

Summary
Ferroptosis is a recently described form of cell death driven by iron-dependent lipid peroxidation. This type of cell death was first observed in response to treatment of tumor cells with a small molecule chemical probe named erastin. Most subsequent advances in understanding the mechanisms governing ferroptosis involved the use of genetic screens and small molecule probes. We describe herein the utility and limitations of chemical probes that have been used to analyze and perturb ferroptosis, as well as mechanistic studies of ferroptosis that benefitted from the use of these probes and genetic screens. We also suggest probes for ferroptosis and highlight mechanistic questions surrounding this form of cell death that will be a high priority for exploration in the future.
Keywords: ferroptosis, iron, metabolism, ROS, cancer, lipid peroxidation, chemical probe, cell death, glutathione, cysteine
eTOC Blurb
Ferroptosis is a type of cell death first observed upon treatment of cancer cells with chemical probes, and now seen in diverse contexts. Stockwell and Jiang describe probes for perturbing ferroptosis, current understanding of the biology of ferroptosis, and therapeutic applications of controlling this form of cell death.
Graphical Abstract
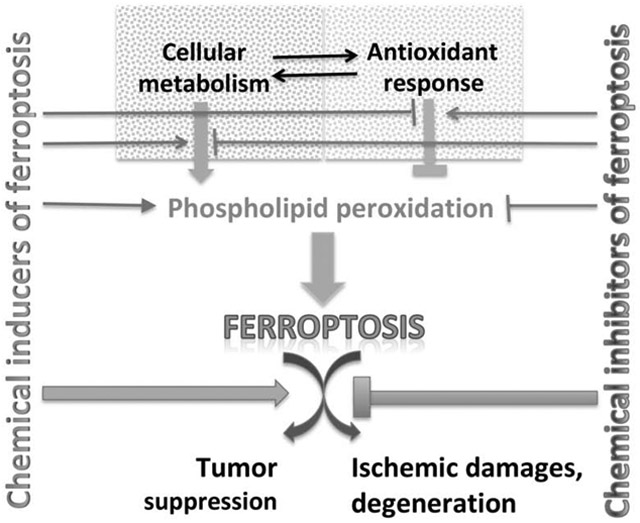
Using chemical probes to illuminate ferroptosis
Small molecule probes are valuable tools both for interrogating and perturbing biological systems (Stockwell, 2000). Such tools have been valuable in the study of different types of regulated cell death, including apoptosis and necroptosis (Gangadhar and Stockwell, 2006). Most recently, a new mode of cell death, ferroptosis, was elucidated and studied through the use of small molecule chemical probes (Dixon et al., 2012). We consider here some of the features and limitations of these ferroptosis probes, and suggest potential future probes of interest.
Ferroptosis is driven by the peroxidation of phospholipids that contain polyunsaturated fatty acyl tails (Stockwell et al., 2017). The induction of this form of cell death requires three hallmarks (see Figure 1)— (i) the presence of redox-active iron, in the form of the labile iron pool (Yang and Stockwell, 2008), and iron-dependent peroxidation enzymes, such as lipoxygenases (Wenzel et al., 2017; Yang et al., 2016) and cytochrome P450s (Zou et al., 2020), (ii) the presence of the key substrates that undergo peroxidation, namely phospholipids that have polyunsaturated fatty acyl tails with bis-allylic carbons that are prone to undergo peroxidation (Dixon et al., 2015; Yang et al., 2016), and (iii) failure of the complex lipid peroxide repair network, involving pathways such as glutathione-GPX4 (Yang et al., 2014), GCH1-BH4 (Kraft et al., 2020), and NADPH-FSP1-CoQ10 (Bersuker et al., 2019a; Dixon and Stockwell, 2019; Doll et al., 2019a; Shimada et al., 2016a; Shimada et al., 2016b).
Figure 1. The ferroptosis pathway and relevant chemical probes.

Molecules including cellular metabolites and protein factors that promote ferroptosis are in red, and those that prevent ferroptosis are in blue. Boxes show sets of chemical probes that inhibit system xc−, GPX4, lipid peroxidation, and CoQ10 production. Glu, glutamate; Cys, cysteine; GCL, glutamate-cysteine ligase; GSS, glutathione synthase; GSH, reduced glutathione; GSSG, oxidized glutathione; PUFA, polyunsaturated fatty acid; PL, phospholipid; LOX, lipoxygenase; R; free radicals; Et, ethyl. See Table for additional information.
Chemical probes for system xc−
The first discovered inducer of ferroptosis, and which illuminated the phenomenon of ferroptosis itself, was a novel small molecule identified from a diverse chemical library that was termed erastin (Dolma et al., 2003). Subsequent studies revealed that erastin induces ferroptosis by inhibiting the activity of the cystine-glutamate antiporter known as system xc− (Dixon et al., 2012; Dixon et al., 2014) (Table 1) . In cell types that obtain cysteine in the form of cystine supplied by system xc−, inhibition of this antiporter results in depletion of the intracellular pools of both the reduced and oxidized forms of glutathione (GSH and GSSG) (Dixon et al., 2014). This depletion in glutathione impairs the activity of glutathione peroxidase 4 (GPX4), which normally suppresses the formation of phospholipid hydroperoxides; the end result is accumulation of peroxidized phospholipids, which leads to ferroptotic death (Yang et al., 2014).
Table 1. Ferroptosis-relevant chemical probes.
Compounds that have been reported to inhibit functions relevant to ferroptosis are listed, along with Pubchem CID numbers, if known, chemical class, growth inhibitory potency (GI50) in a relevant cell line, and PMID for the relevant reference.
| Compound | Pubchem CID | Class | GI50 (nM) | Cell line | Reference (PMID) |
|---|---|---|---|---|---|
| System xc- inhibitors | |||||
| Erastin | 11214940 | Chlorophenoxyacetamide | 2000 | BJeLR | 12676586 |
| Piperazine erastin (PE) | 72710858 | Chlorophenoxyacetamide | 900 | BJeLR | 24439385 |
| Imidazole ketone erastin (IKE) | 91824786 | Chlorophenoxyacetamide | 3 | BJeLR | 26231156 |
| Sorafenib | 216239 | Diarylurea | 18000 | HT-1080 | 24844246 |
| Sulfasalazine | 5339 | Diazo | 20000 | U87G | 21889337 |
| DPI2 | 5728915 | Rhodanine | 2000 | BJeLR | 24439385 |
| RSL5 | 2863472 | Hexahydroquinolinone | 10000 | IBS CSC121707693 | 18355723 |
| Glutamate | 23672308 | amino acid | 5000000 | N18-RE-105 | 2576375 |
| GPX4 inhibitors | |||||
| (1S, 3R)-RSL3 | 1750826 | Chloroacetamide | 200 | BJeLR | 18355723 |
| DPI3 | 3689415 | Chloroacetamide | 20 | BJeLR | 24439385 |
| DPI4 | 3689416 | Chloroacetamide | 20 | BJeLR | 24439385 |
| DPI7/ML162 | 3689413 | Chloroacetamide | 20 | BJeLR | 24439385 |
| DPI6 | 4381125 | Chloroacetamide | 200 | BJeLR | 24439385 |
| DPI8 | 4230741 | Chloroacetamide | 200 | BJeLR | 24439385 |
| DPI9 | 12004949 | Chloroacetamide | 200 | BJeLR | 24439386 |
| DPI12 | 2416356 | Chloroacetamide | 100 | BJeLR | 24439385 |
| DPI13 | 2449454 | Chloroacetamide | 800 | BJeLR | 24439385 |
| DPI18 | 932657 | Chloroacetamide | 200 | BJeLR | 24439385 |
| DPI19 | 1637653 | Chloroacetamide | 200 | BJeLR | 24439385 |
| DPI15 | 6545175 | chloromethyltriazine | 400 | BJeLR | 24439385 |
| DPI17 | 932617 | chloromethyltriazine | 80 | BJeLR | 24439385 |
| Altretamine | 2123 | chloromethyltriazine | 250000 | U-2932 | 26186195 |
| DPI10 | 15945537 | nitroisoxazole | 300 | BJeLR | 22297109 |
| ML210 | 49766530 | nitroisoxazole | 70 | BJeLR | 22297109 |
| JKE-1674 | N/A | masked nitrile oxide | 30 | LOX-IMVI | biorxiv: doi: https://doi.org/10.1101/376764 |
| NSC144988 | 286532 | furoxan | 1700 | LOX-IMVI | 31841309 |
| Withaferin A | 265237 | steroidal lactone | 1000 | IMR32 | 29939160 |
| RTAs | |||||
| Ferrostatin-1 | 4068248 | ortho-phenylenediamine | 45 | MEFs | 28386601 |
| Liproxstatin-1 | 135735917 | spiroquinoxalinamine | 38 | MEFs | 28386601 |
| Butylated hydroxytoluene (BHT) | 31404 | phenol | 10000 | BJeLR | 22632970 |
| alpha-tocopherol | 86472 | dihydrochromene | 20000 | BJeLR | 22632970 |
| Phenoxazine | 67278 | Phenoxazine | 9 | MEFs | 28837769 |
| CoQ10 pathway | |||||
| Idebenone | 3686 | 1,4-benzoquinone | 10000 | HT-1080 | 27159577 |
| 4-chlorobenzoic acid | 6318 | Benzoid acid | 3000000 | U2OS | 31634900 |
| Cerivastatin | 446156 | Statin | 1000 | HT-180 | 27159577 |
| FIN56 | N/A | Fluorene | 100 | BJeLR | 27159577 |
| iFSP1 | N/A | pyridobenzimidazole | 100 | Pfa1 | 31634899 |
| Iron Chelators and sources | |||||
| Deferoxamine (DFO) | 2973 | polyamide | 100000 | HT-1080 | 22632970 |
| Ciclopirox | 2749 | hydroxamic acid | 5000 | HT-1080 | 22632970 |
| Endoperoxides | |||||
| FINO2 | N/A | endoperoxide | 15000 | BJeLR | 26797166 |
Erastin was subsequently modified to create more potent and drug-like system xc− inhibitors for inducing ferroptosis: piperazine erastin (PE) is slightly more potent than erastin (Table 1), but has substantially improved water solubility and metabolic stability (Yang et al., 2014). Imidazole ketone erastin (IKE) has substantially improved potency and metabolic stability (Table 1), with moderately improved aqueous solubility (Larraufie et al., 2015). Both PE (Yang et al., 2014) and IKE (Zhang et al., 2019a) are suitable for use in cell culture and animal studies, but IKE is generally more effective due to its greater potency.
Additional compounds have also been found to inhibit system xc− and induce ferroptosis, but with lower potency and less selectivity than erastin, PE, and IKE. The approved drug and multi-targeted kinase inhibitor sorafenib blocks system xc− function, likely indirectly as a result of inhibiting one of its kinase targets, but also induces a necrotic death mechanism at slightly higher concentrations (Dixon et al., 2014). Sorafenib has also been reported to inhibit necrosome assembly and necroptosis (Feldmann et al., 2017; Martens et al., 2017). Therefore, sorafenib (Figure 1) can be used to inhibit system xc− and to induce ferroptosis in cell culture and in animals, but its necrotic activity, multi-targeted kinase activity, and anti-necroptotic activity also need to be considered in evaluating the mechanistic implications of sorafenib’s effects. Simultaneous pharmacodynamic studies testing for markers of ferroptosis with sorafenib are one means of testing whether sorafenib induces ferroptosis under specific conditions. The approved drug sulfasalazine (Figure 1) has also been used as a system xc− inhibitor (Gout et al., 2001). However, sulfasalazine has very low potency, and is metabolically unstable in vivo, making it difficult to use in animal studies; it is best used as a confirmatory tool in cell culture studies. The compounds DPI2 (Yang et al., 2014) and RSL5 (Yang and Stockwell, 2008) have similar effects as erastin, and may also be system xc− inhibitors, although this potential mechanism has not been tested directly (Figure 1, Table 1).
The amino acid and neurotransmitter glutamate has also been shown to inhibit system xc− and to induce ferroptosis in some cellular contexts (Murphy et al., 1989). Glutamate has a variety of other effects, including the induction of Ca++-mediated excitotoxicity via NMDA receptors, implying that it is difficult to use glutamate as a chemical probe per se for inducing ferroptosis. However, glutamate-induced ferroptosis may be a relevant model for some neurological conditions (Dixon et al., 2012).
The effects of all system xc− inhibitors should depend entirely on inhibition of cystine uptake; this can be tested experimentally by co-treating cells in culture with β-mercaptoethanol or other reducing agents that reduce extracellular cystine to cysteine, resulting in uptake through transporters independent of system xc−. The effects of erastin, for example, are entirely reversed by co-treatment with β-mercaptoethanol, demonstrating that erastin blocks cystine import, but not cysteine import (Dixon et al., 2014). Of note, erastin affinity analogs bind directly to VDAC2 and VDAC3 in a pulldown assay, and VDAC2/3 are require for erastin lethality (Yagoda et al., 2007), but the relationship of this observation to system xc− inhibition is not clear.
Chemical probes for GPX4
Shortly after erastin was discovered, another small molecule inducer of ferroptosis was identified that acted independently of system xc− (Yang and Stockwell, 2008). This compound, named RSL3, was found to covalently inhibit GPX4, blocking one of the key repair systems for phospholipid peroxides in cells (Yang et al., 2014). RSL3 is a potent and irreversible inhibitor of GPX4, due to its reactive chloroacetamide moiety (Yang et al., 2016), but is not generally suitable for in vivo use due to poor solubility and unfavorable absorption, distribution, metabolism and excretion (ADME) properties.
Additional chloroacetamide-containing inhibitors of GPX4 have subsequently been identified, including DPI7/ML162, DPI6, DPI8, DPI9, DPI12, DPI13, DPI15, and DPI19 (Yang et al., 2014). All of these compounds have been shown to exhibit butylated-hydroxytoluene-(BHT)-sensitive cell killing, and buthionine-sulfoximine-(BSO)-enhanced cell killing in the BJ-derived cell lines, along with the induction of C11-BODIPY oxidation, all hallmarks of ferroptosis (Yang et al., 2014). DPI7/ML162, DPI12, DPI13, and DPI19 have been confirmed to inhibit GPX4 activity in BJeLR cell lysates (Yang et al., 2014). These compounds are not as well characterized as RSL3; DPI7/ML162, the best characterized among these additional chloroacetamide GPX4 inhibitors, can be used to verify the GPX4 dependence of RSL3. RSL3 and ML162 have substantially different structures, aside from the chloroacetamide moiety, and thus likely have different off-target effects; use of both compounds increases the probability that effects observed with the two probes are due to GPX4 inhibition.
Three additional structural classes of GPX4 inhibitors have been reported (Figure 1). First, DPI17, DPI18 (Yang et al., 2014), and altretamine (Woo et al., 2015) are chloromethyltriazines that also act as covalent GPX4 inhibitors. DPI17 and DPI18 are structurally similar compounds and have been shown, like the chloroacetamide compounds above, to exhibit cell killing activity with hallmarks of ferroptosis. DPI17 has been confirmed to inhibit GPX4 activity in BJeLR cell lysates. DPI17 and DPI18 are thus likely to be covalent GPX4 inhibitors (Yang et al., 2014). Second, DPI10 and ML210 (Weiwer et al., 2012) contain a nitroisoxazole moiety that has been reported to generate a nitrile oxide electrophile that reacts with GPX4 in a cellular context (Eaton et al., 2018); the related compounds JKE-1674 and JKE-1716, and the furoxan-containing NSC144988, as well as related diacylfuroxans, also generate nitrile oxide electrophiles that inhibit GPX4 (Eaton et al., 2019). Finally, the natural product withaferin A, which is a steroidal lactone and epoxide, has been reported to act as a GPX4 inhibitor, likely through its electrophilic groups (Hassannia et al., 2018).
Inhibitors of ferroptosis
A number of small molecule radical trapping agents (RTAs) suppress ferroptosis through their ability to interrupt the lipid peroxidation process (Figure 1). These RTAs include the potent RTAs ferrostatin-1 (Dixon et al., 2012), liproxstatin-1 (Friedmann Angeli et al., 2014), phenoxazine (Shah et al., 2017), and the less potent butylated hydroxytoluene (BHT), and alpha-tocopherol (Dixon et al., 2012) (Table 1). Ferrostatin-1 has low metabolic stability (Hofmans et al., 2016), and therefore in vivo experiments with this compound should be undertaken with caution. More stable ferrostatins are needed for in vivo studies. Some improved analogs have been developed and tested in vivo (Linkermann et al., 2014), although additional improvements are needed. Liproxstatin-1, on the other hand, is as potent as ferrostatin-1 and has suitable properties for use in animal models (Friedmann Angeli et al., 2014). Both ferrostatin-1 and liproxstatin-1 have been used in numerous cell culture experiments and to date have been verified to selectively suppress ferroptosis and not other cell death processes, nor to exhibit any obvious off-target effects. Phenoxazine is also potent, but needs to be more thoroughly characterized in terms of its selectivity and in vivo suitability. BHT and alpha-tocopherol have moderate potencies as ferroptosis suppressors and are less likely to be selective and effective in vivo for this purpose (Table 1). Moreover, since all of these RTAs act with widely different potencies, the concentration necessary in each cell system should be examined carefully. In addition, these compounds can also suppress lipid peroxidation that does not lead to ferroptotic death, so they should be used in combination with other probes of ferroptosis to verify that their activity in any given system is due to suppressing ferroptosis, versus other lipid-peroxidation-dependent events. Interestingly, a recent screen for genes that suppress ferroptosis when overexpressed identified GCH1 as a top hit (Kraft et al., 2020). The protective effect of GCH1 expression against ferroptosis was due to the ability of GCH1 to stimulate the biosynthesis of tetrahydrobiopterin, a natural radical-trapping agent that also suppresses ferroptosis by driving enhanced levels of reduced CoQ10 (see below). Cu(II)atsm has also been reported to inhibit ferroptosis, probably through radical-trapping activity (Southon et al., 2020). Finally, deuterated polyunsaturated fatty acids (PUFAs) also suppress lipid peroxidation (Gaschler et al., 2018; Hatami et al., 2018; Hill et al., 2012; Yang et al., 2016).
Iron chelators have also been used to suppress ferroptosis, to test whether a suspected cell death process proceeds via ferroptosis. Two common tools for this purpose are deferoxamine (DFO), which is a polyamide that chelates extracellular iron, making it inaccessible to intracellular processes, and ciclopirox, which is a simpler compound with better cell penetration that also chelates iron, suppressing its role in driving ferroptosis (Dixon et al., 2012). Adding excess iron to cells can sensitize to ferroptosis (Dixon et al., 2012) or even drive ferroptosis under some conditions (Huang et al., 2019).
There are a number of other reagents and mechanisms for suppressing ferroptosis that have been reported. Incorporation of PUFAs into phospholipids is required for ferroptosis, as peroxidation of free fatty acids does not lead to ferroptosis, for unknown reasons. Knockdown of LPCAT3 or ACSL4 suppresses this incorporation of PUFAs into phospholipids and suppresses ferroptosis; thiazolidinediones were reported to inhibit ACSL4 (Doll et al., 2017), although they have additional activities that need to be considered. Multiple inhibitors of autophagy-lysosomal process, such as lysosomal blockers chloroquine and bafilomycin A1, can also suppress cysteine deprivation-induced ferroptosis, at least partially through preventing the degradation of cellular iron-storage protein ferritin by autophagy, i.e., ferritinophagy (see later in “Ferroptosis and iron signaling” for more detail).
Lipoxygenase inhibitors also suppress ferroptosis, as lipoxygenases contribute to the lipid peroxidation that drives ferroptosis, especially its initiation (Yang et al., 2016). However, many lipoxygenase inhibitors also act as radical trapping agents, so they need to be used with care and evaluated for RTA activity (Shah et al., 2018a). Selenium is normally limiting for the biosynthesis of the selenoprotein GPX4, and thus treatment of cells with selenium or selenium-containing compounds can in some cases stimulate GPX4 expression and suppress ferroptosis (Alim et al., 2019).
Chemical probes for the CoQ10 pathway
Several reports have demonstrated that the lipophilic antioxidant ubiquinol/coenzyme Q10 (CoQ10) suppresses lipid peroxidation and ferroptosis in some contexts (Bersuker et al., 2019b; Doll et al., 2019b; Shimada et al., 2016b). There are several chemical probes that have been reported to modulate this effect of CoQ10 on ferroptosis (Table 1). First, the water-soluble CoQ10 analog idebenone mimics the antioxidant effect of CoQ10 when added exogenously to cells, suppressing lipid peroxidation with moderate potency (Shimada et al., 2016b). Second, 4-chlorobenzoate can inhibit CoQ10 production in cells with low potency, sensitizing to ferroptosis in contexts where endogenous CoQ10 suppresses lipid peroxidation (Bersuker et al., 2019b). Third, since CoQ10 is derived from the mevalonate pathway, statins such as cerivastatin, which inhibit HMG-CoA reductase, can also sensitize cells to ferroptosis in some contexts (Shimada et al., 2016b). Fourth, the compound FIN56 was reported to act through the mevalonate pathway to drive ferroptosis, presumably through depleting CoQ10 (Shimada et al., 2016b).
Finally, the CoQ10 reductase known as Ferroptosis Suppressor Protein 1 (FSP1, formerly AIFM2) was recently reported to suppress ferroptosis through regenerating reduced CoQ10. An inhibitor of FSP1 was reported, named iFSP1 (Bersuker et al., 2019b)—this compound may be useful for testing the relevance of FSP1 and reduced CoQ10 in various contexts.
Other inducers of ferroptosis
In 2015, a novel inducer of ferroptosis was reported that did not act through direct targeting of GPX4, inhibiting system xc−, or depletion of CoQ10 (Abrams et al., 2016). This compound, named FINO2, was found to oxidize iron, resulting in inactivation of GPX4 enzymatic activity (Gaschler et al., 2018). This compound can be used to test whether loss of GPX4 activity drives a phenotype independent of direct GPX4 targeting or depletion of glutathione or CoQ10.
Recently, several groups discovered that radiation can induce ferroptosis, that ferroptosis inducers can act as radiosensitizers, and that ferroptosis inhibitors can block the effects of radiation (Lei et al., 2020; Li et al., 2019a; Li et al., 2019b; Ye et al., 2020; Zhang et al., 2020). This provides a new approach to treat both radiosensitive and radioresistant cancers, as well as side effects of radiation on normal tissues.
In summary, there are a variety of probes for inducing and inhibiting ferroptosis. These probes have different potencies, selectivities, and ADME properties, suggesting they must be carefully used for applications of interest. In general, using multiple probes can increase the confidence of drawing robust conclusions, assuming the concentrations and specificity are relevant to the tested conditions.
Detecting ferroptosis
There are a number of assays in use to detect discrete events that occur in ferroptosis. First, inhibitors of system xc− itself can be detected by measuring either import of radiolabeled cystine or export of glutamate (Dixon et al., 2012; Dixon et al., 2014). Second, inhibitors of system xc− commonly induce depletion of glutathione (Dixon et al., 2012), which can be measured using the thiol-reactive Ellman’s reagent, or by liquid chromatography–mass spectrometry (LC-MS) (Yang et al., 2014). Third, lipid peroxidation can be measured with surrogate fluorescent dyes that partition into membranes, such as C11-BODIPY (Dixon et al., 2012) or Liperfluo (Alborzinia et al., 2018); by detecting end products of lipid peroxidation, such as malondialdehyde (MDA) with the TBARS assay (Zhang et al., 2019a) or with antibodies that detect MDA adducts on proteins (Yamada et al., 2001); or using LC-MS-based detection of lipids and their oxidation products (Kagan et al., 2017). Finally, downstream gene expression changes in ferroptosis can be detected, such as increases in CHAC1, PTGS2, and SLC7A11 (Dixon et al., 2012; Yang et al., 2014).
Connections between ferroptosis and other cellular processes
It has been demonstrated that cellular metabolism, redox, iron homeostasis, and various signaling pathways all impinge on ferroptosis (Gao and Jiang, 2018; Stockwell et al., 2017). Here, we discuss the regulation of ferroptosis by these central processes. Studies leading to such mechanistic understanding would not have been possible without the use of the ferroptosis-modulating chemical probes as described above. Conversely, as numerous pharmacological agents have been developed as specific inhibitors of these ferroptosis-regulatory signaling and metabolic pathways, these agents can be used as chemical probes for the investigation of ferroptosis, especially in its interplay with these pathways.
Ferroptosis and metabolism
Energy metabolism
Several recent studies have linked energy metabolism to ferroptosis regulation. On the one hand, energy stress activates 5'-adenosine-monophosphate-activated protein kinase (AMPK), which suppresses ferroptosis through effects on lipid metabolism (Lee et al., 2020). On the other hand, mitochondrial respiration facilitates ferroptosis in response to cysteine deprivation (Gao et al., 2019).
As described above, early research in ferroptosis developed pharmacological tools as triggers of ferroptosis, such as small molecule inhibitors of the system xc− cystine-glutamate antiporter (e.g., erastin) and GPX4 (e.g., RSL3) (Dixon et al., 2012; Yang et al., 2014). These compounds, by ablating cellular reducing agents or inhibiting the enzymatic activity required for countering lipid peroxides, drive ferroptotic death. Since exogenous oxidizing agents are not used in these experimental systems, one may wonder about the origin of the endogenous oxidative stress that is unleashed by erastin and RSL3.
As the center of cellular metabolism, mitochondrial activity contributes significantly to the generation of cellular reactive oxygen species (ROS), and thus may drive ferroptosis in some contexts. Mitochondria generate various forms of ROS: for example, superoxide radical (O2−) is a byproduct of electron transport chain activity, and it can be converted into hydrogen peroxide (H2O2) by superoxide dismutase (SOD) enzymes (Zorov et al., 2014). Both superoxide and hydrogen peroxide can react with lipids to generate lipid peroxides.
In a recent study, through a series of systematic pharmacological and molecular experiments, it was demonstrated that normal mitochondrial function, including that carried out by both the TCA cycle and electron transport chain, is crucial for ferroptosis induced by cellular cysteine deprivation (Gao et al., 2019). Interestingly, before the term ferroptosis was coined, a pro-death function of mitochondria was reported in a form of oxidative cell death known as oxytosis (Lewerenz et al., 2018; Tan et al., 1998), which may overlap with ferroptosis. Mechanistically, the pro-death function of mitochondria in ferroptosis is unrelated to that in apoptosis. In mitochondria-mediated apoptosis, mitochondria function as a storage for caspase-activating factors, such as cytochrome c and Smac, and thus apoptotic death requires damage of mitochondrial membrane (Jiang and Wang, 2004). In contrast, the normal metabolic function of mitochondria promotes cysteine-deprivation-induced ferroptosis, and a sharp increase in mitochondrial membrane potential, the indicator of mitochondrial ATP-generating activity, precedes eventual ferroptotic death (Gao et al., 2019).
It should be noted that although mitochondrial activity is a major source of cellular ROS production, there are other sources of cellular ROS generation that can also lead to lipid peroxidation. This may explain why blocking mitochondrial activity ablates cysteine-deprivation-induced ferroptosis, but not that induced by GPX4 inhibition; upon GPX4 inhibition, lipid peroxides from mitochondria-independent events can be amplified by Fenton chemistry, leading to ferroptosis (Gao et al., 2019).
The role of mitochondria in ferroptosis also provides a mechanistic explanation for an early observation showing that glutamine and glutaminolysis drive this mode of cell death (Gao et al., 2015). Glutamine, via glutaminolysis, generates α-ketoglutarate αKG), serving as a crucial anaplerotic source for the mitochondrial TCA cycle, especially when glucose is limited (Owen et al., 2002). Consistently, αKG and other downstream metabolites of the TCA cycle can replace glutamine in supporting cysteine deprivation-induced ferroptosis (Gao et al., 2019).
Given the key role of the metabolic function of mitochondria in ferroptosis, one can speculate that other nutrients that fuel the TCA cycle, such as glucose and fatty acid (via β-oxidation), may also promote cysteine-deprivation-induced ferroptosis. Indeed, both glucose depletion and inhibition of β-oxidation can decrease cysteine-deprivation-induced ferroptosis (modest for β-oxidation inhibition and more potent for glucose depletion), likely due to their function in the TCA cycle. These observations are satisfying, but they also raise intriguing mechanistic questions. For example, if the anaplerotic role of glutamine is the sole basis underlying its ferroptotic function, then why is this amino acid essential for cysteine-deprivation-induced ferroptosis under the condition that glucose is abundant (Gao et al., 2015)? Further studies are needed to tackle this question.
Lipid biosynthesis
It appears that only certain specific types of phospholipid peroxides can lead to the execution of ferroptosis. For example, hydroperoxidation of phosphatidyl ethanolamine (PE) has been suggested to be preferentially associated with ferroptosis (Kagan et al., 2017), and the enzymes involved in this process, including Acyl-CoA Synthetase Long Chain Family Member 4 (ACSL4) (Dixon et al., 2015; Doll et al., 2017; Yuan et al., 2016) and iron-dependent lipoxygenases (LOX) (Chu et al., 2019; Friedmann Angeli et al., 2014; Kenny et al., 2019; Seiler et al., 2008; Shintoku et al., 2017; Wenzel et al., 2017; Yang et al., 2016; Ye and Stockwell, 2017), as well as cytochrome P450 oxidoreductase (POR) (Zou et al., 2020) have been demonstrated to be required for ferroptosis (Doll et al., 2017; Yang et al., 2016). Since arachidonic acid and adrenic acid are the preferred substrates of ACSL4, presumably these specific PUFA species are the major acyl chains on phospholipids that undergo peroxidation for ferroptosis initiation. For various LOX members, it is still under debate which specific member is more relevant to ferroptosis—some confusion was due to the use of certain LOX inhibitors that possess free radical-trapping activity and thus can inhibit ferroptosis independent of LOX (Shah et al., 2018b). More potent and selective LOX inhibitors without antioxidant activity will be important for resolving this issue.
Intriguingly, monounsaturated fatty acid (MUFA) molecules have been demonstrated to be inhibitors of ferroptosis (Yang et al., 2016): exogenously added MUFAs can protect cultured cells from ferroptosis (Magtanong et al., 2019). Consistently, stearoyl-CoA desaturase 1 (SCD1), the enzyme that converts stearic acid to MUFA oleic acid, negatively regulates ferroptosis in an enzymatic-activity-dependent manner (Tesfay et al., 2019). Mechanistically, as both PUFAs and MUFAs can be the substrates for peroxidation, how cells distinguish them for peroxidation and how MUFAs inhibit ferroptosis warrant further investigation.
Experimentally, as phospholipid peroxidation is a crucial event for ferroptosis, methods for its accurate and quantitative measurement are needed. As described earlier in this review, fluorogenic lipophilic probes, coupled with flow cytometry or microscopic imaging, are routinely used for this purpose. It should be cautioned that these lipophilic probes are surrogates instead of direct readouts of cellular phospholipid peroxidation. To directly measure and quantify ferroptosis-associated phospholipid peroxidation, methods based on mass spectrometric lipidomic analysis were developed (Tyurina et al., 2019). Theoretically, this approach is more quantitative and able to profile the specific molecular identity of lipids as well. However, at current stage this approach has not been widely used in the field of ferroptosis, due to poor stability of peroxides (thus low signal and poor reproducibility) and complexity of the experimental procedure.
Ferroptosis and iron signaling
Although ferroptosis has been known to be iron-dependent (and named based on this property), the precise function of iron in ferroptosis has not become clear until recently. It is now believed that iron plays multiple roles in ferroptosis. First, as a key component of a cohort of metabolic enzymes and energy-generating protein complexes, iron is indispensable for these oxidation-based metabolic processes, the source of cellular ROS generation. Second, iron is a cofactor of LOXs and P450s, essential enzymes for the biosynthesis of phospholipid peroxides. Third, iron can propel lipid peroxidation by catalyzing non-enzymatic Fenton reaction (Braughler et al., 1986). When GPX4, FSP1, and/or GCH1 function is inhibited or exhausted, this chain reaction leads to rapid production of phospholipid peroxides, and ultimately, ferroptosis.
Because of the crucial physiological role of iron and detrimental consequences associated with both scarcity and excess of cellular iron, the quantity of cellular labile iron (i.e., freely available iron) needs to be closely regulated. Indeed, in mammalian cells, there is an elegant and complicated signaling network dedicated to the regulation of cellular iron homeostasis. This network includes factors that sense iron concentrations (IRP1 and IRP2), as well as proteins regulated by IRP1 and IRP2 at the mRNA level, such as transferrin receptor (TFRC; it functions to import iron into cells), ferritin (it stores excessive cellular iron), and others; see (Andrews and Schmidt, 2007) for an extensive review of this topic.
In the process of ferroptosis, this iron-regulatory pathway is hijacked through multiple mechanisms to increase cellular labile iron. First, ferroptosis can engage autophagy, a stress-responsible catabolic process that mediates lysosomal degradation of selected intracellular contents. Upon cysteine deprivation, autophagy is activated in such a way that it degrades ferritin (a process also referred to as ferritinophagy (Dowdle et al., 2014; Mancias et al., 2014)), thus releasing free iron into the cytoplasm (Gao et al., 2016; Hou et al., 2016). Second, ferroptosis-inducing conditions can increase cellular free iron contents via dysregulating IRP1 (Andrews and Schmidt, 2007). In normal cells, the iron-promoting activity of IRP1 is controlled by Fe-S clusters. When cellular iron is low, IRP1 is free of Fe-S clusters, and thus increase cellular iron contents by binding to a variety of mRNA molecules and altering the synthesis of the corresponding proteins. When cellular iron is high, there will be sufficient Fe-S clusters associate with IRP1, thus suppressing its mRNA-binding/iron-promoting activity. However, Fe-S clusters are highly vulnerable to peroxides. Therefore, upon oxidative stress, such as conditions that trigger ferroptosis, even when cellular iron is not low, IRP1 will still promote cellular iron increase due to the depletion of Fe-S clusters by peroxides.
Ferroptosis and cell signaling
In theory, signal transduction pathways regulating cellular metabolism, redox homeostasis, and iron homeostasis may all impact ferroptosis. One might generalize that if a signaling pathway promotes cell growth (such as most tumorigenic signaling pathways), it might then increase cellular metabolism and consequently oxidative burden, leading to an increased tendency of ferroptosis. However, such a generalization is far from the reality. For example, the Myc oncogene, which promotes cell growth and upregulates a variety of cellular metabolic machines, has been reported to suppress, instead of potentiate, ferroptosis (Jiang et al., 2017). Oncogenic Ras signaling has been reported to both sensitize (Dixon et al., 2012) and downregulate (Schott et al., 2015) ferroptosis. The p53 tumor suppressor has been reported to sensitize to ferroptosis when p53 is expressed at basal levels in non-transformed cells (Jennis et al., 2016; Jiang et al., 2015) but to inhibit ferroptosis when p53 is stimulated pharmacologically (Tarangelo et al., 2018; Xie et al., 2017) or when p53 is expressed in some, but not all, transformed cells (Xie et al., 2017).
The following reasons may explain how a specific signaling pathway exerts distinctive and sometime opposite effects on ferroptosis, barring some rare cases in which the discrepancy was due to experimental errors, such as the use of pharmacological agents with off-target effect. First, the function of a signaling process may be transient and context-dependent; thus, its effect on ferroptosis could also be varied under different contexts. Second, a signaling pathway may activate multiple downstream events with opposing effect on ferroptosis; e.g., while it may increase cellular metabolism and thus oxidative burden, it could also simultaneously activate an antioxidant response, and thus its eventual impact on ferroptosis would be determined by how the counterbalance of these two responses plays out under the specific condition. Therefore, when analyzing the functional communication between a signal transduction pathway and ferroptosis, specific biological or pathological contexts, including cell or tissue types, their genetic background, and growth/nutrient status, should all be considered.
Another important parameter for consideration is intercellular communication. It was reported recently that ferroptosis can be regulated in a non-cell-autonomous manner via cadherin-mediated cell-cell contacts (Wu et al., 2019). Such intercellular interaction suppresses ferroptosis by transducing signals to intracellular merlin/NF2-Hippo-YAP pathway. As a transcription co-activator, YAP stimulates the expression of multiple ferroptosis mediators, including TFRC and ACSL4. Therefore, inhibition of YAP activity by cadherin-induced Hippo pathway renders cells more resistant to ferroptosis. Consistent with this finding, TAZ, a homolog of YAP frequently overexpressed in renal cancer, was recently reported to sensitize renal cancer cells to ferroptosis induction (Yang et al., 2019).
Regulation of programmed cell death by intercellular signaling is not new, with death receptor-mediated apoptosis, or extrinsic apoptosis, as a classic example (Jiang and Wang, 2004). However, unlike the case of extrinsic apoptosis in which one cell (expressing death ligand) induces the death of the other (expressing death receptor), cadherin-mediated intercellular signaling protects all involved cells. Speculatively, this might be an ancient mechanism protecting the cellular community and multicellular organisms from oxidative stress and ferroptosis, the adverse and inevitable side effects of redox-based cellular metabolism (ancestral metazoan species such as cnidarians and placozoans encode cadherins (Gul et al., 2017)).
Potential physiological functions of ferroptosis
As side products of redox-based metabolism, ROS can also play physiologically beneficial roles in mediating signaling events (Holmstrom and Finkel, 2014). We can wonder if the more devastating cell lethal effect of redox metabolism, i.e., ferroptosis, can also be harnessed for specific physiological functions that benefit the organism. Direct proof for this is lacking, due to the lack of biomarkers for monitoring ferroptosis in vivo, and that ferroptosis mediators identified to date are multifunctional, making unambiguous genetic validation impossible. On the other hand, mounting evidence, although indirect, suggests that tumor suppression might be a physiological function of ferroptosis.
The potential role of ferroptosis as a native tumor suppressive mechanism is based on the functional investigation of several tumor suppressor molecules, including p53, BAP1, and fumarase. p53 can potentiate ferroptosis by transcriptionally downregulating SLC7A11, a subunit of system xc− cysteine/glutamate antiporter, and upregulating glutaminase-2 (GLS2), a mitochondrial enzyme involved in glutaminolysis (Jennis et al., 2016; Jiang et al., 2015). Strikingly, a p53 mutant defective in stimulating apoptosis and growth arrest but still capable of potentiating ferroptosis can still function to prevent tumorigenesis in vivo (Jiang et al., 2015), suggesting that the ferroptosis-promoting activity of p53 contributes to its in vivo tumor suppressive function. Similarly, BAP1, an epigenetic regulator and tumor suppressor mutated in various types of cancer, can downregulate the transcription of SLC7A11 and thus potentiate ferroptosis (Zhang et al., 2018). As one of the metabolic enzymes driving the TCA cycle, fumarase surprisingly also acts as a tumor suppressor (Tomlinson et al., 2002). The recent finding that the mitochondrial TCA cycle promotes cysteine-deprivation-induced ferroptosis provides a plausible explanation for this observation (Gao et al., 2019).
Importantly, recent work indicates that ferroptosis might also contribute to the immune surveillance-mediated tumor suppression. IFNγ secreted by CD8+ T cells can downregulate the expression of system xc− antiporter in tumor cells and thus potentiate tumor cell ferroptosis (Wang et al., 2019). This finding lends further support to the hypothesis that one of the physiological functions of ferroptosis is to suppress tumorigenesis.
Diseases
Cancer
Although the initial development of pharmacological inducers of ferroptosis was for the purpose of cancer treatment (Dixon et al., 2012), the concept that ferroptosis induction is a promising cancer therapeutic approach was solidified more recently by the observations that a plethora of therapy-resistant cancer cells, including those with strong mesenchymal and metastatic properties, are more susceptible to GPX4 loss or inhibition (Hangauer et al., 2017; Viswanathan et al., 2017). These findings suggest that ferroptosis induction may provide a unique avenue for the treatment of certain highly malignant cancers. Mechanistically, the recent finding that the E-cadherin-NF2-Hippo-YAP signaling regulates ferroptosis explains how mesenchymal cancer cells are more sensitive to ferroptosis induction (Wu et al., 2019), because loss of expression of epithelial markers, such as E-cadherin, is a hallmark of epithelial-to-mesenchymal transition (EMT) (Dongre and Weinberg, 2019). Furthermore, as E cadherin, NF2, and Hippo components Lats1 and Lats2 are tumor suppressors whose loss of function mutation lead to malignancy and metastasis, these mutations can serve as biomarkers predicting cancer cell responsiveness to ferroptosis-inducing therapies.
Which molecules can be targeted to induce cancer cell ferroptosis with a sufficient therapeutic index? GPX4 is an obvious candidate. However, since GPX4 knockout causes embryonic lethality in mice (Friedmann Angeli et al., 2014), potential adverse side effects associated with GPX4 targeting need to be considered. The system xc− cystine-glutamate antiporter is another candidate. Considering that its knockout does not cause significant abnormality in mice and that its subunit SLC7A11 is overexpressed in various cancers (McCullagh and Featherstone, 2014; Zhang et al., 2019b), this molecule might be a more attractive therapeutic target. The newly discovered ferroptosis suppressor FSP1, noted above, is a potential target as well, as it is highly expressed in some cancers and is an enzyme with a small molecule-binding pocket (Bersuker et al., 2019b; Doll et al., 2019b). However, more potent and selective inhibitors of FSP1 are needed to address its suitability as a drug target, as well as its pharmacological tractability.
Numerous proof-of-principle experiments have been conducted demonstrating the anticancer effect of ferroptosis induction in animal models. Some of these experiments, using the erastin derivative IKE to induce tumor cell ferroptosis in vivo, showed that certain cancer types and cancers with specific biomarkers are responsive to ferroptosis induction as a monotherapy (Wu et al., 2019; Zhang et al., 2019b); and some other experiments showed synergistic effects of ferroptosis induction in combination with other approaches, including radiation and immune checkpoint blockade (Lang et al., 2019; Wang et al., 2019). Collectively, ferroptosis-inducing cancer therapy is a newly emerged exciting area that needs to be further explored.
Other diseases
Mounting evidence indicates that ferroptosis is the major cell death modality associated with ischemic organ injuries, including ischemic heart disease and kidney failure (Friedmann Angeli et al., 2014; Gao et al., 2015; Linkermann et al., 2014). Ferroptosis has also been implicated as a pathological contributor to neurodegenerative diseases (DeGregorio-Rocasolano et al., 2019; Pena-Bautista et al., 2019). In addition, the death of oligodendrocytes in models of Pelizaeus-Merzbacher Disease is due to ferroptosis (Nobuta et al., 2019). Ferroptosis has also been linked to an autoinflammatory disease (Tao et al., 2020), chronic obstructive pulmonary disease caused by cigarette smoke (Yoshida et al., 2019), stroke (Alim et al., 2019; Karuppagounder et al., 2018; Tuo et al., 2017; Zille et al., 2017), cardiomyopathy (Fang et al., 2019), and Mycobacterium-tuberculosis-induced cell death and tissue necrosis (Amaral et al., 2019). Therefore, ferroptosis inhibition should be considered as a potential therapeutic intervention for these diseases, many of which lack effective treatment currently. Ferroptosis inhibitors amenable for in vivo use, such as liproxstatin-1, can be considered as proto-compounds for the development of such therapies.
Summary
While a variety of tools exist to induce and inhibit ferroptosis, and to monitor its execution, additional tools would be valuable. Many of the regulators mentioned, have insufficiently potent and specific small molecule modulators available, or none at all, to probe their function. In addition, many of the tools that exist would benefit from improved potency, selectivity, and ADME properties compatible with use in animals. Such improved tools would allow rapid and reliable testing of the potential relevance of ferroptosis to diverse physiological and pathophysiological processes in animal models. Given the expanding impact of ferroptosis, such improved tools would make a significant impact in accelerating our understanding of this process and in the development of ferroptosis-modulating therapies.
Significance
Ferroptosis is a form of regulated cell death involving oxidative damage to membrane lipids that is of increasing relevance to a diverse array of physiological and disease processes. Here, we describe how small molecule chemical probes were used to discover the phenomenon of ferroptosis and illuminate some of its key regulatory mechanisms. In addition, we describe the latest advances in the biology of ferroptosis, and its relevance to specific diseases. These latest findings open new questions and provide new opportunities for future research directions.
Acknowledgements
The research of B.R.S. is supported by grants from the National Cancer Institute (R35CA209896 and P01CA087497) and the National Institute for Neurological Disorders and Stroke (1R61NS109407). X.J. is supported by National Cancer Institute (R01CA204232 and R01CA166413).
Footnotes
Declaration of interests
B.R.S. holds equity in and serves as a consultant to Inzen Therapeutics and is an inventor on patents and patent applications related to ferroptosis. X.J. is an inventor on patents related to cell death and autophagy.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrams RP, Carroll WL, and Woerpel KA (2016). Five-Membered Ring Peroxide Selectively Initiates Ferroptosis in Cancer Cells. ACS Chem Biol 11, 1305–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alborzinia H, Ignashkova TI, Dejure FR, Gendarme M, Theobald J, Wolfl S, Lindemann RK, and Reiling JH (2018). Golgi stress mediates redox imbalance and ferroptosis in human cells. Commun Biol 1, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, Seravalli J, Ai Y, Sansing LH, Ste Marie EJ, et al. (2019). Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell 177, 1262–1279 e1225. [DOI] [PubMed] [Google Scholar]
- Amaral EP, Costa DL, Namasivayam S, Riteau N, Kamenyeva O, Mittereder L, Mayer-Barber KD, Andrade BB, and Sher A (2019). A major role for ferroptosis in Mycobacterium tuberculosis-induced cell death and tissue necrosis. J Exp Med 216, 556–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews NC, and Schmidt PJ (2007). Iron homeostasis. Annual review of physiology 69, 69–85. [DOI] [PubMed] [Google Scholar]
- Bersuker K, Hendricks J, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al. (2019a). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al. (2019b). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braughler JM, Duncan LA, and Chase RL (1986). The involvement of iron in lipid peroxidation. Importance of ferric to ferrous ratios in initiation. J Biol Chem 261, 10282–10289. [PubMed] [Google Scholar]
- Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, Song S, Tavana O, and Gu W (2019). ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol 21, 579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGregorio-Rocasolano N, Marti-Sistac O, and Gasull T (2019). Deciphering the Iron Side of Stroke: Neurodegeneration at the Crossroads Between Iron Dyshomeostasis, Excitotoxicity, and Ferroptosis. Front Neurosci 13, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Patel DN, Welsch ME, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti N, Slusher BS, et al. (2014). Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife, doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, and Stockwell BR (2019). The Hallmarks of Ferroptosis. Annual Review of Cancer Biology 3, 35–54. [Google Scholar]
- Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, Superti-Furga G, and Stockwell BR (2015). Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem Biol 10, 1604–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Grocin AG, Xavier da Silva TN, Panzilius E, Scheel C, et al. (2019a). FSP1 is a glutathione-independent ferroptosis suppressor. Nature. [DOI] [PubMed] [Google Scholar]
- Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Grocin AG, Xavier da Silva TN, Panzilius E, Scheel CH, et al. (2019b). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698. [DOI] [PubMed] [Google Scholar]
- Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolma S, Lessnick SL, Hahn WC, and Stockwell BR (2003). Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 3, 285–296. [DOI] [PubMed] [Google Scholar]
- Dongre A, and Weinberg RA (2019). New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol 20, 69–84. [DOI] [PubMed] [Google Scholar]
- Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, Menon S, Wang Z, Honda A, Pardee G, et al. (2014). Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol 16, 1069–1079. [DOI] [PubMed] [Google Scholar]
- Eaton JK, Furst L, Ruberto RA, Moosmayer D, Hillig RC, Hilpmann A, Zimmermann K, Ryan MJ, Niehues M, Badock V, et al. (2018). Targeting a Therapy-Resistant Cancer Cell State Using Masked Electrophiles as GPX4 Inhibitors. bioRxiv. [Google Scholar]
- Eaton JK, Ruberto RA, Kramm A, Viswanathan VS, and Schreiber SL (2019). Diacylfuroxans Are Masked Nitrile Oxides That Inhibit GPX4 Covalently. J Am Chem Soc 141, 20407–20415. [DOI] [PubMed] [Google Scholar]
- Fang X, Wang H, Han D, Xie E, Yang X, Wei J, Gu S, Gao F, Zhu N, Yin X, et al. (2019). Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A 116, 2672–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann F, Schenk B, Martens S, Vandenabeele P, and Fulda S (2017). Sorafenib inhibits therapeutic induction of necroptosis in acute leukemia cells. Oncotarget 8, 68208–68220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, et al. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16, 1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangadhar NM, and Stockwell BR (2006). Chemical genetic approaches to probing cell death. Curr Opin Chem Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, and Jiang X (2018). To eat or not to eat-the metabolic flavor of ferroptosis. Curr Opin Cell Biol 51, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Monian P, Pan Q, Zhang W, Xiang J, and Jiang X (2016). Ferroptosis is an autophagic cell death process. Cell Res 26, 1021–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Monian P, Quadri N, Ramasamy R, and Jiang X (2015). Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell 59, 298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, and Jiang X (2019). Role of Mitochondria in Ferroptosis. Mol Cell 73, 354–363 e353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA, Heindel DW, Zuckerman DS, Bos PH, Reznik E, et al. (2018). FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol 14, 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gout PW, Buckley AR, Simms CR, and Bruchovsky N (2001). Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia 15, 1633–1640. [DOI] [PubMed] [Google Scholar]
- Gul IS, Hulpiau P, Saeys Y, and van Roy F (2017). Evolution and diversity of cadherins and catenins. Exp Cell Res 358, 3–9. [DOI] [PubMed] [Google Scholar]
- Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, Galeas J, Dhruv HD, Berens ME, Schreiber SL, et al. (2017). Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassannia B, Wiernicki B, Ingold I, Qu F, Van Herck S, Tyurina YY, Bayir H, Abhari BA, Angeli JPF, Choi SM, et al. (2018). Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J Clin Invest 128, 3341–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatami A, Zhu C, Relano-Gines A, Elias C, Galstyan A, Jun M, Milne G, Cantor CR, Chesselet MF, and Shchepinov MS (2018). Deuterium-reinforced linoleic acid lowers lipid peroxidation and mitigates cognitive impairment in the Q140 knock in mouse model of Huntington's disease. Febs J. [DOI] [PubMed] [Google Scholar]
- Hill S, Lamberson CR, Xu L, To R, Tsui HS, Shmanai VV, Bekish AV, Awad AM, Marbois BN, Cantor CR, et al. (2012). Small amounts of isotope-reinforced polyunsaturated fatty acids suppress lipid autoxidation. Free Radic Biol Med 53, 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmans S, Vanden Berghe T, Devisscher L, Hassannia B, Lyssens S, Joossens J, Van Der Veken P, Vandenabeele P, and Augustyns K (2016). Novel Ferroptosis Inhibitors with Improved Potency and ADME Properties. J Med Chem 59, 2041–2053. [DOI] [PubMed] [Google Scholar]
- Holmstrom KM, and Finkel T (2014). Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol 15, 411–421. [DOI] [PubMed] [Google Scholar]
- Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, Kang R, and Tang D (2016). Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang KJ, Wei YH, Chiu YC, Wu SR, and Shieh DB (2019). Assessment of zero-valent iron-based nanotherapeutics for ferroptosis induction and resensitization strategy in cancer cells. Biomater Sci 7, 1311–1322. [DOI] [PubMed] [Google Scholar]
- Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, Scott JP, Cai KQ, Campbell MR, Porter DK, et al. (2016). An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev 30, 918–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, and Gu W (2015). Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang XJ, and Wang XD (2004). Cytochrome C-mediated apoptosis. Annual Review of Biochemistry 73, 87–106. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Mao C, Yang R, Yan B, Shi Y, Liu X, Lai W, Liu Y, Wang X, Xiao D, et al. (2017). EGLN1/c-Myc Induced Lymphoid-Specific Helicase Inhibits Ferroptosis through Lipid Metabolic Gene Expression Changes. Theranostics 7, 3293–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karuppagounder SS, Alin L, Chen Y, Brand D, Bourassa MW, Dietrich K, Wilkinson CM, Nadeau CA, Kumar A, Perry S, et al. (2018). N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E2 to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann Neurol 84, 854–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny EM, Fidan E, Yang Q, Anthonymuthu TS, New LA, Meyer EA, Wang H, Kochanek PM, Dixon CE, Kagan VE, et al. (2019). Ferroptosis Contributes to Neuronal Death and Functional Outcome After Traumatic Brain Injury. Crit Care Med 47, 410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Muller C, Zandkarimi F, Merl-Pham J, Bao X, Anastasov N, Kossl J, et al. (2020). GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent Sci 6, 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, Liao P, Zhou J, Zhang Q, Dow A, et al. (2019). Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discov. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larraufie MH, Yang WS, Jiang E, Thomas AG, Slusher BS, and Stockwell BR (2015). Incorporation of metabolically stable ketones into a small molecule probe to increase potency and water solubility. Bioorg Med Chem Lett, July 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, Tyagi S, Ma L, Westbrook TF, Steinberg GR, et al. (2020). Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol 22, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, Ajani JA, Xiao Q, Liao Z, Wang H, et al. (2020). The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res 30, 146–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewerenz J, Ates G, Methner A, Conrad M, and Maher P (2018). Oxytosis/Ferroptosis-(Re-) Emerging Roles for Oxidative Stress-Dependent Non-apoptotic Cell Death in Diseases of the Central Nervous System. Front Neurosci 12, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Duan L, Yuan S, Zhuang X, Qiao T, and He J (2019a). Ferroptosis inhibitor alleviates Radiation-induced lung fibrosis (RILF) via down-regulation of TGF-beta1. Journal of inflammation 16, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhuang X, and Qiao T (2019b). Role of ferroptosis in the process of acute radiation-induced lung injury in mice. Biochem Biophys Res Commun 519, 240–245. [DOI] [PubMed] [Google Scholar]
- Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz PS, et al. (2014). Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A 111, 16836–16841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, Ward CC, Cho K, Patti GJ, Nomura DK, et al. (2019). Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem Biol 26, 420–432 e429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancias JD, Wang X, Gygi SP, Harper JW, and Kimmelman AC (2014). Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens S, Jeong M, Tonnus W, Feldmann F, Hofmans S, Goossens V, Takahashi N, Brasen JH, Lee EW, Van der Veken P, et al. (2017). Sorafenib tosylate inhibits directly necrosome complex formation and protects in mouse models of inflammation and tissue injury. Cell death & disease 8, e2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullagh EA, and Featherstone DE (2014). Behavioral characterization of system xc- mutant mice. Behav Brain Res 265, 1–11. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Miyamoto M, Sastre A, Schnaar RL, and Coyle JT (1989). Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 2, 1547–1558. [DOI] [PubMed] [Google Scholar]
- Nobuta H, Yang N, Ng YH, Marro SG, Sabeur K, Chavali M, Stockley JH, Killilea DW, Walter PB, Zhao C, et al. (2019). Oligodendrocyte Death in Pelizaeus-Merzbacher Disease Is Rescued by Iron Chelation. Cell stem cell 25, 531–541 e536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen OE, Kalhan SC, and Hanson RW (2002). The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem 277, 30409–30412. [DOI] [PubMed] [Google Scholar]
- Pena-Bautista C, Vento M, Baquero M, and Chafer-Pericas C (2019). Lipid peroxidation in neurodegeneration. Clin Chim Acta 497, 178–188. [DOI] [PubMed] [Google Scholar]
- Schott C, Graab U, Cuvelier N, Hahn H, and Fulda S (2015). Oncogenic RAS Mutants Confer Resistance of RMS13 Rhabdomyosarcoma Cells to Oxidative Stress-Induced Ferroptotic Cell Death. Frontiers in oncology 5, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Radmark O, Wurst W, et al. (2008). Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell metabolism 8, 237–248. [DOI] [PubMed] [Google Scholar]
- Shah R, Margison K, and Pratt DA (2017). The Potency of Diarylamine Radical-Trapping Antioxidants as Inhibitors of Ferroptosis Underscores the Role of Autoxidation in the Mechanism of Cell Death. ACS Chem Biol 12, 2538–2545. [DOI] [PubMed] [Google Scholar]
- Shah R, Shchepinov MS, and Pratt DA (2018a). Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent Sci ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah R, Shchepinov MS, and Pratt DA (2018b). Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent Sci 4, 387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Hayano M, Pagano NC, and Stockwell BR (2016a). Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity. Cell Chem Biol 23, 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ, and Stockwell BR (2016b). Global Survey of Cell Death Mechanisms Reveals Metabolic Regulation of Ferroptosis. Nat Chem Biol 12, 497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintoku R, Takigawa Y, Yamada K, Kubota C, Yoshimoto Y, Takeuchi T, Koshiishi I, and Torii S (2017). Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci 108, 2187–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southon A, Szostak K, Acevedo KM, Dent KA, Volitakis I, Belaidi AA, Barnham KJ, Crouch PJ, Ayton S, Donnelly PS, et al. (2020). Cu(II) (atsm) inhibits ferroptosis: Implications for treatment of neurodegenerative disease. Br J Pharmacol 177, 656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell BR (2000). Chemical genetics: ligand-based discovery of gene function. Nature reviews Genetics 1, 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE, et al. (2017). Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S, Sagara Y, Liu Y, Maher P, and Schubert D (1998). The regulation of reactive oxygen species production during programmed cell death. J Cell Biol 141, 1423–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao P, Sun J, Wu Z, Wang S, Wang J, Li W, Pan H, Bai R, Zhang J, Wang Y, et al. (2020). A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1. Nature 577, 109–114. [DOI] [PubMed] [Google Scholar]
- Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, and Dixon SJ (2018). p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell reports 22, 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesfay L, Paul BT, Konstorum A, Deng Z, Cox AO, Lee J, Furdui CM, Hegde P, Torti FM, and Torti SV (2019). Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res 79, 5355–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, et al. (2002). Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 30, 406–410. [DOI] [PubMed] [Google Scholar]
- Tuo Q-Z, Lei P, Jackman KA, Li X-I, Xiong H, Li X-L, Liuyang Z-Y, Roisman L, Zhang S-T, Ayton S, et al. (2017). Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Molecular Psychiatry, in press. [DOI] [PubMed] [Google Scholar]
- Tyurina YY, Tyurin VA, Anthonymuthu T, Amoscato AA, Sparvero LJ, Nesterova AM, Baynard ML, Sun W, He R, Khaitovich P, et al. (2019). "Redox lipidomics technology: Looking for a needle in a haystack". Chem Phys Lipids 221, 93–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. (2017). Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, et al. (2019). CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiwer M, Bittker JA, Lewis TA, Shimada K, Yang WS, MacPherson L, Dandapani S, Palmer M, Stockwell BR, Schreiber SL, et al. (2012). Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg Med Chem Lett 22, 1822–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel SE, Tyurina YY, Zhao J St., Croix CM, Dar HH, Mao G, Tyurin VA, Anthonymuthu TS, Kapralov AA, Amoscato AA, et al. (2017). PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo JH, Shimoni Y, Yang WS, Subramaniam P, Iyer A, Nicoletti P, Rodriguez Martinez M, Lopez G, Mattioli M, Realubit R, et al. (2015). Elucidating Compound Mechanism of Action by Network Perturbation Analysis. Cell 162, 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, Chen ZN, and Jiang X (2019). Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 572, 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, Zhong M, Yuan H, Zhang L, Billiar TR, et al. (2017). The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell reports 20, 1692–1704. [DOI] [PubMed] [Google Scholar]
- Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, et al. (2007). RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S, Kumazawa S, Ishii T, Nakayama T, Itakura K, Shibata N, Kobayashi M, Sakai K, Osawa T, and Uchida K (2001). Immunochemical detection of a lipofuscin-like fluorophore derived from malondialdehyde and lysine. J Lipid Res 42, 1187–1196. [PubMed] [Google Scholar]
- Yang WH, Ding CC, Sun T, Rupprecht G, Lin CC, Hsu D, and Chi JT (2019). The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell reports 28, 2501–2508 e2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, and Stockwell BR (2016). Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 113, E4966–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Sriramaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. (2014). Regulation of ferroptotic cancer cell death by Gpx4. Cell 156, 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, and Stockwell BR (2008). Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 15, 234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye LF, Chaudhary KR, Zandkarimi F, Harken AD, Kinslow CJ, Upadhyayula PS, Dovas A, Higgins DM, Tan H, Zhang Y, et al. (2020). Radiation-Induced Lipid Peroxidation Triggers Ferroptosis and Synergizes with Ferroptosis Inducers. ACS Chem Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye LF, and Stockwell BR (2017). Transforming Lipoxygenases: PE-Specific Enzymes in Disguise. Cell 171, 501–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Minagawa S, Araya J, Sakamoto T, Hara H, Tsubouchi K, Hosaka Y, Ichikawa A, Saito N, Kadota T, et al. (2019). Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat Commun 10, 3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Li X, Zhang X, Kang R, and Tang D (2016). Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun 478, 1338–1343. [DOI] [PubMed] [Google Scholar]
- Zhang X, Xing X, Liu H, Feng J, Tian M, Chang S, Liu P, and Zhang H (2020). Ionizing radiation induces ferroptosis in granulocyte-macrophage hematopoietic progenitor cells of murine bone marrow. Int J Radiat Biol, 1–12. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, Sirohi K, Li X, Wei Y, Lee H, et al. (2018). BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol 20, 1181–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, Uchida K, O'Connor OA, and Stockwell BR (2019a). Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, Uchida K, O'Connor OA, and Stockwell BR (2019b). Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem Biol 26, 623–633 e629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zille M, Karuppagounder SS, Chen Y, Gough PJ, Bertin J, Finger J, Milner TA, Jonas EA, and Ratan RR (2017). Neuronal Death After Hemorrhagic Stroke In Vitro and In Vivo Shares Features of Ferroptosis and Necroptosis. Stroke 48, 1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, and Sollott SJ (2014). Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 94, 909–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, Sandoval-Gomez G, Clish CB, Doench JG, and Schreiber SL (2020). Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol 16, 302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]