

Abstract
Cell therapy for spinal cord injury offers the possibility of replacing lost cells after trauma to the central nervous system (CNS). In preclinical studies, synthetic hydrogels are often co-delivered to the injury site to support survival and integration of the transplanted cells. These hydrogels ideally mimic the mechanical and biochemical features of healthy CNS extracellular matrix while also providing the possibility of localized drug delivery to promote healing. In this work, we synthesize peptide-functionalized polymers that contain both a peptide sequence for incorporation into self-assembled peptide hydrogels along with bioactive peptides that inhibit scar formation. We demonstrate that peptide hydrogels formulated with the peptide-functionalized polymers possess simliar mechanical properties (soft and shear-thinning) as peptide-only hydrogels. Small angle neutron scattering analyses reveal that polymer-containing hydrogels possess larger inhomogeneous domains but that small-scale features such as mesh size remain the same as peptide-only hydrogels. We further confirm that the integrated hydrogels containing bioactive peptides exhibit thrombin inhibition activity, which has previously shown to reduce scar formation in vivo. Finally, while survival of encapsulated cells was poor, cells cultured on the hydrogels exhibited good viability. Overall, the described composite hydrogels formed from self-assembling peptides and peptide-modified polymers are promising, user-friendly materials for CNS applications in regeneration.
Graphical Abstract
Integration of a peptide-modified with self-assembling peptide hydrogels imparts bioactivity to provides a potential platform for cell therapy applications
Introduction
Spinal cord injuries (SCI) can be calamitous due to the severe potential outcomes (paralysis) and the limited regeneration that occurs post-injury. The average incidence rate of SCI this past decade was reported to be around 54 per million in the United States, 60 per million in China and 5 to 150 per million in Europe.1, 2 While vehicular crashes remain the leading cause of SCI in the United States, accounting for around 40% of SCIs, the growing elderly population has contributed to significant increases in SCIs resulting from falls (>20% of SCIs).3 Treatments for SCIs are typically multifaceted, expensive and long in duration and include pharmacological agents, surgery, electrical stimulation and rehabilitation therapy.4
Cell therapy has been proposed as a potential regenerative treatment approach for SCI.5 In concert with this approach, biomaterials that support the survival and integration of transplanted cells are being developed, and have included implantable scaffolds (hollow, porous, or fiber-based) and injectable hydrogels.6–11 Injectable hydrogels, crosslinked networks composed primarily of water, can conform to the injury site after injection, thus avoiding the need for surgical implantation. Examples of injectable hydrogels applied for SCI therapy include synthetic polymers12–16 and natural materials such as hyaluronic acid,17–20 agarose,21 and fibrin.22 Furthermore, therapeutic drugs, ranging from small molecule drugs to growth factors, can be incorporated within the hydrogels for localized delivery, thus improving drug access to the central nervous system while minimizing offsite effects.7
The inflammatory response that occurs after SCI, including the obstructive glial scar, can impede natural and cell therapy-based regeneration.23 For example, thrombin that extravasates within the CNS after injury has been reported to trigger local inflammation.24 We have demonstrated that local inhibition of thrombin through delivery of inhibitors such as hirudin and bivalirudin reduces gliosis in both mouse and rat models of spinal cord injury.12, 14 Injectable hydrogels that (i) support the viability of transplanted cells, (ii) locally deliver drugs to improve integration of transplanted cells, and (iii) are scalable and reproducible for potential clinical translation are critical in the development of cell therapies for SCI.
Zhang and colleagues reported a class of peptides with alternating hydrophilic and hydrophobic residues that self-assemble and form hydrogels in physiological conditions.25–27 The peptides assemble in β-sheet structures that interact through ionic pair formation between acidic and basic amino acids.27 While the individual peptides are susceptible to protease degradation, the assembled structures are resistant to proteolytic enzymes.26 These materials support cell attachment, including primary neurons, and are non-immunogenic in vivo, as assessed by antibody induction.27–29 Miller and colleagues optimized peptide sequences and reported that the octapeptide FEFEFKFK forms hydrogels at relatively low concentrations (8 mg/mL).30, 31
We hypothesized that self-assembling hydrogels based on the FEFEFKFK peptide (FEFK) are a potential platform for neural cell transplantation applications. Further, we proposed to integrate drug conjugated polymers with the FEFK peptides for local thrombin inhibition. In this work, we synthesize FEFK-conjugated polymers functionalized with bivalirudin, an FDA-approved thrombin inhibitor.32 We demonstrate that FEFK polymers successfully self-assemble with FEFK peptides and display active peptide drug, inhibiting thrombin (Figure 1). The resulting hydrogels support culture of neural progenitor cells when seeded on the hydrogel surface.
Figure 1.
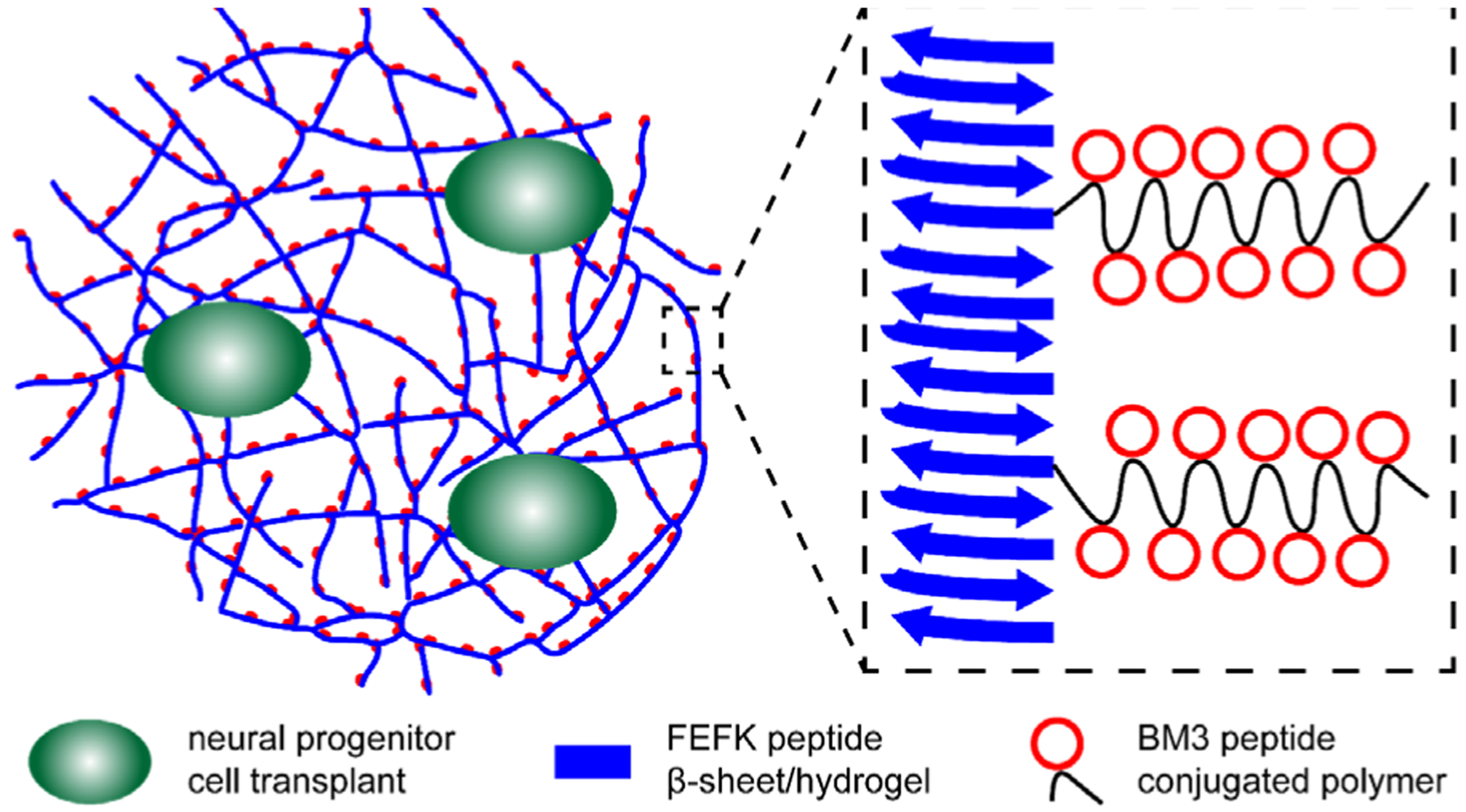
Schematic depicting hydrogel formulation with magnification showing integration of peptide-containing polymers displaying a cleavable, thrombin-inhibiting peptide (BM3, red) with self-assembling beta sheet peptides (blue) that form nanofibers. Neural progenitor cells (green) could be co-delivered with this injectable formulation that locally inhibits thrombin for better engraftment of transplanted cells.
Results and Discussion
We designed the peptide-containing polymer to include three components: a water-soluble polymer backbone, multiple bivalirudin peptides conjugated to the polymer backbone for localized thrombin inhibition and an FEFK peptide functionalized on the polymer terminus (Figure 2). The polymer was synthesized by reversible addition-fragmentation chain-transfer (RAFT) polymerization of triethylene glycol methyl ether methacrylate (TEGMA), to impart water solubility, with pyridyl disulfide ethyl methacrylate (PDSEMA), for peptide conjugation, at a 9:1 ratio using 4-cyano-4-(ethylsulfanylthiocarbonyl)sulfanylpentanoic acid (ECT) as a chain transfer agent. Control homopolymers of TEGMA were also synthesized (pTEGMA). We obtained copolymers of p(TEGMA-co-PDSEMA) with average molecular weight (MW) of 27.4 kDa and dispersity (D) of 1.17 with desired TEGMA to PDSEMA ratios confirmed by NMR and homopolymers of pTEGMA with MW of 28 kDa and D = 1.12. (Supplemental Figure 1). Bivalirudin peptide fused with a matrix metalloproteinase-3 substrate sequence (BM3) was prepared by solid phase peptide synthesis with a C-terminal cysteine for conjugation to the PDSEMA units in the copolymer. Peptide was successfully grafted to 86% of pyridyl disulfide groups, as determined by UV absorbance readings. Finally, cysteine-functionalized FEFK (sequence: NH2-CGGFEFEFKFK-CONH2) was conjugated to the polymer by radical-catalyzed reaction of the cysteine thiol with the trithiocarbonate RAFT agent, yielding dual-peptide functionalized polymer p(TEGMA-co-BM3)-FEFK or control polymer pTEGMA-FEFK.33, 34 Conjugation efficiency of the FEFK was >90%, as determined by UV measurement of a DNP-labeled FEFK and by HPLC (Supplemental Figure 2).
Figure 2.
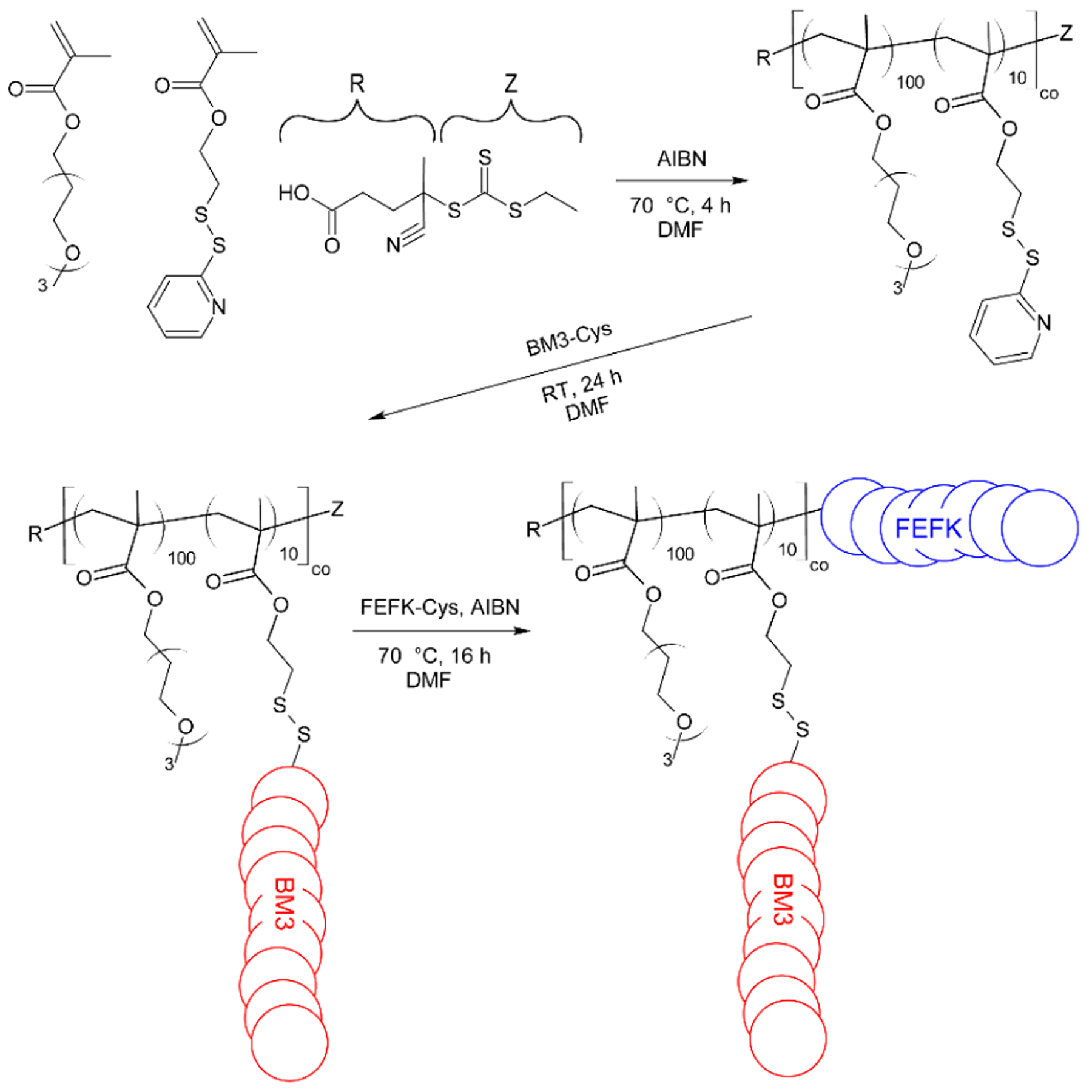
Schematic of p(TEGMA-co-BM3)-FEFK synthesis. The polymer backbone is synthesized by RAFT copolymerization of a water-soluble triethylene glycol methyl ether methacrylate monomer with a pyridyl disulfide ethyl methacrylate monomer to which the thrombin inhibitor peptide, BM3, is grafted post-polymerization via disulfide exchange. The self-assembling peptide FEFK is then conjugated to the polymer terminus by radical end-capping.
Circular dichroism analysis confirms that the FEFK peptide forms β-sheet structures with minimum ellipticity around 218 nm and maximum ellipticity around 190 nm, as previously reported for similar AEAEAKAK peptides (Figure 3A–C).27 p(TEGMA-co-BM3)-FEFK also forms beta sheets at concentrations ≥ 5 μM (Figures 3A–C). Mixtures of FEFK peptide with p(TEGMA-co-BM3)-FEFK polymer where the total FEFk peptide concentration is held constant at 5 μM also retain β-sheet structure (Figure 3D).
Figure 3.

(A-C) Circular dichroism of FEFK peptide p(TEGMA-co-BM3)-FEFK (FEFK-Polymer) at various peptide concentrations. D. Circular dichroism of mixtures FEFK peptide and p(TEGMA-co-BM3)-FEFK (FEFK-Polymer) FEFK-polymer at total peptide concentration of 5 μM.
Hydrogels were formed by dissolving peptides and polymers at 99:1 molar ratio and heating to 90 °C. Upon cooling, the materials self-assembled into free-standing gels (Figure 4A). Softer materials (<1 kPa) have been shown to promote neuronal and astrocytic differentiation of neural stem cells cultured on top of hydrogels of varying stiffnesses, whereas stiffer materials (> 7 kPa) encourage oligodendrocyte differentiation.35 Rheology studies were conducted with FEFK and mixtures of FEFK and p(TEGMA-co-BM3)-FEFK over a frequency range of 0.1–628 rad/s or 0.01–100 Hz. The storage (G’) and loss (G”) moduli were relatively constant as oscillatory frequency increased, and storage modulus was nearly one order of magnitude greater than loss modulus, confirming gelation under these conditions (Figure 4B). The Miller group previously demonstrated that the thermoresponsive polymer poly(N-iso-propylacrylamide (polyNIPAAm) conjugated to FEFK peptides also form hydrogels.36 Importantly, the Miller group has demonstrated that the stiffness of FEFK gels can be increased by the addition of sodium hydroxide during formulation.37 We observed a similar phenomenon. In the absence of sodium hydroxide during formulation (pH ~4), the hydrogels that formed were very soft (Supplemental Figure 3), and the average G’ ± 1SD were as follows: FEFK 0.027 kPa ± 0.006 kPa, pTEGMA-FEFK 0.027 kPa ± 0.007 kPa, and p(TEGMA-co-BM3)-FEFK 0.094 kPa ± 0.012 kPa. With addition of sodium hydroxide (pH ~7.4), average G’ ± 1SD were as follows - FEFK 18.8 kPa ± 3.5 kPa, pTEGMA-FEFK 16.8 kPa ± 3.3 kPa and p(TEGMA-co-BM3)-FEFK 15.0 kPa ± 2.7 kPa. Addition of pTEGMA-FEFK and p(TEGMA-co-BM3)-FEFK did not significantly affect the storage modulus. The viscosity of the FEFK and FEFK/FEFK-Polymer gels are inversely proportional to shear rate, showing characteristic ‘shear-thinning’ behaviour (Figure 4C). These shear-thinning materials therefore have suitable properties for application as injectable hydrogels into the spinal cord.
Figure 4.

A. Hydrogels formed from FEFK, FEFK with pTEGMA-FEFK polymers and FEFK and p(TEGMA-co-BM3)-FEFK polymers. B. Frequency sweep of 20 mg/mL solutions of FEFK, p(TEGMA)-FEFK and p(TEGMA-co-BM3)-FEFK at 2.5% wt. For each gel the data represents three replicates averaged together. C. Viscosity profile of 20 mg/mL solutions of p(TEGMA-co-BM3)-FEFL,p(TEGMA)-FEFK, and pure FEFK gels at 2.5% wt. All three hydrogels exhibited similar shear-thinning behavior. For each gel, the data represents three replicates averaged together.
The morphology of the FEFK, FEFK/Polymer (using control p(TEGMA-co-BM3) polymer without FEFK functionalization) and FEFK/FEFK-Polymer (FEFK mixed with p(TEGMA-co-BM3)-FEFK) hydrogels in D2O was also characterized by small angle neutron scattering (SANS) (Figure 5). SANS data on all FEFK peptide gels (19.8 mg/mL FEFK and 10 mg/mL of either p(TEGMA-co-BM3) or p(TEGMA-co-BM3)-FEFK) show a characteristic correlation peak (Q ~ 0.035Å−1) that is indicative of regular fiber meshes (D ~ 18 nm) found in fiber-forming FEFK hydrogels at higher concentrations.30 Notably, this peak feature is present in the pure FEFK hydrogel as well as in all hydrogels containing the p(TEGMA-co-BM3)-FEFK conjugates and in control samples containing p(TEGMA-co-BM3), which indicates that the small-scale features of the network, such as the mesh size, are preserved even after the incorporation of the polymer. However, SANS data at low-q values also shows significant differences between FEFK/FEFK-Polymer hydrogels compared to either pure FEFK hydrogels or FEFK/Polymer hydrogels with control polymers that are not expected to interact with peptide fibers. These changes are observed as variations in the power-law scattering behavior of FEFK hydrogels at low-q, which varies from a power-law exponent of −2.5 for the FEFK peptide hydrogel to −3 for FEFK with incorporated p(TEGMA-co-BM3)-FEFK. Such changes in scattering occurring at low-q are suggestive of the formation of larger inhomogeneous domains that frequently develop in hydrogels. It is hypothesized that the presence of the polymer may influence the kinetics of peptide self-assembly during sample gelation. Thus, though the small-scale hydrogel mesh is preserved in all samples, the larger-scale domains can be affected by the inclusion of p(TEGMA-co-BM3)-FEFK polymers with conjugated FEFK peptide domains. The fibrous structure of the FEFK-based hydrogels was further confirmed by transmission electron microscopy (TEM) (Supplemental Figure 4). Consistent with previous reports of similar self-assembling peptides, fiber lattices with diameters ~ 10–20 nm are observed.26, 38 Inclusion of FEFK-Polymer does not significantly alter the overall fiber structure over these length scales.
Figure 5.
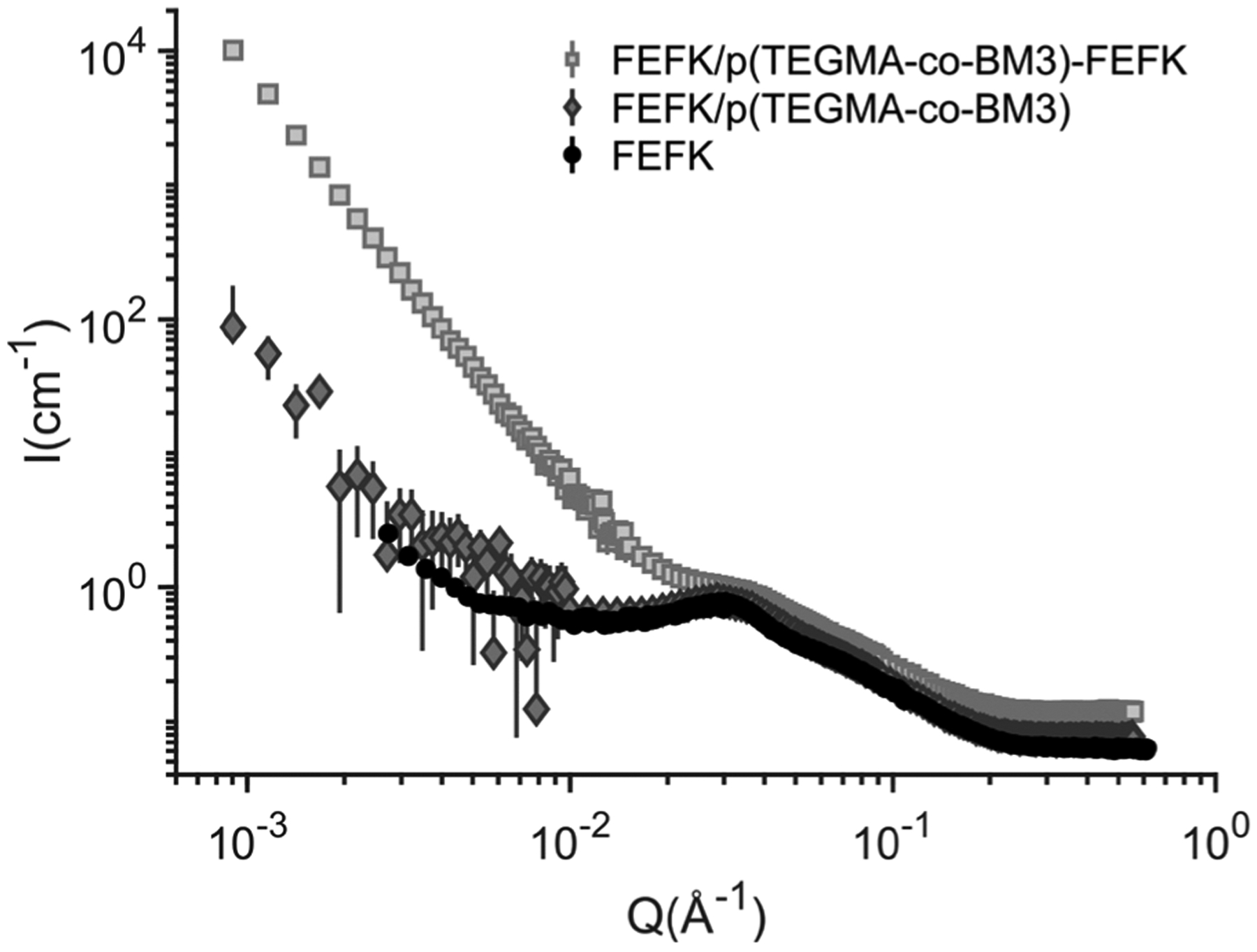
Neuron scattering of FEFK gels (20 mg/mL FEFK) formed from FEFK peptide alone and formulated with either control polymer pTEGMA-co-BM3 or with p(TEGMA-co-BM3)-FEFK.
Finally, the bioactivity and biocompatibility of the integrated material was tested. The activity of hydrogel-incorporated bivalirudin was assessed using a colorimetric thrombin inhibition assay and compared to both bivalirudin peptide and unconjugated bivalirudin peptide fused with the MMP3 substrate linker (BM3). The peptide-conjugated hydrogel showed similar thrombin inhibition to the free peptide-linker (Figure 6) but lower than the unmodified bivalirudin. We have also previously observed that bivalirudin-linker fusions show slightly compromised thrombin inhibition compared to unmodified bivalirudin (data not shown), but that activity of bivalirudin-conjugated to polymers is similar to that of the bivalirudin-linker.12 The linker provides a spacing between the hydrogel and bivalirudin so that binding to thrombin can occur without steric hindrance. Importantly, we previously reported that the level of activity observed with the bivalirudin-linker is sufficient to reduce gliosis at the spinal cord lesion site in vivo.12
Figure 6.
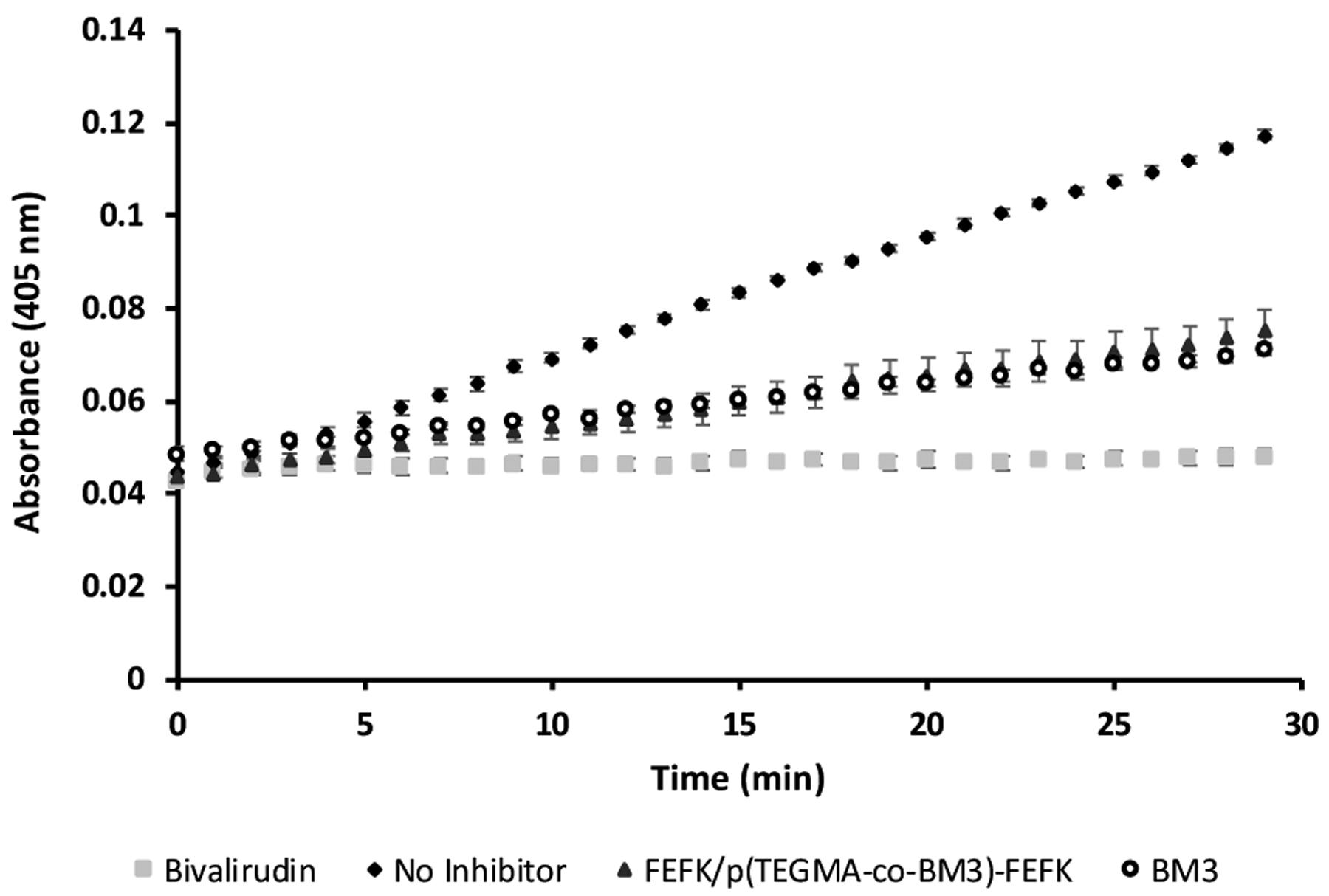
Thrombin activity over time determined by a colorimetric assay. Thrombin activity is inhibited in the presence of bivalirudin, BM3 peptide and FEFK/p(TEGMA-co-BM3)-FEFK polymers.
Murine neural progenitor cells were cultured both on top of and encapsulated within FEFK hydrogels. Cell viability, as assessed by Calcein AM (for cell viability) and ethidium bromide homodimer staining (for cell death), of encapsulated cells was poor, but cells seeded on top of FEFK hydrogels survived well (Supplemental Figure 5). This is in contrast to previous reports of extended chondrocyte culture by encapsulation within FEFK hydrogels,36 but consistent with our previous report of poor viability of cells encapsulated within oligo(ethylene glycol) methacrylate-based hydrogels.39 Based on these results, NPC viability 24h after seeding on top of FEFK/FEFK-Polymer hydrogels was assessed by Live/Dead assay with imaging by confocal microscopy (Figure 7A). Cell viability negatively correlated with increasing FEFK peptide concentration in all hydrogels tested (Figure 7B: ~70% at 5 mg/mL; ~50% at 10 mg/mL; ~20% at 20 mg/mL). The addition of p(TEGMA-co-BM3)-FEFK and pTEGMA-FEFK did not increase hydrogel toxicity beyond that observed for the FEFK peptide-only gels. Indeed, the addition of p(TEGMA-co-BM3)-FEFK actually increased viability compared to both pTEGMA-FEFK or FEFK only when [FEFK] = 20 mg/mL (50% vs. 20%). This finding was unexpected and unlikely to be attributed to the thrombin-inhibiting activity of bivalirudin, since heparin was also included in the neural progenitor cell culture media. Regardless, the FEFK/p(TEGMA-co-BM3)-FEFK formulation can support the growth of neural progenitor cells and is therefore a promising drug depot formulation to inhibit thrombin and reduce glial scarring with cell therapy applications in spinal cord injury.
Figure 7.
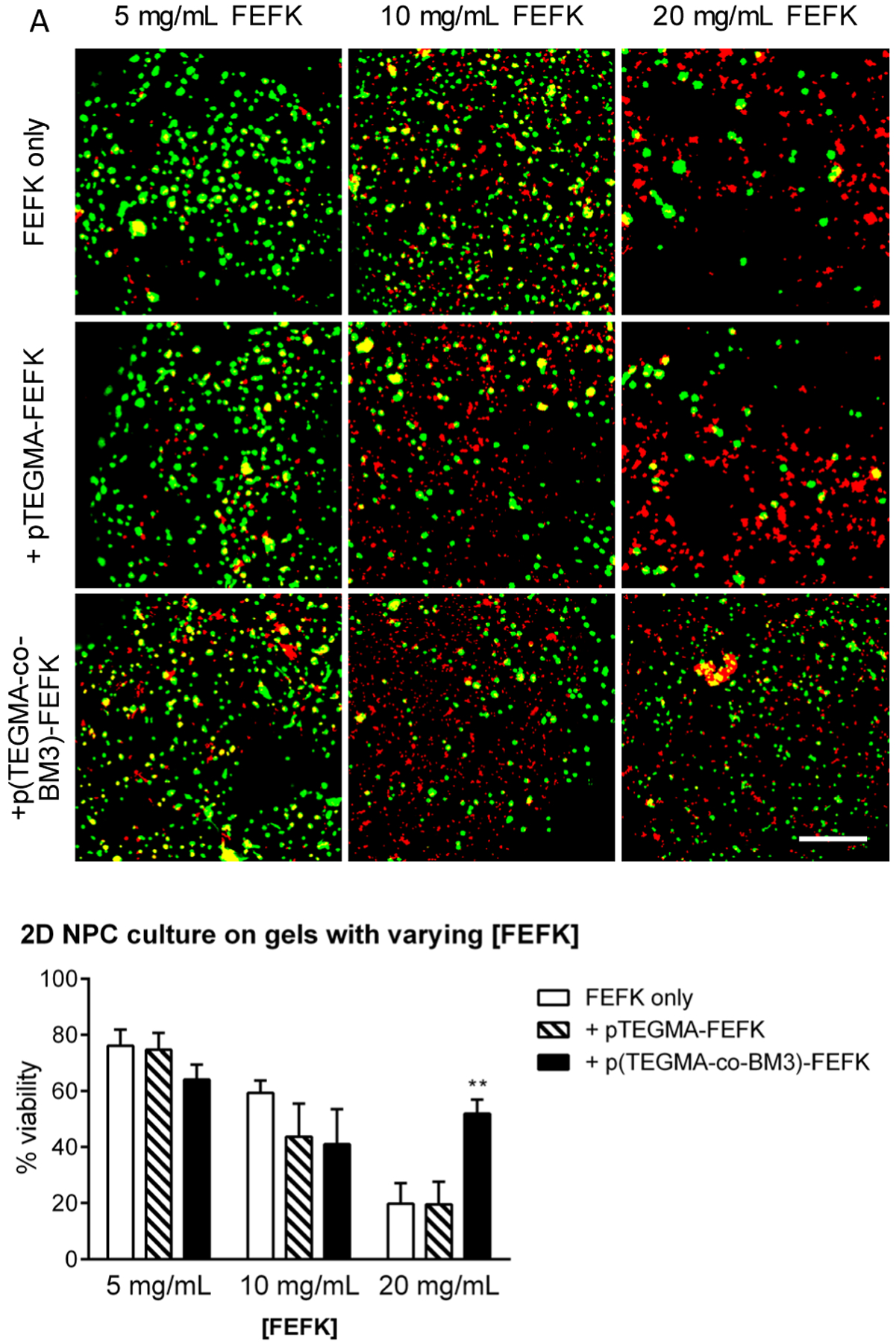
A. Confocal laser scanning microscopy images of NPCs cultured on top of various FEFK gels for 24h before staining with Calcein (green; live cells) and EtBr (red; dead cells). Each image is a maximum projection z-stack of ≥ 100 μm depth; scale bar = 100 μm. B. Quantification of cell viability in representative micrographs of 2D cultured NPC averaged over four technical replicates. Viability = (calcein+ cell count) / (total cell count). **=p<0.01 by T-test.
Conclusions
In this work, we synthesized a peptide-based polymer functionalized with a self-assembling peptide (FEFK) and thrombin-inhibiting peptides that can be released enzymatically by MMP3 (BM3). The polymer can be incorporated into FEFK peptide hydrogels with minimal impact on hydrogel properties as assessed by rheological studies, small angle neutron scattering experiments, and transmission electron microscopy. Functionalized hydrogels are equipped with the ability to locally inhibit thrombin activity and neural progenitor cells cultured in the presence of these hydrogels have good viability. Thus, these shear-thinning, self-assembled hydrogels are promising materials for serial application with cell transplantation into the spinal cord.
Experimental
Peptide synthesis.
Peptides were synthesized by FMOC solid phase peptide synthesis using a Liberty Blue microwave synthesizer using a rink-amide resin and analyzed by high performance liquid chromatography (HPLC). Peptide sequences were confirmed by MALDI mass spectrometry analysis. The peptides synthesized include: NH2-FEFEFKFK-CONH2 (FEFK), NH2-CGGFEFEFKFK-CONH2 (C-FEFK), NH2-FPRPGGGNGDFEEIPEEYLGGGRPKPVE(Nva)WRKGC-COHN2 (BM3), and NH2-CGK(DNP)GFEFEFKFK-CONH2 (C-DNP-FEFK).
Polymer synthesis.
The polymer backbone was synthesized by reversible addition fragmentation chain transfer (RAFT) copolymerization of triethylene glycol methyl ether methacrylate (TEGMA) and pyridyl disulfide ethyl methacrylate (PDSEMA) monomers using 4-cyano-4-(ethylsulfanylthiocarbonyl) sulfanylvpentanoic acid (ECT) as a chain transfer agent and azobisisobutyronitrile (AIBN as initiator at 135:15:1:0.15 molar ratio and overall monomer concentration of 5 M in dimethylformamide (DMF). The reaction flask was sealed and purged with argon for 10 minutes before being stirred at 70 °C for 18 hours in a preheated oil bath. The reaction was quenched by exposure to atmosphere and precipitated into a large excess of cold diethyl ether, and then centrifuged for 5 minutes at 5000 rpm. The supernatant ether solution was removed in order to purify the solution of monomers. The precipitation and centrifugation were repeated, and the polymer dried overnight in a vacuum chamber at room temperature. The polymer’s molecular weight and polydispersity were analyzed using gel permeation chromatography (GPC). As a control, pTEGMA homopolymer was synthesized and purified in the same manner as the co-polymer. Polymers were modified with BM3 peptide by reaction of cysteine thiols with pyridyl disulfide in the PDSEMA units of the polymer at a 15:1 ratio in DMF overnight at room temperature. The polymer was purified by dialysis against deionized water for 24 h with repeated buffer exchanges. The contents of the dialysis bag were collected, frozen, and then lyophilized. Conjugation was confirmed by reducing the resulting polymer a 10 mM solution of DTT at 37 °C for 30 min and quantifying released, unreacted 2-pyridone by absorbance measurements at 343 nm. FEFK was conjugated to the drug loaded polymer by an end capping reaction that resulted in the removal of the trithiocarbonate group as previously reported.22 The polymer-peptide conjugate was purified by dialysis against ethanol (to prevent peptide self-assembly) for 36 h with repeated dialysate changes. The final dialyzed polymer was collected by rotary evaporation to remove solvent followed by drying under vacuum.
Hydrogel formulation.
All peptide solutions were prepared at 2.5 wt% by dissolving FEFK and FEFK-polymer in doubly distilled water (ddH2O) at 99:1 molar ratio. As controls, pure FEFK peptide hydrogels were also prepared. This was the equivalent of 66.4% weight FEFK and 33.6% weight p(TEGMA-co-BM3)-FEFK. This ratio was the maximum amount of p(TEGMA-co-BM3)-FEFK that could be added while maintaining the gel structure, as determined by a series of titrations. The solutions were then vortexed and placed in an oil bath at 90 °C for 2 hours to ensure solutions were completely dissolved.
Transmission electron microscopy.
Transmission electron microscopy (TEM) was used to image the polymer/peptide solutions as previously reported.23 Peptide and polymer/peptide solutions were formed as previously described, however at a concentration of 0.5 mg/ml. Carbon-coated copper grids (400 mesh, Agar scientific) were glow discharged and placed shiny side down on the surface of a 10 ul droplet of sample for 10 seconds. The grids were placed on a 10 μl droplet of double deionized water for 10 seconds and blotted. Washed grids were placed on a 10 μl droplet of prepared and filtered uranyl acetate solution (4% w/v) for 60 seconds for the purposes of negative staining. This was blotted against double folded filter paper. Images were taken with a Tecnai G2 F20 Supertwin TEM operating at 200 kV.
Rheological studies.
Hydrogels with 20 mg/mL concentration of FEFK were formed with or without the inclusion of FEFK-polymer conjugates at 5 mg/mL by dissolving the components in diluted pH 7.4 PBS (3:7 PBS:H2O) and heating in an oven at 90 °C for 2 hr, similar to previous work.36 For every 1 mL of gel, 5 uL of 10N NaOH was added prior to the oven step. The gels were checked with pH strips before and after the addition of NaOH to confirm a pH 7 was acheived. An Anton Paar MCR302 rheometer was used with a 50 mm cone-plate setup with a 100 um gap (~800uL of sample). An environmental chamber was used to maintain temperature at 37 degrees Celsius and to prevent evaporation of water during testing. Amplitude sweeps were conducted to identify the linear viscoelastic region, and 0.1% strain was chosen for the frequency sweeps. Frequency sweeps were completed from 0.1–628 rad/s (0.01–100 Hz). An average of three replicates for each gel were used in Figures 4B and 4C.
Small angle neutron scattering.
FEFK, FEFK-p(TEGMA-co-BM3)/FEFK, and p(TEGMA-co-BM3)-FEFK were prepared as 2.5% wt hydrogels, as described in the gel formation section. D2O was used instead of H2O to increase the contrast between the sample and solvent. Samples were loaded into standard SANS cells with quartz windows. SANS measurements were completed on the NG7 beamline at the NIST Center for Neutron Research using 6 and 8.1 Å neutrons.40 The SANS data was reduced onto absolute scale using the standard NIST reduction protocols.41
Thrombin inhibition.
The activity of the BM3 conjugated onto the polymer-peptide conjugate was assayed with an in vitro thrombin colorimetric activity assay previously reported.14 Bivalirudin solution in thrombin reaction buffer (100 mM Tris, 150 mM NaCl, 0.1% PEG-8000, pH 7.5) was formed as a positive control, with a bivalirudin molar equivalent of p(TEGMA-co-BM3) in solution being formed as well. Thrombin reaction buffer alone was used as a negative control. 95.75 ul of each solution was placed into well plates, and 1 ul of thrombin (20 ug/mL) was added. The mixtures were incubated for 10 minutes at room temperature. After the 10 minutes, 6.5 uL of thrombin substrate S-2238 (1 mg/ml) was added to each mixture, and the absorbance at 405 nm was read every minute for 30 minutes using a Tecan Safire2 plate reader. To assay the activity of the gel formulation, 2.5% wt FEFK/p(TEGMA-co-BM3)-FEFK gels were formed as described in the gel formation section, 25 uL (equivalent moles of bivalirudin to the positive control and p(TEGMA-co-BM3)) of gel placed into well plates and immersed with 70.75 uL of thrombin reaction buffer. 1 uL of the thrombin solution was added and the mixture incubated for 10 minutes, after which 6.5 uL of the S-2238 solution was added and the absorbance measured as described above.
Cell viability studies.
Neural progenitor cells (NPCs) were dissociated from the forebrains of 8 week old female mice and cultured in serum-free growth media (high glucose DMEM supplemented with N2, Glutamax, heparin sulfate, FGF2, and EGF). Hydrogels with varying concentrations of FEFK (5, 10, or 20 mg/mL) were formed with or without the inclusion of FEFK-polymer conjugates at 5 mg/mL by dissolving the components in diluted pH 7.4 PBS (3:7 PBS:H2O) and heating in an oven at 90 °C for 2 hr, similar to previous work.36 Fully dissolved hydrogels were pipetted (50 μL) into wells of a 96-well plate and allowed to cool for 10 minutes at room temperature. For 2D culture, NPC growth media containing antibiotics was added to the gels and exchanged once an hour for a total of three exchanges. After the gels were equilibrated with media, NPCs were seeded onto the gels at a density of 4×104 cells/well and cultured for 24 hours. For 3D culture, 4×104 cells in 5 μL were mixed into the cooled gels using a pipette tip, immediately covered with media, and cultured for 24 hours. After 24 hours of culture, media was aspirated and gels were washed once with HBSS. Gels were then incubated in 1X LIVE/DEAD viability stain (Invitrogen) in HBSS for 30 minutes at 37 °C/5% CO2 according to the manufacturer’s instructions. Viability stain was aspirated, gels were washed once with HBSS, and then imaged in HBSS using a custom Leica SP8X confocal laser scanning microscope (Keck Microscopy Center, University of Washington). Cell counting was performed in MATLAB using a basic disk-shaped watershed segmentation algorithm available upon request.
Supplementary Material
Acknowledgements
This work was supported by a DOD SCIRP Investigator Initiated Award (SC130249) and by NIH R01NS064404. We acknowledge support from the National Institutes of Health (S10 OD016240) to the W.M. Keck Center for Advanced Studies in Neural Signalling and the assistance of Keck Center manager Dr. Nathaniel Peters.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Jain NB, Ayers GD, Peterson EN, Harris MB, Morse L, O’Connor KC and Garshick E, Jama, 2015, 313, 2236–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jazayeri SB, Beygi S, Shokraneh F, Hagen EM and Rahimi-Movaghar V, European spine journal, 2015, 24, 905–918. [DOI] [PubMed] [Google Scholar]
- 3.Chen Y, He Y and DeVivo MJ, Archives of physical medicine and rehabilitation, 2016, 97, 1610–1619. [DOI] [PubMed] [Google Scholar]
- 4.Ahuja CS, Wilson JR, Nori S, Kotter MR, Druschel C, Curt A and Fehlings MG, Nature reviews Disease primers, 2017, 3, 17018. [DOI] [PubMed] [Google Scholar]
- 5.Assinck P, Duncan GJ, Hilton BJ, Plemel JR and Tetzlaff W, Nature neuroscience, 2017, 20, 637. [DOI] [PubMed] [Google Scholar]
- 6.Führmann T, Anandakumaran PN and Shoichet MS, Advanced healthcare materials, 2017, 6, 1601130. [DOI] [PubMed] [Google Scholar]
- 7.Ziemba AM and Gilbert RJ, Frontiers in pharmacology, 2017, 8, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hassannejad Z, Zadegan SA, Vaccaro AR, Rahimi-Movaghar V and Sabzevari O, Injury, 2019, 50, 278–285. [DOI] [PubMed] [Google Scholar]
- 9.Marchini A, Raspa A, Pugliese R, El Malek MA, Pastori V, Lecchi M, Vescovi AL and Gelain F, Proceedings of the National Academy of Sciences, 2019, 116, 7483–7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raspa A, Pugliese R, Maleki M and Gelain F, Biotechnology and bioengineering, 2016, 113, 253–259. [DOI] [PubMed] [Google Scholar]
- 11.Tran KA, Partyk PP, Jin Y, Bouyer J, Fischer I and Galie PA, Acta Biomaterialia, 2020. [DOI] [PubMed] [Google Scholar]
- 12.Chu DS, Sellers DL, Bocek MJ, Fischedick AE, Horner PJ and Pun S, Biomaterials science, 2015, 3, 41–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piantino J, Burdick J, Goldberg D, Langer R and Benowitz L, Experimental neurology, 2006, 201, 359–367. [DOI] [PubMed] [Google Scholar]
- 14.Sellers DL, Kim TH, Mount CW, Pun SH and Horner PJ, Biomaterials, 2014, 35, 8895–8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Estrada V, Brazda N, Schmitz C, Heller S, Blazyca H, Martini R and Müller HW, Neurobiology of disease, 2014, 67, 165–179. [DOI] [PubMed] [Google Scholar]
- 16.Woerly S, Petrov P, Sykova E, Roitbak T, Simonova Z and Harvey A, Tissue engineering, 1999, 5, 467–488. [DOI] [PubMed] [Google Scholar]
- 17.Baumann MD, Kang CE, Tator CH and Shoichet MS, Biomaterials, 2010, 31, 7631–7639. [DOI] [PubMed] [Google Scholar]
- 18.Gupta D, Tator CH and Shoichet MS, Biomaterials, 2006, 27, 2370–2379. [DOI] [PubMed] [Google Scholar]
- 19.Khaing ZZ, Milman BD, Vanscoy JE, Seidlits SK, Grill RJ and Schmidt CE, Journal of Neural Engineering, 2011, 8, 046033. [DOI] [PubMed] [Google Scholar]
- 20.Mothe AJ, Tam RY, Zahir T, Tator CH and Shoichet MS, Biomaterials, 2013, 34, 3775–3783. [DOI] [PubMed] [Google Scholar]
- 21.Jain A, Kim Y-T, McKeon RJ and Bellamkonda RV, Biomaterials, 2006, 27, 497–504. [DOI] [PubMed] [Google Scholar]
- 22.Wilems TS, Pardieck J, Iyer N and Sakiyama-Elbert SE, Acta biomaterialia, 2015, 28, 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fitch MT and Silver J, Experimental neurology, 2008, 209, 294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nishino A, Suzuki M, Ohtani H, Motohashi O, Umezawa K, Nagura H and Yoshimoto T, Journal of neurotrauma, 1993, 10, 167–179. [DOI] [PubMed] [Google Scholar]
- 25.Zhang S, Nature biotechnology, 2003, 21, 1171. [DOI] [PubMed] [Google Scholar]
- 26.Zhang S, Holmes T, Lockshin C and Rich A, Proceedings of the National Academy of Sciences, 1993, 90, 3334–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang S, Holmes TC, DiPersio CM, Hynes RO, Su X and Rich A, Biomaterials, 1995, 16, 1385–1393. [DOI] [PubMed] [Google Scholar]
- 28.Holmes TC, de Lacalle S, Su X, Liu G, Rich A and Zhang S, Proceedings of the National Academy of Sciences, 2000, 97, 6728–6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Semino CE, Kasahara J, Hayashi Y and Zhang S, Tissue engineering, 2004, 10, 643–655. [DOI] [PubMed] [Google Scholar]
- 30.Saiani A, Mohammed A, Frielinghaus H, Collins R, Hodson N, Kielty C, Sherratt M and Miller A, Soft Matter, 2009, 5, 193–202. [Google Scholar]
- 31.Mohammed A, Miller AF and Saiani A, 2007.
- 32.Warkentin TE, Greinacher A and Koster A, Thrombosis and haemostasis, 2008, 99, 830–839. [DOI] [PubMed] [Google Scholar]
- 33.Harrisson S, Macromolecules, 2009, 42, 897–898. [Google Scholar]
- 34.Lamm RJ, Lim EB, Weigandt KM, Pozzo LD, White NJ and Pun SH, Biomaterials, 2017, 132, 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leipzig ND and Shoichet MS, Biomaterials, 2009, 30, 6867–6878. [DOI] [PubMed] [Google Scholar]
- 36.Maslovskis A, Tirelli N, Saiani A and Miller AF, Soft Matter, 2011, 7, 6025–6033. [Google Scholar]
- 37.Mujeeb A, Miller AF, Saiani A and Gough JE, Acta biomaterialia, 2013, 9, 4609–4617. [DOI] [PubMed] [Google Scholar]
- 38.Leon EJ, Verma N, Zhang S, Lauffenburger DA and Kamm RD, Journal of Biomaterials Science, Polymer Edition, 1998, 9, 297–312. [DOI] [PubMed] [Google Scholar]
- 39.Elias PZ, Liu GW, Wei H, Jensen MC, Horner PJ and Pun SH, Journal of controlled release, 2015, 208, 76–84. [DOI] [PubMed] [Google Scholar]
- 40.Glinka C, Barker J, Hammouda B, Krueger S, Moyer J and Orts W, Journal of applied crystallography, 1998, 31, 430–445. [Google Scholar]
- 41.Kline SR, Journal of applied crystallography, 2006, 39, 895–900. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.