lamellar units that were continuous around the majority (> 50%) of the circumference were counted in the H&E-stained sections. The transverse VSMC diameter was measured across the nucleus using image analysis software and was averaged from 20 cells in sections that were immunostained for α-SMA10, 37. The medial area that stained positive for α-SMA, elastin Van Gieson, and Sirius red (to measure VSMC contractile protein, extracellular elastin, and extracellular collagen levels, respectively) was calculated using image analyses software. The vessel area measurements of the lumen (within the endothelium), the intima (between the endothelium and the IEL), the media (between the IEL and the EEL), and the whole vessel (within the EEL) were calculated from 5 serial cross-sections that were 150 μm apart. Computer-assisted image analysis and NIH Image 1.60 (http://rsbweb.nih.gov/nih-image/) were used to complete these measurements10, 37.
Statistical analysis.
Statistical analyses were performed using GraphPad Prism 6.0 software (GraphPad software). All figures are representative of at least three experiments unless otherwise noted. Normality of the data (using Shapiro-Wilk test) and the equality of group variance (using Brown-Forsythe test) were performed on all data using SigmaPlot 14. Data are presented as the mean ± SEM values of biological replicates. Comparisons between two groups were performed by unpaired, two tailed t-test, between more than two groups by one-way ANOVA followed by Bonferroni’s post-hoc test or by two-way ANOVA. P values were two-tailed and values < 0.05 were considered to indicate statistical significance. P < 0.05, P < 0.01 and P < 0.001 are designated in all figures with *, **, ***, respectively.
RESULTS
AIP1A was downregulated and AIP1B was increased in diseased aortae.
During our study of AIP1 regulation in atherosclerosis and graft arteriosclerosis, we detected a shorter form of AIP1, named AIP1B, in the diseased vessels. Using antibodies that specifically recognized the N-terminal PH domain (anti-AIP1A) or an internal PER domain (anti-pan-AIP1), we showed that AIP1B lacked the N-terminal PH domain (Fig.1A). AIP1A was the predominant protein form found in healthy aortae, but was drastically reduced in diseased vessels, including those from ApoE-KO mice fed a high-fat diet12 and from human patients with atherosclerosis. In contrast, AIP1B expression was not detectable in healthy vessels, but was increased in the diseased vasculature (Fig.1B). Immunofluorescence staining indicated that AIP1A was present in the healthy aortic endothelium and intima, which was consistent with previous findings that revealed AIP1 expression in vascular endothelial and smooth muscle cells10, 17. AIP1A downregulation was evident in ECs from diseased human coronary arteries that had atheroma and graft rejection (Fig.1C–D, with quantification in Fig.1E). However, AIP1A mRNA was not significantly altered when healthy and diseased vessels were compared (not shown), which suggested that AIP1A was regulated at a post-transcriptional level.
Fig.1. AIP1A was downregulated and AIP1B was upregulated in diseased aortae.
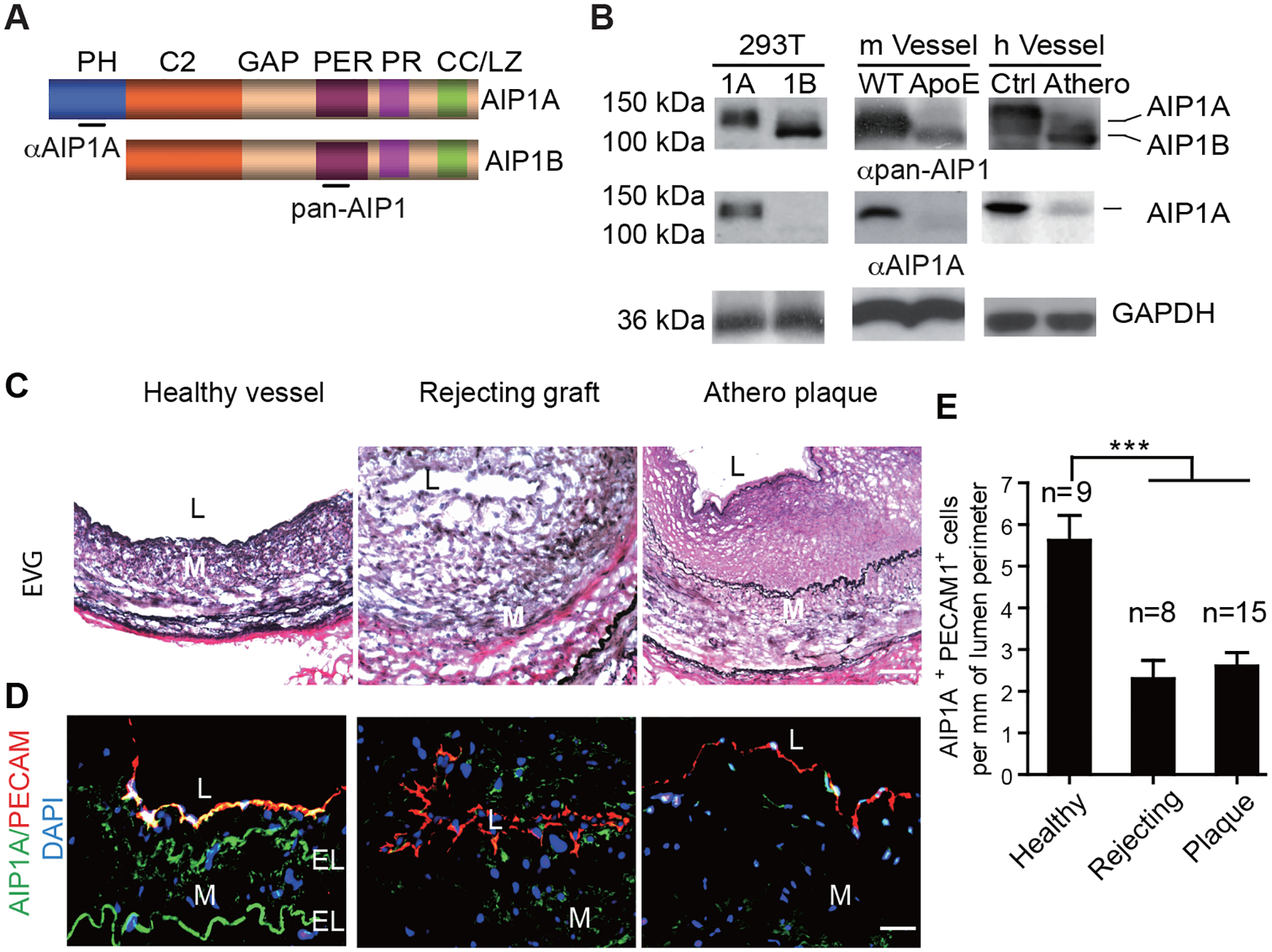
A. A schematic diagram of AIP1 structural domains. PH, PH domain; C2, PKC conserved domain; GAP, GTPase-activating protein domain; PER, period-like domain; PR: proline-rich region; CC/LZ, coiled coil/leucine-zipper domain. Antibodies that recognize the PH domain (αAIP1A) or the PER domain (αpan-AIP1) are indicated. B. Expression of AIP1A and AIP1B in the aorta. 293T: Verification of antibody specificity using lysates from 293T cells that overexpressed AIP1A or AIP1B. m Vessel: Aortae from WT and ApoE−/− mice after 20 weeks on a high-fat diet. h Vessel: human coronary artery specimens from individuals with atheroma or no disease were collected. Cell and tissues lysates were analyzed by western blotting with anti-AIP1A and anti-pan-AIP1 antibodies. n=3. C-E. Human coronary artery specimens with no disease, with graft arteriosclerosis (GA) from chronically rejecting heart allografts, or with atherosclerotic plaques were collected. EVG staining (C) and immunostaining for AIP1A expression with antibodies against AIP1A (green) and the endothelial cell marker, PECAM-1 (red). Nuclei are labelled with DAPI. Vessel lumen (L), media (M) and elastic lamina (EL) are indicated. Representative images are presented in (C) and (D) with quantification in (E). All data are presented as the mean ± SEM from independent clinical specimens of varying number per group as indicated, *** P<0.001, one-way ANOVA followed by Bonferroni’s post-hoc test. Scale bar: 50 μm (C,D).
AIP1A was downregulated by Smurf1-mediated proteolytic degradation in response to inflammation.
To define the mechanism of AIP1A degradation under inflammatory conditions, human umbilical vein ECs (HUVECs and human aortic ECs (HAECs) were treated with the proinflammatory cytokine, tumor necrosis factor-α (TNF). We noticed that AIP1A was drastically decreased in HUVECs after TNF treatment, and that the change occurred concomitant with an increase in AIP1B expression (Fig.2A). However, RT-PCR with AIP1A 5’ UTR-specific primers indicated that AIP1A mRNA levels were not altered (Supplemental Fig.1). These results suggested that AIP1A was regulated at the protein level. Similar results were obtained in HAECs. To further define the mechanism by which TNF induces AIP1A degradation, HUVECs were treated with TNF (10 ng/ml) in the presence of the proteasome inhibitor, MG132. TNF-induced AIP1A downregulation was attenuated by MG132, which suggested that inflammation specifically induced AIP1A degradation (Fig.2A).
Fig.2. AIP1A was degraded by the Smurf1-mediated proteolytic pathway in response to TNF.
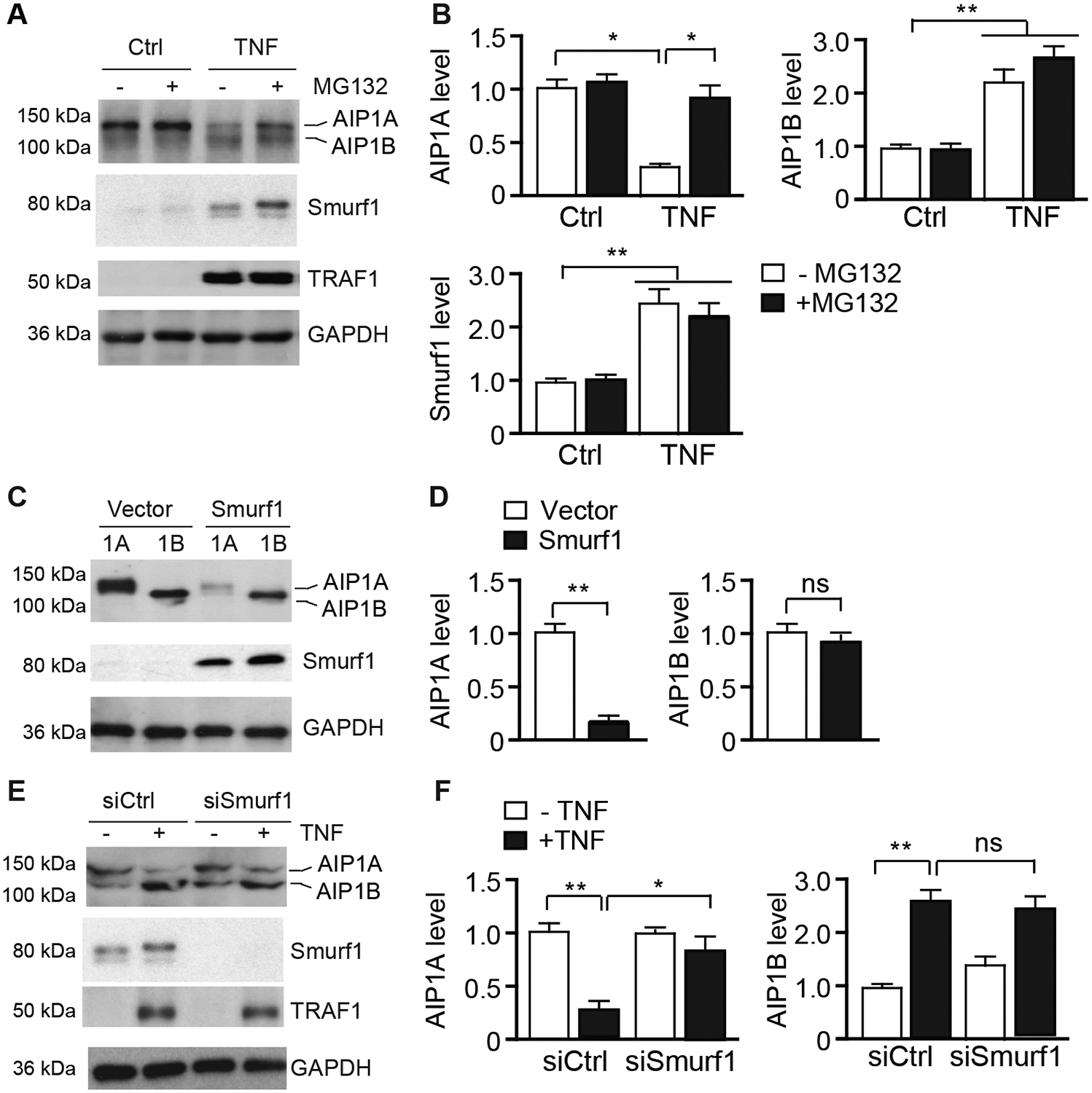
A-B. HUVECs were untreated or treated with TNF (10 ng/ml) for 16 h followed by incubation in the presence or absence of MG132 (20 μM) for 8 h. AIP1, Smurf1, and TNF-inducible TRAF1 proteins were detected by western blotting respective antibodies (A). Protein bands were quantified by densitometry and fold changes are presented by taking untreated group as 1.0 (B). n=3. C-D. HUVECs were transfected with an expression plasmid for AIP1A or AIP1B in the presence or absence of Smurf1 expression plasmid for 48 h. AIP1 expression was determined by western blotting with anit-pan-AIP1 (C). AIP1 protein bands were quantified by densitometry and fold changes are presented by taking vector group as 1.0 (D). n=3. E-F. HUVECs were transfected with control or Smurf1 siRNA for 48 h. Cells were then untreated or treated with TNF (10 ng/ml) for 8 h. AIP1, Smurf1, and TRAF1 proteins were detected by western blotting (E). AIP1 protein bands were quantified by densitometry and fold changes are presented by taking untreated group as 1.0 (F). All data are presented as the mean ± SEM. * P<0.05, ** P<0.01, one-way ANOVA followed by Bonferroni’s post-hoc test (B) or unpaired, two tailed t-test (D,F).
AIP1A is known to be degraded by several proteolytic pathways in a ubiquitination-dependent manner39, 40. Interestingly, it has also been reported that SMAD ubiquitylation regulatory factor-1 (Smurf1), which is a member of the Nedd4-like E3 ligase family, binds to the PH domain of AIP1A and can induce AIP1A degradation40. Moreover, we found that Smurf1 expression in ECs was increased in response to TNF (see Fig.2A). Therefore, we examined if Smurf1 mediated TNF-induced AIP1A degradation in ECs. Consistent with previous reports, overexpression of Smurf1 in HUVECs induced protein degradation of AIP1A, but not of AIP1B (Fig.2C, with quantification in Fig.2D). Conversely, Smurf1 knockdown with siRNA blocked TNF-induced AIP1A degradation but had no effect on AIP1B expression (Fig.2E, with quantification in Fig.2F). These data suggested that TNF-induced AIP1A degradation was regulated via a Smurf1-mediated proteolytic pathway.
AIP1B transcription was epigenetically regulated by RIF1/H3K9 trimethylation.
Although AIP1A mRNA was not altered in response to TNF in HUVECs, the total AIP1 mRNA, which included both AIP1A and AIP1B, was increased by 3-fold in response to TNF (Supplemental Fig.I). These data suggested that AIP1B was induced by TNF at the transcriptional level. It has also been shown that the histone H3K27 methyltransferase, EZH2, silences AIP1A expression in human cancer22. To determine the regulation of AIP1B transcription in ECs, we examined the expression of histone modifications in HUVECs that occurred in response to TNF. We found that EZH2 and H3K27me3 were not affected by TNF in HUVECs. Interestingly, we observed that TNF induced a drastic reduction in RIF1 and H3K9me3 in ECs. Notably, the reduced expression of RIF1 and H3K9me3, which is normally functions as transcriptional repressor, was accompanied by increased AIP1B protein expression (Fig.3A with quantifications in 3B). To determine if AIP1B was regulated by a RIF1/H3K9 trimethylation complex in vascular ECs, we examined the effects of RIF1 siRNA and H4K9 methyltransferase inhibitors on AIP1B expression. RIF1 knockdown or inhibition of H4K9 methyltransferase by BIX-01294 (5 μM for 24 h) in ECs significantly increased AIP1B expression (Fig.3C–D with quantifications in 3E). In contrast to the TNF treatment, AIP1A was not altered by RIF1 knockdown or H4K9 methyltransferase inhibition. These data indicated that TNF, but not RIF1 knockdown, induced AIP1A protein degradation. We also examined effects of other proinflammatory cytokines on AIP1 expression. Results showed that TNF and IL-1β, but not IFN-γ or IL-6, induced downregulation of AIP1A, RIF1 and H3K9me3 concomitant with upregulation of AIP1B (Supplemental Fig.II). H3K9 methyltransferase can directly bind to NF-κB41, therefore it is possible that TNF and IL-1β-activated NF-κB signaling is involved in the regulation of AIP1 expression. In contrast to AIP1A, TNF-induced reduction of RIF1 was not affected by Smurf1 siRNA (Supplemental Fig.III). How TNF induces RIF1 downregulation needs more investigation.
Fig.3. AIP1B transcription was epigenetically regulated by RIF1/H3K9 methylation.
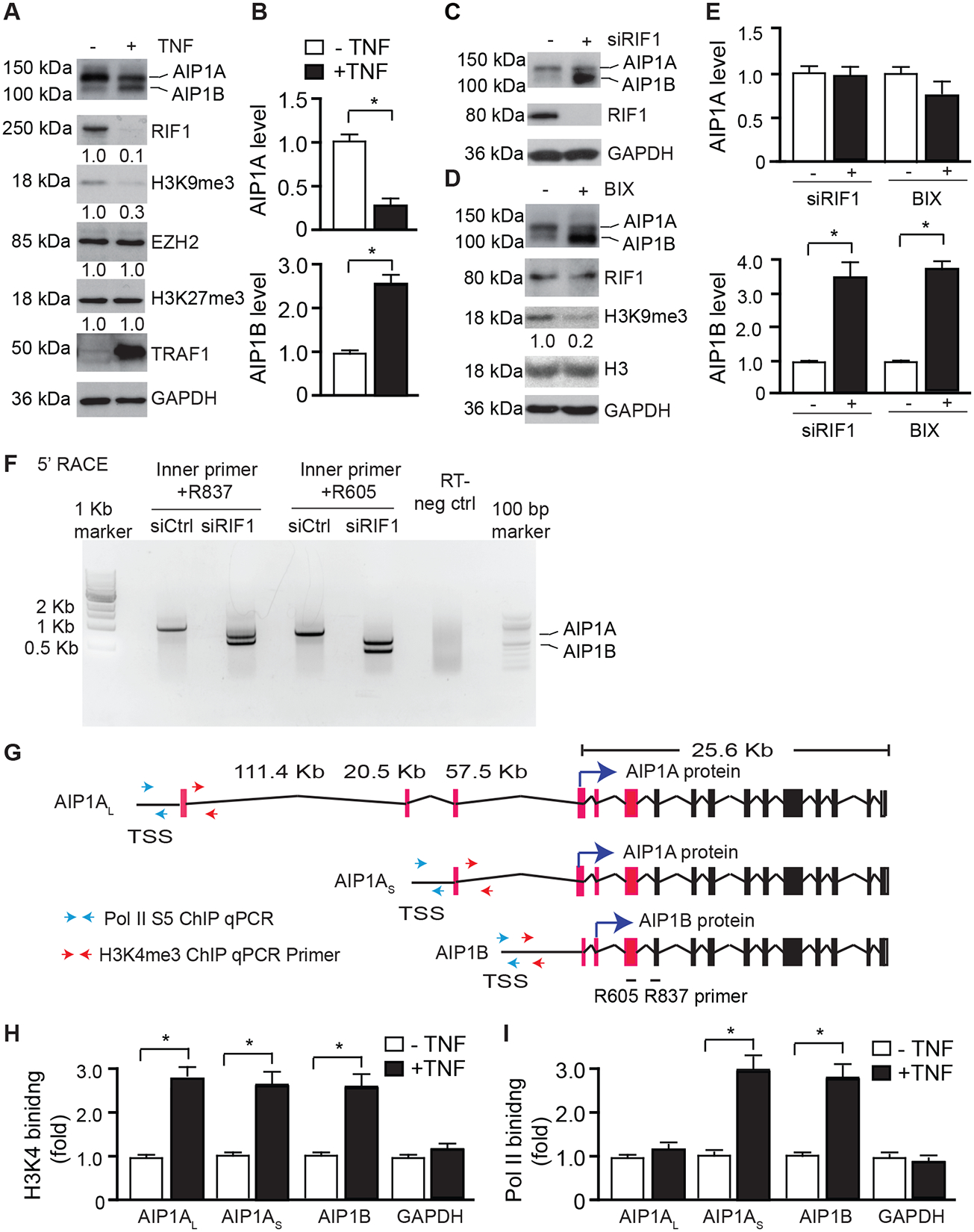
A-B. TNF induced downregulation of RIF1/H3K9me3. HUVECs were untreated or treated with TNF (10 ng/ml) for 24 h. AIP1, EZH2/H3K27me3, and RIF1/H3K9me3 were detected by western blotting with the respective antibodies (A). AIP1 protein bands were quantified by densitometry and fold changes are presented by taking untreated group as 1.0 (B). n=3. C-E. RIF1/H3K9me3 repressed AIP1B expression. HUVECs were transfected with control or RIF1 siRNA for 48 h (C), or treated with the H3K9 methyltransferase inhibitor, BIX-01294 (5 μM for 24 h (D). AIP1, RIF1/H3K9me3, and total H3 were detected by western blotting with the respective antibodies. AIP1 protein bands were quantified by densitometry and fold changes are presented by taking control group as 1.0. n=3. F. RIF1 repressed AIP1B transcription. HUVECs were transfected with control or RIF1 siRNA for 48 h. The 5’ RACE was performed with AIP1-specific nested PCR with two sets of internal reverse primers (R837 and R605). The PCR products were analyzed by agarose gel electrophoresis, followed by extraction for DNA sequencing. G. Diagram of the AIP1 transcripts. Exon sequences from the 5’ RACE are depicted in red. Potential translational start sites for AIP1A and AIP1B proteins are indicated by blue arrows. Paired primers near the TSS used for H3K4me3 and Pol II ChIP assays (H-I) are indicated by red and green arrows, respectively. H-I. ChIP assay for the promoter occupancy by H3K4me3 and Pol II phosphor-Ser5 (Pol II S5). HUVECs were untreated or treated with TNF (10 ng/ml) for 24 h and genomic DNAs were used for ChIP assays with antibodies against H3K4me3 and Pol II S5 followed by qPCR with selected primers near the TSS regions of AIP1 transcripts. GAPDH transcript was used as an internal control. The specific binding was quantified by normalization with each input and fold changes are presented by taking untreated group as 1.0. n=3. All data are presented as the mean ± SEM. ** P<0.01, ** P<0.001, unpaired, two tailed t-test.
To determine if RIF1/H3K9me3 indeed regulated AIP1B at a transcriptional level, we performed 5’ rapid amplification of cDNA ends (RACE) in human ECs that were transfected with control or RIF1 siRNA. In control ECs, we detected a long transcript (AIP1AL) that could encode AIP1A. However, in RIF1 knockdown ECs, the long transcript disappeared and two different transcripts were present (Fig.3F). DNA sequence analyses indicated that one transcript (AIP1AS) encoded both AIP1A and AIP1B, and the other one (AIP1B) only encoded AIP1B (Fig.3G). Moreover, TNF-treated HUVECs expressed the same AIP1 transcripts AIP1AS and AIP1B as those detected in the RIF1KD cells. More importantly, expression of the AIP1B transcript was induced in atherosclerotic aortae (ApoE−/− mice) compared to aortae from WT after 20 weeks on high-fat diet (Supplemental Fig.IV A–C).
These data suggested that AIP1B expression might utilize an alternative promoter. We searched the Encyclopedia of DNA Elements (ENCODE) for H3K9me3 ChIP-seq results from HUVECs and found that two H3K9me3 peaks were detected at the AIP1 locus. One peak was located at the 3’ terminus of the AIP1 locus and the other peak was located at the transcriptional initiation site (TSS) of AIP1. H3K4me3 peaks are known to be highly enriched at active promoters near the TSS and are positively correlated with transcription. Interestingly, several H3K4me3 peaks were detected within the AIP1B upstream region (Supplemental Fig.IV D). To determine whether or not H3K4me3 regulates AIP1B expression, we performed ChIP-PCR analyses with specific primers designed within the predicted promoter region. Specifically, we designed the ChIP PCR primers for the H3K4me3- and Pol II-binding site from the public database, and performed the ChIP-PCR using anti-H3K4me3 (an indicative for an open promoter/enhancer region) and anti-Pol II S5 (an active form of Pol II, indicate of active transcription initiation site) (Fig.3G). Results showed that TNF significantly increased the occupancy of H3K4me3 on the AIP1AL, AIP1AS and AIP1B promoters. TNF also significantly increased the Pol II binding to the AIP1AS and AIP1B promoters. However, TNF had no effect on the binding of Pol II to the AIP1AL promoter (Figure 3H–I). Taken together, these data suggest that a potential promoter is located at upstream of AIP1B TSS region; TNF regulates AIP1B expression by releasing RIF1/K4K9me3 from, while enhancing the bindings of H3K4me3/Pol II to, the AIP1B enhancer/promoter regions.
AIP1B was localized to the mitochondria, which augmented TNF-induced ROS production and EC activation.
Previously, we showed that AIP1A binds to PI4P via its PH domain9. PI4P is a lipid component that localizes proteins to the cytoplasmic membrane and early endocytic vesicles42. To investigate the mechanism by which AIP1B enhanced EC activation and immunogenicity, we performed a membrane lipid strip assay using the recombinant proteins, GST-AIP1A and GST-AIP1B9. As a control, AIP1A binding to PI4P and AIP1B binding to sulfatide, which we previously reported for the C2 domain of AIP1, were evaluated18. To our surprise, AIP1B exhibited the strongest binding to cardiolipin, a mitochondrial lipid (Fig.4A–B). We then examined the cellular localization of endogenous AIP1B by cellular fractionation of RIF1-knockdown ECs. Consistently, AIP1B induced by RIF1 knockdown was found in the mitochondrial fraction (Fig.4C).
Fig.4. AIP1B localized to the mitochondria where it augmented TNF-induced ROS production and EC activation.
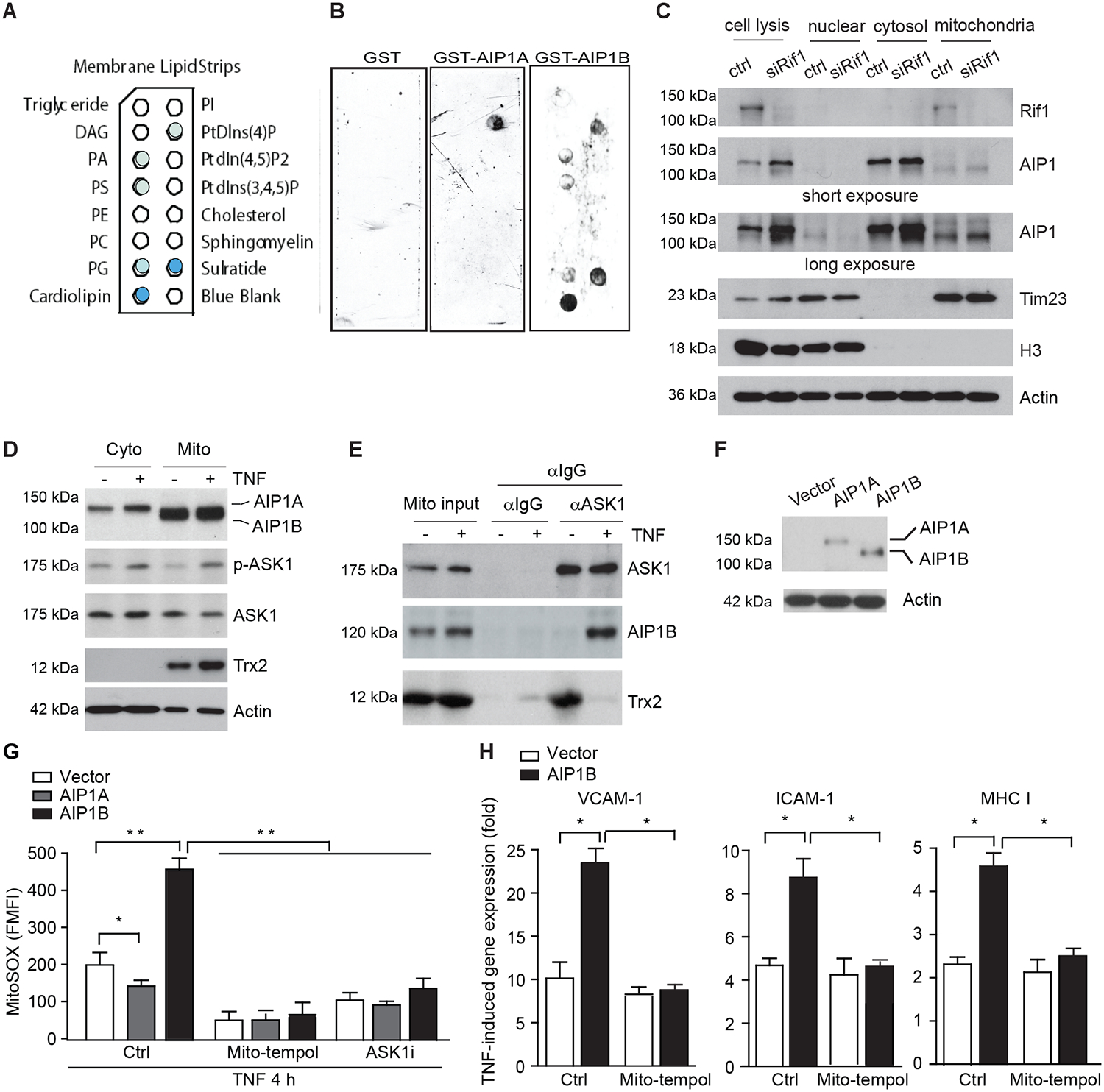
A-B. AIP1B bound to the mitochondrial lipid, cardiolipin, in a membrane lipid strip assay that used recombinant GST, GST-AIP1A, and GST-AIP1B. The identity of each lipid spot (A) and images of the strip (B) are presented. The positive spots are marked by solid colors where cardiolipin exhibited the strongest binding. n=2. C. Endogenous AIPB localized to the mitochondria. HUVECs were transfected with control or RIF1 siRNA. Cellular fractionation was performed to isolate nuclear, cytosolic, and mitochondrial fractions. Levels of RIF1, AIP1, the mitochondrial marker, Tim23, and the nuclear marker, histone H3, were determined by western blotting. n=4. D-E. RIF1 siRNA-transfected HUVECs were untreated or treated with TNF (10 ng/ml for 4 h). (D) Cytoplasm and mitochondrial fractions were isolated and phosphor-ASK1 (p-T845) and protein expression were determined by immunoblotting. (E) Mitochondrial fractions were subjected to co-immunoprecipitation assay with anti-ASK1 followed by immunoblotting with anti-ASK1 and anti-Trx2. n=2. F-H. AIP1B enhanced ROS production and inflammation. HUVECs were transfected with siRNA against the AIP1 3’UTR to knockdown endogenous AIP1 and then infected with lentivirus expressing EGFP (Vector), AIP1A or AIPB. (F) Western blotting with anti-pan-AIP1. (G) Cells were pre-treated with mitoROS scavenger mito-tempol (10 μM) or ASK1 inhibitor (10 μM) for 1 h, followed by TNF treatment for 4 h. Intracellular ROS were detected by flow cytometry with a specific probe MitoSOX. (H) Cells were treated with TNF (10 ng/ml) in the absence (Ctrl) or presence of mito-tempol (10 μM) for 12 h, and TNF-induced VCAM-1, ICAM-1 and MHC class I expression was measured with RT-PCR that was normalized to GAPDH. TNF-induced fold changes compared to the untreated controls are presented. n=3. All data are presented as the mean ± SEM. * P<0.05, ** P<0.01, two-way ANOVA (G) or unpaired, two tailed t-test (H).
We have previously shown that AIP1A in the cytoplasm facilitates TNF-induced ASK1 signaling3. Interestingly, ASK1 can be detected in the mitochondria where it is suppressed by redox protein thioredoxin-2 (Trx2) but is activated in response to stresses33. We determined if AIP1B associated with mitochondrial ASK1 to augment TNF-induced ASK1 activation. To this end, RIF1 siRNA-transfected HUVECs were untreated or treated with TNF (10 ng/ml for 4 h). As reported previously, ASK1 was detected in both cytosolic and mitochondria, and TNF induced ASK1 activation in both compartments (Fig.4D). Co-immunoprecipitation assay with anti-ASK1 showed that Trx2 and ASK1 formed a complex in resting ECs, but the Trx2-ASK1 complex was disrupted in response TNF. However, AIP1B bound to ASK1 in TNF-treated ECs but not in resting EC (Fig.4E). These results suggested that AIP1B formed a complex with ASK1 in the mitochondria in response to TNF.
Since mitochondrial ASK1 directly induces mitochondrial ROS production38, we examined if AIP1B augments ASK1-mediated ROS accumulation. To this end, HUVECs were transfected with siRNA against the AIP1 3’UTR to knockdown total endogenous AIP1, and AIP1A and AIP1B was then individually re-expressed by lentiviruses (Fig.4F). Consistent with our previous report that AIP1A inhibited NOX2-dependent cytosolic ROS generation and subsequently attenuated mitoROS14, AIP1A re-expression reduced TNF-induced mtROS using MitoSOX as a mitoROS probe. However, AIP1B enhanced TNF-induced mitoROS production in ECs. Moreover, AIP1B-induced ROS were blocked by the mitochondrial ROS scavenger mito-tempol and by an ASK1 inhibitor38(Fig.4G). Taken together, these data suggest that AIP1B is localized in mitochondria and increased mitochondrial ASK1-mediated ROS in EC.
AIP1A is a potent anti-inflammatory protein that suppresses TNF-induced EC inflammation10, 17. Based on the shift from AIP1A to AIP1B in chronic inflammation, we reasoned that AIP1A and AIP1B have different functions in inflammatory signaling. To test this, we determined the effects of AIP1A and AIP1B on TNF-induced EC activation. More specifically, the gene expression of inflammatory molecules was measured with RT-PCR. Under inflammatory conditions, AIP1A expression strongly suppressed, while AIP1B strongly augmented, TNF-induced the gene expression of several proinflammatory molecules including vascular cell adhesion molecule 1 (VCAM-1), intracellular adhesion molecule 1 (ICAM-1), and major histocompatibility (MHC) class I (Supplemental Fig.V). Furthermore, mitoROS scavenger mito-tempol diminished AIP1B-augmented expression of these proinflammatory molecules (Fig.4H). Collectively, these data indicated that AIP1B localized to the mitochondria where it promoted TNF-induced ROS production and EC activation.
An AIP1B transgene increased oxidative stress and vascular remodeling in mouse models.
To determine the in vivo function of AIP1B, we generated EC-specific AIP1B transgenic mice (AIP1B-ECTG) that expressed human AIP1B (with an HA-tag at the C-terminus) and were driven by an EC-specific VE-cadherin promoter30 (Fig.5A). AIP1BETG under wildtype (WT) (AIP1+/+) background were backcrossed to C57BL/6J for 6 generations. The AIP1B transgene was specifically detected in the vascular endothelium by immunostaining with anti-HA. Western blotting with anti-AIP1 showed that AIP1B protein levels from the transgene mice were similar to endogenous AIP1A levels (Fig.5B–C), which mimicked the RIF1 knockdown in HUVECs.
Fig.5. The AIP1B transgene increased carotid ligation-induced oxidative stress and inflammation.
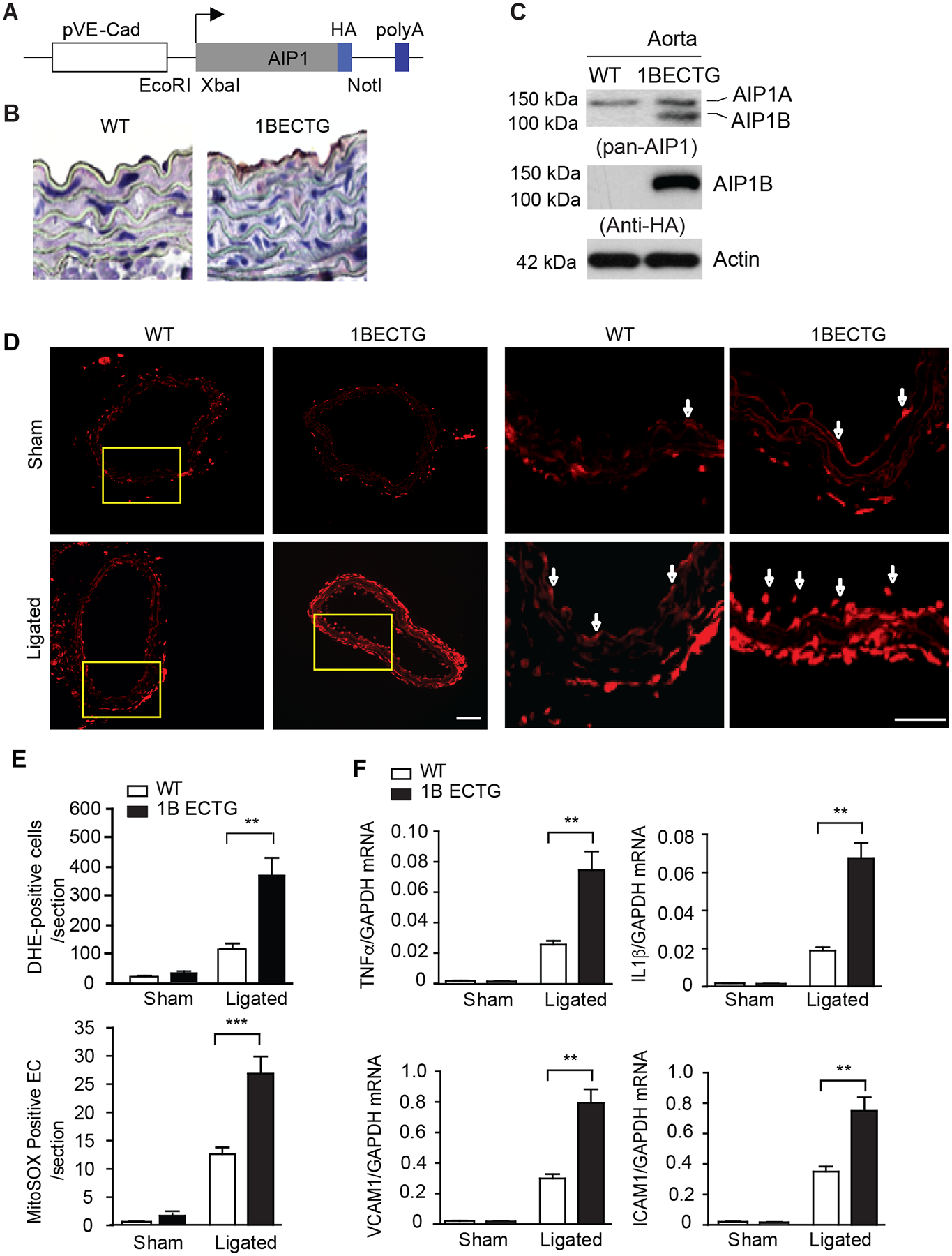
A-C. Characterization of AIP1B-ECTG mice. (A) A schematic of the transgenic vector for HA-tagged human AIP1B under the EC-specific VE-cadherin gene promoter. Restriction sites for cloning XbaI and NotI are indicated. (B) AIP1B expression in the aorta. Representative mouse aortae from WT and AIP1B-ECTG mice were collected as frozen sections, and the transgene was detected by immunohistochemistry with an anti-HA antibody. (C) Aortic AIP1 expression was detected by western blotting with anti-pan-AIP1 and anti-HA antibodies. n=2. D-F. ROS production and inflammation in AIP1B-ECTG mice. Carotid arteries and tissue from WT and AIP1B-ECTG mice with complete ligation near the carotid bifurcation on the left common carotid artery were harvested at day 3 post-ligation. Oxidative stress in the common carotid arteries was determined by in situ detection of superoxide with MitoSOX fluorescence. Representative images are shown in (D) with high magnification images of the boxed area on the right. Quantification of MitoSOX positive cells/sections are presented in (E). n=4. (F) Gene expression of inflammatory molecules was determined by RT-PCR with normalization to GAPDH. Relative expression levels are presented. n=6. All data are presented as the mean ± SEM. ** P<0.01, *** P<0.001, unpaired, two tailed t-test. Scale bar: 200 μm (D).
The carotid ligation model is characterized by neointimal hyperplasia, and the role of ECs in limiting inflammation and remodeling has been well characterized35–37. To determine the function of AIP1 in vascular remodeling, left common carotid artery (LCA) ligation in WT and AIP1B-ECTG mice was performed. The mouse LCA was ligated before the carotid bifurcations of the external, internal carotid, and occipital arteries. The right common carotid artery (RCA) remained un-ligated and typically sections at 200–800 μm from the ligation were examined (Supplemental Fig.VI). Due to the effect of AIP1B on ROS generation and EC inflammation in vitro, we first measured ROS production in the arteries in response to ligation. At day 3 post-ligation, tissue oxidative stress in the common carotid arteries was evaluated by detecting superoxide with dihydroethidium (DHE) fluorescence in situ. Consistent with the in vitro data that AIP1B was induced to associate with ASK1 activation, basal vascular ROS was not affected by the AIP1B transgene. Ligation increased DHE+ staining, which was strongly enhanced by the AIP1B transgene. DHE+ staining appeared throughout the vessel (Fig.5D, with quantification in Fig.5E). Gene expression of proinflammatory cytokines and EC activation markers were also measured by RT-PCR. The AIP1B transgene had no effect on basal inflammation under the WT background. However, the AIP1B transgene significantly augmented ligation-induced gene expression of TNF-α, IL-β, VCAM-1 and ICAM-1 (Fig.5F).
In response to complete ligation of the vessel near the carotid bifurcation, we found that medial VSMCs rapidly proliferated and that the proliferation was followed by extensive neointima formation along the endothelial lining43. At 3–4 weeks post-ligation, luminal narrowing and vascular luminal remodeling within the LCA that occurred 1–2 mm from the ligation site was visualized. The same vascular remodeling was not observed in the un-ligated RCA. Histological and morphometric analyses indicated that the luminal vessel area was reduced by 50% with neointima formation occurring at 3 weeks in WT vessels. Endothelial deletion of AIP1 significantly enhanced neointima formation and luminal narrowing (90% at 3 weeks) without significantly altering the overall vessel area (Fig.6A, with quantification in Fig.6B for male mice). The AIP1B transgene also augmented vascular remodeling in female mice despite overall reduced responses compared to male mice (Supplemental Fig.VII). These data suggested that AIP1B enhanced oxidative stress and that inflammation promoted vascular remodeling.
Fig.6. The AIP1B transgene promoted vascular remodeling in mouse models.
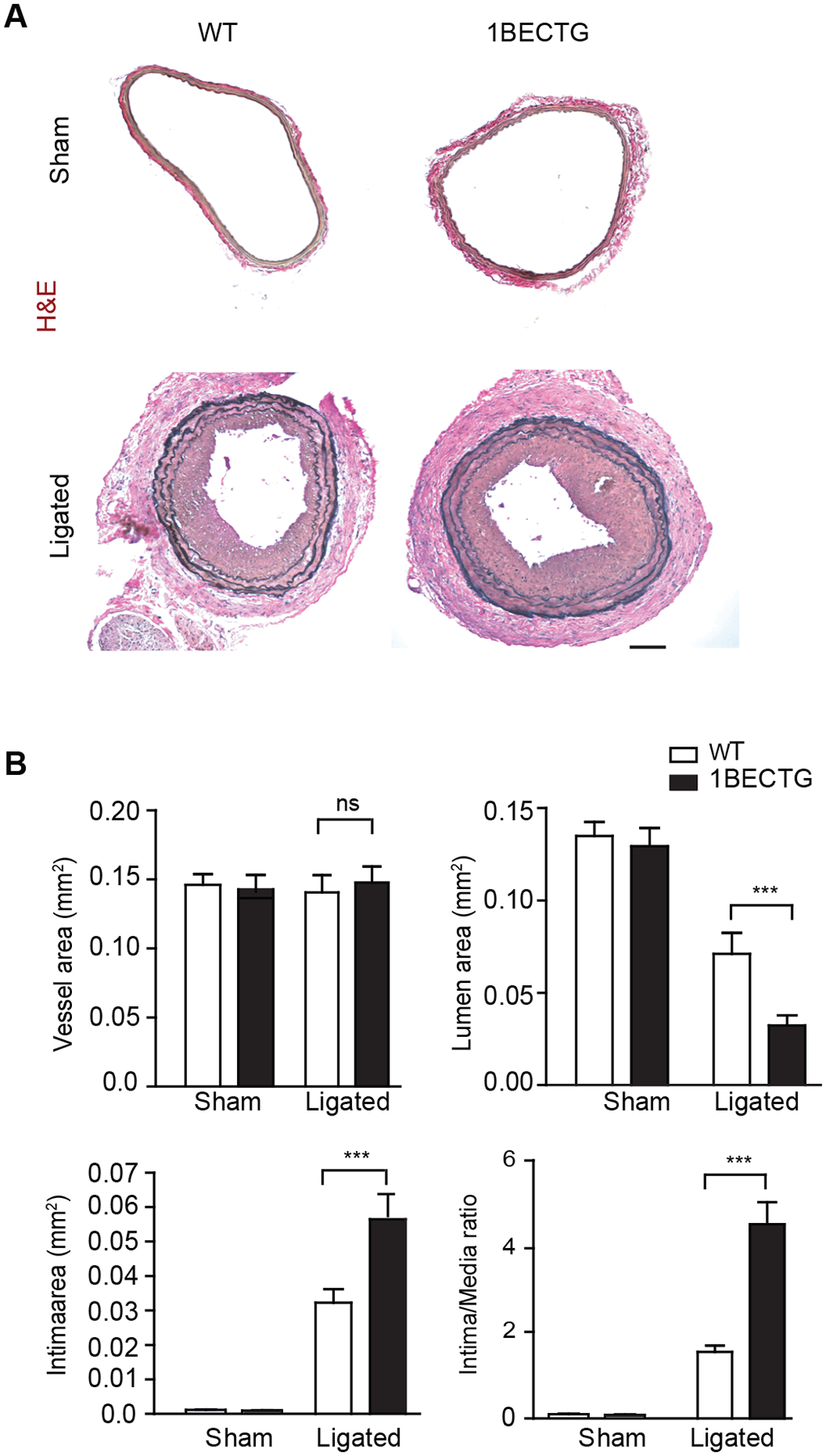
Carotid arteries and tissue from WT or AIP1B-ECTG mice with complete ligation near the carotid bifurcation on the left common carotid artery were harvested at 3 weeks post-ligation. A. Histological analysis of artery cross sections with H&E staining. Representative photomicrographs are presented for sections at 500 μm from the ligation site. B. Morphometric assessment of the whole vessel (within the EEL), the artery lumen (within the endothelium), the intima (between the endothelium and the IEL), the media (between the IEL and the EEL). The ratio of the intima to the media was calculated from 5 serial cross sections from n=6 male mice. All data are presented as the mean ± SEM. ns: non-significant; *** P<0.001, unpaired, two tailed t-test. Scale bar: 200 μm (A).
DISCUSSION
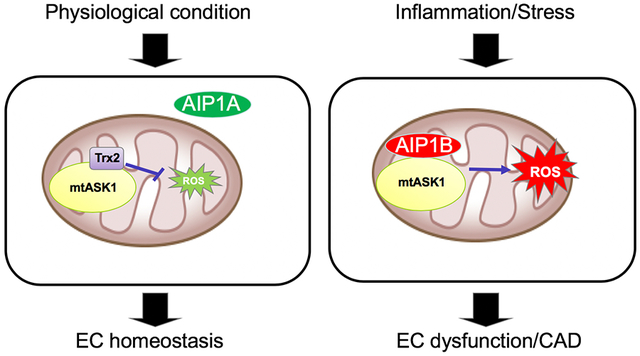
In the present study, we identified a new AIP1 isoform, AIP1B, which lacks an N-terminal PH domain when compared to the previously discovered isoform, AIP1A. In response to the inflammatory cytokine, TNF, increased Smurf1 signaling in ECs mediated the proteolytic degradation of AIP1A via the PH domain. Interestingly, TNF also induced the downregulation of RIF1, which is a complex of H3K9me3 transferase. Moreover, after TNF treatment, RIF1 knockdown or inhibition of H3K9me3 transferase induced AIP1B transcription and AIP1B protein expression in ECs. Contrary to the anti-inflammatory and anti-oxidant function of the AIP1A isoform, AIP1B localized to the mitochondria and enhanced TNF-induced mitoROS generation and EC activation. Consistent with these in vitro data, AIP1A was decreased and AIP1B was increased in diseased aortae that presented with atherosclerotic plaques and graft arteriosclerosis. Importantly, the EC-specific AIP1B transgene in mice augmented mitoROS production, EC dysfunction, and neointima formation in vascular remodeling models. We propose that a shift from an anti-inflammatory AIP1A to a pro-inflammatory AIP1B leads to chronic inflammation and vascular diseases (Fig.7: Model for the AIP1 isoform switch in vascular disease).
Fig.7. Model for the AIP1 isoform switch in vascular disease.

Cytosolic AIP1A forms a complex with NOX2 via its PH domain to block the formation of an active NOX2 complex. In contrast, AIP1B localizes to the mitochondria and enhances TNF-induced mitoROS generation. CVD risk factors and inflammation reduce AIP1A through Smurf1-mediated proteolytic degradation. AIP1B is normally suppressed by RIF1/H3K9me3, but this suppression is released by inflammation, which leads to a shift from an AIP1A to an AIP1B dominated-state in endothelial cells. A shift from AIP1A to AIP1B under chronic inflammation promotes enhanced ROS production and CVD progression.
AIP1A, which acts as a tumor suppressor gene, is silenced by DNA methylation in cancer21. AIP1A is also regulated by histone modifications. Specifically, EZH2-mediated H3K27 methylation represses AIP1A expression in several human cancers22, 44. Moreover, an EZH2 complex associates with the AIP1A gene in cancer cells, but not in normal prostatic epithelial cells. Our current data suggests that AIP1A expression in vascular ECs is not regulated at a transcriptional level. Instead, protein levels are regulated by a proteolytic degradation pathway in response to inflammatory cytokines. Specifically, TNF-induced Smurf1 expression via the PH domain of AIP1A mediates the degradation of anti-inflammatory AIP1A. In contrast to AIP1A, we found that AIP1B was primary regulated at the transcriptional level by cytokines. Our data indicated that TNF reduced RIF1/H3K9me3 but had no effect on EZH2/H3K27me3. H3K9 trimethylation acts as a marker for gene inactivation, but the role of RIF1/H3K9 trimethylation in the vasculature is unknown24–27. We unexpectedly uncovered that AIP1B was epigenetically suppressed by the RIF1/H3K9 methyltransferase complex. Therefore, knockdown of RIF1, which is an adaptor protein that recruits H3K9 methyltransferase to histones, or inhibition of H3K9 trimethylation promoted AIP1B expression in vascular ECs. Furthermore, TNF, via downregulation of RIF1, enhanced AIP1B expression in vascular ECs. In addition to suppressing AIP1B, H3K9 trimethylation has also been reported to repress TNF gene expression and proinflammatory NF-κB signaling45. Collectively, these studies support a novel function for H3K9 trimethylation in the epigenetic regulation of inflammation and vascular diseases.
There are several possibilities that AIP1B could be generated: a limited proteolysis at the N-terminus of AIP1A, an alternatively splicing from the AIP1A mRNA, an alternative translation start site or/and a transcript from an independent promoter. We have provided the following evidence to support that AIP1B is transcribed from its own promoter. 1) The AIP1A transcript is detected in resting ECs. However, the AIP1B transcript, but not the AIP1A transcript, are strongly induced in RIF1 knockdown or TNF-treated ECs. 2) Based on the DNA sequence analyses of the 5’ RACE products, AIP1B is a distinct transcript from AIP1A and the AIP1B 5’ UTR is part of the exon within the AIP1A transcript. Therefore, AIP1B is unlikely generated by an alternative splicing. 3) It is known that histone methylation (such as H3K9me3) plays a critical role in regulating alternative promoter usage46, 47. HUVEC ChIP data showed that there were both H3K9me3 and H3K4me3 peaks at the AIP1 locus. Moreover, our ChIP-PCR analyses by H3K4me3 and Pol II support that AIP1B contains a promoter near its TSS region. Further studies need to characterize the AIP1B promoter activity and regulation under inflammation.
Although AIP1B can be detected in mitochondrial fractions isolated from resting EC, it is unclear how AIP1B is translocated into the mitochondria and how AIP1B activates mitoROS production. Two types of mitochondrial targeting signal have been described: presequences and internal signals48. The C2 domain of AIP1B contains two lysine clusters and could potentially function as a mitochondrial targeting signal48. Moreover, AIP1B via its C2 domain bound to the mitochondrial lipid, cardiolipin, in the in vitro lipid strip assay. Given that AIP1A has an additional N-terminal PH domain, it is likely that the PH domain preferentially binds to PI4P to localize AIP1A to the cytoplasmic membrane and endosomes. Alternatively, the extra PH domain may also block the accessibility of the C2 domain of AIP1A for mitochondrial localization as AIP1A forms a closed conformation by intramolecular interactions between the N-terminal and C-terminal domains3. It is conceivable that deletion of the PH domain leads to exposure of the C2 domain which can bind to cardiolipin and targets AIP1B to mitochondria.
ROS generation in the cytosol occurs primarily through the NADPH oxygenases (NOXs), which catalyze the oxidation of NADPH to NADP+ or NADH to NAD+ and reduce molecular oxygen to the reactive intermediate, superoxide49, 50. Our recent finding indicates that AIP1A is an endogenous protein suppressor of NOX enzymes in ECs. Specifically, AIP1 via its proline-rich region binds to the SH3 domain of the cytosolic subunit, p47phox, to disrupt the formation of an active NOX2 complex, which then attenuates ROS production14. TNF also induces mitochondria-derived ROS51, which subsequently contribute to EC dysfunction and the progression of autoinflammatory disorders and vascular diseases34, 52. We have recently shown that AIP1-interacting kinase (ASK1) can be targeted to the mitochondria3. Furthermore, mitochondrial ASK1 (mtASK1) directly induces mitochondrial dysfunction, which is characterized by reduced respiration, reduced ATP production, and increased ROS production38. Interestingly, the C2 domain is required for the binding and activation of ASK13. It is plausible that AIP1B via its C2 domain binds to and activates mtASK1 to mediate mitoROS production and proinflammatory EC phenotypes (Fig.7).
Mice with an AIP1A global or EC-specific deletion (AIP1-KO and AIP1-ECKO) exhibit enhanced inflammatory angiogenesis13, 18, high-fat diet-induced atherosclerosis12, graft arteriosclerosis10, and neointima formation in the carotid ligation model. These phenotypes in AIP1-KO and AIP1-ECKO mice are largely attributed to enhanced endothelial activation. We generated an EC-specific transgenic AIP1B mouse that expressed AIP1B and was driven by an EC-specific VE-cadherin promoter. The role of the AIP1B transgene in vascular remodeling was first tested in a carotid ligation model under a WT background with normal AIP1 expression (AIP1+/+:AIP1B-ECTG). Aortae from the AIP1B-ECTG mice had normal structures and typical basal vascular function under physiological conditions. However, the AIP1B transgene drastically enhanced ROS production, EC inflammation, and vascular remodeling in the mouse model. This phenotype was similar to what we previously observed in AIP1A deletion mice, which suggests that loss of AIP1A and gain of AIP1B converge at a common pathway: EC dysfunction. AIP1A is a strong anti-inflammatory adaptor molecule that suppresses multiple inflammatory signaling pathways and inhibits cytosolic ROS production14. Current data now shows that AIP1B enhanced TNF-induced ROS production and EC inflammation in the presence (expression of AIP1B in WT EC) or absence of AIP1A (reconstitution of AIP1B in AIP1-KO EC). These results suggest that AIP1B functions independent of AIP1A (i.e., AIP1B does not function as a dominant negative by suppressing AIP1A activity). We postulate that the AIP1 isoform switch from AIP1A to AIP1B increases the production of both cytosolic and mitoROS to synergistically induce EC activation and cardiovascular disease (CVD) (Fig.7). We will test this model in vivo by expressing an AIP1B transgene in the absence of AIP1A (AIP1−/−:AIP1B-ECTG). Furthermore, AIP1+/+:AIP1B-ECTG, AIP1+/+:ECTG, and AIP1−/−:AIP1B-ECTG mice may represent an early stage, a mid-stage, and a late stage of vascular disease, respectively. These models will allow us to define the role of the AIP1 isoform switch during vascular disease progression. The AIP1 (DAB2IP) gene has been implicated in several human CVDs including abdominal aortic aneurysm, peripheral vascular disease, the early onset of myocardial infarction, and pulmonary embolism15. AIP1B-ECTG mice provide an ideal in vivo systems to determine the role of AIP1 isoforms in cardiovascular diseases.
Supplementary Material
HIGHLIGHTS.
A new AIP1 isoform named AIP1B lacks an N-terminal PH domain when comparted to AIP1A and is resistant to TNF-induced proteolytic degradation.
The RIF1/H3K9me3 transferase complex suppresses AIP1B transcription, and TNF releases the repression.
AIP1B localizes to the mitochondria and enhances TNF-induced mitoROS generation and EC activation.
An EC-specific AIP1B transgene in mice augments mitoROS production, EC dysfunction, and neointima formation in vascular remodeling models.
TWEET.
A new AIP1 isoform AIP1B is epigenetically regulated by an RIF1/H3K9me3 complex, and localizes in mitochondria to augment mitochondrial ROS generation, endothelial dysfunction and vascular disease.
ACKNOWLEDGMENTS
b) Sources of Funding: This work was supported by NIH grants HL115148 and R01 HL109420. HJZ is supported by a National Career Development Award from the American Heart Association (19CDA34760284). This work was partly supported by a National Natural Science Foundation of China (U1601219 and 81970420).
c) Disclosure: None.
NON-STANDARD ABBREVIATIONS AND ACRONYMS:
- AIP1
ASK1-interacting protein-1
- CVD
Cardiovascular disease
- GA
graft arteriosclerosis
- EC
endothelial cell
- H3K9me3
histone H3 lysine-9 tri-methylation
- LCA
left carotid artery
- mitoROS
mitochondrial ROS
- PI4P
phosphatidylinositol 4-phosphate
- RIF1
Rap1-interacting factor 1 (or replication timing regulatory factor 1)
- ROS
reactive oxygen species
- SMA
smooth muscle cell actin
- VSMC
vascular smooth muscle cells
REFERENCES
- 1.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol 2007;7:803–815 [DOI] [PubMed] [Google Scholar]
- 2.Pober JS, Min W, Bradley JR. Mechanisms of endothelial dysfunction, injury, and death. Annu Rev Pathol 2009;4:71–95 [DOI] [PubMed] [Google Scholar]
- 3.Zhang R, He X, Liu W, Lu M, Hsieh JT, Min W. Aip1 mediates tnf-alpha-induced ask1 activation by facilitating dissociation of ask1 from its inhibitor 14-3-3. J Clin Invest 2003;111:1933–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang H, Zhang R, Luo Y, D’Alessio A, Pober JS, Min W. Aip1/dab2ip, a novel member of the ras-gap family, transduces traf2-induced ask1-jnk activation. J Biol Chem 2004;279:44955–44965 [DOI] [PubMed] [Google Scholar]
- 5.Zhang H, Zhang H, Lin Y, Li J, Pober JS, Min W. Rip1-mediated aip1 phosphorylation at a 14-3-3-binding site is critical for tumor necrosis factor-induced ask1-jnk/p38 activation. J Biol Chem 2007;282:14788–14796 [DOI] [PubMed] [Google Scholar]
- 6.Zhang H, He Y, Dai S, Xu Z, Luo Y, Wan T, Luo D, Jones D, Tang S, Chen H, Sessa WC, Min W. Aip1 functions as an endogenous inhibitor of vegfr2-mediated signaling and inflammatory angiogenesis in mice. J Clin Inves 2008;118:3904–3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xie D, Gore C, Zhou J, Pong RC, Zhang H, Yu L, Vessella RL, Min W, Hsieh JT. Dab2ip coordinates both pi3k-akt and ask1 pathways for cell survival and apoptosis. Proc Natl Acad Sci U S A 2009;106:19878–19883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie D, Gore C, Liu J, Pong RC, Mason R, Hao G, Long M, Kabbani W, Yu L, Zhang H, Chen H, Sun X, Boothman DA, Min W, Hsieh JT. Role of dab2ip in modulating epithelial-to-mesenchymal transition and prostate cancer metastasis. Proc Natl Acad Sci U S A 2010;107:2485–2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wan T, Liu T, Zhang H, Tang S, Min W. Aip1 functions as arf6-gap to negatively regulate tlr4 signaling. J Biol Chem 2010;285:3750–3757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu L, Qin L, Zhang H, He Y, Chen H, Pober J, Tellides G, Min W. Aip1 prevents graft arteriosclerosis by inhibiting ifn-γ-dependent smooth muscle cell proliferation and intimal expansion. Cir Res 2011;109:418–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Min W, Pober JS. Aip1 in graft arteriosclerosis. Trends Cardiovasc Med 2011;21:229–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang Q, Qin L, Dai S, Zhang H, Pasula S, Zhou H, Chen H, Min W. Aip1 suppresses atherosclerosis by limiting hyperlipidemia-induced inflammation and vascular endothelial dysfunction. Arterioscler Thromb Vasc Biol 2013;33:795–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang J, Zhou HJ, Ji W, Min W. Aip1-mediated stress signaling in atherosclerosis and arteriosclerosis. Curr Atheroscler Rep 2015;17:503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J, Chen C, Li L, Zhou HJ, Li F, Zhang H, Yu L, Chen Y, Min W. Endothelial aip1 regulates vascular remodeling by suppressing nadph oxidase-2. Front Physiol 2018;9:396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gretarsdottir S, Baas AF, Thorleifsson G, et al. Genome-wide association study identifies a sequence variant within the dab2ip gene conferring susceptibility to abdominal aortic aneurysm. Nat Genet 2010;42:692–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harrison SC, Cooper JA, Li K, Talmud PJ, Sofat R, Stephens JW, Hamsten A, Consortium H, Sanders J, Montgomery H, Neil A, Simon Broome Research C, Humphries SE. Association of a sequence variant in dab2ip with coronary heart disease. Eur Heart J 2012;33:881–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qin L, Min W, Xin S. Aip1 suppresses transplant arteriosclerosis through inhibition of vascular smooth muscle cell inflammatory response to ifngamma. Anat Rec (Hoboken) 2018 [DOI] [PubMed] [Google Scholar]
- 18.Ji W, Li Y, He Y, Yin M, Zhou HJ, Boggon TJ, Zhang H, Min W. Aip1 expression in tumor niche suppresses tumor progression and metastasis. Cancer Res 2015;75:3492–3504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park JY. Promoter hypermethylation in prostate cancer. Cancer control : journal of the Moffitt Cancer Center 2010;17:245–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bird A DNA methylation patterns and epigenetic memory. Genes Dev 2002;16:6–21 [DOI] [PubMed] [Google Scholar]
- 21.Chen H, Toyooka S, Gazdar AF, Hsieh JT. Epigenetic regulation of a novel tumor suppressor gene (hdab2ip) in prostate cancer cell lines. J Biol Chem 2003;278:3121–3130 [DOI] [PubMed] [Google Scholar]
- 22.Chen H, Tu SW, Hsieh JT. Down-regulation of human dab2ip gene expression mediated by polycomb ezh2 complex and histone deacetylase in prostate cancer. J Biol Chem 2005;280:22437–22444 [DOI] [PubMed] [Google Scholar]
- 23.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol 2005;6:838–849 [DOI] [PubMed] [Google Scholar]
- 24.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006;125:315–326 [DOI] [PubMed] [Google Scholar]
- 25.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Mendenhall E, O’Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007;448:553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohtani K, Dimmeler S. Epigenetic regulation of cardiovascular differentiation. Cardiovasc Res 2011;90:404–412 [DOI] [PubMed] [Google Scholar]
- 27.Ohtani K, Vlachojannis GJ, Koyanagi M, Boeckel JN, Urbich C, Farcas R, Bonig H, Marquez VE, Zeiher AM, Dimmeler S. Epigenetic regulation of endothelial lineage committed genes in pro-angiogenic hematopoietic and endothelial progenitor cells. Circ Res 2011;109:1219–1229 [DOI] [PubMed] [Google Scholar]
- 28.Dan J, Liu Y, Liu N, Chiourea M, Okuka M, Wu T, Ye X, Mou C, Wang L, Wang L, Yin Y, Yuan J, Zuo B, Wang F, Li Z, Pan X, Yin Z, Chen L, Keefe DL, Gagos S, Xiao A, Liu L. Rif1 maintains telomere length homeostasis of escs by mediating heterochromatin silencing. Dev Cell 2014;29:7–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eid RE, Rao DA, Zhou J, Lo SF, Ranjbaran H, Gallo A, Sokol SI, Pfau S, Pober JS, Tellides G. Interleukin-17 and interferon-gamma are produced concomitantly by human coronary artery-infiltrating t cells and act synergistically on vascular smooth muscle cells. Circulation 2009;119:1424–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun JF, Phung T, Shiojima I, Felske T, Upalakalin JN, Feng D, Kornaga T, Dor T, Dvorak AM, Walsh K, Benjamin LE. Microvascular patterning is controlled by fine-tuning the akt signal. Proc Natl Acad Sci U S A 2005;102:128–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dai S, He Y, Zhang H, Yu L, Wan T, Xu Z, Jones D, Chen H, Min W. Endothelial-specific expression of mitochondrial thioredoxin promotes ischemia-mediated arteriogenesis and angiogenesis. Arterioscler Thromb Vasc Biol 2009;29:495–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Min W, Lin Y, Tang S, Yu L, Zhang H, Wan T, Luhn T, Fu H, Chen H. Aip1 recruits phosphatase pp2a to ask1 in tumor necrosis factor-induced ask1-jnk activation. Circ Res 2008;102:840–848 [DOI] [PubMed] [Google Scholar]
- 33.Zhang R, Al-Lamki R, Bai L, Streb JW, Miano JM, Bradley J, Min W. Thioredoxin-2 inhibits mitochondria-located ask1-mediated apoptosis in a jnk-independent manner. Circ Res 2004;94:1483–1491 [DOI] [PubMed] [Google Scholar]
- 34.Zhang H, Luo Y, Zhang W, He Y, Dai S, Zhang R, Huang Y, Bernatchez P, Giordano FJ, Shadel G, Sessa WC, Min W. Endothelial-specific expression of mitochondrial thioredoxin improves endothelial cell function and reduces atherosclerotic lesions. Am J Pathol 2007;170:1108–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rudic RD, Bucci M, Fulton D, Segal SS, Sessa WC. Temporal events underlying arterial remodeling after chronic flow reduction in mice: Correlation of structural changes with a deficit in basal nitric oxide synthesis. Circ Res 2000;86:1160–1166 [DOI] [PubMed] [Google Scholar]
- 36.Rudic RD, Shesely EG, Maeda N, Smithies O, Segal SS, Sessa WC. Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J Clin Invest 1998;101:731–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang PC, Qin L, Zielonka J, Zhou J, Matte-Martone C, Bergaya S, van Rooijen N, Shlomchik WD, Min W, Sessa WC, Pober JS, Tellides G. Myd88-dependent, superoxide-initiated inflammation is necessary for flow-mediated inward remodeling of conduit arteries. J Exp Med 2008;205:3159–3171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang Q, Zhou HJ, Zhang H, Huang Y, Hinojosa-Kirschenbaum F, Fan P, Yao L, Belardinelli L, Tellides G, Giordano FJ, Budas GR, Min W. Thioredoxin-2 inhibits mitochondrial ros generation and ask1 activity to maintain cardiac function. Circulation 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dai X, North BJ, Inuzuka H. Negative regulation of dab2ip by akt and scffbw7 pathways. Oncotarget 2014;5:3307–3315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li X, Dai X, Wan L, Inuzuka H, Sun L, North BJ. Smurf1 regulation of dab2ip controls cell proliferation and migration. Oncotarget 2016;7:26057–26069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ea CK, Hao S, Yeo KS, Baltimore D. Ehmt1 protein binds to nuclear factor-kappab p50 and represses gene expression. J Biol Chem 2012;287:31207–31217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Di Febbo C, Baccante G, Reale M, Castellani ML, Angelini A, Cuccurullo F, Porreca E. Transforming growth factor beta1 induces il-1 receptor antagonist production and gene expression in rat vascular smooth muscle cells. Atherosclerosis 1998;136:377–382 [DOI] [PubMed] [Google Scholar]
- 43.Korshunov VA, Berk BC. Flow-induced vascular remodeling in the mouse: A model for carotid intima-media thickening. Arterioscler Thromb Vasc Biol 2003;23:2185–2191 [DOI] [PubMed] [Google Scholar]
- 44.Min J, Zaslavsky A, Fedele G, McLaughlin SK, Reczek EE, De Raedt T, Guney I, Strochlic DE, Macconaill LE, Beroukhim R, Bronson RT, Ryeom S, Hahn WC, Loda M, Cichowski K. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating ras and nuclear factor-kappab. Nat Med 2010;16:286–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levy D, Kuo AJ, Chang Y, Schaefer U, Kitson C, Cheung P, Espejo A, Zee BM, Liu CL, Tangsombatvisit S, Tennen RI, Kuo AY, Tanjing S, Cheung R, Chua KF, Utz PJ, Shi X, Prinjha RK, Lee K, Garcia BA, Bedford MT, Tarakhovsky A, Cheng X, Gozani O. Lysine methylation of the nf-kappab subunit rela by setd6 couples activity of the histone methyltransferase glp at chromatin to tonic repression of nf-kappab signaling. Nat Immunol 2011;12:29–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pal S, Gupta R, Davuluri RV. Alternative transcription and alternative splicing in cancer. Pharmacol Ther 2012;136:283–294 [DOI] [PubMed] [Google Scholar]
- 47.Pal S, Gupta R, Kim H, Wickramasinghe P, Baubet V, Showe LC, Dahmane N, Davuluri RV. Alternative transcription exceeds alternative splicing in generating the transcriptome diversity of cerebellar development. Genome Res 2011;21:1260–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Truscott KN, Brandner K, Pfanner N. Mechanisms of protein import into mitochondria. Curr Biol 2003;13:R326–337 [DOI] [PubMed] [Google Scholar]
- 49.Lassegue B, Griendling KK. Nadph oxidases: Functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol.30:653–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Konior A, Schramm A, Czesnikiewicz-Guzik M, Guzik TJ. Nadph oxidases in vascular pathology. Antioxid Redox Signal 2014;20:2794–2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quintero M, Colombo SL, Godfrey A, Moncada S. Mitochondria as signaling organelles in the vascular endothelium. Proc Natl Acad Sci U S A 2006;103:5379–5384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hansen JM, Zhang H, Jones DP. Mitochondrial thioredoxin-2 has a key role in determining tumor necrosis factor-alpha-induced reactive oxygen species generation, nf-kappab activation, and apoptosis. Toxicol Sci 2006;91:643–650 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.