

Abstract
Proptosis, the protrusion of the eyeball from the orbit, results from a wide variety of pathologies that can be vision- or life-threatening. Clinical history, associated physical exam findings, and imaging features are all crucial in establishing the underlying etiology. The differential diagnosis is broad, and includes infectious, inflammatory, vascular, and neoplastic entities that range from benign and indolent, to malignant and aggressive. While treatment varies significantly based on the disease process, all are aimed at preserving vision, salvaging the globe, preventing disfigurement, and reducing mortality. Both internists and general ophthalmologists should be familiar with the causes of proptosis in order to initiate the work-up for, and appropriately triage, affected patients.
Keywords: Proptosis, orbital tumors, orbital inflammation, orbital infection, orbital vascular anomalies
Introduction
Proptosis, also known as exophthalmos, is a clinical exam finding in a wide variety of ocular conditions. Generally, a 2 mm or greater asymmetry between the protrusion of a patient’s eyes is considered abnormal.1 Recognizing the various causes of proptosis is critical as many can be vision threatening (Table 1). This review will focus on select orbital processes and their presentations.
Table 1.
Differential diagnosis of proptosis by laterality and age.
| Differential diagnosis by laterality | |||
|---|---|---|---|
| Unilateral Proptosis | Bilateral Proptosis | ||
| Orbital cellulitis | Thyroid eye disease | ||
| Developmental anomalies | Orbital myositis | ||
| Inflammatory conditions | Cavernous sinus thrombosis | ||
| Vascular anomalies | Metastatic neuroblastoma | ||
| Neoplasms | |||
| Metabolic disease | |||
| Differential diagnosis by age | |||
| 1 – 5 years of age | 5-10 years of age | 10 – 30 years of age | 30 – 50 years of age |
| Dermoid Metastatic neuroblastoma Rhabdomyosarcoma Epithelial cyst Glioma of the optic nerve Sphenoid wing meningioma Hemangioma |
Pseudotumor, orbital extension of retinoblastoma, malignant lymphoma and leukemias, dermoid, hemangioma | pseudotumor, mucocele, meningioma, TED, lacrimal gland tumor, malignant lymphoma/leukemia, dermoid, hemangioma | Pseudotumor, mucocele, malignant lymphoma/leukemia, hemangioma, TED, lacrimal gland tumors |
Over 70: melanoma, pseudotumor, lymphoma, metastatic tumor, mucocele
History and Exam
A systematic approach to the history and physical exam (Table 2) is crucial in constructing a relevant differential diagnosis. Remembering the 6 P’s of the orbital history and physical exam can be helpful to create a quick and easy framework for the medical provider.
Table 2.
Systematic Evaluation to Orbital Disease
| History of Present
Illness | |
| • History of present illness | • Past Medical History |
| ο What is the problem? | ο Thyroid? |
| ο When did it start? | ο Cancer? |
| ▪ Who noticed? | ο Diabetes? |
| ▪ Trauma related? | ο HIV? |
| • Progression? | • Medications |
| • Pain? | ο Steroids? |
| ο Quality | ο Other immunosuppressants? |
| ο Severity | • Social History |
| • Changes in Vision? | ο IV drug use? |
| • Diplopia? | • Family History |
| ο Monocular vs. binocular | ο Cancer? |
| ο Horizontal vs. vertical | ο Thyroid disease? |
| • Skin changes? | • Review of Systems |
| ο Edema | ο Constitutional B symptoms (fever, malaise, weight loss, night sweats)? |
| ο Erythema | |
| • Has it been evaluated in the past? | ο GI, breast, prostate, and endocrine |
| Physical Examination | |
| • Vital signs | • Listen for bruits |
| ο Visual acuity - hyperopic shift | • Palpate |
| ο Pupils | ο Along orbital rim (mobile or fixed |
| ο Intraocular Pressure - attempt also | nodules) |
| in upgaze | ο Along anterior orbit |
| • Lid vital signs | ▪ Trochlea |
| ο Palpebral fissure | ▪ Orbital lobe of lacrimal gland |
| ο Levator function | • Anterior segment |
| ο Margin Reflex Distance 1/2 | ο Conjunctiva |
| • Markers of optic nerve function | ▪ Dilated/tortuous vessels |
| ο Visual acuity | ▪ Chemosis |
| ο Pupils | ▪ Injection |
| ο Color plates/color desaturation | ο Cornea |
| ο Nerve appearance (pallor, edema) | ▪ Exposure keratopathy |
| ο 30–2 Visual Field | ▪ Superior Limbic Keratitis |
| • Observe | ο Gonioscopy |
| ο Pulsation | ο Blood in Schlemm’s canal |
| ο Proptosis (>2 mm asymmetry) | • Posterior Segment |
| ▪ Hertel exophthalmeter | ο Nerve |
| ▪ Ant’s eye view | ▪ Optociliary shunt vessels |
| ▪ Beware of pseudoproptosis | ▪ Edema |
| ο Globe displacement | ▪ Pallor |
| ▪ Ductions | ο Choroidal folds |
| ο Periorbital changes | |
HISTORY:
1. Pain.
Is there pain at rest? Is there pain with eye movements? What is the distribution of pain? Is it constant or intermittent? Pain is usually indicative of inflammation of a sensory nerve within the orbit. These nerves are typically branches off the ophthalmic and maxillary divisions of the trigeminal nerve. These nerves can be inflamed from an adjacent process (eg. thyroid eye disease or orbital inflammatory pseudotumor aka idiopathic orbital inflammation), nerve infiltration by a tumor (eg. adenoid cystic carcinoma of the lacrimal gland), scar (post trauma), or it can be idiopathic.
2. Progression.
Have the changes occurred quickly (hours to days) or over a longer period of time? Inflammatory and infectious processes typically have a much quicker progression than solid tissue tumors and/or vascular tumors.
EXAM:
3. Proptosis.
Is the globe misplaced superiorly, inferiorly, medially, laterally, or straight out (aka axially)? An exophthalmometer is used to quantify the distance from the orbital rim to the corneal surface of each eye in order to assess the symmetry of globe protrusion. An orbital mass will typically displace the globe opposite the direction it originates from, unless there is axial proptosis (eye protrudes directly forward), indicating a lesion within the muscle cone of the orbit/directly behind the globe.
4. Pulsation.
Does the globe pulsate? Are bruits auscultated when applying the bell of the stethoscope to the globe? Vascular tumors, particularly those with an arterial component (eg. arteriovenous fistula) will pulsate and have an associated bruit.
5. Palpation.
Is a mass palpable along the orbital rim or within the anterior orbit? Is it firm? Is it mobile? Is there a focal area of tenderness? Palpation of a lesion in a particular area will direct the provider to an area of focus when reviewing an imaging study and determining an approach to biopsy of the lesion. Firm, immobile lesions are usually of more grave concern than soft, mobile lesions.
6. Periocular Changes.
Are there changes in the color of the skin of the eyelids? Is there swelling of the eyelids? Is there an eyelid lesion? Tumors in the orbit often originate from a previously unrecognized skin tumor (basal or squamous cell carcinoma). Swelling of the eyelids can be indicative of an orbital mass or congestion from compression of the venous circulation or elevated pressure within the venous circulation within the orbit (which can be from local congestion in the orbit/cavernous sinus or systemic as in the case of pulmonary hypertension). Enlargement of the vessels on the conjunctiva may also be an indication of the same.
Prior photographs also prove to be a very helpful component of gathering a history on a patient with new onset proptosis as it can help determine the degree of change from baseline.
A vital component of the ophthalmic examination is an assessment of the function of the optic nerve. The provider should be familiar in taking the “Optic Nerve Vital Signs” (David Tse, personal communication). These include measurement of the patient’s best corrected visual acuity, assessing the pupils for presence of a relative afferent pupillary defect, performing confrontational visual fields, assessing color vision (either through use of color plates or by red color desaturation) and by looking at the optic nerve for signs of edema and/or pallor through direct ophthalmoscopy. Lesions compressing the optic nerve place the individual at risk of permanent vision loss if not addressed in an expeditious fashion. A full cranial nerve exam should always be performed when evaluating a patient with proptosis as this may aid in localizing a lesion. Further ophthalmologic examination should then be performed with a slit lamp to assess the anterior segment and direct or indirect ophthalmoscopy to evaluate the retina.
Orbital imaging allows for further characterization of proptosis. Computed tomography (CT) scans provide detailed images of bony structures and adjacent soft tissue, and is particularly useful in cases of thyroid eye disease or orbital trauma. Magnetic resonance imaging (MRI) allows for better soft tissue differentiation in the periorbital, orbital, and intracranial spaces.2,3 On T1 weighted imaging, fat, choroid, hemorrhage, and white matter will normally be bright. On T2 weighted imaging, choroid, vitreous, grey matter, cerebrospinal fluid, and subacute hemorrhage will normally be bright.4,5 Some causes of gadolinium enhancement on MRI include choroidal detachments, choroidal melanomas, uveitic processes, inflammation of the extraocular muscles as in thyroid eye disease, optic neuritis, lacrimal gland tumors, solid tumors, and capsules or septae of lesions.5,6 Arteriography and venography are useful imaging techniques, particularly in cases of aneurysmal lesions, arteriovenous malformations, and carotid-cavernous sinus fistulas.7 Orbital ultrasonography can be particularly useful in the diagnosis and characterization of vascular lesions, other intra and extraconal masses, and extraocular muscle enlargement.8, 9 Basic laboratory evaluation in the work up of proptosis should include CMP, CBC, PT/PTT, and a thyroid panel.
Infectious etiologies
Orbital Cellulitis
Preseptal cellulitis is characterized by infection anterior to the orbital septum while orbital cellulitis involves infection posterior to the orbital septum. Infection most commonly arises from adjacent spread of the sinuses, but may also be seen after trauma to the skin, where infection gets beyond the barrier of the orbital septum, posteriorly into the orbit. A rarer cause of cellulitis can arise in a bacteremic patient where micro-organisms can seed intraorbital tissues. In the case of the most common etiology of orbital cellulitis, adjacent sinusitis, infection is polymicrobial in adults and monomicrobial in children. The most commonly implicated organisms in these cases are Staphylococcus species, Streptococcus species and Haemophilus species. Clinical features that distinguish orbital cellulitis from preseptal cellulitis are proptosis, restricted extraocular motility, conjunctival injection, and signs of optic nerve compromise which include a relative afferent pupillary defect, optic nerve edema and/or pallor, diminished color vision and visual acuity, and constricted visual fields, though these may not all be present on exam (Figure 1).10 Classic radiographic features on CT scan of the orbits and paranasal sinuses in cases of orbital cellulitis are orbital fat stranding, anterior displacement of the globe, inflammation of the extraocular muscles, thickened sinus mucosa, and sometimes the presence of sub-periosteal abscesses (particularly in the case of adjacent sinusitis).11
Figure 1. Orbital cellulitis.
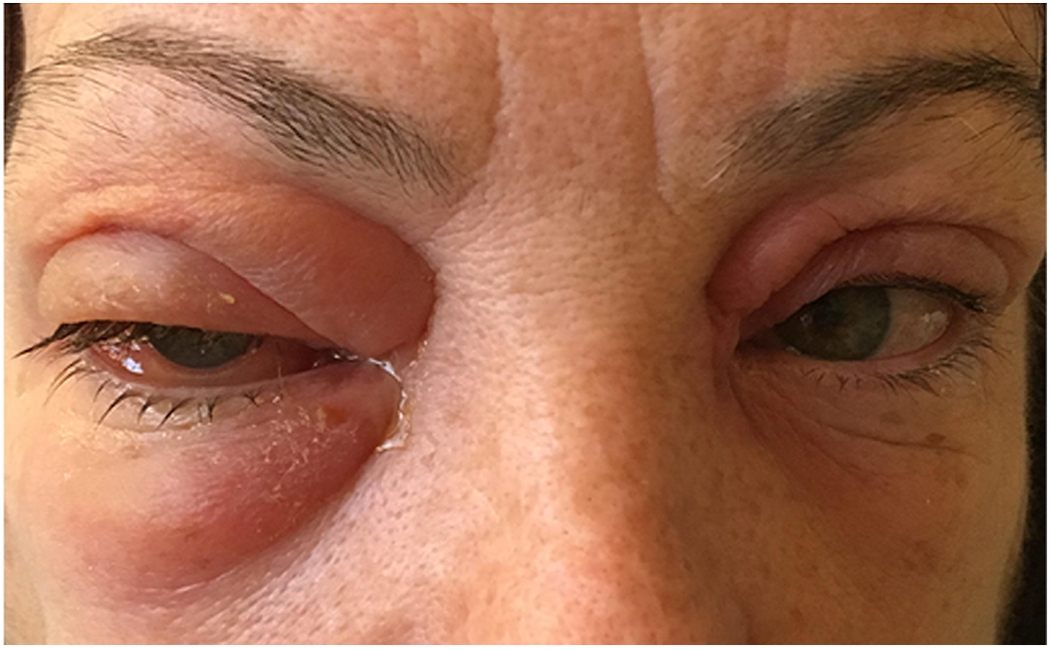
64-year-old healthcare worker found to be MRSA positive who presented with 4-day history of periorbital edema, lid swelling and chemosis.
Urgent orbital imaging, hospitalization, and broad-spectrum intravenous antibiotics administration are important in all cases of orbital cellulitis. Close monitoring with frequent ophthalmologic examinations is crucial to assess response to antibiotic therapy. Empiric intravenous antibiotic therapy typically includes vancomycin for MRSA coverage, plus a third-generation cephalosporin or ampicillin-sulbactam or piperacillin-tazobactam. Fluoroquinolones can be used in cases of penicillin or cephalosporin allergies. Transition to oral therapy should be handled with care in cases where a culture has not been obtained, given emergence of resistant strains of organisms. If significant improvement is not noted within 48-72 hours, repeat imaging should be performed to evaluate for abscess formation. In the case of adjacent sinusitis, abscesses typically are seen in a subperiosteal location. Abscesses that fail to respond to intravenous therapy within 48 hours, large abscesses greater than 10 mm in diameter, and abscesses associated with visual compromise meet criteria for immediate surgical drainage along. Consultation with ENT is also appropriate as drainage of the involved sinsus(es) may be indicated at the same time. Complications from untreated orbital cellulitis can be vision or life threatening. These include cavernous sinus thrombosis, central retinal artery or vein thrombosis, intracranial extension of abscess and optic neuropathy.12
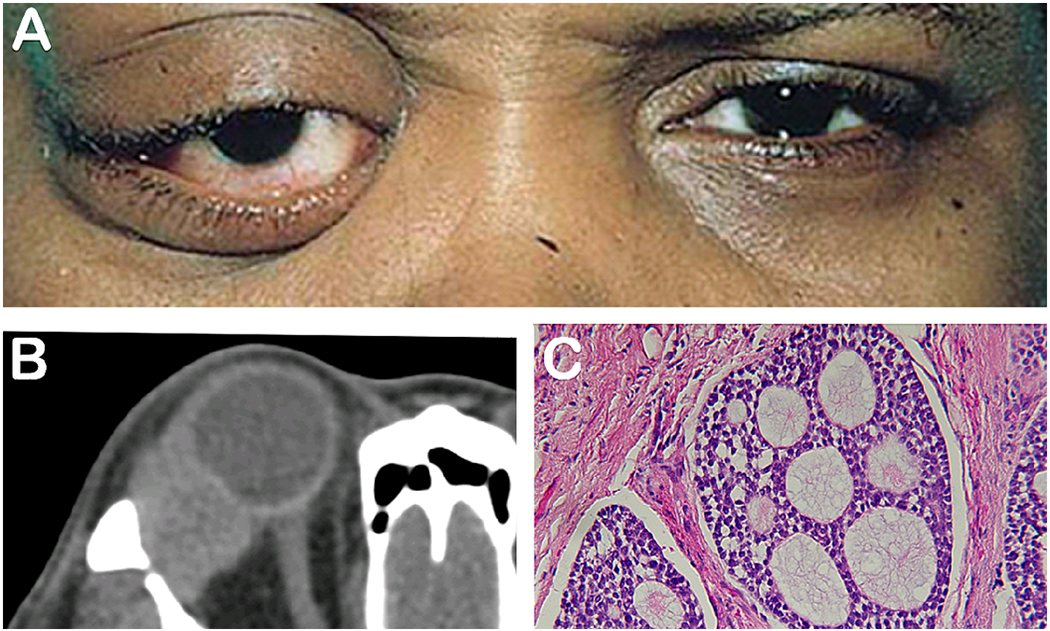
Mucormycosis
Rhino-orbital mucormycosis is an invasive, opportunistic fungal infection with high morbidity and mortality that can affect immunocompromised patients, diabetic patients, patients undergoing deferoxamine therapy, and patients with many other associated conditions. In most cases, mucormycosis is caused by fungi in the order Mucorales, such as Rhizopus, Mucor, and Rhizomucor. Infection originating in the sinuses can extend directly to the brain, the orbit, and the cavernous sinus. Orbital involvement can present clinically with proptosis, periorbital edema, extraocular motility restriction, facial numbness, and visual compromise. (Figure 2). Though considered a form of orbital cellulitis, the typical inflammatory features (eg. redness of the conjunctiva and erythema of eyelids) are blunted due to the diminished immunological response by the patient. Cranial nerve palsies may indicate cavernous sinus involvement and carries a poor prognosis. Examination of the nasal cavity or the oropharynx is an important diagnostic step, and often reveals necrotic eschars. Samples of these tissues in the nasal cavity or oropharynx can be swabbed/scraped and sent immediately to the microbiology lab for Gram staining to rapidly confirm the diagnosis. The seasoned microbiologist will quickly identify the characteristic non-septate hypha with right angle branching. If the specimen is negative in the context of high clinical suspicion, a larger specimen can be obtained to improve the yield in finding hyphae while empiric treatment is continued.13 Sinus symptoms include epistaxis, tenderness and headaches. Signs of intracranial spread include obtundation, hemiparesis, and seizures. CT and MRI imaging show the extent of disease and can aid in surgical planning which may involve ENT and Neurosurgery.
Figure 2. Mucormycosis.
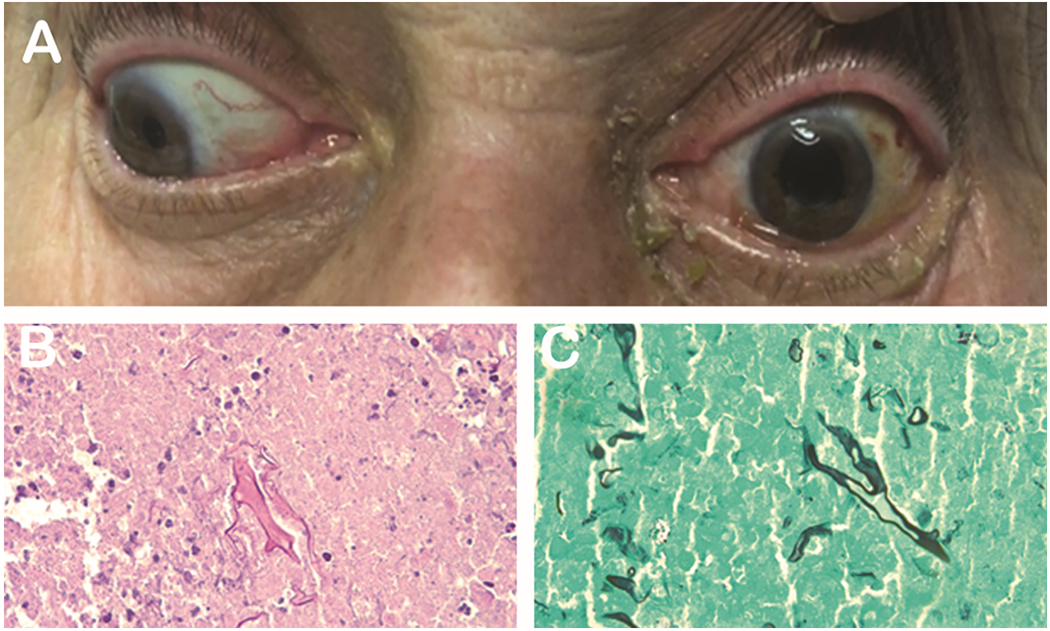
72-year-old man with a past medical history of diabetes and COPD on steroids presented with 3 day history of mild proptosis, ophthalmoplegia and decrease in vision (A). H&E (B) and GMS (C) show growth of mucormycosis. Photo courtesy of Kenneth Fan, MD, MBA.
Treatment includes a multidisciplinary approach with antifungal therapy and surgical debridement along with efforts to rapidly address the underlying immunosuppressed state. Patients in diabetic ketoacidosis require expeditious normalization of their electrolytes, glucose and pH. Immunocompromised patients on chemotherapeutic agents and/or immune modulating agents will need to be optimized by their respective providers as well. The lipid formulation of the polyene amphotericin B is the initial antifungal agent of choice. Salvage therapy with posaconazole can be considered for patients who are not responding to amphotericin B or cannot tolerate it. Fluconazole and voriconazole do not have reliable activity against the culprit fungi. Adjunctive treatment with hyperbaric oxygen can be considered in select patients.14,15 Surgical treatment for disease limited to the orbit involves orbital exenteration with repeat debridement if margins are positive. Frozen sections or specimens stained with calcofluor help assess margins.16,17 Globe sparing treatments have been described in case reports, showing success with intraorbital amphotericin injections, however, not all cases are amenable to this approach and this modality should be approached with caution given the high mortality associated with this disease process.
Mucocele
Paranasal sinus mucoceles are benign, encapsulated, epithelium-lined lesions that are commonly caused by obstruction of sinus ostia and frequently invade into the orbit, causing periorbital swelling and proptosis. The frontal and ethmoid sinuses are the most commonly affected locations.18,19 Risk factors include inflammation leading to closure of the sinus ostia (allergies, infection, sinus tumors, trauma, and prior surgery), as well as anatomic abnormalities. Once the ostia are obstructed, mucous secretions accumulate, causing expansion of the mucocele.20
Expansion into the orbit can result in progressive periorbital swelling, pain, proptosis and diplopia (Figure 3). Visual acuity can be compromised as a result of compressive optic neuropathy.21 CT imaging shows a homogenous mass lined by a contrast enhancing membrane and adjacent bony remodeling. When there is uncertainty in the diagnosis, MRI can be performed and will demonstrate varying T1 and T2 signal strength; gadolinium can be helpful in determining the underlying etiology in secondary mucocele formation.22
Figure 3. Mucocele.
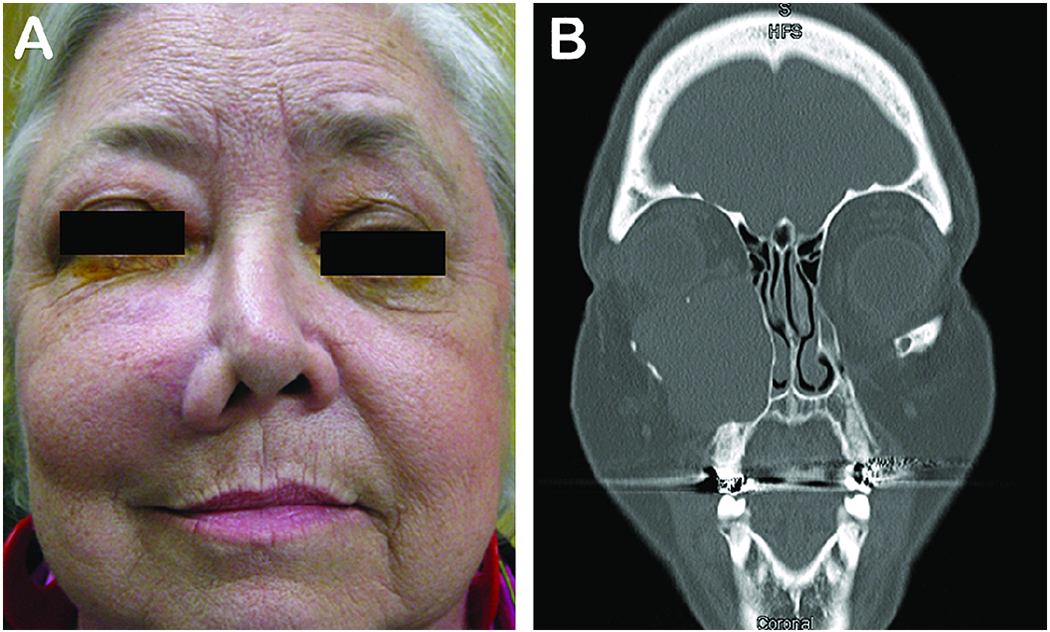
A 63-year-old woman who presented with a slowly progressing mass on the check with diplopia and upward displacement of the left globe (A). CT scan revealed an enlarged encapsulated left maxillary sinus mucocele (B).
Early surgical marsupialization or excision with endoscopic sinus surgery, and less commonly with an external approach, are the standard treatments for symptomatic paranasal sinus mucoceles. Vision loss is more common with posterior ethmoid, clinoid and sphenoid mucoceles, given their proximity to the optic nerve, and with infected mucoceles (mucopyoceles). Recurrence is lower with an endoscopic versus external surgical approach, lower with marsupialization versus total excision, and higher in patients with predisposing factors for the development of mucoceles.23,24,25,26
Orbital Inflammatory Conditions
Thyroid eye disease
Estimated to have an annual incidence of 16 women and 3 men per 100,000 people,27 thyroid eye disease (TED) is the most common cause of unilateral or bilateral proptosis.28 An autoimmune inflammatory condition, the disease is driven by the production of autoantibodies against the thyrotropin receptor, which stimulates the proliferation of orbital fibroblasts and adipocytes and upregulates other inflammatory mediators.29 TED is more common in women than men, and has a bimodal distribution, with peaks of incidence in the 5th and 7th decades of life.30 Thyroid eye disease is largely associated with autoimmune thyroid processes (95%) such as Graves Disease (90%) and Hashimoto’s thyroiditis (5%), however there is a small subset of patients (5%) that are euthyroid.31 Approximately 25% of patients with Graves disease will develop thyroid eye disease.32 Risk factors for thyroid eye disease include smoking, older age, extreme physical or psychological stress, prior treatment with radioactive iodine, and increased titers of antithyroid stimulating hormone receptor antibodies.33,34
The most common ocular manifestation of TED is eyelid retraction, seen in 90% of patients. In fact, eyelid retraction may manifest prior to symptoms of hyperthyroidism or serologic evidence of hyperthyroidism by as many as 6-12 months. The internist and general ophthalmologist should remain vigilant in a patient who presents with asymptomatic unilateral eyelid retraction and maintain a low threshold for serial serologic thyroid evaluations.
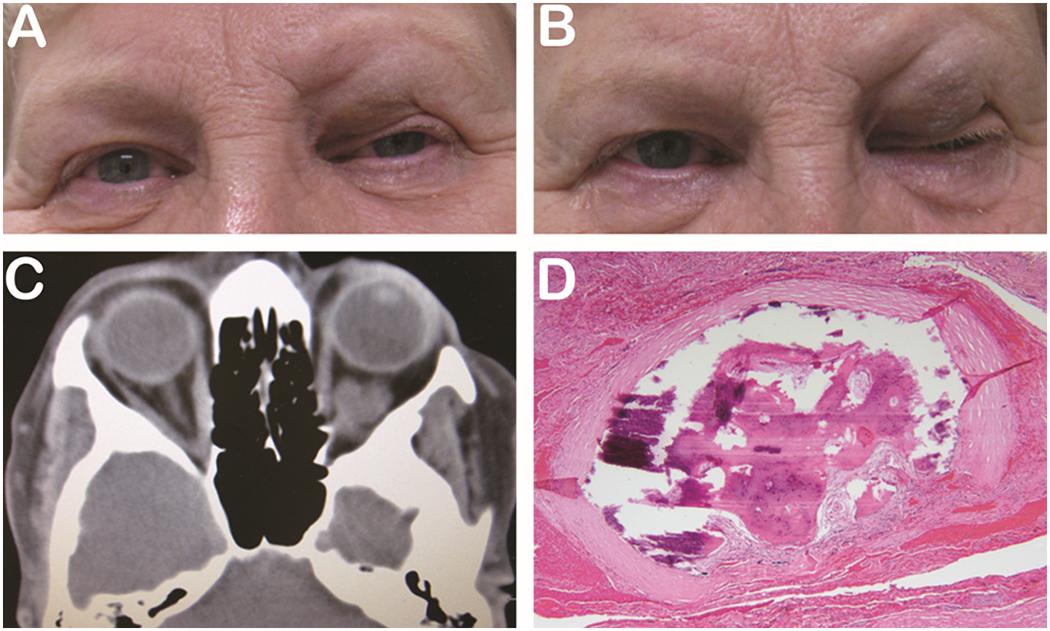
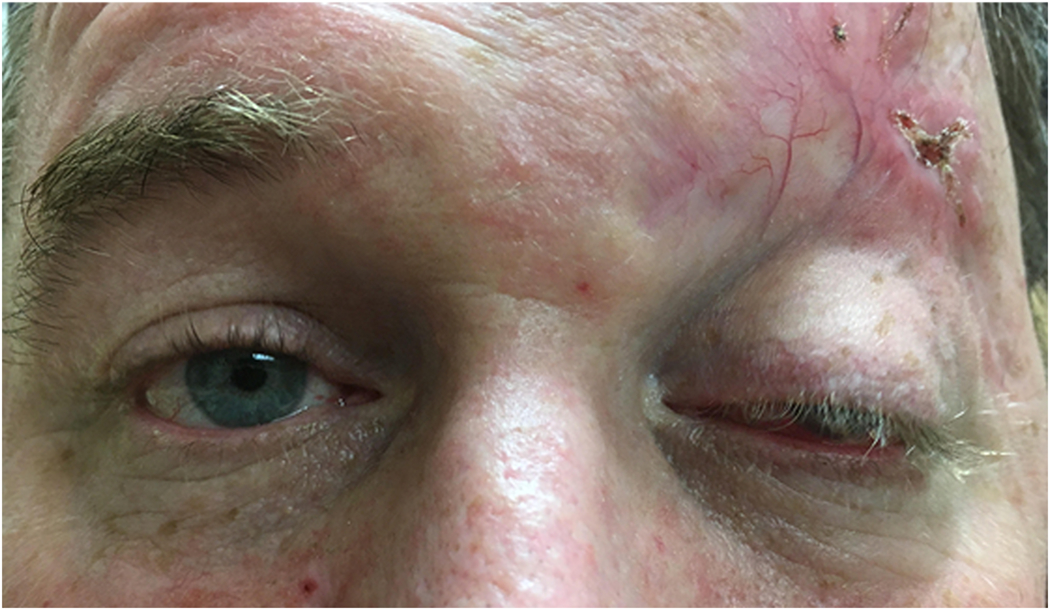
Other common clinical findings include proptosis, ocular misalignment, and lagophthalmos. Common ophthalmologic symptoms include dull orbital pain with or without eye movements, ocular surface discomfort and excessive tearing. Severe disease can result in vision loss through a variety of mechanisms including compressive optic neuropathy, corneal ulceration, globe subluxation, and choroidal folds. (Figure 4). The natural course of the disease consists of an initial inflammatory phase lasting 6–24 months, followed by a quiescent/cicatricial phase marked by scar formation within the orbit and eyelids.35 Grading the level of inflammation can be achieved using the Clinical Activity Score (CAS), which is useful in helping gauge level of activity between visits and guiding treatment of the orbital component of the disease.36
Figure 4. Thyroid eye disease.
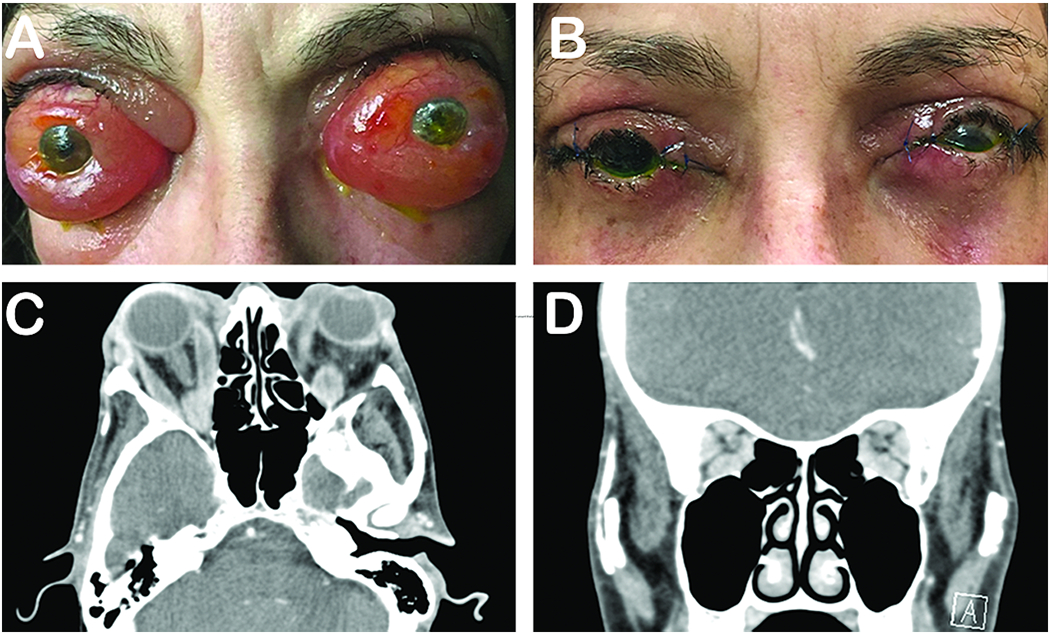
57-year-old woman presented with a long-standing history of proptosis and chemosis with acute decrease in vision (A). The patient underwent a bilateral 3-wall orbital decompression (B). Pre-operative CT axial (C) and coronal (D) illustrate substantial orbital apex crowding. Photo courtesy of Alexandra Levitt, MD.
The diagnosis of TED can be confirmed with the presence of various clinical and radiographic signs. Laboratory evaluation should include T3, free T4, TSH levels, thyroid peroxidase, TSH receptor antibody, and thyroid stimulating immunoglobulins.37 CT and MRI are the preferred modalities for detecting orbital fat expansion and fusiform rectus muscle enlargement with sparing of muscle tendons.38,39 The appearance of the muscle helps distinguish this process from other muscle involving processes (idiopathic orbital inflammation, lymphoma, etc.) where the muscle tendons are not spared.
Ophthalmologists and endocrinologists must work together to treat patients with thyroid eye disease. In all thyroid eye disease patients, smoking cessation and control of thyroid hormone levels is critical. For mild disease, treatments can include lubrication for ocular surface disease, transconjunctival botulinum toxin injections for eyelid retraction, and selenium supplementation (100 micrograms twice daily) for those with orbital pain. For moderate to severe TED, IV and oral corticosteroids are the mainstay of treatment. One recommended treatment includes 500mg IV methylprednisolone weekly for 6 weeks, followed by 250mg weekly for 6 weeks,40 however relapse rates are approximately 20% after complete treatment.41 Radiotherapy in combination with systemic steroids has also been shown to be an effective treatment approach. In the case of treatment failure or intolerance, immunosuppressive therapies such as methotrexate or mycophenolate, or biologic therapy with rituximab, can be considered. Azathioprine, while not effective as a single agent, can be used in combination with steroids or radiotherapy. Two promising new biologic agents, teprotumumab42 and tocilizumab43, are currently being studied with regards to their efficacy in the treatment of moderate, non sight threatening thyroid eye disease with extremely promising results. For severe, sight-threatening, and disfiguring thyroid eye disease, a combination of therapies including immunosuppression, radiation therapy, and urgent surgical decompression should be considered. In stable patients with inactive disease, disfiguring proptosis should be addressed first with orbital decompression, followed by strabismus surgery for patients with diplopia, and lastly, surgical lid retraction repair for those with corneal exposure from lid retraction.44,45,46
Granulomatosis with polyangiitis
Granulomatosis with polyangiitis (GPA; formerly, Wegener’s granulomatosis), is a rare autoimmune small vessel vasculitis with a prevalence in the United States of 3 per 100,000. The disease most commonly occurs in the fifth and sixth decades of life, and affects Caucasians more than other races. Its pathogenesis is linked to the generation of anti-neutrophil cytoplasmic antibodies (ANCA). The disease is characterized by necrotic granulomatous inflammation described in various organ systems including skin, kidneys, heart, upper and lower airways, and ears.47,48
Signs and symptoms of ocular and orbital GPA include pain, erythema and edema of the eyelids, conjunctival injection, nasolacrimal duct obstruction, ophthalmoplegia, diplopia, proptosis, and vision loss. Vision loss can be caused by necrotizing scleritis, peripheral ulcerative keratitis (melting of the cornea), and compressive optic neuropathy caused by orbital inflammation and granulomatous orbital masses. Diagnosis of GPA is confirmed with laboratory and clinicopathologic correlation. Although ANCA positivity is very helpful in establishing the diagnosis, it is not required. Nonspecific acute phase reactants like ESR and CRP will often be elevated, and there is frequently a leukocytosis. CT with contrast can show orbital inflammation, granulomatous lesions, obliteration of tissue planes, and bony erosion. On MRI imaging intraorbital GPA lesions are usually T2 hypointense with variable enhancement with gadolinium. Radiologic findings, however, are often non-specific. Treatment options include systemic glucocorticoids, immunomodulatory agents such as cyclophosphamide, methotrexate, mycophenolate mofetil, and azathioprine, and biologies like rituximab. Rarely, there is severe orbital inflammation requiring surgical decompression.49,50
Sarcoidosis
Sarcoidosis is a systemic inflammatory disease of unknown etiology that commonly affects the lungs, skin, and eyes. Histopathologically, it is characterized by non-caseating granulomatous inflammation. In patients with biopsy proven sarcoidosis, African Americans are more likely to develop ocular manifestations than Caucasians.51 Ocular involvement is the presenting manifestation in approximately 20 to 30% of sarcoidosis patients, and can affect all intraocular and adnexal tissues. Eyelid granulomas, conjunctival granulomas, scleritis, iris nodules, anterior uveitis, intermediate uveitis, retinal vasculitis, choroiditis, choroidal granulomas, and papillitis are just some of the many forms of intraocular involvement of sarcoidosis. Neurosarcoidosis often results in cranial neuropathies, including vision loss from optic nerve or chiasmal lesions. In the orbit, sarcoidosis can involve the lacrimal gland, orbital fat, extraocular muscles, and optic nerve sheath, and can cause proptosis as well as pain, diplopia, vision loss, and globe displacement.52
On imaging, orbital and adnexal sarcoidosis demonstrates circumscribed masses in approximately 85% of patients, and diffuse infiltration in about 15%. Orbital and adnexal sarcoidosis is most conclusively diagnosed with biopsy, though in cases where biopsy is not feasible, chest CT or chest x-ray demonstrating hilar adenopathy, elevated serum angiotensin converting enzyme, and elevated serum lysozyme can aid in the diagnosis. Of note, in one study, systemic disease was diagnosed before the diagnosis of orbital/adnexal disease in 37%, at the time of diagnosis in 27%, and after diagnosis in 7% of patients. Treatment options include observation, intralesional steroid injections, systemic corticosteroid therapy, surgical excision.53 For refractory disease, other steroid sparing agents, such as methotrexate, hydroxychloroquine, azathioprine, and mycophenolate can be considered.54,55
Idiopathic Orbital Inflammation
Idiopathic orbital inflammation (IOI), also known as orbital pseudotumor and nonspecific orbital inflammation, is an inflammatory condition of the orbit with no known etiology. Histopathologic analysis of IOI tissue reveals a nonspecific, polymorphous inflammatory infiltrate and sometimes demonstrates dense fibrosis. It can present with myositis, dacryoadenitis, scleritis, diffuse inflammation, apical inflammation, and it can occasionally present with a focal mass. Disease is most commonly unilateral, but can also be bilateral. Clinical signs of IOI vary greatly based on the tissues involved. In dacryoadenitis (inflammation of the lacrimal gland), the most common subset of IOI, a painful mass in the lateral upper eyelid is seen. In anterior orbital involvement and myositis, pain, periorbital edema, ptosis, chemosis, decreased ocular motility can be seen. In diffuse disease, pain and proptosis are typically seen in addition to other findings. Posterior orbit or orbital apex involvement is more often associated with vision loss, as well as orbital pain, decreased ocular motility, and proptosis. Optic perineuritis is a rare manifestation of IOI characterized by inflammation of the optic nerve sheath and typically presents with pain and visual field defects. Inflammation can extend intracranially through the superior and inferior orbital fissures and optic canal, causing cavernous sinus, pterygopalatine fossa, or middle cranial fossa involvement.
CT and MRI findings will similarly vary based on the tissues involved, and may show enhancing lesions, lacrimal gland or rectus muscle enlargement, scleral enhancement, or inflammation of the eyelids and orbital fat. Tendon involvement helps to distinguish IOI from TED with similar radiographic features. Laboratory testing is unremarkable. Diagnosis is typically made on the basis of clinical presentation, orbital imaging, and prompt response to systemic steroid administration. In cases of failed or incomplete response to steroids or recurrence, orbital biopsy can be considered to establish a diagnosis and rule out other processes. Alternative therapies for refractory IOI cases include external beam radiotherapy and immunomodulatory agents such as methotrexate, azathioprine, cyclosporine, and cyclophosphamide.56,57,58
IgG4 serves as a newly characterized fibro-inflammatory disease along the spectrum of orbital inflammation. First related ophthalmic cases occurred in chronic sclerosing dacryoadenitis, however, infiltration within the extraocular muscles, infraorbital nerve, surrounding orbital tissue and nasal lacrimal duct, and sclera has been described. The gold standard of diagnosis requires biopsy with immunohistochemical staining of increased IgG4+ plasma cells. Treatment usually consists of systemic prednisone. Other immunosuppressive agents including azathioprine, methotrexate and mycophenolate have been used in steroid refractory treatment. Rituximab an anti-CD20 inhibitor serves as a third line of treatment by depleting CD20-positive B-cells, impeding the ability to create IgG4+ cells, has been particularly successful in treatment of the disease.59,60,61
Vascular Causes
Orbital hematoma/retrobulbar hemorrhage
Retrobulbar hemorrhage (RBH) or orbital hematoma is a rare and rapidly progressive cause of orbital compartment syndrome that can result in severe vision loss due to compression of the optic nerve, occlusion of the central retinal artery, or both. It is caused by trauma in the majority of cases, but can also occur postoperatively after repair of orbital fractures, endoscopic sinus surgery, and blepharoplasty. Presenting signs and symptoms include pain, proptosis, diminished vision, pupillary disturbances, reduced ocular motility, elevated intraocular pressure, and subconjunctival hemorrhage. Lateral canthotomy with inferior and superior cantholysis is the mainstay of treatment for symptomatic RBH, but urgent surgical evacuation of the hematoma should be pursued if canthotomy fails to provide sufficient relief. Shorter time to treatment is associated with a lower likelihood of blindness.62,63
Carotid-cavernous fistulas
Carotid-cavernous fistulas (CCF) are anomalous communications between the carotid artery and the cavernous sinus that can occur spontaneously (~25-30%) or following trauma (70-75%), classically in cases of basilar skull fractures. They can be classified hemodynamically as low flow or high flow. Anatomic classifications are based on whether there is a “direct” connection between the cavernous sinus and internal carotid artery, or an “indirect” connection between the cavernous sinus and branches of the internal or external carotid artery. Presenting signs and symptoms are often more pronounced in the high flow variants, and can include proptosis, ocular bruit, chemosis, corkscrew episcleral vessels, pain, ophthalmoplegia, and decreased vision (Figure 6).
Figure 6. Carotid-cavernous fistulas.
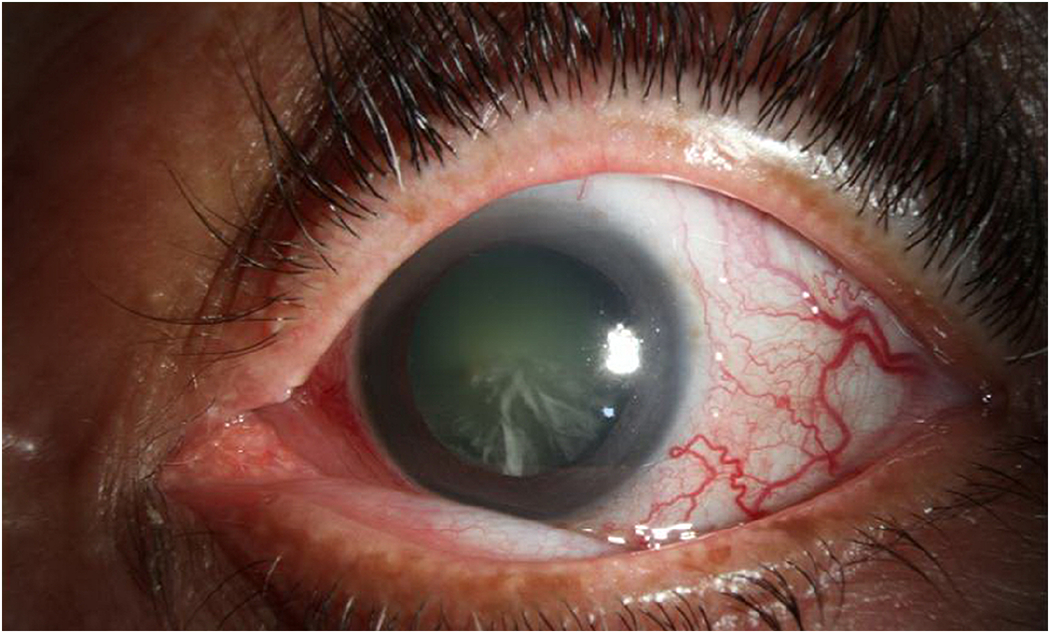
An 83-year-old woman who presented with bilateral orbital congestion syndrome and prominent corkscrew episcleral vessels was found to have a carotid cavernous fistula and underwent coil embolization.
CT, CTA, MRI, and MRA can show a dilated superior ophthalmic vein, orbital congestion, proptosis, enlarged extraocular muscles, cavernous sinus enlargement, and associated fractures. Cerebral angiogram is the gold standard for diagnosis of CCFs. Some slow flow lesions can spontaneously resolve. Treatment options for symptomatic lesions threatening vision include surgical clipping or suturing of the fistula, surgical ligation of the carotid artery, endovascular embolization, endovascular placement of covered stent grafts in the ICA, or endovascular arterial sacrifice. Radiosurgery can be utilized in low flow fistulas, but will not result in complete obliteration until months or years after treatment. Manual compression of the ipsilateral cervical carotid is another option for indirect, low flow CCFs.64,65
Orbital varix
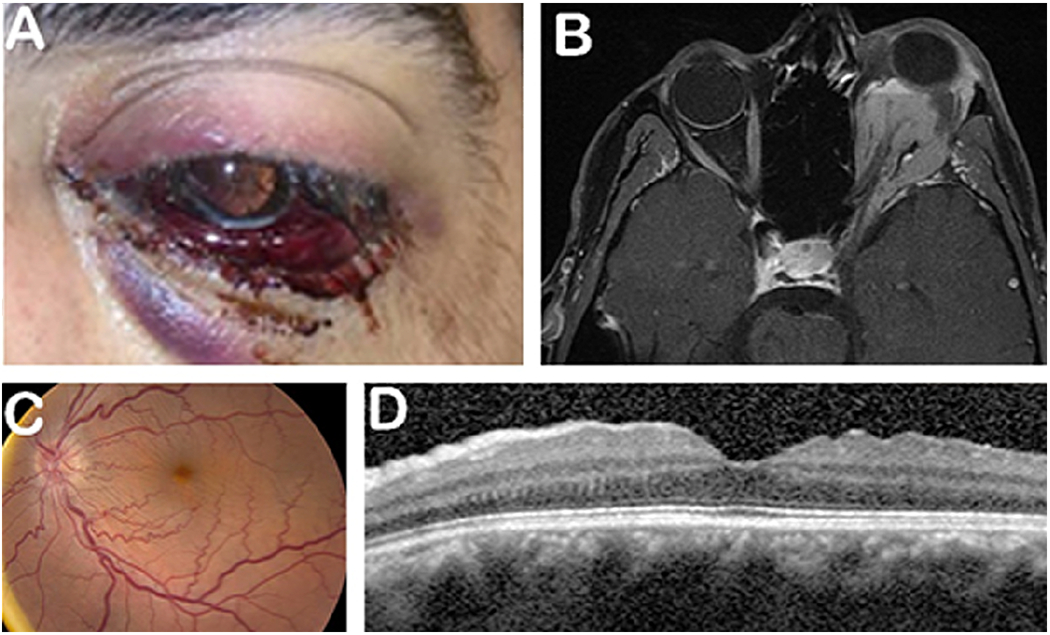
Orbital varices are venous malformations consisting of a network of thin-walled, distensible low-flow vessels. These lesions typically manifest in the 2nd or 3rd decade of life, and are usually unilateral, although cases of bilateral varices have been reported. Patients complain of intermittent positional proptosis that occurs whenever venous pressure increases, such as with Valsalva maneuvers, coughing, and forward bending (Figure 7). In cases of thrombosis or hemorrhage, proptosis can develop rapidly and painfully, and can result in orbital compartment syndrome and potential vision loss. Lesions are better seen on CT and MRI when imaging is done in a position that increases venous pressure and distends the varix (prone or Valsalva maneuver), and typically enhance with contrast administration.66 Doppler ultrasound can also be performed dynamically to demonstrate distensibility of lesions. Treatment of orbital varices is typically conservative, however for symptomatic lesions, surgical resection, carbon dioxide laser ablation, sclerosing therapy, and transcatheter or percutaneous embolization can be considered.67,68,69 Over time, patients can develop enophthalmos from fat atrophy.70
Figure 7. Orbital varix.
63-year-old woman presents with left bluish upper eyelid lesion (A) that increases in size with Valsalva (B). MRI shows isointense vascular lesion (C) and H&E shows thrombosed orbital varix (D).
Orbital lymphatic malformations
Orbital lymphatic malformations, previously known as lymphangiomas, are vascular malformations comprised of dilated sacs and lobules lined by endothelial cells that typically present in childhood. Pure lymphatic malformations are classified as type 1 orbital vascular malformations indicating no flow, however, there can be combined lymphaticovenous lesions with low venous flow, which are classified as type 2 lesions. They often have both intraorbital and periorbital/eyelid components; they do not respect tissue planes, and they can also be associated with intracranial vascular malformations. They are classified as macrocystic (cysts larger than 2 cm3 in volume), microcystic (cysts less than 2 cm in volume3), or mixed (multiple large and small cysts). Many of these lesions are small and asymptomatic, only coming to attention as an incidental finding when an imaging study is performed assessing something else. Larger lesions manifest by proptosis, ptosis, periorbital edema, pain, and limitation in ocular motility (Figure 8). Lesions can enlarge acutely in the setting of a respiratory tract infection (due to a robust immune response) or from a spontaneous hemorrhage into the lesion. These can be sight threatening if they cause optic nerve compression and require careful monitoring and possible surgical intervention.
Figure 8. Orbital lymphangioma.
25-year-old man presents with intermittent proptosis and subconjunctival hemorrhage (A). MRI showsw an intraconal multi-cystic lesion (B). Posterior exam shows optic nerve edema and tortuous vessels (C) and retinal folds (D).
On CT and MRI imaging, lymphatic malformations will appear as a mix of solid and cystic components, sometimes with fluid-fluid levels (if there is stasis) and with variable enhancement with contrast. Doppler ultrasound can be used to assess for flow. If there is a venous component to the lesion, it may demonstrate distensibility with Valsalva on dynamic imaging.
Observation can be appropriate for asymptomatic lesions. Indications for intervention include threat for development of amblyopia (ptosis blocking the pupil, or large lesion causing refractive error from misshapened globe), compressive optic neuropathy, exposure keratopathy, and severe disfigurement. Surgical excision can be difficult because of the lesion’s irregular and non-encapsulated nature and high risk of recurrence with incomplete excision. Intralesional injections of liquid polymers like cyanoacrylate or fibrin glue can be performed to facilitate excision. Alternatively, non-surgical approaches can be considered with percutaneous injection of sclerosants such as bleomycin, tetradecyl sulfate, ethanol and doxycycline, percutaneous drainage with chemoablation, and adjunctive medical management with systemic corticosteroids or sildenafil. Orbital exenteration may be appropriate in some cases of severe disfigurement and blind painful eye.71,72
Capillary Hemangioma
Capillary or infantile hemangiomas, are benign vascular neoplasms that represent the most common eyelid and orbital tumors of infancy. Systemic associations include PHACES and Kassabach-Merritt syndromes. They typically present within the first few weeks to months of life, with 1/3 present at birth. Complete resolution occurs in about ¾ of untreated patients by age 7. A rapid proliferative phase typically occurs until 6–18 months of age, during which endothelial cells multiple and form clusters, followed by an involutional phase, in which fibrosis predominates. Risk factors include female sex, prematurity, low birth weight, advanced maternal age. Lesions can be superficial, deep, or both. Superficial lesions are red in color and blanch on compression. Subcutaneous lesions are usually bluish in color and spongy-feeling (Figure 9). Large orbital lesions present with unilateral proptosis without skin discoloration. Orbital lesions are most commonly seen in the superior orbit. Valsalva maneuvers may result in enlargement of the mass lesion.
Figure 9. Capillary hemangioma.
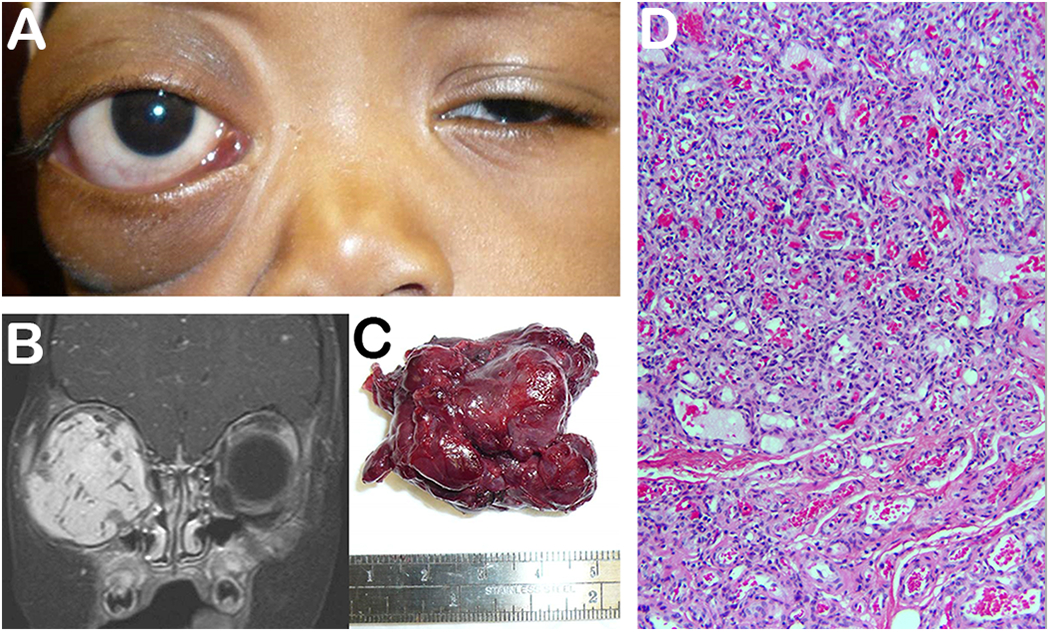
4-year-old boy presents with gradual right orbital proptosis over past few months (A). MRI imaging reveals a large homogenous hyperintense bright lesion that enhances with gadolinium (B). An encapsulated vascular lesion was removed (C). H&E illustrates a tumor composed of capillary sized blood vessels (D) (Photo courtesy of David Tse and Sara Wester).
Other clinical findings include ptosis, strabismus, and compressive optic neuropathy. Any changes in visual acuity or ocular alignment places them at risk of developing amblyopia. The same can be said if there is a ptosis that is blocking light from entering the eye. A treatment strategy that quickly addresses these clinical manifestations is paramount in allowing for full maturation of the visual circuit.
On CT imaging, orbital capillary hemangiomas appear as homogeneous, enhancing soft tissue masses that can either be well-circumscribed or irregular. On MRI, they appear hyperintense on T2 weighted images with fat suppression. Internal lobulations and signal flow voids can be seen as well.
Treatment is indicated for symptomatic lesions that threaten vision loss or disfigurement. Historically, treatment options included injection of local corticosteroid, laser, and surgical excision. More recently, oral beta blockers have been found to be highly effective in treating capillary hemangiomas during the proliferative phase of tumor growth, and they have become the first line treatment. A typical dose of oral propranolol ranges from 2 to 3 mg/kg/day. This treatment is usually coordinated with a pediatrician. Topical beta blockers can be used for superficial lesions. Surgical excision can be considered for well-circumscribed lesions and for those with an inadequate response to medical therapy.73,74
Cavernous hemangioma
Cavernous hemangioma, more recently labeled as cavernous venous malformation, is the most common benign orbital lesion in adults. Women are affected more often than men, and presentation typically occurs in the 4th or 5th decades of life. The most common presentation is progressive axial proptosis, but other presenting signs and symptoms are diplopia, globe displacement, pain, and vision impairment as a result of compressive optic neuropathy or hyperopic shift from flattening of the posterior globe by tumor. On ophthalmological exam, afferent pupillary defect, choroidal folds, optic disc swelling or atrophy, visual field deficits, and extraocular motility disturbances may be seen. Systemic associations with cavernous hemangiomas include Maffucci syndrome and blue rubber bleb nevus syndrome. Like lymphatic venous malformations, most of these lesions are asymptomatic and identified as an incidental finding when neuroimaging is performed to assess an unrelated symptom.
Ultrasound of cavernous hemangiomas will show encapsulated masses with medium to high internal reflectivity without signs of internal vascular flow. CT or MRI imaging demonstrates well-circumscribed lesions that can indent the globe, mold to bony surfaces, and show variable enhancement. MRI is most useful in surgical planning as it better delineates anatomic relationships between the tumor and surrounding structures. T1-weighted MRI shows lesions that are isointense to muscle and hypointense to fat, while T2-weighted MRI shows lesions that are hyperintense to fat with variable contrast enhancement. Lesions most commonly occur in the lateral aspect of the intraconal space. Histopathology reveals encapsulated lesions with fibrous septae separating ectatic vessels lined by endothelial cells. There can also be areas of intralesional thrombosis.
Asymptomatic cavernous hemangiomas can often be observed. Symptomatic or disfiguring lesions can be treated with surgical excision through orbitotomy, endoscopic resection, or neurosurgical approach. If surgery poses too much risk, given the proximity to critical anatomical structures or lesion size, or if the patient is unable or unwilling to undergo surgery, intralesional sclerotherapy can be considered.75,76
Orbital Tumors and other malignancies
Orbital rhabdomyosarcoma
Rhabdomyosarcoma, a rare malignant neoplasm arising from mesenchymal cells, is the most common soft tissue sarcoma in children, and accounts for about 5% of all childhood cancers, occurring in the first decade of life. Forty percent involve the head and neck region. Orbital cases account for 25% of the head and neck rhabdomyosarcoma, and have increased survival when compared to other primary head and neck sites, possibly because they typically present with more localized disease. Histopathologically, the four major types include pleomorphic, embryonal, alveolar, and botryoid. Embryonal rhabdomyosarcoma is the most common subtype and has the best prognosis, while alveolar and pleomorphic rhabdomyosarcomas portend a worse prognosis. 77,78,79
Ocular involvement can be primary, secondary or metastatic. Primary ocular rhabdomyosarcoma typically presents with rapidly progressing proptosis and globe displacement, and can be misdiagnosed as orbital cellulitis or nonspecific orbital inflammation. The most common presenting signs and symptoms of orbital rhabdomyosarcoma other than proptosis and globe displacement include ptosis, conjunctival and eyelid swelling, palpable mass, and pain. The superonasal aspect of the orbit is most commonly involved. Posterior lesions can cause ophthalmoplegia, optic nerve compression, and choroidal folds. Intracranial extension and invasion of the paranasal sinus can occur. Secondary orbital rhabdomyosarcoma occurs from direct extension form the paranasal sinus or nasopharynx, and initially presents with nasal congestion or epistaxis followed later by the orbital signs mentioned above (Figure 10). Metastatic disease to the orbit, while uncommon, can occur from other primary sites.
Figure 10. Rhabdomyosarcoma.

9-year-old girl presents with an 8-day history of progressive periorbital swelling and decreased vision (A). MRI demonstrates a large, enhancing, infiltrative intraconal mass (B). Orbital biopsy shows a small blue cell tumor staining for Myod1, desmin, and myogenin consistent with embryonal rhabdomyosarcoma (C).
CT typically demonstrates a moderately well-defined, enhancing, homogenous mass that is isodense to extraocular muscles and may demonstrate bony erosion. T1-weighted MRI demonstrates an enhancing lesion that is hypointense to orbital fat and iso- or hyperintense to extraocular muscles.80,81
Management of suspected orbital rhabdomyosarcoma begins with either an incisional or excisional biopsy to establish the diagnosis. Orbital imaging can help determine whether complete surgical removal is possible, or whether surgical debulking of the lesion is more appropriate. Orbital radiation (4,000 to 5,000 Gy) and chemotherapy (typically vincristine and actinomycin) are mainstays of treatment. Occasionally, orbital exenteration is required due to recurrence of the lesion after treatment or because of extensive orbital invasion. There should be frequent follow up visits and repeat orbital imaging after treatment is completed to monitor for disease recurrence.82
Orbital Lymphoma
Lymphoma, a malignant tumor arising from B-lymphocytes, T-lymphocytes, or natural killer cells, is the most common malignancy of the ocular adnexa, and can arise in the conjunctiva, eyelids, orbit, and lacrimal gland. T-cell lymphomas are rare in the orbit, and more commonly affect the eyelids. Lymphoma typically affects elderly patients. About ¾ of orbital B-cell lymphomas occur as primary lesions, while the remaining 1/4 occur secondarily from local invasion into the orbit or metastatic disease. About 56% of T-cell lymphomas are primary while about 44% are secondary. The most common subtype of orbital lymphoma is extranodal marginal zone B-cell lymphoma (EMZL), followed by diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL), mantle cell lymphoma, and natural killer T-cell lymphoma. Patients with HIV, other causes of immunosuppression, and autoimmune diseases such as Sjogren’s syndrome, systemic lupus erythematosus, rheumatoid arthritis, and Hashimoto’s thyroiditis are at increased risk of developing lymphoma. There may be an association between development of lymphoma and infection with Helicobacter pylori, Borrelia burgdorferi, Chlamydia psittaci, and viruses including human T-cell leukemia virus-type 1, hepatitis C, human herpes virus-8, and Epstein Barr virus.
The most common clinical presentations of orbital lymphomas are proptosis, extraocular motility restriction, periorbital swelling, pain, ptosis, decreased vision, double vision, globe displacement, chemosis, and presence of a palpable orbital mass (Figure 11). Systemic B-symptoms can occur in a small subset of patients. While the vast majority of orbital B-cell lymphomas present unilaterally, almost half of orbital mantle cell lymphomas present with bilateral involvement.
Figure 11. Orbital lymphoma.
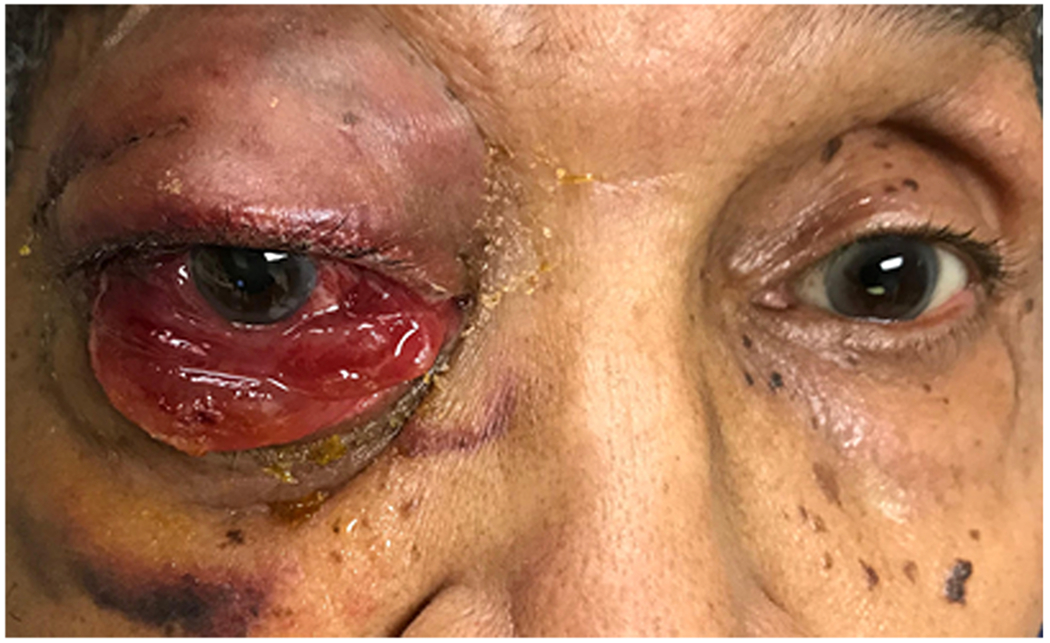
86-year-old man with multiple myeloma status post 3 weeks of chemotherapy presents with 2 months worsening right eye pain, vision loss, proptosis and complete ophthalmoplegia. Photo courtesy of Zubair Ansari, MD.
Diagnosis of orbital lymphoma is made with biopsy and subsequent histopathological, immunohistochemical, and molecular analysis. CT imaging will typically demonstrate a high-density, homogenous mass with mild contrast enhancement, while MRI imaging typically demonstrates a mass that is isointense to the brain on T1 with variable contrast enhancement. On both CT and MRI imaging, orbit lymphomas characteristically mold to the globe with minimal bony erosion. Systemic evaluation with laboratory work-up and radiological imaging is crucial in staging the disease. Staging can be performed according to the TNM system and must be performed prior to treatment.
Radiotherapy can be used as monotherapy in patients with low-grade and localized primary lymphomas, and can be used in conjunction with chemotherapy in cases of systemic spread or high-grade lymphoma. Surgical de-bulking is typically performed in conjunction with other treatments including chemotherapy and radiation therapy. Surgical approach depends on tumor location and size. One of the more commonly employed chemotherapy regimens is CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone). The monoclonal antibody, Rituximab, which binds the CD20 antigens on normal B-cells and tumor cells, is often used in combination with chemotherapy and has improved survival rates in patients with lymphoma. Doxycycline has been proposed as a treatment option for EMZL, given the possible association with C. psittaci infection, but is not typically a first line therapy. Prognosis depends largely on histological subtype of the lymphoma, as well as clinical stage of disease. Low-grade lymphomas like EMZL and FL have high remission rates while high-grade lymphomas like natural killer T-cell lymphoma and DLBCL have higher mortality rates.83,84,85,86
Lacrimal gland lesions
Lesions of the lacrimal gland include a wide spectrum of tumors, inflammatory processes, and infiltrative processes. Primary tumors can be categorized as epithelial (~1/2), lymphoid (~1/3), or mesenchymal. There can also be secondary invasion of the lacrimal gland from adjacent structures or metastases. Pleomorphic adenoma, which is an epithelial tumor, is the most common of the benign lacrimal gland lesion, and represents about 20% of all lacrimal gland tumors. Other less common benign tumors include oncocytoma and myoepithelioma. The most common malignant lesion is the epithelial tumor, adenoid cystic carcinoma. Other less common malignant lesions include carcinoma ex pleomorphic adenoma, mucoepidermoid carcinoma, ductal carcinoma, lymphoid tumors (discussed in the Orbital Lymphoma section above), neuroendocrine carcinoma, secondary invading tumors, and metastases.
Typical clinical presentations include downward and medial displacement of the globe, palpable mass, ptosis, proptosis, periocular edema (Figure 12). Pain from perineural invasion is usually seen in adenoid cystic carcinoma, but is not common for other lacrimal gland lesions. Malignant lesions typically have a more aggressive course and more rapid onset of symptoms while benign tumors typically have a more indolent course and slower growth.
Figure 12. Adenoid cystic carcinoma.
53-year-old woman presents with a painful supratemporal mass (A). Imaging reveals lacrimal gland mass with irregular margins and bony erosion (B). Histopathological analysis shows cribriform “Swiss cheese” pattern (C) (photo courtesy of David Tse).
CT and MRI imaging is useful in characterizing lacrimal gland lesions. While benign tumors like pleomorphic adenoma are typically well circumscribed round or oval lesions, malignant lesions usually have irregular margins, calcification, and can cause destruction to surrounding tissue and bone.87,88
Treatment of benign tumors of the lacrimal gland typically involves surgical resection of the lesions. Pleomorphic adenoma, the most common benign lacrimal gland tumor, must be resected completely as this tumor may undergo malignant transformation over time. Malignant lesions, such as adenoid cystic carcinoma, are typically resected completely through an orbital exenteration. Many recommend chemoreduction of the tumor prior to resection, as chemoreduction may offer an opportunity to perform successful tumor resection through a globe sparing technique. Adjuvant radiation therapy is also performed for malignant lesions. The average survival rate of adenoid cystic carcinoma is 1.5 years, with a 10-year survival rate of 20-30% and a high recurrence rate of 75%. Recent studies have shown that complete exenteration does not prolong survival. The use of neoadjuvant intra-arterial chemotherapy with orbital exenteration and radiation treatment with cycles of adjuvant chemotherapy has had marked success in a subset of patients.89 However, a standard, globally accepted treatment protocol for this disease does not exist at this time.
Optic Nerve Glioma
Optic nerve gliomas (ONG) are most common in children and adolescents and represent about 25% of all optic pathway gliomas, which can also affect the optic chiasm, tracts, radiations, or hypothalamus. ONGs can occur sporadically, but most are associated with neurofibromatosis type 1 (NF1). About 20% of children with NF1 have an ONG. The natural history is widely variable and can include stable tumors, tumors with slow growth, or rapidly growing tumors with severe progression of symptoms. These lesions are rarely malignant. Clinical presentation can include decreased vision, color vision deficit, visual field defects, afferent pupillary defect, nystagmus, strabismus, proptosis, and optic disc pallor on fundus exam. Determination of vision loss, and monitoring of vision to document disease progression in young children or infants can be technically challenging, and should be performed by an experienced ophthalmologist. MRI of the brain and orbits is the best modality for diagnosing and monitoring progression of ONGs. They can appear as diffuse enlargement of the optic nerve, sometimes with a fusiform appearance, and are typically isointense on T1 and iso- to hyperintense on T2. Gadolinium enhancement may be demonstrated.
Clear indications for treatment can be controversial given the difficulty in defining meaningful disease progression, but typically include progressive vision loss, progressive visual field defects on perimetry, and sometimes significant progression on neuroimaging. Large disfiguring ONGs and lesions that extend beyond the orbital apex and encroach on the optic chiasm are managed through surgical resection in an effort to avoid compromise of vision in the contralateral eye. Malignant optic nerve gliomas are managed with a combination of chemotherapy, radiation and surgical resection. Surgical resection is otherwise not ndicated given high risk of vision loss.90,91 Current therapies include systemic chemotherapy for first line treatment, MEK inhibitors and bevacizumab for salvage therapy, and radiotherapy as a last resort for refractory disease in older patients. Radiotherapy can be associated with secondary malignant neoplasms and cerebrovascular disease, and as these risks are already elevated in patients with NF1, its use is in this population is avoided unless absolutely necessary.
Optic Nerve Sheath Meningioma
Optic nerve sheath meningiomas (ONSM) are rare, benign neoplasms of the meninges surrounding the optic nerve, representing about 2% of all orbital tumors and 1–2% of all meningiomas. There is a female predilection, and presentation occurs most commonly during middle age. ONSMs are associated with neurofibromatosis type 2.92
Clinical presentation of ONSMs commonly includes vision loss of variable degrees, visual field defects, proptosis (59%), and strabismus. On fundus exam, the optic disc can demonstrate swelling, optic atrophy, or optociliary shunt vessels which are dilated anastomoses between the retinal and choroidal venous systems. MRI, particularly T1-weighted with fat suppression, is the most useful imaging technique employed in the diagnosis of ONSMs. The most common imaging appearances is tubular expansion of the meninges around the nerve or “tram track” appearance, followed by globular, fusiform, and focal enlargement of the nerve.
While clinical course varies, ONSMs are typically slow growing with progressive vision loss. Biopsy is not commonly employed, but can be considered when the diagnosis is uncertain. Observation can be considered in patients without vision loss. Radiotherapy, while associated with side effects such as radiation retinopathy and vascular occlusion, has significantly lower risk of complications compared with surgical resection, and is the mainstay of treatment for ONSMs. With radiotherapy treatment for ONSMs, stability or improvement in vision has been reported in over 80% of patients. Surgical resection of ONSMs often results in vision loss, and can be considered in patients with no useful vision on the affected side in order to prevent contralateral spread, or in cases of severe disfigurement.93,94
Metastatic tumors to the orbit
Orbital metastasis has an estimated prevalence in 2 to 4.7%% of cancer patients. The rise of orbital metastatic lesions can be related to an increase in median survival rate of cancer patients, improved diagnostic capability and treatment. Predominantly in adults over the age of 75 years, many of these patients have known systemic involvement; however, 1/4 of patients have no systemic diagnosis and require a systemic work up. The most common etiologies include breast carcinoma (28.5% - 58.8%), lung carcinoma (8-12%), prostate carcinoma (3-10%), cutaneous melanoma (5.3 – 15%), carcinoids (4-5%) and other malignancies. Metastasis secondary to breast carcinoma and melanoma have a delayed presentation due to a latency period while lung cancers occur earlier in the disease course.95,96
Common presenting symptoms include proptosis, globe displacement, pain, diplopia or ptosis. Enophthalmos can occur with sclerotic tumors such as certain subtypes of breast and gastric carcinoma (Figure 13). Facial flushing or vasomotor disturbances may be seen in carcinoid tumors due to secreted bioactive amines. MRI imaging can illustrate well-defined soft-tissue discrete lesions with possible extraocular muscle involvement. Prostate carcinoma shows osteoblastic lesions. Cutaneous melanoma has a high involvement of the extraocular muscles. An orbital biopsy is required to establish a diagnosis in all cases of metastatic disease.
Figure 13. Invasive squamous cell into the orbit.
51-year-old man with prior history of squamous cell carcinoma of the brow status post excision who presents with 2 month history of worsening proptosis of the left eye.
Treatment goals for orbital metastasis depend on the patient’s overall health and ability to maintain quality of life or preserve vision. Surgical resection can be appropriate in cases of metastatic cutaneous melanoma or sarcomas. Radiotherapy is useful for orbital lesions with rapid expansion. Additional chemotherapy may be required. Melanomas may warrant immunotherapy. Combined hormonal therapy can be useful in prostate cancer. Orbital metastasis has a poor prognosis: breast cancer (31 months), lung cancer (16 – 22 months) and melanoma (12 months).
Other causes of “proptosis”: Pseudoproptosis
Pseudoproptosis occurs when the eye has a proptotic anatomic appearance, not due from a mass effect from a lesion displacing the globe. Considerations include a buphthalmic eye, such as might be seen as a result of congenital glaucoma or high myopia, contralateral enophthalmos (from a contralateral orbital fracture), asymmetric bony orbital size/volume, or asymmetric eyelid anatomy.
Conclusion
Causes of proptosis range with a wide breadth of pathology. A systematic approach when evaluating proptosis can narrow down the differential diagnosis. The internist and ophthalmologist should be familiar with the range of disease so as to appropriately triage these patients to an oculoplastic specialist. An understanding of the vision threatening etiologies will improve patient case and outcomes.
Figure 5. Non-specific orbital inflammatory syndrome.
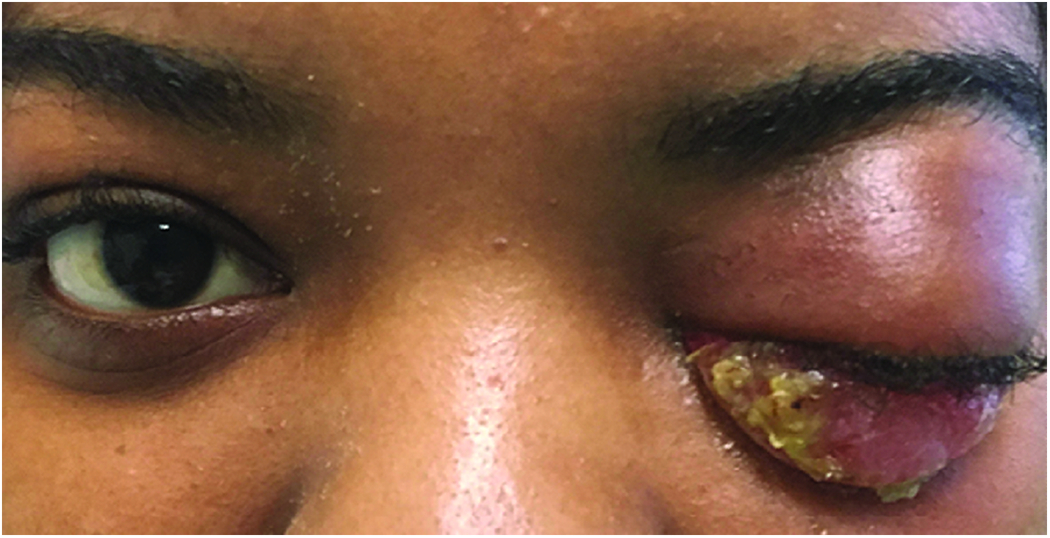
26-year-old Haitian woman who presented with 1-year history of loss of vision and proptosis. Patient did not respond to intravenous steroids. Biopsy revealed non-specific inflammation, negative for IgG4.
References:
- 1.Rootman J Diseases of the Orbit: A Multidisciplinary Approach. 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 2003. [Google Scholar]
- 2.Dutton JJ. Atlas of Clinical and Surgical Orbital Anatomy. 2nd ed. Philadelphia: Elsevier Saunders; 2011. [Google Scholar]
- 3.Aviv RI, Casselman J. Orbital imaging: Part 1. Normal anatomy Clin Radiol. 2005. March;60(3):279–87. [DOI] [PubMed] [Google Scholar]
- 4.Conneely MF, Hacein-Bey L, Jay WM. Magnetic resonance imaging of the orbit. Semin Ophthalmol. 2008. May-Jun;23(3): 179–89. [DOI] [PubMed] [Google Scholar]
- 5.Hallman JT, Pillay P, Koh LH, Goh KY, Yu WY. Eye Globe Abnormalities on MR and CT in adults: An Anatomical Approach. Korean J Radiol. 2016. Sep-Oct; 17(5): 664–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aviv RI1, Miszkiel K. Orbital imaging: Part 2. Intraorbital pathology. Clin Radiol. 2005. March;60(3):288–307. [DOI] [PubMed] [Google Scholar]
- 7.Rootman J1, Heran MK, Graeb DA. Vascular malformations of the orbit: classification and the role of imaging in diagnosis and treatment strategies. Ophthalmic Plast Reconstr Surg. 2014. Mar-Apr;30(2):91–104. [DOI] [PubMed] [Google Scholar]
- 8.Fledelius HC. Ultrasound in ophthalmology. Ultrasound Med Biol. 1997;23(3):365–75. [DOI] [PubMed] [Google Scholar]
- 9.Yan L, He G, Zhou X, Zheng Y, Zhu Y, Yang J, Zhang M, Zhou Y. Clin Radiol. 2017. September;72(9):798 Contrast-enhanced ultrasound in the diagnosis of orbital space-occupying lesions. [DOI] [PubMed] [Google Scholar]
- 10.Ekhlassi T, Becker N. Preseptal and orbital cellulitis. Dis Mon. 2017. February;63(2):30–32. [DOI] [PubMed] [Google Scholar]
- 11.Tsirouki T, Dastiridou AI, Ibanez Flores N, Cerpa JC, Moschos MM, Brazitikos P, Androudi S. Orbital cellulitis. Surv Ophthalmol. 2018. Jul-Aug;63(4):534–553. [DOI] [PubMed] [Google Scholar]
- 12.Danishyar A, Sergent SR. Orbital Cellulitis. StatPearls Publishing. 2019. [Google Scholar]
- 13.Yohai RA, Bullock JD, Aziz AA, Markert RJ. Survival factors in rhino-orbital-cerebral mucormycosis. Surv Ophthalmol. 1994. Jul-Aug;39(l):3–22. [DOI] [PubMed] [Google Scholar]
- 14.Bl Spellberg, Walsh TJ, Kontoyiannis DP, Edwards J Jr, Ibrahim AS. Recent advances in the management of mucormycosis: from bench to bedside. Clin Infect Dis. 2009. 0;48(12): 1743–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greenberg RN, Mullane K, van Burik JA, Raad I, Abzug MJ, Anstead G, Herbrecht R, Fangston A, Marr KA, Schiller G, Schuster M, Wingard JR, Gonzalez CE, Revankar SG, Corcoran G, Kryscio RJ, Hare R. Posaconazole as salvage therapy for zygomycosis. Antimicrob Agents Chemother. 2006. January;50(l): 126–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nithyanandam S, Jacob MS, Battu RR, Thomas RK, Correa MA, D’Souza O. Rhino-orbito-cerebral mucormycosis. A retrospective analysis of clinical features and treatment outcomes. Indian J Ophthalmol. 2003. September;51(3):231–6. [PubMed] [Google Scholar]
- 17.Hirabayashi KE, Kalin-Hajdu E, Brodie FL, Kersten RC, Russell MS, Vagefi MR. Retrobulbar Injection of Amphotericin B for Orbital Mucormycosis. Ophthalmic Plast Reconstr Surg. 2017. Jul-Aug;33(4):e94–e97. [DOI] [PubMed] [Google Scholar]
- 18.Har-El G Endoscopic management of 108 sinus mucoceles. Laryngoscope. 2001. December;lll(12):2131–4. [DOI] [PubMed] [Google Scholar]
- 19.Capra GG, Carbone PN, Mullin DP. Paranasal Sinus Mucocele. Head Neck Pathol. 2012. September; 6(3): 369–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Devars du Mayne M, Moya-Plana A, Malinvaud D, Laccourreye O, Bonfds P. Sinus mucocele: natural history and long-term recurrence rate. Eur Ann Otorhinolaryngol Head Neck Dis. 2012. June; 129(3): 125–30. [DOI] [PubMed] [Google Scholar]
- 21.Zukin LM, Hink EM, Liao S, Getz AE, Kingdom TT, Ramakrishnan V. Endoscopic Management of Paranasal Sinus Mucoceles: Meta-analysis of Visual Outcomes. Otolaryngol Head Neck Surg. 2017. November;157(5):760–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lloyd G, Lund VJ, Savy L, Howard D. Optimum imaging for mucoceles. J Laryngol Otol. 2000. March;1 14(3):233–6. [DOI] [PubMed] [Google Scholar]
- 23.Capra GG, Carbone PN, Mullin DP. Head and Neck Pathol. 2012. September; 6(3): 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loo JL, Looi AL, Seah LL. Visual outcomes in patients with paranasal mucoceles. Ophthalmic Plast Reconstr Surg. 2009. Mar-Apr;25(2): 126–9. [DOI] [PubMed] [Google Scholar]
- 25.Kim YS, Kim K, Lee JG, Yoon JH, Kim CH. Paranasal sinus mucoceles with ophthalmologic manifestations: a 17-year review of 96 cases. Am J Rhinol Allergy. 2011. Jul-Aug;25(4):272–5 [DOI] [PubMed] [Google Scholar]
- 26.Obeso S, Llorente JL, Rodrigo JP, Sanchez R, Mancebo G, Suarez C. Paranasal sinuses mucoceles. Our experience in 72 patients. Acta Otorrinolaringol Esp. 2009;60(5):332–9. [DOI] [PubMed] [Google Scholar]
- 27.Bartley GB. The epidemiologic characteristics and clinical course of ophthalmopathy associated with autoimmune thyroid disease in Olmsted County, Minnesota. Trans Am Ophthalmol Soc. 1994;92:477–588. [PMC free article] [PubMed] [Google Scholar]
- 28.Kamminga N, Jansonius NM, Pott JR, Links TP. Unilateral proptosis: the role of medical history. Br J Ophthalmol. 2003. March; 87(3): 370–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weiler DL. Thyroid eye disease: a review. Clin Exp Optom. 2017. January;100(1):20–25. [DOI] [PubMed] [Google Scholar]
- 30.Wiersinga WM, Bartalena L. Epidemiology and prevention of Graves’ ophthalmopathy. ypoer [DOI] [PubMed] [Google Scholar]
- 31.Bartley GB, Fatourechi V, Kadrmas EF, Jacobsen SJ, Ilstrup DM, Garrity JA, Gorman CA. Clinical features of Graves’ ophthalmopathy in an incidence cohort. Am J Ophthalmol. 1996. March;121(3):284–90. [DOI] [PubMed] [Google Scholar]
- 32.Tanda ML, Piantanida E, Liparulo L, Veronesi G, Lai A, Sassi L, Pariani N, Gallo D, Azzolini C, Ferrario M, Bartalena L. Prevalence and natural history of Graves’ orbitopathy in a large series of patients with newly diagnosed graves’ hyperthyroidism seen at a single center. J Clin Endocrinol Metab. 2013. April;98(4)T443–9. [DOI] [PubMed] [Google Scholar]
- 33.Khong JJ, Finch S, De Silva C, Rylander S, Craig JE, Selva D, Ebeling PR. Risk Factors for Graves’ Orbitopathy; the Australian Thyroid-Associated Orbitopathy Research (ATOR) Study. J Clin Endocrinol Metab. 2016. July;101(7):2711–20. [DOI] [PubMed] [Google Scholar]
- 34.Kung AW, Yau CC, Cheng A. The incidence of ophthalmopathy after radioiodine therapy for Graves’ disease: prognostic factors and the role of methimazole. J Clin Endocrinol Metab. 1994. August;79(2):542–6. [DOI] [PubMed] [Google Scholar]
- 35.Li Z, Cestari DM, Fortin E. Thyroid eye disease: what is new to know? Curr Opin Ophthalmol. 2018. November;29(6):528–534. [DOI] [PubMed] [Google Scholar]
- 36.Mouritis M, et al. Clinical activity score as a guide in the management of patients with Graves’ ophthalmopathy. Clinical Endocrinology 1997. 47; 9–14 [DOI] [PubMed] [Google Scholar]
- 37.Bahn RS. Graves’ ophthalmopathy. N Engl J Med. 2010. Feb 25;362(8):726–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trokel SL, Hilal SK. Recognition and differential diagnosis of enlarged extraocular muscles in computed tomography. Am J Ophthalmol. 1979. April;87(4):503–12. [DOI] [PubMed] [Google Scholar]
- 39.Bailey CC, Kabala J, Laitt R, Goddard P, Hoh HB, Potts MJ, Harrad RA. Magnetic resonance imaging in thyroid eye disease. Eye (Lond). 1996; 10(5):617–9. [DOI] [PubMed] [Google Scholar]
- 40.Bartalena L, Baldeschi L, Boboridis K, et al. The 2016 European Thyroid Association/European Group on Graves’ Orbitopathy Guidelines for the Management of Graves’ Orbitopathy. Eur Thyroid J. 2016. March;5(1):9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zang S, Ponto KA, Kahaly GJ. Clinical review: intravenous glucocorticoids for graves’ orbitopathy: efficacy and morbidity. Journal of Clinical Endocrinology and Metabolism. 2011;96(2):320–332. [DOI] [PubMed] [Google Scholar]
- 42.Smith TJ, Kahaly GJ, Ezra DG. Teprotumumab for Thyroid-Associated Ophthalmopathy. N Engl J Med. 2017; 4;376( 18): 1748–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sy A, Eliasieh K, Silkiss RZ. Clinical Response to Tocilizumab in Severe Thyroid Eye Disease. Ophthalmic Plast Reconstr Surg. 2017. May-Jun;33(3):e55–e57. [DOI] [PubMed] [Google Scholar]
- 44.Strianese D Update on Graves disease: advances in treatment of mild, moderate and severe thyroid eye disease. Curr Opin Ophthalmol. 2017. September;28(5):505–513. [DOI] [PubMed] [Google Scholar]
- 45.Gillespie EF, Smith TJ, Douglas RS. Thyroid eye disease: towards an evidence base for treatment in the 21st century. Curr Neurol Neurosci Rep. 2012. June;12(3):318–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith TJ, Kahaly GJ, Ezra DG, Fleming JC, Dailey RA, Tang RA, Harris GJ, Antonelli A, Salvi M, Goldberg RA, Gigantelli JW, Couch SM, Shriver EM, Hayek BR, Hink EM, Woodward RM, Gabriel K, Magni G, Douglas RS. [Google Scholar]
- 47.Lutalo PM, D’Cruz DP. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener’s granulomatosis). J Autoimmun. 2014. Feb-Mar;48–49:94–8. [DOI] [PubMed] [Google Scholar]
- 48.Cotch MF, Hoffman GS, Yerg DE, Kaufman GI, Targonski P, Kaslow RA. The epidemiology of Wegener’s granulomatosis. Estimates of the five-year period prevalence, annual mortality, and geographic disease distribution from population-based data sources. Arthritis Rheum. 1996. January;39(l):87–92. [DOI] [PubMed] [Google Scholar]
- 49.Muller K, Lin JH. Orbital granulomatosis with polyangiitis (Wegener granulomatosis): clinical and pathologic findings. Arch Pathol Lab Med. 2014. August;138(8): 1110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sfiniadaki E, Tsiara I, Theodossiadis P, Chatziralli E Ocular Manifestations of Granulomatosis with Polyangiitis: A Review of the Literature. Ophthalmol Ther. 2019. June;8(2):227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Evans M, Sharma O, LaBree L, Smith RE, Rao NA. Differences in clinical findings between Caucasians and African Americans with biopsy-proven sarcoidosis. Ophthalmology. 2007. Feb;l 14(2):325–33. [DOI] [PubMed] [Google Scholar]
- 52.Pasadhika S, Rosenbaum JT. Ocular sarcoidosis. Clin Chest Med. 2015. December;36(4):669–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Demirci H, Christianson MD. Orbital and adnexal involvement in sarcoidosis: analysis of clinical features and systemic disease in 30 cases. Am J Ophthalmol. 2011. June;151(6):1074–1080.el. [DOI] [PubMed] [Google Scholar]
- 54.Beegle SH, Barba K, Gobunsuy R, Judson MA. Current and emerging pharmacological treatments for sarcoidosis: a review. Drug Des Devel Ther. 2013. Apr 12;7:325–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mukheqee B, Ambreen A, Alam MS. Sarcoidosis of the ocular adnexa. Ann Allergy Asthma Immunol. 2017. April;118(4):512–513. [DOI] [PubMed] [Google Scholar]
- 56.Ycsiltas YS, Giinduz AK. Idiopathic Orbital Inflammation: Review of Literature and New Advances. Middle East Afr J Ophthalmol. 2018. Apr-Jun;25(2):71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ding ZX, Lip G, Chong V. Idiopathic orbital pseudotumour. Clin Radiol. 2011. September;66(9):886–92. [DOI] [PubMed] [Google Scholar]
- 58.Mombaerts I, Goldschmeding R, Schlingemann RO, Koomneef L. What is orbital pseudotumor? Surv Ophthalmol. 1996. Jul-Aug;41(1):66–78. [DOI] [PubMed] [Google Scholar]
- 59.Carruthers MN, Topazian MD, Khosroshahi A, et al. Rituximab for lgG4-related disease: a prospective, open-label trial. Ann Rheum Dis 2015; 74:1171–1177. [DOI] [PubMed] [Google Scholar]
- 60.Wallace Z, Mattoo H, Mahajan V, et al. Predictors of relapse in lgG4-related disease following rituximab. Rheumatology 2016; 55:1000–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Derzko-Dzulynsky L IgG4-related disease in the eye and ocular adnexa. Curr Opin Ophthalmol. 2017. November;28(6):617–622. [DOI] [PubMed] [Google Scholar]
- 62.Christie B, Block L, Ma Y, Wick A, Afifi A. Retrobulbar hematoma: A systematic review of factors related to outcomes. J Plast Reconstr Aesthet Surg. 2018. February;71(2): 155–161. [DOI] [PubMed] [Google Scholar]
- 63.Fattahi T, Brewer K, Retana A, Ogledzki M. Incidence of retrobulbar hemorrhage in the emergency department. J Oral Maxillofac Surg. 2014. December;72(12):2500–2. [DOI] [PubMed] [Google Scholar]
- 64.Kohli GS, Patel BC. Carotid Cavernous Fistula. StatPearls Publishing; 2018. December. [PubMed] [Google Scholar]
- 65.Ellis JA, Goldstein H, Connolly ES Jr, Meyers PM. Carotid-cavernous fistulas. Neurosurg Focus. 2012. May;32(5):E9. [DOI] [PubMed] [Google Scholar]
- 66.Smoker WR, Gentry LR, Yee NK, Reede DL, Nerad JA. Vascular lesions of the orbit: more than meets the eye. Radiographics. 2008. Jan-Feb;28(l): 185–204. [DOI] [PubMed] [Google Scholar]
- 67.Kumar RR, Singh A, Singh A; Abhishek. Embolization of a deep orbital varix through endovascular route. Indian J Ophthalmol. 2015. March;63(3):270–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vadlamudi VI, Gemmete JJ2, Chaudhary N2, Pandey AS2, Kahana A. Transvenous sclerotherapy of a large symptomatic orbital venous varix using a microcatheter balloon and bleomycin. J Neurointerv Surg. 2016. August;8(8):e30. [DOI] [PubMed] [Google Scholar]
- 69.Arat YO, Mawad ME, Boniuk M. Orbital venous malformations: current multidisciplinary treatment approach. Arch Ophthalmol. 2004. August;122(8):1151–8. [DOI] [PubMed] [Google Scholar]
- 70.Hamedani M, Poumaras JA, Goldblum D. Diagnosis and management of enophthalmos. Surv Ophthalmol. 2007. Sep-Oct;52(5):457–73. [DOI] [PubMed] [Google Scholar]
- 71.Nassiri N, Rootman J, Rootman DB, Goldberg RA. Orbital lymphaticovenous malformations: Current and future treatments. Surv Ophthalmol. 2015. Sep-Oct;60(5):383–405. [DOI] [PubMed] [Google Scholar]
- 72.Saha K, Leatherbarrow B. Orbital lymphangiomas: a review of management strategies. Curr Opin Ophthalmol. 2012. September;23(5):433–8. [DOI] [PubMed] [Google Scholar]
- 73.Haik BG, Karcioglu ZA, Gordon RA, Pechous BP. Capillary hemangioma (infantile periocular hemangioma). Surv Ophthalmol. 1994. Mar-Apr;38(5):399–426. [DOI] [PubMed] [Google Scholar]
- 74.Jockin YM, Friedlander SF. Periocular infantile hemangioma. Int Ophthalmol Clin. 2010. Fall;50(4): 15–25. [DOI] [PubMed] [Google Scholar]
- 75.Sullivan TJ. Vascular Anomalies of the Orbit—A Reappraisal. Asia Pac J Ophthalmol (Phila). 2018. Sep-Oct;7(5):356–363. [DOI] [PubMed] [Google Scholar]
- 76.Calandriello L, Grimaldi G, Petrone G, Rigante M, Petroni S, Riso M, Savino G. Cavernous venous malformation (cavernous hemangioma) of the orbit: Current concepts and a review of the literature. Surv Ophthalmol. 2017. Jul-Aug;62(4):393–403. [DOI] [PubMed] [Google Scholar]
- 77.Pontes FS, de Oliveira JI, de Souza LL, de Almeida OP, Fregnani ER, Vilela RS, Silva WM, Fonseca FP, Pontes HA. Clinicopathological analysis of head and neck rhabdomyosarcoma: A series of 10 cases and literature review. Med Oral Patol Oral Cir Bucal. 2018. Mar I;23(2):el88–el97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Turner JH, Richmon JD. Head and neck rhabdomyosarcoma: a critical analysis of population-based incidence and survival data. Otolaryngol Head Neck Surg. 2011. December;145(6):967–73. [DOI] [PubMed] [Google Scholar]
- 79.Amer KM, Thomson JE, Congiusta Dl, Dobitsch A Chaudhry A, Li M, Chaudhry A, Bozzo A, Siracuse B, Aytekin MN, Ghert M, Beebe KS. Epidemiology, Incidence, and Survival of Rhabdomyosarcoma Subtypes: SEER and ICES Database Analysis. J Orthop Res. 2019. October;37(10):2226–2230. [DOI] [PubMed] [Google Scholar]
- 80.Karcioglu ZA, Hadjistilianou D, Rozans M, DeFrancesco S. Orbital Rhabdomyosarcoma. Cancer Control. 2004. Sep-Oct;ll(5):328–33. [DOI] [PubMed] [Google Scholar]
- 81.Sohaib SA, Moseley I, Wright JE. Orbital rhabdomyosarcoma — the radiological characteristics. Clinical Radiology. 1998. May;53(5):357–362. [DOI] [PubMed] [Google Scholar]
- 82.Shields JA, Shields CL. Rhabdomyosarcoma: Review for the Ophthalmologist. Survey of Ophthalmology. 2003. Jan-Feb;48(l):39–57. [DOI] [PubMed] [Google Scholar]
- 83.Olsen TG. Heegaard S. Orbital Lymphoma. Surv Ophthalmol. 2019. Jan-Feb;64(l):45–66. [DOI] [PubMed] [Google Scholar]
- 84.Olsen TG, Holm F, Mikkelsen LH, Rasmussen PK, Coupland SE, Esmaeli B, Finger PT, Graue GF, Grossniklaus HE, Honavar SG, Khong JJ, McKelvie PA, Mulay K, Sjo LD, Vemuganti GK, Thuro BA, Heegaard S. Orbital Lymphoma-An International Multicenter Retrospective Study. Am J Ophthalmol. 2019. March; 199:44–57. [DOI] [PubMed] [Google Scholar]
- 85.Decaudin D, de Cremoux P, Vincent-Salomon A, Dendale R, Rouic LL. Ocular adnexal lymphoma: a review of clinicopathologic features and treatment options. Blood. 2006. Sep 1; 108(5): 1451–60. [DOI] [PubMed] [Google Scholar]
- 86.Gerbino G, Boffano P, Benech R, Baietto F, Gallesio C, Arcuri F, Benech A. Orbital lymphomas: clinical and radiological features. J Craniomaxillofac Surg. 2014. July;42(5):508–12. [DOI] [PubMed] [Google Scholar]
- 87.von Holstein SL, Therkildsen MH, Prause JU, Stenman G, Siersma VD, Heegaard S. Lacrimal gland lesions in Denmark between 1974 and 2007. Acta Ophthalmol. 2013. June;91(4):349–54. [DOI] [PubMed] [Google Scholar]
- 88.Andreasen S, Esmaeli B, Holstein SL, Mikkelsen LH, Rasmussen PK, Heegaard S. An Update on Tumors of the Lacrimal Gland. Asia Pac J Ophthalmol (Phila). 2017. Mar-Apr;6(2): 159–172. [DOI] [PubMed] [Google Scholar]
- 89.Tse DT, Kossler AL, Feuer WJ, et al. Long-term outcomes of neoadjuvant intra-arterial cytoreductive chemotherapy for lacrimal gland adenoid cystic carcinoma. Ophthalmology. 2013. July;120(7):1313–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Farazdaghi MK, Katowitz WR, Avery RA. Current treatment of optic nerve gliomas. Curr Opin Ophthalmol. 2019. September;30(5):356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Avery RA1, Fisher MJ, Liu GT. Optic pathway gliomas. J Neuroophthalmol. 2011. September;31(3):269–78. [DOI] [PubMed] [Google Scholar]
- 92.Dutton JJ. Optic nerve sheath meningiomas. Surv Ophthalmol. 1992. Nov-Dec;37(3): 167–83. [DOI] [PubMed] [Google Scholar]
- 93.Parker RT, Ovens CA, Fraser CL, Samarawickrama C. Optic nerve sheath meningiomas: prevalence, impact, and management strategies. Eye Brain. 2018. Oct 24;10:85–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Eddleman CS, Liu JK. Optic nerve sheath meningioma: current diagnosis and treatment. Neurosurg Focus. 2007;23(5):E4. [DOI] [PubMed] [Google Scholar]
- 95.Ahmad SM, Esmaeli B. Metastatic tumors of the orbit and ocular adnexa. Curr Opin Ophthalmol. 2007. September;18(5):405–13. [DOI] [PubMed] [Google Scholar]
- 96.Goldberg RA, Rootman J, Cline RA. Tumors metastatic to the orbit: a changing picture. Surv Ophthalmol. 1990. Jul-Aug;35(1):1–24. [DOI] [PubMed] [Google Scholar]