

Abstract
Breast cancer is the most commonly diagnosed type of cancer and one of the leading causes of cancer-associated mortality in women. In addition, the underlying molecular mechanisms of the occurrence and development of breast cancer requires further investigation. In the present study, bioinformatics analysis was performed to identify differentially expressed genes (DEGs) between breast cancer and normal breast tissues to investigate the underlying molecular mechanisms. In addition, reverse transcription-quantitative PCR and immunohistochemistry (IHC) were performed to investigate the protein and mRNA expression levels of a specific DEG, discs large-associated protein 5 (DLGAP5). A Cell Counting Kit-8 assay and flow cytometry analysis were used to assess the effects of DLGAP5 on cell proliferation. In total, 85 DEGs were identified in the three Gene Expression Omnibus datasets, including 40 upregulated and 45 downregulated genes. In addition, 30 hub genes were identified following the construction of a protein-protein interaction network, and 28 of the 30 hub genes were established to be indicators of breast cancer prognosis. DLGAP5 was highly expressed in breast cancer specimens, and its expression levels were correlated with clinical stage and lymph node status. In addition, downregulation of DLGAP5 repressed the proliferation of breast cancer MDA-MB-231 cells and induced cell cycle arrest. Additionally, DLGAP5 was identified to be localized in the mitochondria, and the presence of a conserved microtubule-associated proteins 1A/1B light chain 3B-interacting region motif suggested that DLGAP5 may serve a role in mitophagy. The present results demonstrated an association between DLGAP5 expression levels and the clinicopathological characteristics of patients with breast cancer using IHC. In conclusion, DLGAP5 may be a promising target in the diagnosis and treatment of breast cancer.
Keywords: breast cancer, bioinformatics analysis, discs large-associated protein 5, mitophagy
Introduction
Breast cancer (BC) is the most commonly diagnosed cancer and the leading cause of cancer-associated mortality in women; according to data published by the Global Cancer Statistics in 2018, there were >2.1 million new cases that year and 0.6 million BC-associated mortalities worldwide (1). Despite recent advances in surgery, radiotherapy, chemotherapy, neoadjuvant chemotherapy and endocrine therapy, the incidence of BC has increased in developed (54.4 per 100,000, female by age-standardized in 2018) and developing (31.3 per 100,000, female by age-standardized in 2018) countries (1,2). The incidence of BC in China has increased from 75.3 to 127.55 per 100,000 between 2005–2015, respectively (3). Therefore, it is important to identify novel specific targets to improve the currently available therapeutic strategies and to clarify the underlying molecular mechanisms of BC.
In the past decade, various studies have investigated gene expression levels in BC. These studies have screened numerous differentially expressed genes (DEGs) that may be involved in the development and progression of BC (4,5). However, the results have been largely inconsistent due to the heterogeneity of specimens across experiments and the use of different detection platforms and data processing methods (6,7). To identify novel DEGs associated with BC, the Robust Rank Aggregation (RRA) package was used to integrate multiple gene expression profiles in the present study. The RRA package uses P-values to compare each ranked gene against a randomly ranked gene and then re-ranks the genes (8). Therefore, the RRA package can be used to improve the current understanding of the mechanism underlying BC.
The human discs large-associated protein 5 (DLGAP5) gene, which is mapped to chromosome 14q22.3, is a cell cycle regulator involved in carcinogenesis (9). Wang et al (10) reported that DLGAP5 was upregulated in non-small cell lung cancer (NSCLC) and was associated with a shorter survival time. In cytological experiments, the knockdown of DLGAP5 caused inhibition of proliferation, migration and invasion of NSCLC cells (10). Previous studies have also shown that the expression levels of DLGAP5 are upregulated in bladder (11), prostate (12) and liver cancer (13), as well as leukemia (14) were associated with poor prognosis. In the present study, DLGAP5 was identified to be a hub gene in GEO and Gene Expression-Based Outcome for BC Online (GOBO) databases. Furthermore, DLGAP5 expression levels were confirmed in 24 paired tumor and normal samples, and in 160 paraffin-embedded BC specimens. In vitro functional analysis, a Cell Counting Kit (CCK)-8 assay and cell cycle analysis were performed to determine the underlying molecular mechanism of DLGAP5 in the progression of BC. The present study aimed to identify potential novel prognosis biomarkers and potential novel targets, to facilitate the development of novel drugs for the treatment of BC.
Materials and methods
Identification of DEGs from the GEO database
In total, three BC datasets, including GSE21422 (15), GSE29431 (Lopez et al, unpublished data, 2011) and GSE61304 (16), were obtained from the GEO database (http://www.ncbi.nlm.nih.gov/geo) (Table I). These three datasets were generated using the GPL570 platform of the (HG-U133_Plus_2) Affymetrix Human Genome U133 Plus 2.0 array (http://www.affymetrix.com/index.affx). GSE21422 contained five healthy breast specimens and 14 BC specimens. GSE29431 contained 12 healthy breast specimens and 54 primary BC specimens. GSE61304 contained four healthy breast specimens of an epithelial origin and 58 BC specimens of an epithelial origin. Subsequently, the platform and matrix files were downloaded from GEO. The dataset information is presented in Table I. The downloaded files were processed with R software [version 2.6.2 (17)], calibrated, standardized and log2-converted as necessary. To identify the DEGs in each dataset, the limma package in R was used with the cutoff criteria of |log2-fold-change (FC)|>1 and P<0.05 (18).
Table I.
Details for Gene Expression Omnibus breast cancer datasets.
Integration of the microarray data
To identify significant and reliable DEGs, the Robust Rank Aggregation (RRA) package (19) was used. The RRA package assumes that each gene is randomly ordered within each dataset (20). A gene that was ranked high had a low P-value, following correction and had a greater probability of being considered a DEG. This was used to identify DEGs and to rank them consistently and reliably without the influence of noise using R (8). This approach allowed an improved understanding of the molecular mechanisms involved in cancer (21). Meanwhile, the heatmap package [version 1.0.12 (22)] was used to generate the heatmap of these DEGs.
GO and KEGG pathway analysis
To define the biological functions of the DEGs, GO term enrichment and KEGG pathway analysis were performed using DAVID (23). P<0.05 was considered to indicate a statistically significant difference.
Establishment of the protein-protein interaction (PPI) network
STRING (https://string-db.org/) is an online tool designed to collect and integrate information from known and predicted PPI in a large number of organisms (24). The DEGs were analyzed using STRING to construct the PPI network. A confidence score >0.4 was set as the cutoff value. Data was subsequently visualized using Cytoscape software [version 3.7.1 (23)]. Molecular Complex Detection (MCODE) was performed within Cytoscape to screen significant modules of PPI network. The criteria default parameters were as follows: Degree cut-off=10, node score cut-off=0.2, k-core=2 and max. depth=100 (25).
Online database extraction
Kaplan-Meier (KM) plots were used to analyze the overall survival time of patients with BC. The hazard ratios with 95% confidence interval and log-rank P-values were calculated using the KM plotter (26). GOBO (co.bmc.lu.se/gobo/), an online tool that contains 1,881 BC specimens, was used to identify genes that co-expressed with DLGAP5, and determine the clinical characteristics of DLGAP5 in patients with BC (27,28). Furthermore, GOBO also supports the applications of PAM50 gene expression subtype analysis, which is recognized as a prognostic gene signature assay by the National Comprehensive Cancer Network (29). To analyze the associations between DLGAP5 expression levels and the clinicopathological characteristics (ER, PR, Her-2, basal-like status, and tumor grade), the GEO datasets GSE12093, GSE7390, GSE6532 (30), GSE3494, GSE1456, GSE2603 (31), GSE2034 (32), GSE11121 (33), GSE4922 and GSE5327 (34) were combined. One-way ANOVA with Tukey's post-hoc test was conducted when making comparisons in datasets containing multiple groups (27,35). Genes that correlated with DLGAP5 expression, with a Pearson correlation coefficient >0.4 were also selected from the GOBO database.
Tissue samples
The present study was approved by The Ethics Committee of Tongji Hospital and written informed consent was provided by all patients prior to the study start. A total of 24 BC tissues and matched normal samples were obtained from patients with BC (median age, 46 years; age range, 30–60 years), following surgery at Tongji Hospital, Tongji Medical College of Huazhong University of Science and Technology (Wuhan, China) between May 2019 and June 2019. All samples were preserved in RNAlater Stabilization Solution (Qiagen GmbH) overnight at 4°C, and subsequently stored at −80°C.
Reverse transcription-quantitative (RT-q)PCR
Total RNA was extracted from tissues using TRIzol® reagent (Invitrogen; Thermo Fisher Scientific, Inc.). RT was performed using HiScript®II RT SuperMix (Vazyme Biotech Co., Ltd.). The protocol for RT was as follows: 42°C for 2 min, 50°C for 15 min and 85°C for 5 sec. qPCR was performed using SYBR Green qPCR Mix (Toyobo Co., Ltd.). The following primer sequences were used for qPCR: DLGAP5 forward, 5′-AAGTGGGTCGTTATAGACCTGA-3′ and reverse, 5′-TGCTCGAACATCACTCTCGTTAT-3′; GAPDH forward, 5′-GGAGCGAGATCCCTCCAAAAT-3′ and reverse, 5′-GGCTGTTGTCATACTTCTCATGG-3′. The following thermocycling conditions were used for qPCR: Initial denaturation for 3 min at 95°C, 45 cycles of 10 sec at 95°C, 30 sec at 60°C and 15 sec at 95°C, and a final extension at 95°C for 15 sec and 65°C for 5 sec. Melt curve analysis was subsequently performed. Relative mRNA expression levels were calculated using the 2−ΔΔCq method (36). All experiments were performed in triplicate.
Immunohistochemistry (IHC)
A tissue microarray (cat. no. BR1921c; Alenabio) was used to validate the expression levels of DLGAP5 in healthy breast and BC specimens. A total of 192 breast specimens were analyzed, of which 80 samples were invasive ductal carcinoma, 80 samples were invasive lobular carcinoma, 21 samples were adjacent healthy tissues, four samples were cancer adjacent breast tissue and seven samples were healthy tissues. Additionally, 8 samples were missing estrogen receptor (ER) status and progesterone receptor (PR) status, while 12 samples were missing human epidermal growth factor receptor 2 (HER2) status information. Tumors were staged based on tumor-node-metastasis (TNM) system (7th edition) (37). Antigens were retrieved in citrate buffer at 95°C (pH 6) for 15 min, and 3% hydrogen peroxide was used for endogenous peroxidase blocking at 37°C for 30 min, followed by incubation with 10% goat serum (Abcam) at room temperature for 1 h. IHC was performed using a rabbit polyclonal anti-human DLGAP5 primary antibody (1:200; cat. no. A13575; Abclonal), and the slides were incubated overnight at 4°C. The sections were subsequently treated with a horseradish peroxidase-conjugated anti-rabbit immunoglobulin G secondary antibody (1:500; cat. no. bs-0295D-HRP; BIOSS), at 37°C for 30 min and the signal was visualized by staining with 3,3′-diaminobenzidine at room temperature for 1 min. After washing the slides with water, the sections were counterstained with hematoxylin at room temperature for 2 min. Corresponding negative control samples (cat. no. BR1921c Trail; Alenabio) were incubated with PBS overnight at 4°C instead of the primary antibody, and the subsequent steps were consistent with the conditions of the DLGAP5 primary antibody test group. The samples were examined using an Olympus BX-51 light microscope (Olympus Corporation; magnification, ×400).
Scoring of the staining results
The staining intensity and the percentage of positive cells were determined by two experienced pathologists who were blinded to the experimental groups. The staining intensity was scored as: i) 0, colorless; ii) 1, light yellow; iii) 2, brown/yellow; and iv) 3, brown. The percentage of positive cells was scored as: i) 0, no cell staining; ii) 1, >25% of cells stained; iii) 2, 25–50% of cells stained; iv) 3, 51–75% of cells stained; and v) 4, >75% of cells stained. The two scores were multiplied to generate values ranging between 0 and 12. The strong expression group (++/+++) was defined as a score ≥5. The weak expression group (−/+) was defined as a score <5.
Cell culture and transfection
The human BC cell line MDA-MB-231 was obtained from The Cell Bank of Type Culture Collection of Chinese Academy of Sciences. Cells were cultured in L-15 medium (HyClone; GE Healthcare Life Sciences) supplemented with 1% penicillin/streptomycin (Invitrogen; Thermo Fisher Scientific, Inc.) and 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.) at 37°C with 5% CO2. Cells were seeded into 6-well plates until they reached 50% confluence and Lipofectamine® 3000 (Invitrogen; Thermo Fisher Scientific, Inc.) was subsequently used to transfect the small interfering RNAs (siRNAs) with 100 nM per well according to the manufacturer's protocol. Subsequent experimentation was conducted after transfection for 48 h. The DLGAP5 siRNA (si-DLGAP5-1 and si-DLGAP5-2) and nontargeting negative control (si-NC) were purchased from Guangzhou RiboBio Co., Ltd. The sequences were as follows: si-DLGAP5-1, 5′-GAATCCAGATGGAGTCTTA-3′; si-DLGAP5-2, 5′-GAAGTCCCATCACTTGAAA-3′; and si-NC, 5′-TTCTCCGAACGTGTCACGTdTdT-3′. Knockdown efficiency of DLGAP5 siRNA was assessed using RT-qPCR.
CCK-8 and cell cycle assays
For the CCK-8 assay (cat. no. CK04, Dojindo Molecular Technologies, Inc.), after transfection for 48 h, ~3×103 MDA-MB-231 cells were resuspended in 100 µl L-15 medium and plated into 96-well plates. After cultured for 24, 48, 72 and 96 h, cells were incubated with CCK-8 solution (10 µl/well) for 2 h at 37°C. The optical density was measured at 450 nm using a microplate reader (http://www.moleculardevices.com).
For the cell cycle assay, cells were harvested after transfection for 72 h and fixed with pre-cooled 70% ethanol at −20°C overnight. Cells were washed twice with PBS to remove all ethanol. RNase A enzyme (Thermo Fisher Scientific, Inc.) and propidium iodide (0.05 mg/ml) were added and incubated for 30 min at room temperature in the dark before analyzing the samples using a FACSCalibur system (Beckman Coulter). FlowJo software version 7.6 (FlowJo LLC) was used for the cell cycle analysis. Each experiment was performed >2 times.
Prediction of mitophagy receptors
GeneCards is an integrative database that provides comprehensive information on all annotated and predicted human genes; it can also provide information on the localization of the protein of interest (38). In addition, the iLIR database is an online tool that is used to identify microtubule-associated proteins 1A/1B light chain 3B (LC3)-interacting region (LIR) motifs within eukaryotic proteins (39,40). These two databases were used to determine whether DLGAP5 may function in mitophagy as a potential autophagy receptor.
Statistical analysis
The GSE29431 and GSE61304 datasets were selected in the following analysis due to a larger sample size and unpaired Student's t-test was used. Paired Student's t-test was applied to detect DLGAP5 expression in BC tissues and matched normal samples. χ2 test were performed to analyze the IHC results. One-way analysis of variance (ANOVA), followed by Tukey's post-hoc test were performed when making comparisons in the GOBO datasets containing multiple groups. One-way ANOVA and Dunnett's post-hoc test were applied when multiple groups were compared with the si-NC group. The receiver operating characteristic curves were plotted to identify the diagnostic value of DLGAP5 in BC. Statistical analyses were performed using SPSS version 22.0 (IBM Corp.) and GraphPad Prism 8 (GraphPad Software, Inc.). All data are presented as the mean ± standard deviation of three independent experiments and P<0.05 was considered to indicate a statistically significant difference.
Results
Identification of DEGs in BC
The BC expression microarray datasets GSE21422, GSE29431 and GSE61304 were standardized, and the results are presented in Fig. S1. In the GSE21422 dataset, 946 upregulated and 1,380 downregulated genes were identified by the limma package using an adjusted P<0.05 and |logFC|>1 as the cutoff criteria; in the GSE29431 dataset, 497 upregulated and 948 downregulated genes were identified; in the GSE61304 dataset, 326 upregulated and 511 downregulated genes were identified (Fig. S2).
Identification of DEGs in BC using integrated bioinformatics
The three datasets were screened using the limma package and analyzed using the RRA package (P<0.05 after correction; |logFC|>1). Using this approach, 85 DEGs that included 40 upregulated and 45 downregulated genes were identified (Table SI). The heatmap package was used to generate the heatmap of the top 25 upregulated and downregulated genes (Fig. 1).
Figure 1.
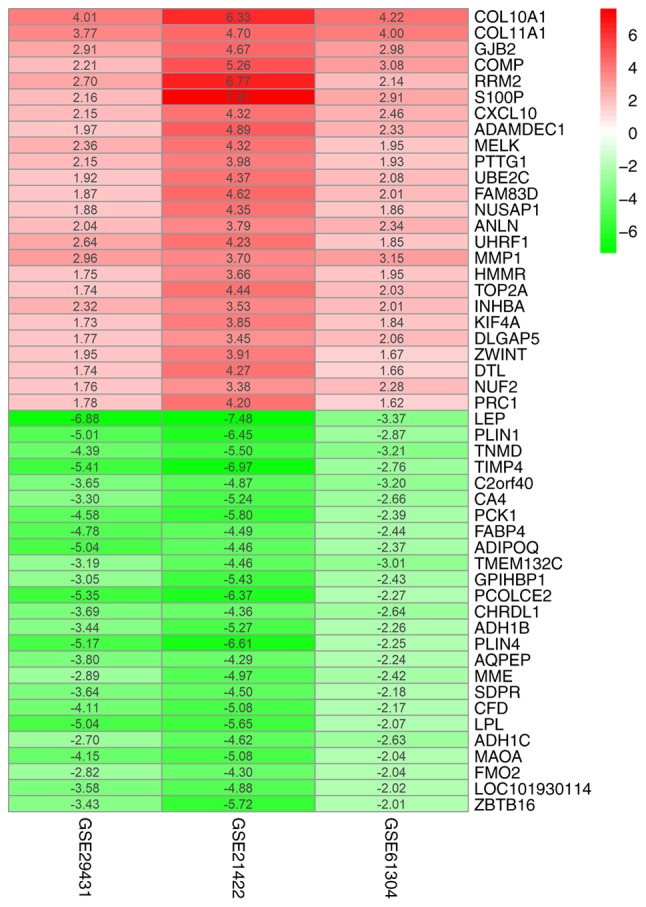
Log2(FC) heatmap of the 50 most significantly differentially regulated genes in the three microarrays. Red and green represent up- and downregulated genes, respectively. The values in the columns represent the log2FC value of each gene. FC, fold change.
GO term enrichment and KEGG pathway analysis
GO term enrichment and KEGG pathway analyses were performed using DAVID. In the biological function group, the upregulated DEGs were involved in ‘cell division’, ‘mitotic nuclear division’, ‘cell proliferation’ and ‘mitotic cytokinesis’ (Table SII), whereas the downregulated DEGs were involved in ‘cellular response to tumor necrosis factor’ and ‘response to linoleic acid’ (Table SIII). In the cell component group, the upregulated DEGs were associated with processes that involved the ‘spindle’, the ‘spindle microtubule’ and the ‘midbody’ (Table SII), whereas the downregulated genes were associated with processes involved in the ‘cell surface’ and ‘extracellular space’ (Table SIII). In the molecular function group, the upregulated DEGs were involved in ‘microtubule binding’ and ‘protein kinase binding’ (Table SII), whereas the downregulated DEGs were involved in ‘heparin binding’ (Table SIII) (Fig. 2A).
Figure 2.
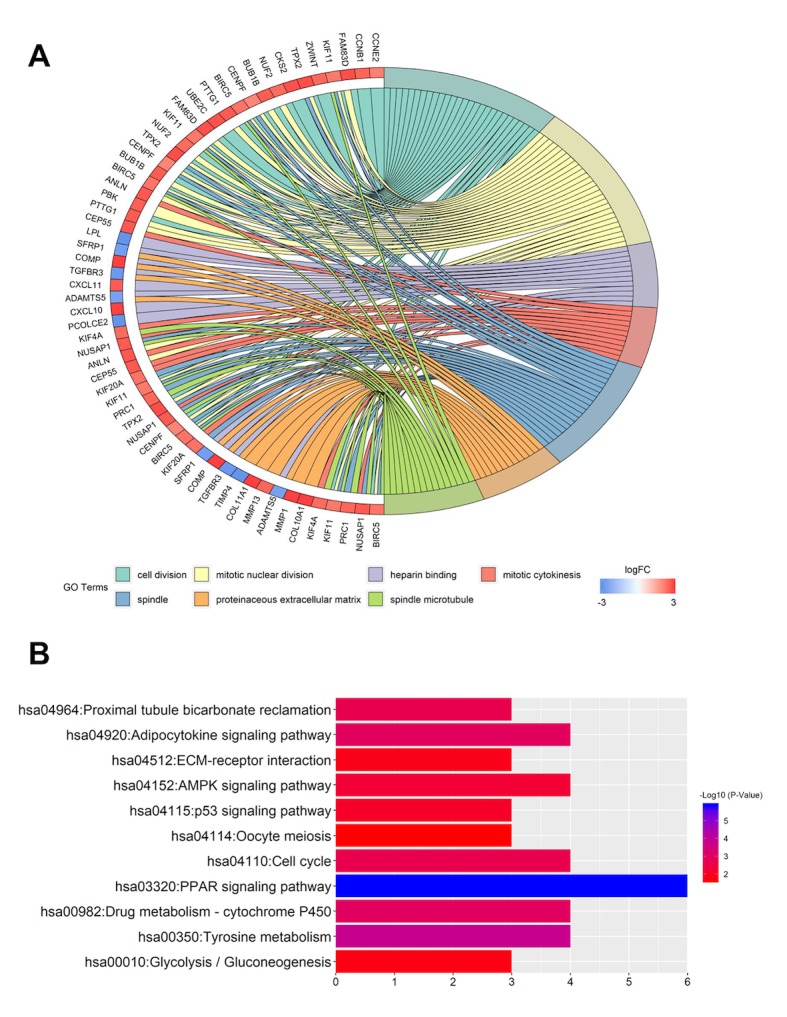
GO and KEGG enrichment analysis of DEGs in patients with breast cancer. (A) GO terms of the DEGs. (B) KEGG pathway enrichment of the DEGs. GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; DEGs, differentially expressed genes.
A total of 11 KEGG pathways were associated with these DEGs. The upregulated genes were particularly enriched in the ‘cell cycle’, ‘p53 signaling pathway’, ‘ECM-receptor interaction’ and ‘oocyte meiosis’ (Fig. 2B). The downregulated genes were enriched in the ‘PPAR signaling pathway’, ‘tyrosine metabolism’, ‘drug metabolism-cytochrome P450’, ‘adipocytokine signaling pathway’, ‘proximal tubule bicarbonate reclamation’, ‘AMPK signaling pathway’ and ‘glycolysis/gluconeogenesis’ (Fig. 2B).
Analysis of the DEGs in BC using the PPI network
The STRING database was used to construct the PPI network of 85 DEGs that included 40 upregulated and 45 downregulated genes, and the results were analyzed using the Cytoscape software (Fig. 3A). A total of 30 hub genes were identified, including ubiquitin-conjugating enzyme E2 C (UBE2C), nucleolar and spindle-associated protein 1 (NUSAP1), baculoviral IAP repeat-containing protein 5 (BIRC5), mitotic checkpoint serine/threonine-protein kinase BUB1 beta (BUB1B), G2/mitotic-specific cyclin-B1 (CCNB1), centrosomal protein of 55 kDa (CEP55), disks large-associated protein 5 (DLGAP5), denticleless protein homolog (DTL), kinesin-like protein KIF11 (KIF11) and kinesin-like protein KIF20A (KIF20A), which were the top 10 DEGs with the highest degrees of protein-protein connectivity.
Figure 3.

PPI network of the DEGs and the forest plot of 28 hub genes associated with breast cancer prognosis in the PPI network. (A) PPI network of the DEGs. Nodes represent proteins, and edges represent connections between proteins. Red and blue circles represent upregulated and downregulated genes, respectively. (B) HRs of the hub genes. Each line in the graph represents a 95% CI, and the middle bar represents the HR. PPI, protein-protein interaction; DEGs, differentially expressed genes; HR, hazard ratio; CI, confidence interval.
Using MCODE, the two most significant functional modules were investigated (Fig. S3). KEGG pathway analysis was performed using DAVID software. The genes in module 1 were mainly involved in the ‘cell cycle control’, ‘p53 signaling’ and ‘oocyte meiosis’, whereas the genes in module 2 were mainly involved in ‘PPAR signaling’, ‘adipocytokine signaling’, ‘AMPK signaling’ and ‘ECM-receptor interaction’ (Table II).
Table II.
Kyoto Encyclopedia of Genes and Genomes pathway analysis of two key modules selected from the protein-protein interaction network.
| A, Module 1 | |||
|---|---|---|---|
| Term | Description | Count | P-value |
| hsa04110 | Cell cycle | 4 | 3.00×10−04 |
| hsa04115 | p53 signaling pathway | 3 | 2.52×10−03 |
| hsa04114 | Oocyte meiosis | 3 | 6.78×10−03 |
| B, Module 2 | |||
| Term | Description | Count | P-value |
| hsa03320 | PPAR signaling pathway | 6 | 1.83×10−08 |
| hsa04920 | Adipocytokine signaling pathway | 4 | 1.15×10−04 |
| hsa04152 | AMPK signaling pathway | 4 | 6.11×10−04 |
| hsa04512 | ECM-receptor interaction | 3 | 6.66×10−03 |
Survival analysis
Information of the 30 hub genes was available in the KM plotter database. Among these, 28 genes were identified as indicators of BC prognosis. The random survival forest map comparing the genes is presented in Fig. 3B.
Analysis of DLGAP5 expression levels using RT-qPCR and online databases
To investigate the role of DLGAP5 in BC, the expression levels of DLGAP5 were examined in BC tissues. The results of the RT-qPCR analysis suggested that DLGAP5 was upregulated in BC tissues compared with normal tissues (P<0.001; Fig. 4A). Due to a relative larger sample size compared with GSE21422, two datasets, GSE29431 and GSE61304 were used in the following analysis. The mRNA expression levels of DLGAP5 in patients with BC were analyzed in the GSE29431 and GSE61304 datasets in order to expand the sample size; the results in these datasets also suggested that DLGAP5 was significantly upregulated in tumor specimens compared with normal breast tissues (P<0.001; Fig. 4B). Furthermore, the receiver operating characteristic (ROC) curves were plotted to identify the diagnostic value of DLGAP5 in BC. DLGAP5 expression levels were associated with tumor classification as assessed by an area under the curve (AUC) >0.8 (GSE29431, AUC=0.821; GSE61304, AUC=0.974; Fig. 4C and D). In detail, the cut-off point for the expression of DLGAP5 to distinguish BC and non-malignant groups using the ROC curve was 5.572, with a sensitivity of 83.3% and a specificity of 75.0% in GSE29431. The cut-off point was 4.573, with a sensitivity of 94.8% and a specificity of 100% in GSE61304. Therefore, high expression of DLGAP5 was associated with a less favorable progression-free (Fig. 4E) and overall survival time (Fig. 4F) using the Kaplan-Meier Plotter online database.
Figure 4.

DLGAP5 expression levels in breast tissues of patients with BC. (A) mRNA expression levels of DLGAP5 in BC tissues compared with normal breast samples. (B) mRNA expression levels of DLGAP5 in an expanded sample size following analysis of the GSE29431 and GSE61304 datasets. (C and D) ROC curve based on DLGAP5 expression levels in (C) GSE29431 and (D) GSE61304 datasets for predicting breast cancer tissue classification. (E) PFS and (F) OS analysis of patients with BC with low and high DLGAP5 expression levels. ***P<0.001. DLGAP5, discs large-associated protein 5; PFS, progression-free survival; OS, overall survival; BC, breast cancer; ROC, receiver operating characteristic; HR, hazard ratio.
Association between DLGAP5 expression levels and the clinicopathological features of BC
To further investigate the association between DLGAP5 expression and the clinicopathological characteristics of the patients with BC, the GOBO online database was used. DLGAP5 expression levels were higher in the basal subtype (n=357) and lower in the normal-like (n=257) and luminal A subtypes (n=482) (Fig. 5A). Similar results were obtained following PAM50 subtype analysis (29) (Fig. 5B). A negative association was observed between the expression levels of DLGAP5 and the ER (Fig. 5C). In addition, DLGAP5 expression levels increased with increasing tumor grade (Fig. 5D).
Figure 5.

Parameters associated with DLGAP5 expression in the GOBO database in patients with breast cancer. (A) Boxplot of DLGAP5 mRNA expression levels stratified based on the molecular subtypes. Boxplot of DLGAP5 mRNA expression levels stratified by (B) PAM50 classification, (C) estrogen receptor status and (D) tumor grade. The expression levels of DLGAP5 were compared among subtypes of breast cancer. P<0.00001 vs. basal, ER-neg and grade 1. DLGAP5, discs large-associated protein 5. HER2, human epidermal growth factor receptor-2; LumA, luminal A subtype; LumB, luminal B subtype; ER, estrogen receptor; -neg, negative; -pos, positive.
IHC was performed to verified the results of online datasets. DLGAP5 staining was brown/yellow in invasive ductal and lobular carcinoma specimens, with expression rate of 98.8% and strong expression rate of 34.4%, which were significantly higher compared with healthy breast tissues (59.4 and 12.5%, respectively; with both comparison P<0.05; Fig. 6). The representative IHC images for DLGAP5 expression levels in BC are presented in Fig. 6, and the information of tissue samples included in the microarray is presented in Table SIV. The 160 cancer specimens were divided into strong (++/+++) or weak (−/+) expression groups, and the association between IHC scores and patient clinicopathological characteristics was investigated. The results revealed that high DLGAP5 expression levels in BC were associated with clinical stage (χ2=4.002; P=0.045) and lymph node status (χ2=5.806; P=0.016) (Table III). No significant associations were present between DLGAP5 expression levels and age, tumor stage, ER, progesterone receptor (PR) or human epidermal growth factor receptor-2 (Her-2) expression (Table III).
Figure 6.

Representative immunohistochemistry images of DLGAP5 expression levels in breast cancer tissue samples. (A and B) Strong, weak and null expression levels of DLGAP5 in (A) breast tumor tissue (n=160) and (B) normal breast tissue (n=32). Magnification, ×400. DLGAP5, discs large-associated protein 5.
Table III.
Association of DLGAP5 expression levels with clinical characteristics of patients with breast cancer.
| DLGAP5 expression levels | ||||
|---|---|---|---|---|
| Clinical characteristics | Strong, n | Weak, n | χ2 | P-value |
| Age, years | 0.639 | 0.420 | ||
| ≤50 | 34 | 58 | ||
| >50 | 21 | 47 | ||
| T stage | 0.420 | 0.520 | ||
| 1-2 | 41 | 83 | ||
| 3-4 | 14 | 22 | ||
| Lymph node | 5.806 | 0.016a | ||
| − | 27 | 72 | ||
| + | 28 | 33 | ||
| Clinical stage | 4.002 | 0.045a | ||
| I–IIB | 38 | 87 | ||
| IIIA-IIIB | 17 | 18 | ||
| ER status | 0.347 | 0.556 | ||
| + | 43 | 84 | ||
| − | 10 | 15 | ||
| PR status | 0.022 | 0.882 | ||
| + | 32 | 61 | ||
| − | 21 | 38 | ||
| Her-2 status | 3.061 | 0.080 | ||
| 3+ | 8 | 6 | ||
| 0-1 | 45 | 89 | ||
P<0.05. DLGAP5, discs large-associated protein 5; T, tumor; ER, estrogen receptor; PR, progesterone receptor; Her-2, human epidermal growth factor receptor-2.
Biological functions of DLGAP5-associated genes
To further investigate the clinical relevance of DLGAP5 in BC, genes that correlated with DLGAP5 expression, with a Pearson correlation coefficient >0.4 were selected from the GOBO database. A total of 292 genes were identified with functions in ‘cell division’, ‘DNA replication’, ‘G1/S transition of mitotic cell cycle’, ‘DNA replication initiation’ and ‘mitotic nuclear division’ (Fig. 7A). KEGG pathways analysis demonstrated that DLGAP5-associated genes were involved in the ‘cell cycle’, ‘DNA replication’, ‘oocyte meiosis’, ‘p53 signaling’ and ‘pyrimidine metabolism’ (Fig. 7B).
Figure 7.

Underlying molecular mechanisms of DLGAP5 activity in breast cancer. (A) Different GO enriched terms associated with genes co-expressed with DLGAP5 in breast cancer. (B) KEGG pathway enrichment of the co-expressed genes. (C) Subcellular localization of DLGAP5. The green intensity represents the localization probability. (D) Domain demonstration of DLGAP5 and its LIR motif (in red) exhibiting conservation across species. GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; DLGAP5, discs large-associated protein 5.
Mitophagy receptor prediction
The predicted subcellular location of DLGAP5 was identified in the GeneCards database. DLGAP5 is primarily localized in the cytosol, cytoskeleton, nuclei and mitochondria (Fig. 7C). Using the iLIR online tool, DLGAP5 was identified to possess a LIR motif, which is conserved in several mammalian species (41) (Fig. 7D).
Effects of DLGAP5 on the proliferation of MDA-MB-231 cells
To further determine the biological functions of DLGAP5 in BC, the MDA-MB-231 cell line was used. siRNA was transfected into BC cells to establish DLGAP5-knockdown cells. RT-qPCR was used to verify the expression levels of DLGAP5 in the transfected cells. Compared with the si-NC-transfected cells, DLGAP5 was significantly downregulated in si-DLGAP5-transfected cells (P<0.001; Fig. 8A). CCK-8 and flow cytometry assays were performed to determine the effects of DLGAP5 knockdown in cell proliferation. The CCK-8 assay demonstrated that knockdown of DLGAP5 repressed the proliferation of MDA-MB-231 cells compared with the si-NC group (P<0.01 at 96 h si-DLGAP5-1 vs. si-NC; P<0.001 at 96 h si-DLGAP5-2 vs. si-NC; Fig. 8B). Cell cycle analysis demonstrated that the cells were predominantly distributed in the G2/M stage in the DLGAP5 knockdown group (si-DLGAP5-1 and si-DLGAP5-2 group, 24.8 and 24.0%, respectively; negative control group, 13.7%; P<0.05; Fig. 8C and D).
Figure 8.

Effects of DLGAP5 on MDA-MB-231 cell proliferation. (A) Expression levels of DLGAP5 in MDA-MB-231 cells transfected with si-NC, si-DLGAP5-1 and si-DLGAP5-2 was determined by reverse transcription-quantitative PCR after transfection for 48 h. (B) Cell Counting Kit-8 assay was used to detect the effects of DLGAP5 on MDA-MB-231 cell proliferation. (C and D) Flow cytometry was used to detect the effects of DLGAP5 on cell cycle regulation in MDA-MB-231 cells. *P<0.05 and ***P<0.001 vs. si-NC; **P<0.01 si-DLGAP5-1 vs. si-NC; ###P<0.001 si-DLGAP5-2 vs. si-NC. si, small interfering; NC, negative control; DLGAP5, discs large-associated protein 5.
Discussion
In the present study, bioinformatics analyses were used to integrate and analyze three BC datasets containing gene expression profiles, GSE21422, GSE29431 and GSE61304. Using the RRA package, 85 DEGs were identified, including 40 upregulated and 45 downregulated genes, which were used to investigate the regulatory mechanisms underlying BC and to generate a PPI network. Using MCODE, two highly connected networks were identified. The first network comprised 27 DEGs that were primarily enriched in ‘cell cycle control’ and ‘p53 signaling and oocyte meiosis’, whereas the second network comprised 13 DEGs that were mainly enriched in ‘PPAR signaling’, ‘adipocytokine signaling’, ‘AMPK signaling’ and ‘ECM-receptor interaction’. Using PPI and KEGG pathway enrichment analysis, various key signaling pathways and hub genes that may serve important roles in the development of BC were identified. The identified pathways and hub genes may represent novel targets to be used in therapy.
In the present study, the STRING database was used to construct the PPI network, in order to detect the hub genes. The genes associated with breast cancer prognosis were selected for further experimentation. Among these 30 hub genes, 28 genes were indicators of BC prognosis, including DLGAP5. The genes with a high degree of protein-protein connectivity in the PPI network play a key role in the network (42). DLGAP5 was one of the top 10 genes with highest degree of protein-protein connectivity. DLGAP5, a microtubule-associated protein, is phosphorylated by Aurora kinase A (AURKA) (43). In addition, DLGAP5 upregulation is associated with poor prognosis of patients with colorectal cancer (44), lung cancer (10), bladder cancer (11) and prostate cancer (12), indicating that DLGAP5 plays an important role in cancer prognosis. Branchi et al (44) reported that DLGAP5 was associated with the nodal status, and high DLGAP5 expression levels were associated with a less favorable overall survival rate in distinct molecular colorectal cancer subtypes. Schneider et al (45) and Shi et al (46) demonstrated that DLGAP5 was a promising diagnostic and prognostic biomarker for lung cancer; high expression levels of DLGAP5 were significantly associated with age, sex, clinical stage, pathological T stage, new tumor event and therapeutic outcome. Tagal et al (47) reported that DLGAP5 was AURKA-dependent and essential for the survival and proliferation of transcription activator BRG1 (SMARCA4/BRG1) mutant NSCLC cells. Furthermore, targeting mitosis-associated genes, including DLGAP5, TPX2 and RAN, has been reported to induce apoptosis of the BRG1 mutant NSCLC cells, both in vitro and in vivo (47). Espinoza et al (12) demonstrated that DLGAP5 was a predictive biomarker for the prognosis of high-risk prostate cancer in addition to functioning as a resistance factor. Furthermore, hypoxia-inducible factor 1-alpha (HIF-1α) binding sites were revealed on the promoter of DLGAP5, indicating that hypoxia plays a modulatory role on DLGAP5 expression (11). These results were further confirmed by Yamamoto et al (48). Eissa et al (11) demonstrated that DLGAP5 was a reliable and promising biomarker for the detection of bladder cancer, and sensitivity of urine cytology was improved when combined with the mRNA expression of DLGAP5. Kim and Cho (49) reported that DLGAP5 was a stem-cell proliferation biomarker.
The age of the patient, size of the tumor and molecular biological factors such as ER, PR or Her-2 expression are notable factors affecting the prognosis of breast cancer (50,51). In addition, whether the tumor was associated with lymph nodes and distant metastases also had an important impact on the patient's prognosis (52). According to the Surveillance, Epidemiology and End Results Program, patients with BC with regional lymph node metastasis exhibit a 5-year survival rate of 85.5%, which is lower compared with the 5-year survival rate in patients with a localized tumor (98.8%) (53). In addition, late-stage breast cancer was also associated with a less favorable prognosis (54). To investigate the association between DLGAP5 expression and the clinicopathological characteristics, the GOBO database was investigated and IHC analysis was performed in the present study. The results demonstrated that high DLGAP5 expression levels in BC were associated with the clinical stage and the lymph node status. These results were consistent with the online survival analysis and suggested that high DLGAP5 expression levels were associated with a poor prognosis in patients with breast cancer.
In the present study, to confirm the biological function of DLGAP5 in BC, 282 DLGAP5-associated genes were selected from the GOBO database. KEGG pathway analysis demonstrated that these genes were mainly involved in ‘cell cycle control’, ‘DNA replication’, ‘oocyte meiosis’, ‘p53 signaling’ and ‘pyrimidine metabolism’. A number of these biological processes have been reported in previous studies. For example, Kuo et al (55) reported that DLGAP5 knockdown inhibited the proliferation of hepatocellular carcinoma cells due to the accumulation of p53 and the downregulation of gankyrin. Zhang et al (56) reported that DLGAP5 was a downstream molecule of NUSAP1 and regulated the cell cycle; in addition, si-NUSAP1 suppressed invasive BC cell proliferation and invasion and enhanced susceptibility to epirubicin by affecting the cell cycle progression. In the present study, a CCK-8 assay and cell cycle analysis were used, and the results demonstrated that knockdown of DLGAP5 induced cell cycle arrest at the G2/M phase and inhibited cell proliferation. The aforementioned processes are important for the functions of normal and tumorigenic cells, suggesting that further studies are required to confirm the role of DLGAP5 in carcinogenesis.
Previously, the clinical significance of autophagy in cancer has been investigated (57,58). Mitochondrial autophagy, or mitophagy, is defined as selective mitochondrial degradation through autophagy (59). Increasing evidence has demonstrated that abnormal mitophagy is associated with cancer development and progression and may be associated with an altered response to cancer therapy (60,61). Meanwhile, mitophagy receptors contain two key features, the mitochondrial localization and the LIR motif, which is required to bind the LC3 ligand (41). Given these characteristics, DLGAP5 may be considered a mitophagy receptor. Of note, DLGAP5 has been reported to be localized in the cytosol, cytoskeleton, nuclei and mitochondria (13). In the present study, DLGAP5 was identified to contain a LIR motif. Although the present results suggested that DLGAP5 may be involved in mitophagy, further studies are required to define the precise role and molecular mechanism of DLGAP5 in BC.
The present study was not without limitations. First, only one cell line was used to detect the role of DLGAP5 in BC. Secondly, the role of DLGAP5 in mitophagy was only predicted using online data instead of validation via patient samples. Thus, further experiments using more cancer cell lines and xenograft models are required, in order to determine the underlying molecular mechanisms of DLGAP5 and other hub genes in the prognosis of BC.
In conclusion, the present study provided evidence to suggest that DLGAP5 acts as a potential oncogene in breast cancer. Knockdown of DLGAP5 largely repressed the proliferation of breast cancer MDA-MB-231 cells and induced cell cycle arrest. Furthermore, the present results provided novel insight into the clinical relevance of DLGAP5 as a biomarker for prognosis and its underlying molecular mechanisms in BC.
Supplementary Material
Acknowledgements
Not applicable.
Funding
The present study was funded by The National Natural Science Foundation of China (grant no. 81802676) and The Wuhan Youth Cadre Project (grant nos. 2017zqnlxr01 and 2017zqnlxr02).
Availability of data and materials
All data generated or analyzed during the present study are included in this published article.
Authors' contributions
TX collected and analyzed the data and was a major contributor of the manuscript. MD acquired and analyzed the clinical characteristics of the breast cancer specimens. TX, HL and RZ performed the experiments. XL designed the present study and critically revised the manuscript. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
The present study was approved by the Ethics Committee of Tongji Hospital and written informed consent was provided by all patients prior to the study start.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.DeSantis CE, Ma J, Gaudet MM, Newman LA, Miller KD, Goding Sauer A, Jemal A, Siegel RL. Breast cancer statistics, 2019. CA Cancer J Clin. 2019;69:438–451. doi: 10.3322/caac.21583. [DOI] [PubMed] [Google Scholar]
- 3.Mubarik S, Malik SS, Wang Z, Li C, Fawad M, Yu C. Recent insights into breast cancer incidence trends among four Asian countries using age-period-cohort model. Cancer Manag Res. 2019;11:8145–8155. doi: 10.2147/CMAR.S208323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang L, Chen Y, Tang X, Wei D, Xu X, Yan F. Long noncoding RNA DCST1-AS1 promotes cell proliferation and metastasis in triple-negative breast cancer by forming a positive regulatory loop with miR-873-5p and MYC. J Cancer. 2020;11:311–323. doi: 10.7150/jca.33982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gruosso T, Gigoux M, Manem VSK, Bertos N, Zuo D, Perlitch I, Saleh SMI, Zhao H, Souleimanova M, Johnson RM, et al. Spatially distinct tumor immune microenvironments stratify triple-negative breast cancers. J Clin Invest. 2019;129:1785–1800. doi: 10.1172/JCI96313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen DQ, Kong XS, Shen XB, Huang MZ, Zheng JP, Sun J, Xu SH. Identification of differentially expressed genes and signaling pathways in acute myocardial infarction based on integrated bioinformatics analysis. Cardiovasc Ther. 2019;2019:8490707. doi: 10.1155/2019/8490707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Võsa U, Kolde R, Vilo J, Metspalu A, Annilo T. Comprehensive meta-analysis of microRNA expression using a robust rank aggregation approach. Methods Mol Biol. 2014;1182:361–373. doi: 10.1007/978-1-4939-1062-5_28. [DOI] [PubMed] [Google Scholar]
- 8.Kolde R, Laur S, Adler P, Vilo J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics. 2012;28:573–580. doi: 10.1093/bioinformatics/btr709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hewit K, Sandilands E, Martinez RS, James D, Leung HY, Bryant DM, Shanks E, Markert EK. A functional genomics screen reveals a strong synergistic effect between docetaxel and the mitotic gene DLGAP5 that is mediated by the androgen receptor. Cell Death Dis. 2018;9:1069. doi: 10.1038/s41419-018-1115-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Q, Chen Y, Feng H, Zhang B, Wang H. Prognostic and predictive value of HURP in non-small cell lung cancer. Oncol Rep. 2018;39:1682–1692. doi: 10.3892/or.2018.6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eissa S, Matboli M, Mansour A, Mohamed S, Awad N, Kotb YM. Evaluation of urinary HURP mRNA as a marker for detection of bladder cancer: Relation to bilharziasis. Med Oncol. 2014;31:804. doi: 10.1007/s12032-013-0804-4. [DOI] [PubMed] [Google Scholar]
- 12.Espinoza I, Sakiyama MJ, Ma T, Fair L, Zhou X, Hassan M, Zabaleta J, Gomez CR. Hypoxia on the expression of hepatoma upregulated protein in prostate cancer cells. Front Oncol. 2016;6:144. doi: 10.3389/fonc.2016.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsou AP, Yang CW, Huang CYF, Yu RC, Lee YC, Chang CW, Chen BR, Chung YF, Fann MJ, Chi CW, et al. Identification of a novel cell cycle regulated gene, HURP, overexpressed in human hepatocellular carcinoma. Oncogene. 2003;22:298–307. doi: 10.1038/sj.onc.1206129. [DOI] [PubMed] [Google Scholar]
- 14.Hatfield KJ, Reikvam H, Bruserud Ø. Identification of a subset of patients with acute myeloid leukemia characterized by long-term in vitro proliferation and altered cell cycle regulation of the leukemic cells. Expert Opin Ther Targets. 2014;18:1237–1251. doi: 10.1517/14728222.2014.957671. [DOI] [PubMed] [Google Scholar]
- 15.Kretschmer C, Sterner-Kock A, Siedentopf F, Schoenegg W, Schlag PM, Kemmner W. Identification of early molecular markers for breast cancer. Mol Cancer. 2011;10:15. doi: 10.1186/1476-4598-10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aswad L, Yenamandra SP, Ow GS, Grinchuk O, Ivshina AV, Kuznetsov VA. Genome and transcriptome delineation of two major oncogenic pathways governing invasive ductal breast cancer development. Oncotarget. 2015;6:36652–36674. doi: 10.18632/oncotarget.5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.R Development Core Team, corp-author. R Foundation Statistical Compututing; Vienna, Austria: 2013. [Nov 1;2013 ]. R: A Language and Environment for Statistical Computing, Version 3.0.1 (2013-05-16) Available online. [Google Scholar]
- 18.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu S, Zhao R, Shen J, Zhang Y, Shi J, Xu C, Chen J, Lin R, Han W, Luo D. Integrated bioinformatics analysis to screen hub genes in the lymph node metastasis of thyroid cancer. Oncol Lett. 2020;19:1375–1383. doi: 10.3892/ol.2019.11188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang J, Han S, Huang W, Chen T, Liu Y, Pan S, Li S. A meta-analysis of microRNA expression in liver cancer. PLoS One. 2014;9:e114533. doi: 10.1371/journal.pone.0114533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li C, Yin Y, Liu X, Xi X, Xue W, Qu Y. Non-small cell lung cancer associated microRNA expression signature: Integrated bioinformatics analysis, validation and clinical significance. Oncotarget. 2017;8:24564–24578. doi: 10.18632/oncotarget.15596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang C, Hu X, Qi F, Luo J, Li X. Identification of CD2, CCL5 and CCR5 as potential therapeutic target genes for renal interstitial fibrosis. Ann Transl Med. 2019;7:454. doi: 10.21037/atm.2019.08.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 24.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–D452. doi: 10.1093/nar/gku1003. (Database Issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4:2. doi: 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagy Á, Lánczky A, Menyhárt O, Győrffy B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci Rep. 2018;8:9227. doi: 10.1038/s41598-018-29514-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ringnér M, Fredlund E, Häkkinen J, Borg Å, Staaf J. GOBO: Gene expression-based outcome for breast cancer online. PLoS One. 2011;6:e17911. doi: 10.1371/journal.pone.0017911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lemler DJ, Lynch ML, Tesfay L, Deng Z, Paul BT, Wang X, Hegde P, Manz DH, Torti SV, Torti FM. DCYTB is a predictor of outcome in breast cancer that functions via iron-independent mechanisms. Breast Cancer Res. 2017;19:25. doi: 10.1186/s13058-017-0814-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loi S, Haibe-Kains B, Desmedt C, Lallemand F, Tutt AM, Gillet C, Ellis P, Harris A, Bergh J, Foekens JA, et al. Definition of clinically distinct molecular subtypes in estrogen receptor-positive breast carcinomas through genomic grade. J Clin Oncol. 2007;25:1239–1246. doi: 10.1200/JCO.2006.07.1522. [DOI] [PubMed] [Google Scholar]
- 31.Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massagué J. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 33.Schmidt M, Böhm D, von Törne C, Steiner E, Puhl A, Pilch H, Lehr HA, Hengstler JG, Kölbl H, Gehrmann M. The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res. 2008;68:5405–5413. doi: 10.1158/0008-5472.CAN-07-5206. [DOI] [PubMed] [Google Scholar]
- 34.Minn AJ, Gupta GP, Padua D, Bos P, Nguyen DX, Nuyten D, Kreike B, Zhang Y, Wang Y, Ishwaran H, et al. Lung metastasis genes couple breast tumor size and metastatic spread. Proc Natl Acad Sci USA. 2007;104:6740–6745. doi: 10.1073/pnas.0701138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu S, Jiang X, Li J, Li C, Guo M, Ye F, Zhang M, Jiao Y, Guo B. Comprehensive analysis of the GATA transcription factor gene family in breast carcinoma using gene microarrays, online databases and integrated bioinformatics. Sci Rep. 2019;9:4467. doi: 10.1038/s41598-019-40811-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 37.Lee SC, Jain PA, Jethwa SC, Tripathy D, Yamashita MW. Radiologists' role in breast cancer staging: Providing key information for clinicians. Radiographics. 2014;34:330–342. doi: 10.1148/rg.342135071. [DOI] [PubMed] [Google Scholar]
- 38.Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, Stein TI, Nudel R, Lieder I, Mazor Y, et al. The genecards suite: From gene data mining to disease genome sequence analyses. Curr Protoc Bioinformatics. 2016;54:1.30.1–1.30.33. doi: 10.1002/cpbi.5. [DOI] [PubMed] [Google Scholar]
- 39.Kalvari I, Tsompanis S, Mulakkal NC, Osgood R, Johansen T, Nezis IP, Promponas VJ. iLIR: A web resource for prediction of Atg8-family interacting proteins. Autophagy. 2014;10:913–925. doi: 10.4161/auto.28260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacomin AC, Samavedam S, Promponas V, Nezis IP. iLIR database: A web resource for LIR motif-containing proteins in eukaryotes. Autophagy. 2016;12:1945–1953. doi: 10.1080/15548627.2016.1207016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Yao Y, Qiu X, Wang G, Hu Z, Chen S, Wu Z, Yuan N, Gao H, Wang J, et al. Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat Immunol. 2019;20:433–446. doi: 10.1038/s41590-019-0324-2. [DOI] [PubMed] [Google Scholar]
- 42.Jian L, Yang G. Identification of key genes involved in diabetic peripheral neuropathy progression and associated with pancreatic cancer. Diabetes Metab Syndr Obes. 2020;13:463–476. doi: 10.2147/DMSO.S235011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu CT, Hsu JM, Lee YC, Tsou AP, Chou CK, Huang CY. Phosphorylation and Stabilization of HURP by Aurora-A: Implication of HURP as a Transforming Target of Aurora-A. Mol Cell Biol. 2005;25:5789–5800. doi: 10.1128/MCB.25.14.5789-5800.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Branchi V, García SA, Radhakrishnan P, Győrffy B, Hissa B, Schneider M, Reißfelder C, Schölch S. Prognostic value of DLGAP5 in colorectal cancer. Int J Colorectal Dis. 2019;34:1455–1465. doi: 10.1007/s00384-019-03339-6. [DOI] [PubMed] [Google Scholar]
- 45.Schneider MA, Christopoulos P, Muley T, Warth A, Klingmueller U, Thomas M, Herth FJ, Dienemann H, Mueller NS, Theis F, Meister M. AURKA, DLGAP5, TPX2, KIF11 and CKAP5: Five specific mitosis-associated genes correlate with poor prognosis for non-small cell lung cancer patients. Int J Oncol. 2017;50:365–372. doi: 10.3892/ijo.2017.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shi YX, Yin JY, Shen Y, Zhang W, Zhou HH, Liu ZQ. Genome-scale analysis identifies NEK2, DLGAP5 and ECT2 as promising diagnostic and prognostic biomarkers in human lung cancer. Sci Rep. 2017;7:8072. doi: 10.1038/s41598-017-08615-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tagal V, Wei S, Zhang W, Brekken RA, Posner BA, Peyton M, Girard L, Hwang T, Wheeler DA, Minna JD, et al. SMARCA4-inactivating mutations increase sensitivity to Aurora kinase A inhibitor VX-680 in non-small cell lung cancers. Nat Commun. 2017;8:14098. doi: 10.1038/ncomms14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamamoto S, Takayama K, Obinata D, Fujiwara K, Ashikari D, Takahashi S, Inoue S. Identification of new octamer transcription factor 1-target genes upregulated in castration-resistant prostate cancer. Cancer Sci. 2019;110:3476–3485. doi: 10.1111/cas.14183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim D, Cho JY. NQO1 is required for β-lapachone-mediated downregulation of breast-cancer stem-cell activity. Int J Mol Sci. 2018;19(pii):E3813. doi: 10.3390/ijms19123813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Freedman RA, Keating NL, Lin NU, Winer EP, Vaz-Luis I, Lii J, Exman P, Barry WT. Breast cancer-specific survival by age: Worse outcomes for the oldest patients. Cancer. 2018;124:2184–2191. doi: 10.1002/cncr.31308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Del Prete S, Caraglia M, Luce A, Montella L, Galizia G, Sperlongano P, Cennamo G, Lieto E, Capasso E, Fiorentino O, et al. Clinical and pathological factors predictive of response to neoadjuvant chemotherapy in breast cancer: A single center experience. Oncol Lett. 2019;18:3873–3879. doi: 10.3892/ol.2019.10729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hueman MT, Wang H, Yang CQ, Sheng L, Henson DE, Schwartz AM, Chen D. Creating prognostic systems for cancer patients: A demonstration using breast cancer. Cancer Med. 2018;7:3611–3621. doi: 10.1002/cam4.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Howlader N, Noone AM, Krapcho M, Miller D, Brest A, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, et al. National Cancer Institute; Bethesda, MD, USA: SEER Cancer Statistics Review, 1975–2016. [Google Scholar]
- 54.Nechuta S, Lu W, Zheng Y, Cai H, Bao PP, Gu K, Zheng W, Shu XO. Comorbidities and breast cancer survival: A report from the Shanghai breast cancer survival study. Breast Cancer Res Treat. 2013;139:227–235. doi: 10.1007/s10549-013-2521-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuo TC, Chang PY, Huang SF, Chou CK, Chao CC. Knockdown of HURP inhibits the proliferation of hepacellular carcinoma cells via downregulation of gankyrin and accumulation of p53. Biochem Pharmacol. 2012;83:758–768. doi: 10.1016/j.bcp.2011.12.034. [DOI] [PubMed] [Google Scholar]
- 56.Zhang X, Pan Y, Fu H, Zhang J. Nucleolar and spindle associated protein 1 (NUSAP1) inhibits cell proliferation and enhances susceptibility to epirubicin in invasive breast cancer cells by regulating cyclin D kinase (CDK1) and DLGAP5 expression. Med Sci Monit. 2018;24:8553–8564. doi: 10.12659/MSM.910364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, inflammation, and immunity: A Troika governing cancer and its treatment. Cell. 2016;166:288–298. doi: 10.1016/j.cell.2016.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rybstein MD, Bravo-San Pedro JM, Kroemer G, Galluzzi L. The autophagic network and cancer. Nat Cell Biol. 2018;20:243–251. doi: 10.1038/s41556-018-0042-2. [DOI] [PubMed] [Google Scholar]
- 59.Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. Elegans. Nature. 2015;521:525–528. doi: 10.1038/nature14300. [DOI] [PubMed] [Google Scholar]
- 60.Lu H, Li G, Liu L, Feng L, Wang X, Jin H. Regulation and function of mitophagy in development and cancer. Autophagy. 2013;9:1720–1736. doi: 10.4161/auto.26550. [DOI] [PubMed] [Google Scholar]
- 61.Chourasia AH, Macleod KF. Tumor suppressor functions of BNIP3 and mitophagy. Autophagy. 2015;11:1937–1938. doi: 10.1080/15548627.2015.1085136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during the present study are included in this published article.