

Abstract
Nighttime lighting is one of the great conveniences of modernization; however, there is mounting evidence that inopportune light exposure can disrupt physiological and behavioral functions. Hospital patients may be particularly vulnerable to the consequences of light at night due to their compromised physiological state. Cardiac arrest/cardiopulmonary resuscitation (CA) was used to test the hypothesis in mice that exposure to dim light at night impairs central nervous system (CNS) recovery from a major pathological insult. Mice exposed to dim light at night (5 lux) had higher mortality in the week following cardiac arrest compared to mice housed in dark nights (0 lux). Neuronal damage was significantly greater in surviving mice exposed to dim light at night after CA versus those housed in dark nights. Dim light at night may have elevated neuronal damage by amplifying pro-inflammatory pathways in the CNS; Iba1 immunoreactivity (an indication of microglia activation) and pro-inflammatory cytokine expression were elevated in mice exposed to dim light at night post-CA. Furthermore, selective inhibition of IL-1β or TNFα ameliorated damage in mice exposed to dim light at night. The effects of light at night on CA outcomes were also prevented by using a wavelength of nighttime light that has minimal impact on the endogenous circadian clock, suggesting that replacing broad-spectrum nighttime light with specific circadian-inert wavelengths could be protective. Together, these data indicate that exposure to dim light at night after global cerebral ischemia increases neuroinflammation, in turn exacerbating neurological damage and potential for mortality.
Keywords: circadian, light pollution, cardiac arrest, microglia, neuroinflammation, cytokines
Introduction
The ability to artificially illuminate the night is one of the greatest conveniences of modernization. However, there is mounting evidence that chronic exposure to light at night can have negative health consequences including increasing the risk for developing cancer, diabetes, obesity, and heart disease (Bedrosian et al., 2016; Fonken and Nelson, 2014; Lunn et al., 2017; Stevens and Zhu, 2015). In contrast, the potential health consequences of acute exposure to light at night remain underexplored. Patients recovering in hospitals may be particularly vulnerable to inopportune light exposure as circadian disruption and sleep/wake disturbances are common after trauma (for examples see (Ayalon et al., 2007; Gaudet et al., 2018a; Gaudet et al., 2018b; Giannoccaro et al., 2013; Jain et al., 2004). We selected the cardiac arrest/cardiopulmonary resuscitation (CA) mouse model of global cerebral ischemia to test the hypothesis that light at night increases neuronal vulnerability because light at night creates a pro-inflammatory neuroenvironment (Bedrosian et al., 2013b; Fonken and Nelson, 2013) and inflammation is an important factor influencing the evolution of neurological damage after cerebral ischemia (Eltzschig and Eckle, 2011).
Global cerebral ischemia damages the central nervous system (CNS), affecting patient survival, as well as long-term cognitive performance and psychological health in survivors. Reducing mortality and minimizing CNS damage to improve patient outcome are essential goals of cardiovascular and cerebrovascular research. Importantly, the majority of permanent CNS damage resulting from cerebral ischemia is mediated by secondary processes that evolve over the first several hours to days after ischemic injury (Eltzschig and Eckle, 2011; Fu et al., 2015; Iadecola and Anrather, 2011). Although several mechanisms contribute to damage following ischemic injury, including energetic failure, excitotoxicity, and oxidative damage (reviewed in (Eltzschig and Eckle, 2011)) manipulation of inflammatory responses are considered a prime target for improving recovery. Following cerebral ischemia, an inflammatory response is triggered by activation of microglia and astrocytes with a corresponding upregulation of pro-inflammatory cytokines (Iadecola and Anrather, 2011). In particular, interleukin-1beta (IL1β) and tumor necrosis factor alpha (TNFα) are pro-inflammatory cytokines that are upregulated within hours following global cerebral ischemia (Saito et al., 1996; Weil et al., 2008) and may remain elevated for weeks (Espinosa-Garcia et al., 2017; Norman et al., 2011), with the potential to exacerbate injury (Fu et al., 2015). The hippocampus is particularly vulnerable to delayed damage following global cerebral ischemia in both animal models (Kirino, 1982) and humans (Horn and Schlote, 1992; Petito et al., 1987). Hippocampal damage is a proxy for overall recovery, as increased hippocampal damage is associated with elevated mortality and greater behavioral deficits following global ischemia (Espinosa-Garcia et al., 2017; Neigh et al., 2009; Neigh et al., 2004; Norman et al., 2010). The trajectory of damage is likely determined within the first several hours after resuscitation, as the inflammatory response takes shape (Eltzschig and Eckle, 2011). Thus, if the recovery environment alters the inflammatory response to cerebral ischemia, then it may play an important role in influencing the extent of neuronal damage that develops following cerebral ischemia.
In particular, hospital lighting warrants examination as a factor that may influence patient recovery because light at night, to which ICU patients are commonly exposed (Durrington et al., 2017), has the potential to dysregulate circadian rhythms and thereby shift the CNS inflammatory mileu (Fonken et al., 2015; Fonken and Nelson, 2013). Light is the most potent entraining signal for the mammalian circadian clock (suprachiasmatic nuclei; SCN): extrinsic light information is conveyed from the retina to the SCN via the retinohypothalamic tract. Light input to the SCN coordinates internal signals, synchronizing daily physiological rhythms to the external light-dark cycle (Reppert and Weaver, 2002). Circadian rhythms are important for optimizing homeostatic functions, including those associated with the immune system (Lange et al., 2010). Indeed, several weeks of exposure to 5 lux of dim light at night (dLAN) elevates expression of pro-inflammatory cytokines in the brains of otherwise healthy rodents (Bedrosian et al., 2013b). However, environmental factors may influence inflammation much more rapidly; a recent laboratory study of healthy, young adults demonstrated that inverting behavioral and environmental cycles for as little as three days was sufficient to significantly increase serum concentrations of high sensitivity c-reactive protein (hsCRP; a marker of systemic inflammation) and pro-inflammatory cytokines (Morris et al., 2016). Therefore, if dim light at night promotes inflammation, then it could exacerbate outcome from CA. We hypothesized that light exposure at night would potentiate injury following CA in male mice. Consistent with our hypotheses, exposure to light at night following CA enhanced acute cytokine responses and increased neuronal death. Moreover, the effects of light at night were ameliorated by selective inhibition of IL1β or TNFα or the use of a wavelength of nighttime light that does not activate photosensitive retinal ganglion cells.
METHODS
Animals.
8-week old male Swiss Webster mice (~30g; Charles River, Kingston, NY) were housed in a temperature- and humidity-controlled vivarium and provided ad libitum access to food and water. Mice were left unmanipulated for > 1 week to recover from the effects of shipping and adjust to a 14:10 light/dark (LD) cycle prior to experimental manipulations. All experimental procedures were conducted in accordance with Guide for the Care and Use of Laboratory Animals and approved by the Ohio State University Institutional Animal Care and Use Committee. Efforts were made to minimize animal use and discomfort.
Cardiac arrest and cardiopulmonary resuscitation procedure.
Mice were anesthetized with 3% isoflurane in air, intubated, and maintained thereafter on 1.5% isoflurane. Mice were ventilated a tidal volume of 150 μL at a respiratory rate of 160 breaths/min. Head and body temperature were monitored with temperature probes. A PE10 catheter was placed into the right jugular vein for epinephrine (EPI) and potassium chloride (KCl) administration. Blood pressure was monitored through a cannula inserted into the right femoral artery and connected to a blood pressure transducer (Columbus, Instruments). Mice were stabilized for 10 min and blood pressure and temperature recorded at 1 min intervals (Fig. S1). Following the 10 min acclimation, body and tail (but not head) temperature were lowered by circulating cold water through a coil system beneath the mouse to induce peripheral hypothermia restricting damage to the CNS during the CA/CPR procedure. CA was induced with an injection of KCL (50 μl, 0.5 M, 4°C) into the jugular catheter and the mouse was disconnected from the ventilator. Once a body temperature of 27°C was reached after approximately 4 min of arrest slow re-warming via a heat lamp and thermal blanket began. After 7 min 45 sec of arrest mice were reattached to the ventilator and 100 % oxygen at a tidal volume of 150 μL and a respiratory rate of 160 breaths/min was ventilated. After 8 min of arrest CPR was initiated with an injection of EPI (16 μg in 0.6 ml saline, 37°C) into the jugular catheter and chest compressions (300/min); 0.5 μg injections of EPI were administered until the mouse resuscitated (with a maximal dose of 32 μg). Mice were maintained on 100% oxygen for 25 min after return of spontaneous circulation and catheters were removed and incisions sutured.
Lighting manipulations.
Following a monitored post-operative recovery period (approximately 2 h), mice were either placed back in dark night housing room (control LD; 14h 150 lux: 10h 0 lux) or mice were placed in a room with a dim light at night cycle (dLAN; 14 h 150 lux: 10 h 0 lux). Both the bright and dim lights were from fluorescent light sources and consisted of ‘cool white’ light composed of wavelengths distributed across the visible spectrum including blue wavelengths, and light intensity was measured inside the animal cage. In the experiment involving dim red light, 5 lux of 636 nm red light was provided.
Tissue collection for staining.
Seven days following the cardiac arrest/cardiopulmonary resuscitation or sham surgery, surviving mice were individually brought into a procedure room, anesthetized with isoflurane vapors and a blood sample was collected via the retro-orbital sinus. Mice then received a lethal injection of sodium pentobarbital and were perfused transcardially with ice-cold 0.1M PBS followed by 50 ml of 4% paraformaldehyde. Brains were post-fixed overnight, cryoprotected in 30% sucrose, frozen on crushed dry ice, and stored at −80°C. 14 μm brain sections were sliced at −22°C using a cryostat and thaw mounted onto Super Frost Plus slides (Fisher, Hampton, NH). Sections were taken in a series of 10 slides so that all slides contained non-consecutive sections for analysis and slides were stored at −20°C until stained with Fluoro-Jade C (FJ-C) or Iba1 using procedures already established in our lab (Weil et al., 2009). Fluoro-Jade C. Cell death was quantified by labeling degenerating neurons with the fluorescein derivative FJ-C (Millipore, Temecula, CA). Mounted sections were thoroughly dried on a slide warmer, immersed in a basic ethanol solution (80% EtOH with 1% NaOH) and rinsed with 70% ethanol followed by water. Slides were placed in a 0.06% potassium permanganate solution for 10 min and rinsed twice. Sections were simultaneously incubated in FJ-C (0.0001% in a 1% acetic acid solution) and counterstained with DAPI (Sigma, St. Louis). Slides were rinsed, dried, cleared in xylene, and then coverslipped with DPX (Sigma). FJ positive cells were counted in the CA1, CA3, and DG of both hippocampal (approximately −1.70 to −2.30 from Bregma, example of selected regions highlighted Fig. S2) hemispheres by a condition blind experimenter using a Nikon E800 microscope at 20X magnification. Cells were counted in two non-consecutive sections (sections were > 140 μm apart to avoid counting duplicate cells) and averaged. Microglia. Microglia were visualized using an Iba1 directed antibody. Slides were dried, rinsed in PBS, and blocked with bovine serum albumin (BSA). Slides were incubated at room temperature for 24 h with rabbit anti-Iba1 antibody (Wako, Richmond, VA) diluted 1:1000 in PBS containing 0.1% Triton-X and BSA. Slides were rinsed and incubated with biotinylated goat anti-rabbit secondary antibody (1:1000; Vector Labs, Burlingame, CA) for 1 h. Sections were quenched in H2O2 in methanol, rinsed, and treated with Elite ABC reagent for 60 min (Vector). Sections were rinsed, developed with DAB (Vector), rinsed, dehydrated, cleared, and coverslipped. Photomicrographs of the hippocampus (approximately −1.70 to −2.30 from Bregma, see Fig. S2) were taken with a Nikon E800 microscope at 20X using Neurolucida software (Microbrightfield, Burlington, VT). Staining for each experiment was performed in a single batch in order to minimize variation in staining intensity. Immunoreactive regions in the captured images were assessed using thresholding with an ROI box (dimensions and size of ROI box was the same for individual regions) around the region of interest with Image J software (NIH). Thresholding was performed within a limited range on individual slides and no sharpening filters or other image processing methods were applied. The left and right hemisphere were analyzed in two non-consecutive sections for each animal and averaged (total of 4 images per animal). Image analysis was completed by a researcher blind to treatment group.
Cytokine expression.
A separate cohort of mice that underwent CA or sham was used for detection of hippocampal cytokine gene expression using quantitative real-time PCR (qPCR). Mice were anesthetized with isoflurane vapors, blood was drawn via the retro-orbital sinus, and mice were rapidly decapitated. Brains were removed and the hippocampus was dissected out and immersed in RNALater stabilizing solution. RNA was subsequently extracted from hippocampal samples using a homogenizer (Ultra-Turrax T8, IKAWorks, Wilmington, NC) and an RNeasy Mini Kit (Qiagen). RNA from microglia was extracted using a Sonicator (Microson XL2000, Misonix Inc., Farmingdale, NY) and an RNAeasy Micro Kit (Qiagen, Valencia CA). RNA was then reverse transcribed into cDNA with M-MLV Reverse Transcriptase enzyme (Invitrogen) according to the manufacturer’s protocol. Pro-inflammatory cytokine expression for IL1β, IL6, and TNFα was determined using inventoried primer and probe assays (Applied Biosystems, Foster City, CA) on an ABI 7500 Fast Real Time PCR System using Taqman® Universal PCR Master Mix. Relative gene expression of individual samples run in duplicate was calculated by comparison to a relative standard curve and standardized by comparison to 18S rRNA signal.
Administration of cytokine inhibitors.
An indwelling cannula was inserted into the left lateral ventricle (cannula position: +0.02 posterior and −0.95 lateral to bregma, extending 2.75 mm below the skull; Plastics One, Roanoke, VA) of anesthetized mice using a stereotaxic apparatus three days prior to CA or sham surgeries. One hour following CA, mice received a 2 μL injection of either a vehicle solution (artificial cerebral spinal fluid, aCSF), mouse IL6 neutralizing antibody (IL6-na; 10 ng), a monoclonal TNF antibody (Infliximab; IFX; 0.2 μg), or recombinant mouse IL-1 receptor antagonist (IL1-ra; 3.6 μg) based on pre-assigned group and were then placed back in their respective lighting conditions. Doses were based on prior intracerebroventricular administration of these compounds in mouse (Arruda et al., 2011; Craft and DeVries, 2006; Karelina et al., 2009; Sun et al., 2018). Cannula placement was verified on Iba1 stained tissue.
Corticosterone radioimmunoassay.
Within 30 min of collection, blood samples were centrifuged at 3000g for 30 min at 4°C. Plasma was collected and stored at −80°C until assayed. The samples were assayed using and I125 corticosterone kit (MP Biomedicals, Solon, OH). The standard curve was run in triplicate and samples in duplicate. All samples within an experiment were run in a single assay.
Statistical Analyses.
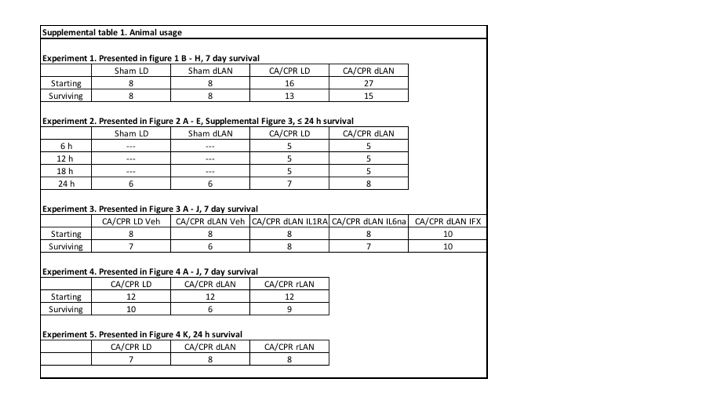
Statistical comparisons among groups were conducted using ANOVA. In the case of significant differences (p<0.05), a post-hoc Tukey’s test was conducted. When conditions of normality or equal variance were not met, the data were log transformed. Above statistical tests were conducted using Prism GraphPad and StatView Software v. 5.0.1. Survival plots were graphed using Kaplan-Meier survival plot in Prism. Differences were considered statistically significant when p < 0.05. Information on experimental group sizes are included in Supplemental Table 1.
RESULTS
Dim light at night increases hippocampal damage following CA
Male Swiss Webster mice were acclimated to a standard light/dark cycle and then underwent 8 min of CA or the sham procedure toward the end of the light phase (see Fig. 1A for experimental outline and Fig. S1 for surgical measures; sham groups n = 8, CA-dLAN n = 27, CA-LD n = 16). A larger number of animals were included in the CA groups to compensate for increased mortality (see Supplemental Table 1 for more information on group sizes). Following the procedures, mice either remained in the LD cycle or were transferred to a dim light at night cycle. Lighting was provided by fluorescent sources and light measurements were taken inside the cage. As anticipated, there was 100% survival in both sham groups. Among the CA mice, mortality in the dLAN group was double that of mice exposed to dark nights, indicating that post-ischemic exposure to dim light at night may compromise cardiac arrest survival (Fig. 1b). Furthermore, cardiac arrest caused a reduction in body weight but only in mice exposed to dim light at night (p < 0.05; Fig 1c).
Figure 1. Dim light at night (dLAN) impairs recovery from a cardiac arrest and cardiopulmonary resuscitation (CA).

(a) Experimental outline for figure 1. Groups included n = 8 – 27 starting and n = 8 – 15 surviving; see supplemental Table 1). (b) Kaplan Meier curve illustrates short-term mortality during the first week following cardiac arrest/CPR (CA). (c) dLAN exposure reduced body mass in mice that underwent the CA procedure. (d) dLAN exacerbates CA induced neuronal damage in the hippocampus, as indicated by increased Fluoro-JadeC (FJC) staining relative to LD. Representative FJC stained sections (20X) from the CA1 region of the hippocampus: (e) Sham-dark night (LD-sham) (f) Sham-dim light at night (dLAN-sham) (g) CA-dark night (CA-LD) and (h) CA-dLAN at one week following the CA/sham procedure. Data were analyzed using a two-way ANOVA with Tukey’s post hoc test. The data in panel b represents mean±SEM; the dagger represents a significant main effect (p<0.05) between sham and CA, while the asterick indicates a significant difference (p<0.05) between CA-LD and CA-dLAN.
Damage to hippocampal neurons was assessed in rodents that survived to one week after the CA or sham procedures (n = 7 – 15/group) via staining for Fluoro-JadeC (FJC), a marker for degenerating neurons. The hippocampus was selected because it is particularly vulnerable to global ischemia damage (Horn and Schlote, 1992; Kirino, 1982; Norman et al., 2010; Petito et al., 1987). FJC staining was uniformly low among Sham mice (Fig. 1d–f), and significantly elevated among mice that underwent CA (p < 0.05; Fig. 1d–h). Moreover, mice exposed to dLAN during recovery from CA had significantly more neuronal damage in the hippocampus than mice exposed to dark nights (p < 0.05; Fig. 1d–h).
Dim light at night alters acute inflammatory status
The neuroinflammatory response following global ischemia is established early and modulates neuronal damage (Fu et al., 2015; Iadecola and Anrather, 2011; Weil et al., 2009). Brain tissue was collected 24 h after CA or sham procedures, i.e., after a single night of post-ischemic dLAN or LD. Hippocampi were collected and processed for quantitative real time PCR (q-PCR) analyses of pro-inflammatory cytokines (n = 6 – 8/ group; see Fig. 2a for experimental outline and supplemental table 1 for information on group sizes). As expected, TNFα, IL1β, and interleukin-6 (IL6) gene expression were elevated among CA as compared to sham mice (p < 0.05; Fig. 2b). Moreover, the single night of exposure to dLAN following CA was sufficient to upregulate the expression of TNFα, compared to mice exposed to a dark night (p < 0.05; Fig. 2b). There were no differences in hippocampal cytokine gene expression between dLAN and LD mice that underwent the sham procedure (p > 0.05).
Figure 2. CA-induced pro-inflammatory cytokine elevations are exacerbated in the hippocampus of dLAN mice.

(a) Outline for experiments in figure 2 (n = 6 – 8/group; see supplemental table 1). (b) Hippocampal TNFα, IL1β, and IL6 mRNA concentrations are increased 24 h following CA relative to sham; furthermore, TNFα expression is potentiated by exposure to dLAN as compared to LD following CA in mice. (c) TNFα expression is elevated as early as 6 hours post-CA in the CA-dLAN group versus the CA-LD group (n = 4/group), this time point coincides with ~4 hours of exposure to dLAN. (d) Serum corticosterone concentrations at 24h were not altered by either CA or light at night (n = 6 – 8/group). There were no significant group differences in IL1β or IL6 at this early time point. Gene expression was assessed via RT-PCR and is presented as fold increase relative to a housekeeping gene (18S). The data are graphed as mean±SEM; a dagger symbol represents a significant main effect (p<0.05) between sham and CA, while an asterisk indicates a significant difference (p<0.05) between CA-LD and CA-dLAN.
Importantly, changes in hippocampal cytokine expression may occur relatively rapidly after placement in dim light at night. The increase in TNFα expression becomes apparent soon after the onset of the dark phase: there was already a significant increase in post-CA TNFα expression among the dLAN cohort compared to all other experimental groups 6 h post-CA and after only 4 h of dim light exposure during the typical dark phase of the light cycle (p < 0.05; Fig. 2c).
The effects of dLAN on CA-induced neuroinflammation do not appear to be due to corticosteroids; there were no differences in corticosterone concentrations between CA or sham groups at 24 h (n = 6 – 8/group; p > 0.05; Fig. 2d). Moreover, an investigation of corticosterone concentration at 6 h intervals following CA did not reveal any significant differences between the CA-dLAN and CA-LD groups during the first 24 h of recovery (n = 4/group; p > 0.05; Fig. S3).
Inhibition of selective pro-inflammatory cytokines ameliorates light-induced damage
Although several mechanisms contribute to damage following ischemic injury, including energetic failure, excitotoxicity, and oxidative stress, manipulation of inflammatory responses are considered a prime target for prevention of damage (Fu et al., 2015; Iadecola and Anrather, 2011). Following ischemic brain damage both selective targeting of specific cytokines and non-selective (e.g., minocycline) inhibition of pro-inflammatory cytokines ameliorate damage improving recovery and behavioral outcomes (Espinosa-Garcia et al., 2017; Mizushima et al., 2002; Neigh et al., 2009; Yrjanheikki et al., 1998). Because our results indicate that dLAN elevates pro-inflammatory cytokines in the hippocampus of mice that have undergone a CA procedure, we hypothesized that selective inhibition of pro-inflammatory cytokines would improve outcome following CA-dLAN. Three days prior to the CA procedure, mice were implanted with a cannula directed at the lateral ventricle. Two hours following CA, mice were administered a single 2μL ICV injection of either vehicle (artificial cerebrospinal fluid; aCSF), mouse IL6 neutralizing antibody (IL6-na; 10 ng), TNF monoclonal antibody (infliximab; IFX; 0.2 μg), or recombinant mouse IL-1 receptor antagonist (IL1-ra; 3.6 μg) (n = 8 –10/group; see experimental outline in Fig. 3A). Doses were based on prior intracerebroventricular administration of these compounds in mouse (Arruda et al., 2011; Craft and DeVries, 2006; Karelina et al., 2009; Sun et al., 2018). Hippocampal damage was evaluated using Fluoro-JadeC as described above. There was no significant effect of the cytokine inhibition on survival (p > 0.05, Fig. 3b), although group numbers may have been insufficient for detecting differences in survival. However, both IL1-ra and IFX decreased hippocampal damage compared to aCSF treatment among CA-dLAN mice (p < 0.05; Fig. 3c), producing levels of FJC staining that were similar to CA-LD mice treated with the vehicle (p>0.05). In contrast, treatment with IL6-na did not ameliorate hippocampal neuronal damage associated with CA-dLAN.
Figure 3. Selective inhibition of specific pro-inflammatory cytokines attenuates inflammation and neuronal cell death following CA and dLAN.

(a) Experimental outline for figure 3 (n = 8 – 10/group, see supplemental table 1). (b) Kaplan Meier curve of mice treated with TNFα, IL1β or IL6 inhibitors 2 h following CA and directly prior to placement in dLAN. (c) Treatment of mice in the dLAN group with the TNFα or IL1β inhibitor reduced neuronal damage (FJC stain) and (d) inhibited proportional area of Iba1 staining at one week after CA relative to the dLAN vehicle treated mice. Representative photomicrographs of microglial staining with Iba1 in the CA1 region of the hippocampus (20X) in mice treated with (e) LD-Veh (f) dLAN-Veh (g) dLAN-IL1ra (h) dLAN-IL6ab or (i) dLAN-IFX. (j) Table provides proportional area of Iba1 staining in subregions of the hippocampus as an indication of microglial activation. The data in panels b, c, and i are provided as mean±SEM; Groups that differ from dLAN-VEH are marked with asterisks in the graphs (p<0.05).
Histological analysis revealed a similar pattern for Iba1 proportional area, which is often used as an index of neuroinflammation. Brain tissue was labeled with Iba-1, an antibody directed against microglia; increased Iba-1 surface area is suggestive of microglial activation (Donnelly et al., 2009). Microglia are the resident CNS myeloid cell and a dominant source of pro-inflammatory cytokines following cerebral ischemia. Perturbations of the neuroenvironment can induce microglial activation, resulting in altered morphology and secretion of pro-inflammatory mediators (Graeber, 2010; Nimmerjahn et al., 2005). Significantly greater Iba1 proportional area in the CA1, CA2, and CA3 subfields of the hippocampus were apparent among the CA-dLAN mice treated with the vehicle (aCSF) relative to the CA-LD mice treated with the vehicle (p < 0.05; Fig. 3d–j and Fig. S4). Furthermore, treatment of CA-dLAN mice with IL1-ra or IFX reduced Iba1 proportional area in the CA1, CA2, and CA3 (p < 0.05; Fig. 3d–j and Fig. S4) relative to the CA-dLAN mice treated with vehicle. Furthermore, Iba1 expression in CA-dLAN mice treated with IL1-ra or IFX did not differ from CA-LD mice treated with the vehicle in any of the hippocampal subfields quantified. In contrast, CA-dLAN mice treated with IL6-na had comparable Iba1 staining in the CA1 as comparable to CA-dLAN mice treated with the vehicle, while levels of Iba1 proportional area in the CA2 and CA3 regions were intermediate between vehicle treated CA-dLAN and CA-LD mice.
Alternative spectra of lighting minimize light-induced damage
The intrinsically photosensitive retinal ganglion cells (ipRGCs) that project to the master circadian pacemaker in the SCN contain melanopsin and are most responsive to the blue region of the visible light spectrum ranging from 450 to 485 nm (Berson et al., 2002); these wavelengths are present in broad spectrum white light, such as natural sunlight and the majority of indoor lighting. Longer wavelengths of lighting, such as red light, do not activate ipRGCs, and therefore minimally influence the circadian system (Brainard et al., 2008; Figueiro and Rea, 2010). Thus, we hypothesized that the use of a wavelength of nighttime light that does not activate the ipRGCs would not exacerbate ischemic outcome. An important caveat to this experiment, however, is mice also lack a long wavelength cone and thus have more limited detection of red wavelength light in general. As in the previous studies the mice were acclimated to the LD light cycle [14h light (150 lux): 10h dark (0 lux)]. Following CA, mice either remained in LD, or were placed in dLAN [14 h light (150 lux): 10 h dim light (5 lux; 6500K cool white light- containing blue wavelengths)] or a dim red light at night cycle [rLAN; 14 h light (150 lux): 10 h dim red (5 lux; 636 nm)] (n = 12/group). This experiment followed the same timeline as Fig. 1a.
Mice exposed to dim white light at night as compared to dark nights had increased mortality (p = 0.05); however, mice exposed to dim red light at night did not differ from LD or dLAN groups (p > 0.05; Fig 4a). FJC staining revealed a significant increase in neuronal damage in the hippocampus among the CA-LAN group compared to both the CA-LD and CA-rLAN groups (p < 0.05; Fig. 4b, d–f). In contrast, hippocampal neuronal damage among CA-rLAN mice was similar to CA-LD mice (p > 0.05). A similar pattern emerged from Iba-1 staining; CA-dLAN mice exhibited an increase in Iba1 proportional area in multiple hippocampal subfields compared to both CA-LD and CA-rLAN conspecifics (p < 0.05; Fig. 4c, g–j and Fig. S5), whereas the rLAN group did not differ from the CA-LD group in any of the comparisons (p > 0.05; Fig. 4c, g–j and Fig. S5).
Figure 4. Manipulation of lighting wavelength minimizes light at night induced damage following CA.

The experimental outline in Figure 4 followed the same timing as Figure 1a (n = 12/group, 6 – 10 surviving, see supplemental table 1). (a) Kaplan Meier curve illustrating survival of mice exposed to dark nights (LD), dim light at night (dLAN), or red light at night (rLAN) following CA. (b) the dLAN group had significantly greater neuronal damage (FJC staining) and (c) increased proportional area of Iba1 staining in the hippocampus at one week after CA than either the CA-LD or CA-rLAN groups; the CA-LD or CA-rLAN groups did not differ significantly in either of these measures. Representative photomicrographs of FJC staining in the CA1 region of the hippocampus (20X) one week after CA in the (d) LD, (e) dLAN, and (f) rLAN groups. Representative photomicrographs of Iba1 staining in the CA1 region of the hippocampus (20X) one week after CA in the (g) LD, (h) dLAN, and (i) rLAN groups. (j) Table providing proportional area of Iba1 staining in subregions of the hippocampus as an indication of microglial activation. (k) Gene expression of TNFα, IL1β, and IL6 is significantly elevated in the hippocampus 24 h following CA in the CA-dLAN group relative to the rLAN group and CA-LD groups (n = 7 – 8/group). Gene expression was assessed via qPCR and is presented as fold increase relative to a housekeeping gene (18S). The data are provided as mean±SEM; an asterisk represents a significant difference between dLAN and both other groups (p<0.05), whereas a pound sign (#) indicates the dLAN group only significantly differs from the rLAN group (p<0.05).
To corroborate the histological assessment of neuroinflammation, hippocampal pro-inflammatory cytokine expression was assessed 24 h after CA (n = 7 – 8/group). This time point allowed exposure to a single night of post-ischemic dark, dim white light or dim red light. Exposure to dLAN significantly elevated hippocampal TNFα and IL6 gene expression compared to both LD and rLAN (p < 0.05; Fig. 4k). dLAN also significantly increased IL1β gene expression compared to rLAN (p < 0.05; Fig 4k). Exposure to rLAN did not significantly alter pro-inflammatory cytokine expression compared to LD (p > 0.05).
DISCUSSION
In terms of productivity, safety, and convenience, there are many tangible benefits to the ability to illuminate the night. However, nighttime light exposure is not innocuous for human health. There is a growing literature linking long-term nighttime light exposure to maladaptive health conditions, including heart disease, metabolic disorders, and cancer (Bedrosian et al., 2016; Fonken and Nelson, 2014; Lunn et al., 2017). The current study provides evidence in a mouse model, that acute exposure to light at night also may be detrimental if it coincides with recovery from brain injury. Indeed, light at night reshapes the physiological response to cerebral ischemia, resulting in increased neuronal damage in mice (Figure 1). Hippocampal cell death is a proxy for overall recovery after global ischemia as increased hippocampal damage is associated with elevated mortality and memory deficits, as well as impaired affective responses (Espinosa-Garcia et al., 2017; Neigh et al., 2009; Neigh et al., 2004; Norman et al., 2010). This is significant because intensive care units, where critically ill cardiovascular patients recover, tend to be well-lit at night. Patients are often exposed to light several times per night (Durrington et al., 2017; Lunn et al., 2017) and even patients whose eyelids are closed may be affected by the light intrusion (Robinson et al., 1991).
Exposure to light at night may exacerbate damage following CA by amplifying pro-inflammatory responses. A single night of dim light exposure directly following resuscitation from CA elevated hippocampal pro-inflammatory cytokine mRNA expression as compared to total darkness during the night. Changes in neuroinflammatory dynamics likely occur rapidly following placement in dim light as TNFα mRNA expression in the hippocampus is elevated as early as 4 h after exposure to dLAN. At these acute time points, dim light exposure appeared to specifically amplify inflammation after injury as there were no significant differences between the sham-LD and sham-dLAN groups. However, chronic exposure to dim light at night may induce neuroinflammatory changes in the absence of injury; several weeks of exposure to dim light at night increases the expression of pro-inflammatory cytokines in the hippocampus of otherwise healthy rodents (Bedrosian et al., 2013b).
Pro-inflammatory cytokines, such as TNFα, are known to contribute to damage after cerebral ischemia (Lambertsen et al., 2012). Thus, these early changes in the inflammatory response caused by dLAN may causally alter the trajectory of recovery, resulting in increased neuronal damage. Indeed, inhibition of either TNFα or IL-1 signaling among CA-dLAN mice significantly reduced Iba1+ proportional area and neuronal damage relative to vehicle treated CA-dLAN mice. Thus, two bodies of evidence point to a role of increased inflammatory responses in mediating elevated neuronal damage among the CA-dLAN mice: (1) both pro-inflammatory cytokine gene expression and neuronal damage were elevated after exposure to dLAN relative to LD, and (2) treatment with IL1ra or IFX prevented the exacerbation of neuronal damage and increases in Iba1+ proportional area observed among vehicle treated CA-dLAN mice. In contrast to inhibition of TNFα or IL-1, blocking IL-6 signaling with an IL-6 neutralizing antibody did not reduce cell death or Iba1 staining. The IL-6 neutralizing antibody dose was based on prior work (Karelina et al., 2009), however, it is possible that higher concentrations of IL-6 neutralizing antibody would prove more effective. Of note, lower levels of mortality occurred in this experiment compared to the other two experiments that measured 7-day survival. This may have occurred for several reasons. For example, there are typically small variations in survival between experimental cohorts that may relate to differences in animals provided by the vendor, variations in reagents used for the procedure, noise levels in the colony/surgical suite, etc. Additionally, the cannula implantation may have induced inflammation in the CNS causing inflammatory pre-conditioning, which is a known protective factor against ischemic brain injury (for example, see (Garcia-Bonilla et al., 2018). Furthermore, this experiment may have included insufficient group sizes for detecting survival. Overall, targeting inflammatory pathways can prevent damage from cerebral ischemia in mice maintained in standard lighting conditions (Lambertsen et al., 2012). Because this is well established, mice maintained in dark nights were not included in these experiments in order to reduce animal use.
Microglia may be a major cellular source of elevated inflammation in mice exposed to dim light at night. The proportional area of Iba1 staining, a marker for microglia activation, was elevated in the hippocampus of mice exposed to light at night as compared to dark nights following CA. Furthermore, inhibiting pro-inflammatory pathways with IFX and IL1ra reduced Iba1 staining throughout the hippocampus and minimized cell death. However, other cellular sources such as neurotoxic reactive astrocytes and/or infiltrating immune cells may contribute to the proinflammatory cytokine response (Liddelow et al., 2017; Lindsberg et al., 2010).
Dim light at night may induce neuroinflammatory changes through several potential mechanisms. First, light at night may suppress nocturnal melatonin secretion. Melatonin exhibits anti-inflammatory properties and can alleviate CNS inflammation (Zhao et al., 2015). However, Swiss Webster mice likely lack appreciable levels of melatonin (Tosini and Menaker, 1998). Second, light at night may elevate neuroinflammation through disturbing sleep (Stenvers et al., 2016). Sleep alterations are associated with CNS inflammation (Bellesi et al., 2017). However, other studies have reported no sleep disturbances in response to dLAN (Borniger et al). Third, nighttime light exposure may alter circadian clock mechanisms in immune cells. Microglia express circadian timekeeping machinery (Fonken et al., 2015) and previous work suggest chronic exposure to dLAN exacerbates microglia pro-inflammatory responses (Fonken and Nelson, 2013). Chronic exposure to dim light at night reduces the amplitude of PER1 and PER2 rhythms in the SCN is mice and hamsters (Bedrosian et al., 2013a; Fonken et al., 2013). Moreover, dim light exposure is associated with disruption in clock gene expression in extra-SCN tissues such as liver (Fonken et al., 2013). The effects of exposure to a single night of 5 lux of dim light at night on clock gene expression has not been evaluated in the SCN or extra-SCN clocks. Future studies could determine whether dim light at night alters clock gene expression and/or behavior, and whether CA renders mice more susceptible to the disruptive effects of dLAN.
Although pharmacological intervention reduced the detrimental effects of dLAN on CA-induced microglial activation and neuronal damage, an alternative approach to improving CA outcome is to modify the physical qualities of the nighttime light to prevent increased neuroinflammation. Indeed, here we show that nighttime red light of the same illuminance as the dim white light did not exacerbate CA-induced neuronal damage or increase Iba1 staining in mice. These data are consistent with studies reporting that red light at night does not affect other aspects of physiology and behavior in humans or other animals to the same extent as broad spectrum white light that contains blue wavelengths (Figueiro and Rea, 2010). The effects of nighttime light may be mediated by the suprachiasmatic nucleus or “master circadian clock”, which receives input from melanopsin containing ipRGCs in the retina. The ipRCGs are activated by blue light (~480nm, found in outdoor and most indoor lighting, especially fluorescent lights), but are generally unaffected by long wavelength light, such as red light. However, a major limitation to this work is that red light at 5 lux is a much lower energy and dimmer than white light. Thus, future experiments should address whether a higher intensity red light source is also protective against light-associated damage following ischemic energy. Furthermore, mice may not be the optimal model to study the effects of red light: the mouse M-cone is most sensitive to light around 510nm, suggesting none of the retinal pigments in mouse are maximally sensitive to the 636nm red light (Hattar et al., 2003).
Although these studies were conducted in nocturnal rodents, they may have important implications for diurnal species. Indeed, the effect of light on entraining the circadian system and inhibition of melatonin is similar between nocturnal and diurnal animals (Challet, 2007). Furthermore, our prior work indicates that exposure to dim light at night is an equally potent facilitator of inflammation in diurnal rodents (Fonken et al., 2011). One benefit to using a nocturnal rodent in these experiments is that sleep disruption was likely not a potential confound (Borniger et al., 2013).
One additional note about this work is that experiments were conducted in young adult mice (8 weeks of age) and cardiac arrest in humans typically occurs in older individuals. The circadian system and immune system change over the course of an animal’s lifespan. Aged animals typically exhibit lower amplitude molecular and behavioral circadian rhythms and slower re-entrainment of rhythms following light induced phase shifts (Davidson et al., 2008; Sellix et al., 2012). This suggests that light at night may have differential effects in a young versus aged animal. Furthermore, the neuroimmune system changes with age. Microglia tend to become more reactive or “primed” with age. Primed microglia exhibit hyperinflammatory responses upon immune activation that can exacerbate pathology (Fonken et al., 2018). Future work should address whether aging leads to increased vulnerability to the disruptive effects of light at night on physiology and behavior.
In sum, the mouse data presented here indicates that exposure to light at night, a common occurrence in hospital rooms, may compromise recovery from cerebral ischemia by exacerbating neuroinflammation. Reducing post-ischemic neuroinflammation among mice exposed to dLAN, prevented the increase in neuronal damage otherwise associated with dLAN. If the effects of white light at night are replicated in cardiovascular patients, then these results could have important implications for the design of lighting in clinical settings and could apply to a broad number of conditions and medical procedures that involve ischemia and inflammation, such as stroke, cardiovascular artery bypass graft, sickle cell disease, sleep apnea, and organ transplant.
Supplementary Material
{kind=link}
Acknowledgements.
The authors thank Kristopher Gaier, Bryan Klein, Dan McCarthy, Kate Karelina, Katie Stuller, and James Walton for technical assistance.
FUNDING SOURCES. This project was supported by a US-Isreali Binational Research Foundation grant to RJN and a National Institute of Health grant (1R01NS092388) to RJN and ACD. LKF was supported by an American Heart Association Pre-doctoral Fellowship.
Footnotes
CONFLICT OF INTEREST STATEMENT. The authors have no conflicts of interest to report. All authors concur with the submission of this manuscript and none of the data have been previously reported or are under consideration for publication elsewhere.
REFERENCES
- Arruda AP, Milanski M, Coope A, Torsoni AS, Ropelle E, Carvalho DP, Carvalheira JB, Velloso LA, 2011. Low-grade hypothalamic inflammation leads to defective thermogenesis, insulin resistance, and impaired insulin secretion. Endocrinology 152, 1314–1326. [DOI] [PubMed] [Google Scholar]
- Ayalon L, Borodkin K, Dishon L, Kanety H, Dagan Y, 2007. Circadian rhythm sleep disorders following mild traumatic brain injury. Neurology 68, 1136–1140. [DOI] [PubMed] [Google Scholar]
- Bedrosian TA, Fonken LK, Nelson RJ, 2016. Endocrine Effects of Circadian Disruption. Annu Rev Physiol 78, 109–131. [DOI] [PubMed] [Google Scholar]
- Bedrosian TA, Galan A, Vaughn CA, Weil ZM, Nelson RJ, 2013a. Light at night alters daily patterns of cortisol and clock proteins in female Siberian hamsters. Journal of neuroendocrinology 25, 590–596. [DOI] [PubMed] [Google Scholar]
- Bedrosian TA, Weil ZM, Nelson RJ, 2013b. Chronic dim light at night provokes reversible depression-like phenotype: possible role for TNF. Mol Psychiatry 18, 930–936. [DOI] [PubMed] [Google Scholar]
- Bellesi M, de Vivo L, Chini M, Gilli F, Tononi G, Cirelli C, 2017. Sleep Loss Promotes Astrocytic Phagocytosis and Microglial Activation in Mouse Cerebral Cortex. J Neurosci 37, 5263–5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berson DM, Dunn FA, Takao M, 2002. Phototransduction by retinal ganglion cells that set the circadian clock. Science 295, 1070–1073. [DOI] [PubMed] [Google Scholar]
- Borniger JC, Weil ZM, Zhang N, Nelson RJ, 2013. Dim light at night does not disrupt timing or quality of sleep in mice. Chronobiol Int 30, 1016–1023. [DOI] [PubMed] [Google Scholar]
- Brainard GC, Sliney D, Hanifin JP, Glickman G, Byrne B, Greeson JM, Jasser S, Gerner E, Rollag MD, 2008. Sensitivity of the human circadian system to short-wavelength (420-nm) light. J Biol Rhythms 23, 379–386. [DOI] [PubMed] [Google Scholar]
- Challet E, 2007. Minireview: Entrainment of the suprachiasmatic clockwork in diurnal and nocturnal mammals. Endocrinology 148, 5648–5655. [DOI] [PubMed] [Google Scholar]
- Craft TK, DeVries AC, 2006. Role of IL-1 in poststroke depressive-like behavior in mice. Biol Psychiatry 60, 812–818. [DOI] [PubMed] [Google Scholar]
- Davidson AJ, Yamazaki S, Arble DM, Menaker M, Block GD, 2008. Resetting of central and peripheral circadian oscillators in aged rats. Neurobiol Aging 29, 471–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly DJ, Gensel JC, Ankeny DP, van Rooijen N, Popovich PG, 2009. An efficient and reproducible method for quantifying macrophages in different experimental models of central nervous system pathology. Journal of neuroscience methods 181, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrington HJ, Clark R, Greer R, Martial FP, Blaikley J, Dark P, Lucas RJ, Ray DW, 2017. ‘In a dark place, we find ourselves’: light intensity in critical care units. Intensive Care Med Exp 5, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltzschig HK, Eckle T, 2011. Ischemia and reperfusion--from mechanism to translation. Nature medicine 17, 1391–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa-Garcia C, Sayeed I, Yousuf S, Atif F, Sergeeva EG, Neigh GN, Stein DG, 2017. Stress primes microglial polarization after global ischemia: Therapeutic potential of progesterone. Brain Behav Immun. [DOI] [PubMed] [Google Scholar]
- Figueiro MG, Rea MS, 2010. The effects of red and blue lights on circadian variations in cortisol, alpha amylase, and melatonin. International journal of endocrinology 2010, 829351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonken LK, Aubrecht TG, Melendez-Fernandez OH, Weil ZM, Nelson RJ, 2013. Dim light at night disrupts molecular circadian rhythms and increases body weight. J Biol Rhythms 28, 262–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonken LK, Frank MG, Gaudet AD, Maier SF, 2018. Stress and aging act through common mechanisms to elicit neuroinflammatory priming. Brain, Behavior, and Immunity In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonken LK, Frank MG, Kitt MM, Barrientos RM, Watkins LR, Maier SF, 2015. Microglia inflammatory responses are controlled by an intrinsic circadian clock. Brain Behav Immun 45, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonken LK, Haim A, Nelson RJ, 2011. Dim light at night increases immune function in nile grass rats, a diurnal rodent. Chronobiol Int 29, 26–34. [DOI] [PubMed] [Google Scholar]
- Fonken LK, Nelson RJ, 2013. Mice exposed to dim light at night exaggerate inflammatory responses to lipopolysaccharide. Brain Behav Immun 34, 159–163. [DOI] [PubMed] [Google Scholar]
- Fonken LK, Nelson RJ, 2014. The effects of light at night on circadian clocks and metabolism. Endocrine reviews 35, 648–670. [DOI] [PubMed] [Google Scholar]
- Fu Y, Liu Q, Anrather J, Shi FD, 2015. Immune interventions in stroke. Nat Rev Neurol 11, 524–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bonilla L, Brea D, Benakis C, Lane DA, Murphy M, Moore J, Racchumi G, Jiang X, Iadecola C, Anrather J, 2018. Endogenous Protection from Ischemic Brain Injury by Preconditioned Monocytes. J Neurosci 38, 6722–6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet AD, Fonken LK, Ayala MT, Bateman EM, Schleicher WE, Smith EJ, D’Angelo HM, Maier SF, Watkins LR, 2018a. Spinal cord injury in rats disrupts the circadian system. eNeuro, ENEURO. 0328–0318.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet AD, Fonken LK, Ayala MT, D’Angelo HM, Smith EJ, Bateman EM, Schleicher WE, Maier SF, Watkins L, 2018b. Spinal cord injury in rats dysregulates diurnal rhythms of fecal output and liver metabolic indicators. J Neurotrauma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannoccaro MP, Moghadam KK, Pizza F, Boriani S, Maraldi NM, Avoni P, Morreale A, Liguori R, Plazzi G, 2013. Sleep disorders in patients with spinal cord injury. Sleep medicine reviews 17, 399–409. [DOI] [PubMed] [Google Scholar]
- Graeber MB, 2010. Changing face of microglia. Science 330, 783–788. [DOI] [PubMed] [Google Scholar]
- Hattar S, Lucas RJ, Mrosovsky N, Thompson S, Douglas RH, Hankins MW, Lem J, Biel M, Hofmann F, Foster RG, Yau KW, 2003. Melanopsin and rod-cone photoreceptive systems account for all major accessory visual functions in mice. Nature 424, 76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn M, Schlote W, 1992. Delayed neuronal death and delayed neuronal recovery in the human brain following global ischemia. Acta Neuropathol 85, 79–87. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Anrather J, 2011. The immunology of stroke: from mechanisms to translation. Nature medicine 17, 796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Namboodri KK, Kumari S, Prabhakar S, 2004. Loss of circadian rhythm of blood pressure following acute stroke. BMC Neurol 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karelina K, Norman GJ, Zhang N, Morris JS, Peng H, DeVries AC, 2009. Social isolation alters neuroinflammatory response to stroke. Proc Natl Acad Sci U S A 106, 5895–5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirino T, 1982. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res 239, 57–69. [DOI] [PubMed] [Google Scholar]
- Lambertsen KL, Biber K, Finsen B, 2012. Inflammatory cytokines in experimental and human stroke. J Cereb Blood Flow Metab 32, 1677–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange T, Dimitrov S, Born J, 2010. Effects of sleep and circadian rhythm on the human immune system. Ann N Y Acad Sci 1193, 48–59. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA, 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsberg PJ, Strbian D, Karjalainen-Lindsberg ML, 2010. Mast cells as early responders in the regulation of acute blood-brain barrier changes after cerebral ischemia and hemorrhage. J Cereb Blood Flow Metab 30, 689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn RM, Blask DE, Coogan AN, Figueiro MG, Gorman MR, Hall JE, Hansen J, Nelson RJ, Panda S, Smolensky MH, Stevens RG, Turek FW, Vermeulen R, Carreon T, Caruso CC, Lawson CC, Thayer KA, Twery MJ, Ewens AD, Garner SC, Schwingl PJ, Boyd WA, 2017. Health consequences of electric lighting practices in the modern world: A report on the National Toxicology Program’s workshop on shift work at night, artificial light at night, and circadian disruption. Sci Total Environ 607–608, 1073–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima H, Zhou CJ, Dohi K, Horai R, Asano M, Iwakura Y, Hirabayashi T, Arata S, Nakajo S, Takaki A, Ohtaki H, Shioda S, 2002. Reduced postischemic apoptosis in the hippocampus of mice deficient in interleukin-1. The Journal of comparative neurology 448, 203–216. [DOI] [PubMed] [Google Scholar]
- Morris CJ, Purvis TE, Hu K, Scheer FA, 2016. Circadian misalignment increases cardiovascular disease risk factors in humans. Proc Natl Acad Sci U S A 113, E1402–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neigh GN, Karelina K, Glasper ER, Bowers SL, Zhang N, Popovich PG, DeVries AC, 2009. Anxiety after cardiac arrest/cardiopulmonary resuscitation: exacerbated by stress and prevented by minocycline. Stroke 40, 3601–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neigh GN, Kofler J, Meyers JL, Bergdall V, La Perle KM, Traystman RJ, DeVries AC, 2004. Cardiac arrest/cardiopulmonary resuscitation increases anxiety-like behavior and decreases social interaction. J Cereb Blood Flow Metab 24, 372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F, 2005. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. [DOI] [PubMed] [Google Scholar]
- Norman GJ, Morris JS, Karelina K, Weil ZM, Zhang N, Al-Abed Y, Brothers HM, Wenk GL, Pavlov VA, Tracey KJ, Devries AC, 2011. Cardiopulmonary arrest and resuscitation disrupts cholinergic anti-inflammatory processes: a role for cholinergic alpha7 nicotinic receptors. J Neurosci 31, 3446–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman GJ, Zhang N, Morris JS, Karelina K, Berntson GG, DeVries AC, 2010. Social interaction modulates autonomic, inflammatory, and depressive-like responses to cardiac arrest and cardiopulmonary resuscitation. Proc Natl Acad Sci U S A 107, 16342–16347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petito CK, Feldmann E, Pulsinelli WA, Plum F, 1987. Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology 37, 1281–1286. [DOI] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR, 2002. Coordination of circadian timing in mammals. Nature 418, 935–941. [DOI] [PubMed] [Google Scholar]
- Robinson J, Bayliss SC, Fielder AR, 1991. Transmission of light across the adult and neonatal eyelid in vivo. Vision Res 31, 1837–1840. [DOI] [PubMed] [Google Scholar]
- Saito K, Suyama K, Nishida K, Sei Y, Basile AS, 1996. Early increases in TNF-alpha, IL-6 and IL-1 beta levels following transient cerebral ischemia in gerbil brain. Neurosci Lett 206, 149–152. [DOI] [PubMed] [Google Scholar]
- Sellix MT, Evans JA, Leise TL, Castanon-Cervantes O, Hill DD, DeLisser P, Block GD, Menaker M, Davidson AJ, 2012. Aging differentially affects the re-entrainment response of central and peripheral circadian oscillators. J Neurosci 32, 16193–16202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenvers DJ, van Dorp R, Foppen E, Mendoza J, Opperhuizen AL, Fliers E, Bisschop PH, Meijer JH, Kalsbeek A, Deboer T, 2016. Dim light at night disturbs the daily sleep-wake cycle in the rat. Scientific reports 6, 35662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens RG, Zhu Y, 2015. Electric light, particularly at night, disrupts human circadian rhythmicity: is that a problem? Philosophical transactions of the Royal Society of London. Series B, Biological sciences 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Zhang G, Chen R, Li R, Wang H, Jiang A, Li Z, Kong L, Fonken LK, Rajagopalan S, Sun Q, Liu C, 2018. Central IKK2 inhibition ameliorates air pollution mediated hepatic glucose and lipid metabolism dysfunction in mice with type II diabetes. Toxicol Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosini G, Menaker M, 1998. The clock in the mouse retina: melatonin synthesis and photoreceptor degeneration. Brain Res 789, 221–228. [DOI] [PubMed] [Google Scholar]
- Weil ZM, Karelina K, Su AJ, Barker JM, Norman GJ, Zhang N, Devries AC, Nelson RJ, 2009. Time-of-day determines neuronal damage and mortality after cardiac arrest. Neurobiol Dis 36, 352–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil ZM, Norman GJ, Barker JM, Su AJ, Nelson RJ, Devries AC, 2008. Social isolation potentiates cell death and inflammatory responses after global ischemia. Mol Psychiatry 13, 913–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J, 1998. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci U S A 95, 15769–15774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, An R, Yang Y, Yang X, Liu H, Yue L, Li X, Lin Y, Reiter RJ, Qu Y, 2015. Melatonin alleviates brain injury in mice subjected to cecal ligation and puncture via attenuating inflammation, apoptosis, and oxidative stress: the role of SIRT1 signaling. J Pineal Res 59, 230–239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.