

Abstract
Background
Glioblastoma (GBM) is the most common and deadliest form of brain cancer in adults. Standard treatment, consisting of surgery and radiochemotherapy, only provides a modest survival benefit and is incapable of combating infiltrating GBM cells in other parts of the brain. New therapies in clinical trials, such as anti-programmed cell death 1 immunotherapy, have so far shown limited success in GBM. Moreover, it is unclear how the growth of GBM suppresses the immune system locally at the site of the brain tumor or if distant sites of tumor cell migration are also involved. Invasive GBM cells in brain tissue beyond the primary tumor limit the use of surgery, thus immunotherapy could be beneficial if activated/suppressed immune cells are present in the contralateral hemisphere.
Methods
Here, we used a syngeneic orthotopic GL26 GBM mouse model and multiparameter fluorescence-activated cell sorting analysis to study the phenotype of resident and infiltrating immune cells in both the brain tumor hemisphere and contralateral hemisphere.
Results
We show that lymphoid cells, including tumor antigen-specific CD8+ tumor-infiltrating lymphocytes (TILs) are present in the tumor and are characterized by a tolerogenic phenotype based on high immune checkpoint expression. Massive infiltration of myeloid cells is observed, expressing immune checkpoint ligands, suggesting an immune-dependent coinhibitory axis limiting TIL responses. Surprisingly, these phenotypes are paralleled in the contralateral hemisphere, showing that infiltrating immune cells are also present at distant sites, expressing key immune checkpoints and immune checkpoint ligands.
Conclusion
Whole-brain analysis indicates active immune involvement throughout the brain, both at the site of the primary tumor and in the contralateral hemisphere. Using the right combination and timing, immune checkpoint blockade could have the potential to activate immune cells at the site of the brain tumor and at distant sites, thereby also targeting diffusely infiltrating GBM cells.
Keywords: neurooncology, immunology
Background
Glioblastoma (GBM) is the most common diffuse infiltrative brain tumor in adults and is considered fatal, with a prognosis of only 15 months after diagnosis.1 The current standard-of-care therapy consists of surgery followed by radiochemotherapy.1 2 Despite this aggressive approach, GBM cannot be effectively treated. Many newly diagnosed patients already have infiltrative growth and tumor recurrence occurs at both primary and secondary sites in the brain.3 Autopsy reports show that 75% of patients have infiltrating GBM tumor cells in the contralateral hemisphere.4 5 Thus, the invasive capacity of GBM cells that migrate beyond the original tumor, throughout the brain, requires a ‘brain-wide’ therapeutic approach. Unfortunately, many clinical trials evaluating novel therapies for patients with GBM have so far not been very successful.6
Because of its success in other tumor types, immunotherapy is considered as an antitumor strategy for GBM. For example, inhibitory receptors on tumor-infiltrating lymphocytes (TILs), termed immune checkpoints, can be blocked by antagonistic antibodies. Indeed, the inhibitory checkpoint receptor programmed cell death 1 (PD-1) was found to dampen tumor-reactive cytotoxic CD8+ T lymphocytes, and antibodies blocking PD-1 have shown clinical benefit to patients with a variety of cancer types.7–10 Mechanistically, anti-PD-1 immune checkpoint blockade (ICB) is dependent on cytotoxic CD8+ T lymphocytes recognizing tumor cells expressing peptide-class I major histocompatibility complexes (MHCs), programmed death-ligand 1 (PD-L1) expression on myeloid cells and local CD28 costimulation by CD80/CD86 on myeloid cells.11–14 Infiltration of lymphoid and myeloid cells in GBM tumors has been reported; however, whether these cells infiltrate to create antitumor immunity or solely facilitate tumor immune evasion is one of the urgent questions to address. The specific number of immune cells that infiltrate the tumor is unknown, although it has been suggested that one-third of GBM tumor bulk consists of myeloid cells.15 16 Less is known about the exact numbers and functions of infiltrating lymphoid cells in the brain tumor and the presence of regulatory T lymphocytes (Tregs).17 18 Although tissue-resident myeloid cells (microglia) have an important function in local homeostasis, they may also alter their function in the context of GBM. Alternatively, infiltrating myeloid cells may accumulate to locally suppress antitumor immunity. The expression of immune checkpoints and their ligands at the tumor site may therefore play a crucial role in local immune suppression.19–21
Since GBM has exclusively an infiltrative non-metastatic growth, it is important to understand if the immune system is armed against the tumor locally or at distant locations in the brain. To date, it is still unclear whether the immune system is suppressed in a similar fashion outside the primary tumor bulk in the brain, where infiltrating tumor-initiating cells migrate, and local brain tumor recurrence often develops. One of the hallmarks to identify is how local immunity evolves at the primary GBM tumor site, and whether adjacent tissues are also accompanied by similar immune cell infiltration. Moreover, it is crucial to understand if infiltrating immune cells coexpress inhibitory immune checkpoints or their ligands, so that immune checkpoint inhibitors can be administered effectively, correlating to local expression. Studies in patients with GBM are limited to the primary tumor site or directly adjacent tissue.22 To investigate whether other brain areas are also inflicted and show immune infiltration and an inhibitory response phenotype, we set out to study both the brain tumor hemisphere and the contralateral hemisphere in a syngeneic orthotopic GBM mouse model. Our study shows defined accumulation of distinct immune subsets locally at the tumor site that coexpress inhibitory immune checkpoints or their ligands, which are partly paralleled in the contralateral hemisphere. This indicates that immune infiltration can be observed in neighboring tissues, potentiating possibilities for future immunotherapy.
Methods
GBM cells, orthotopic mouse model and bioluminescence imaging
GL26-OVA cells were a kind gift from Dr M.G. Castro at the University of Michigan23 and cultured under standard conditions. Briefly, cells were cultured in Dulbecco's Modified Eagle Medium (DMEM; Gibco) supplemented with 10% fetal calf serum (FCS) and 1% penicillin/streptomycin at 37°C under humidified atmosphere with 5% CO2. To generate luciferase-expressing cells for imaging orthotopic tumors, cells were transduced with a lentiviral vector encoding a CSCW-CMV-Fluc-IRES-mCherry plasmid generating GL26-OVA-Fluc/mCherry.
All animal experiments were approved by the VU University Animal Welfare Commission and conducted following national and European regulations and guidelines, according to Directive 2010/63/EU. Female C57BL/6 mice were bred under specific pathogen-free conditions at the Amsterdam Animal Research Center and used in experiments at 8–12 weeks of age. Intracranial injections and tumor monitoring were performed as before.24 Briefly, mice were anesthetized and injected intracranially with 2×105 GL26-OVA-Fluc cells in 2 µL serum-free OptiMEM into the left striatum at the following coordinates from bregma: x+2.0 mm, y+0.5 mm, z −3.0 mm. Sham injections were performed similarly with the injection of 2 µL plain OptiMEM (without cells).
Bioluminescence imaging was used to monitor tumor growth twice a week, after intraperitoneal injection of 200 mg/kg d-luciferin (Gold Biotechnology) and acquisition of photon flux (photons/s) using the Bruker In-Vivo Xtreme system (Bruker) under isoflurane gas anesthesia.
Ex vivo tissue processing, cell preparation and antibody staining
With the onset of symptoms (day 29), all animals were sacrificed. The brain was cut along the sagittal axis and the left and right hemisphere (‘brain tumor hemisphere’ and ‘contralateral hemisphere’, respectively) from the same mouse, as well as a sham-injected hemisphere were stored separately in wells of a 24-well plate containing DMEM that was kept on ice. The hemispheres were cut into small pieces in wells of a 24-well plate containing two working units of Liberase TL (Roche Sigma-Aldrich, 05401020001) and were incubated at 37°C for 30 min. After digestion, enzymes were deactivated using ice-cold RPMI1640 (10% FCS, 1% 50 U/mL penicillin, 50 µg/mL streptomycin, 0.5% N-2-Hydroxyethylpiperazine-N-2-Ethane Sulfonic Acid (HEPES)/EDTA), run through a 70 µm cell strainer, extensively washed and counted before fluorescence-activated cell sorting (FACS) staining.
Equal amounts of cells (5×105) were plated in two 96-well v-bottom plates and stained for FACS analysis. Two different antibody staining panels were used for the lymphoid compartment (online supplementary table 1) and the myeloid compartment (online supplementary table 2). A separate panel was used to confirm Foxp3 staining in a subset of T lymphocytes (online supplementary table 3). OVA257–264(SIINFEKL)-H-2Kb-PE tetramers were a kind gift from Dr J.W. Drijfhout at the Leiden University Medical Center, the Netherlands.
jitc-2019-000323supp001.pdf (212.5KB, pdf)
Flow cytometry and data analysis
Flow cytometry was carried out at the Microscopy and Cytometry Core Facility of the Amsterdam UMC, location VUMC. The BD LSRFortessa X-20 SORP cytometer (BD Biosciences) was calibrated daily using CS&T beads and all samples in were measured using the same CS&T calibration beads lot number. Acquisition was done using an automated plate loader set at 1.0 µL/s acquisition speed.
After acquisition, data were analyzed using FlowJo V.10 analysis software (FlowJo). Raw FCS files were loaded and compensated using UltraComp eBeads (Thermo Fisher) stained with the appropriate fluorochrome-labeled antibodies and then verified using fluorescence-minus one for every single antibody. First, gates were set for stable flow (counts vs time), cells (side scatter-area (SSC-A) vs forward scatter-area (FSC-A)), single cells (forward scatter-height (FSC-H) vs FSC-A) and live cells (fixable viability dye (FVD) negative). Lymphoid cell gates were set for CD45+CD3+ cells, while myeloid cell gates were set for CD45+CD11b+ cells. Subsequently, the resulting number of cells of CD4+, CD8+ or CD11b+ gates of individual samples were concatenated, exported into one FCS file and uploaded to the Cytobank online analysis platform (Danaher, https://www.cytobank.org).
Using the viSNE module25 of the Cytobank platform, t-distributed Stochastic Neighbor Embedding (t-SNE) plots were generated using the following settings: 2500 iterations, perplexity of 50, theta of 0.5 and on up to 30,000 cells. For lymphoid cells, analysis was based on the expression of CD4, PD-1, T cell immunoreceptor with Ig and ITIM domains (TIGIT), Herpes virus entry mediator (HVEM) (CD4+ gate) or the expression of H-2Kb-SIINFEKL tetramer, PD-1, TIGIT (CD8+ gate). For myeloid cells, analysis was based on the expression of CD45, CD11b, MHC-II, Ly6C, PD-L1 and CD155 (CD11b+ gate). In parallel, manual gating based on the abovementioned markers was used to define immune subpopulations, which were color coded and overlaid on the t-SNE plots. Similarly, this manual gating strategy was applied in FlowJo on individual samples for statistical purposes.
Three independent experiments with 10 animals per group (each) were performed. Since the results obtained were comparable, data presented is from one representative experiment. GBM-injected mice, lymphoid n=8 and myeloid n=6 were used. Sham-injected mice, lymphoid n=6 and myeloid n=4 were used.
Statistics
Statistical tests were performed using GraphPad Prism V.8.02. A two-tailed Student’s t-test was used to compare two groups. For more than two groups, analysis of variance was used followed by Tukey post hoc analysis to compare between-group means. A p value less than 0.05 was considered statistically significant. Data are represented as mean±SEM, unless otherwise stated.
Results
We set out to perform an in-depth analysis of the immune composition in an immunocompetent orthotopic GBM mouse model, in which we could compare the immune infiltrates in brain tumor hemisphere versus the contralateral hemisphere and using a medium-only injected sham hemisphere as a control. For this, we used syngeneic GL26-OVA-Fluc/mCherry GBM tumor cells that were intracranially injected into the left striatum of C57BL/6 mice which allowed us to use bioluminescence imaging to monitor tumor growth (figure 1A). Making single-cell suspensions of the separate hemispheres allowed us to determine the lymphocyte and myeloid composition by means of multiparameter flow cytometry (figure 1B). Basic gating for live single immune cells was performed and subsequently for our CD45+ populations of interest (figure 1C). Next, t-SNE plots were generated to visualize lymphoid or myeloid immune subsets within these populations, and their immune checkpoint or immune checkpoint ligand expression, respectively. Finally, defined subsets were manually gated to allow for abundance calculations and statistical analysis (figure 1C, D).
Figure 1.

Experimental set-up of experiment. (A) C57BL/6 mice were intracranially injected with syngeneic GL26-OVA-Fluc tumor cells. Bioluminescence imaging was used to monitor tumor growth and mice were sacrificed on symptoms. (B) The brain of injected mice was harvested and cut along the sagittal axis, separating the brain tumor hemisphere from the contralateral hemisphere. Multiparameter flow cytometry was used to analyze immune cell content. (C) After acquisition, gating was used for selection of stable flow, cells, single cells and live cells. (D) Subsequently, immune populations of interest were selected and exported to generate t-SNE plots, after which these specific immune subsets were manually gated for statistical analysis. GBM, glioblastoma; OVA, ovalbumin.
Tumor growth analysis of GL26-OVA-Fluc showed an initial lag in the first 14 days, followed by a phase in which tumors grew fast. Mice had to be sacrificed 29 days post-tumor inoculation, due to the onset of symptoms (figure 2A). Of each mouse, the brain was harvested and divided into the left ‘brain tumor hemisphere’ and the right ‘contralateral hemisphere’. The left hemispheres of sham-injected mice were taken along as a control to investigate inflammatory alterations due to orthotopic injections (figure 2B). Single-cell suspensions were made of all tissues and multiparameter FACS analysis was performed to quantify frequencies of major immune cell populations (figure 2C). After basic live cell gating in FlowJo, CD45+ immune cells were gated and the total number of immune cells per hemisphere was plotted (figure 2D). In the brain tumor hemisphere, the mean number of isolated immune cells was 54,455±19,010, while from the contralateral hemisphere a mean number of 35,158±19,977 immune cells were isolated. The mean number of immune cells isolated from sham-injected hemisphere was 6848±312. This indicated that the presence of a brain tumor considerably increased the absolute number of immune cells in the brain, compared with the sham injection. However, surprisingly, also the contralateral side of the GL26-injected brains showed a drastically increased number of immune cells compared with sham-injected hemispheres (figure 2D). To analyze the relative proportions of innate versus adaptive immune cells in each hemisphere, gates were drawn for CD3+CD4+ T lymphocytes, CD3+CD8+ T lymphocytes, CD45hiCD11b+ infiltrating myeloid cells and CD45loCD11b+ resident microglia (populations shown relative to the total number of CD45+ cells for each hemisphere; figure 2E). When comparing the contralateral hemisphere with the brain tumor hemisphere and sham hemisphere, we observed that around 20%–25% of CD45+ cells were T lymphocytes, comprising both CD4+ and CD8+ TILs. Most immune cells found in each hemisphere were of myeloid origin (75%). In the brain tumor hemisphere, a large population of myeloid cells were infiltrating CD45hiCD11b+ cells, accounting for almost 50% of myeloid cells present (p<0.001 vs sham), while the other half were resident CD45loCD11b+ microglia (p=0.002 vs sham; p=0.04 vs contralateral). In the contralateral hemisphere, we observed that 25% of myeloid cells were infiltrating cells (p=0.04 vs sham) while 75% consisted of microglia (not significant), indicating that immune cells at the contralateral site also accumulate, with great abundance of infiltrating myeloid cells. In the sham hemisphere, low numbers of immune cells were observed, with most myeloid cells being microglia (90%) and hardly any infiltrating myeloid cells (figure 2E).
Figure 2.

Glioblastoma tumor growth and overall immune cell abundance. (A) Bioluminescence measurements showed intracranial GL26-OVA-Fluc tumor growth in C57BL/6 mice. (B) After sacrificing mice on day 29, the brain was cut along the sagittal axis and separated into the brain tumor hemisphere and contralateral hemisphere. The left hemisphere of a sham-injected mouse was taken along as a control. (C) Gating strategy for different populations of CD45+ immune cells to determine (D) the total number of immune cells per hemisphere and (E) the relative contribution of immune cell populations per hemisphere, distinguishing CD3+ CD4+/CD8+ lymphoid cells and CD11b+ CD45hi/CD45lo myeloid cells in the brains of mice. Data are represented as mean±SEM. Statistical significance was assessed by analysis of variance followed by Tukey multiple comparisons test (ns, p>0.05, *p<0.05, **p<0.005, ***p<0.001). OVA, ovalbumin.
To analyze more in detail whether the subsets of TILs in the tumor microenvironment differed from the T lymphocytes that accumulated in the contralateral hemisphere, we stained isolated immune cells for CD3, CD4 and CD8, and assessed the coexpression of inhibitory immune checkpoints (figure 3A). The relative CD4+ T lymphocyte numbers were not significantly different between the hemispheres, contributing around 10% of alive CD45+ cells (figure 2E). For the t-SNE analysis of the CD4+ population, we included the expression of CD4, PD-1, TIGIT and HVEM. Generated t-SNE visualization showed that the major CD45+CD3+CD4+ populations could be largely defined as either a CD4hi or a CD4lo population and further be divided based on the expression of immune checkpoints PD-1/TIGIT or HVEM, respectively (figure 3B). We then applied manual gating to analyze these CD4+ subsets for their abundance and to perform statistical analysis.
Figure 3.
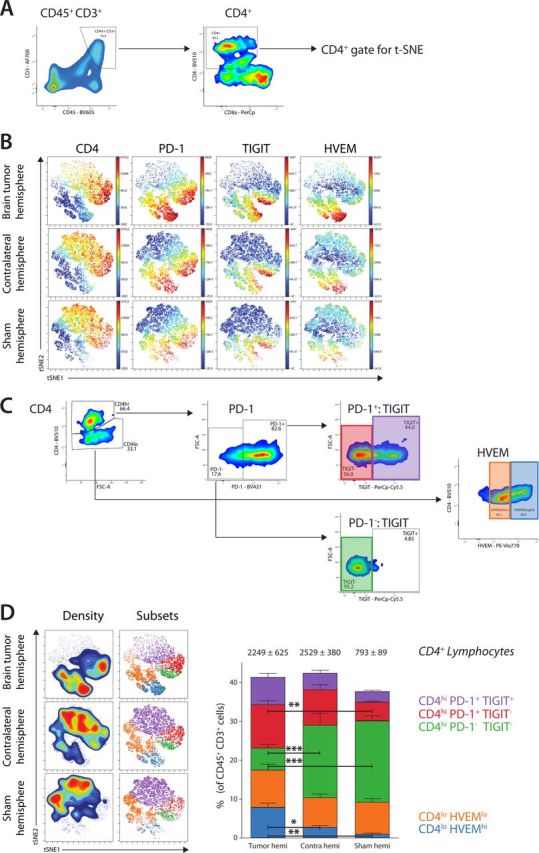
Analysis of the CD4+ T lymphocyte phenotype. (A) Gating strategy for CD45+CD3+ lymphocytes and subsequently CD4+ T lymphocytes. (B) Generated t-SNE plots of CD4+ T lymphocytes based on the expression of CD4, programmed cell death 1 (PD-1), TIGIT, HVEM showed a clear distinction between different subsets and cell abundancies. (C) Gating strategy for different populations of CD4+ T lymphocytes, based on the subsets visualized in the t-SNE plots. Gating was used to determine subset abundance and statistics. (D) Density overlaid on t-SNE plot show the relative cell density in each hemisphere. Manual gating overlaid on t-SNE plots show the location of defined subsets. Bar graphs show the frequency of each CD4+ subset as a percentage of total CD45+CD3+ lymphocytes. Data are represented as mean±SEM. Statistical significance was assessed by analysis of variance followed by Tukey multiple comparisons test (*p<0.05, **p<0.005, ***p<0.001).
When analyzing the CD3+CD4hi population, we could define three different subpopulations based on the expression of PD-1 and TIGIT (figure 3C). In the brain tumor hemisphere, CD4hi TILs represented 24% of the total number of CD3+ lymphocytes. A large population of these cells was CD4hiPD-1+TIGIT+ (purple, 7.1%; p=0.09 vs sham) and a large population was TIGIT negative, expressing only PD-1 (red, 11.3%; p=0.005 vs sham). The smallest population in the brain tumor hemisphere did not express either of these immune checkpoints (green, 5.8%; p<0.001 vs sham; p<0.001 vs contralateral; figure 3D). Analysis of the contralateral hemisphere showed that 32% of CD3+ lymphocytes were CD4hi TILs, displaying similar infiltration as the brain tumor hemisphere. The biggest populations were CD4hiPD-1-TIGIT- TILs (18.8%) and CD4hiPD-1+ TILs (13.7%), present at a higher frequency than in the brain tumor hemisphere. In contrast, the sham hemisphere showed 21% of CD4hiPD-1- cells and only 7.6% CD4hiPD-1+ cells. This indicates that both the brain tumor hemisphere and contralateral hemisphere contained more PD-1+ TIGIT- cells and PD-1+ TIGIT+ cells compared with the sham-injected hemisphere. Expression of these immune checkpoints has previously been shown to characterize exhausted CD4+ T lymphocytes in chronic infections,26 27 and therefore our data suggest that exhausted CD4+ TILs are present throughout the entire brain, due to the presence of a GBM tumor.
In the brain tumor hemisphere, the CD4lo population made up 17.5% of all CD3+ cells. Expression of HVEM was a clearly distinctive factor, with 9.6% of all CD3+ cells being CD4loHVEMlo (orange) and 7.9% having a CD4loHVEMhi (blue; p=0.002 vs sham; p=0.02 vs contralateral) phenotype in the brain tumor hemisphere (figure 3D). In the contralateral hemisphere, frequencies of CD4loHVEMlo (7.8%) and CD4loHVEMhi (2.8%) were clearly lower. Almost all CD4lo cells (9.3% of CD3+) were HVEMlo (8.3%) in the sham hemisphere (HVEMhi: 1.0%). This indicated that two distinct populations of CD4lo could be discriminated based on the expression of HVEM. In addition, t-SNE visualization showed that CD4lo TILs also had high expression of PD-1, particularly in the CD4loHVEMhi population present in the brain tumor hemisphere. However, PD-1 expression was also apparent in the CD4loHVEMlo populations in the contralateral and sham hemispheres (figure 3B).
Together, these data indicate that although CD4+ TILs are present in the contralateral hemisphere and show the same subsets as in the tumor brain hemisphere, the percentage of CD4+ subsets differ. A clear distinction could be made in the CD4hiPD1-TIGIT- population, being the most abundant in the contralateral and sham hemispheres (p<0.001, vs brain tumor hemisphere), and the CD4hiPD1+TIGIT- having the highest presence in the brain tumor hemisphere (p=0.005 vs sham). Moreover, the population of CD4hiPD1+TIGIT- cells in the contralateral hemisphere also approached significance compared with the sham-injected hemisphere (p=0.07). No significant difference between the contralateral and brain tumor hemispheres was observed. As such, higher expression of PD-1 can be attributed to the presence of GBM tumor cells and is a brain-wide phenomenon, not confined to the brain tumor hemisphere. The CD4loHVEMhi population seemed to be mostly present in the brain tumor hemisphere but not so much in the contralateral hemisphere (p=0.02) nor in the sham-injected hemisphere (p=0.002), while no significant differences were observed in the subset of CD4loHVEMlo TILs.
Similar to the CD4+ lymphocytes, we observed that the frequencies of CD8+ lymphocytes between the brain tumor, contralateral and sham-injected hemispheres were similar, around 7% of CD45+ cells (figure 2C). The abundance of subsets, however, differed greatly between the hemispheres. CD8+ effector TILs are crucial for a cytotoxic antitumor response; thus, we analyzed this population including the coexpression of immune checkpoints (figure 4A). As before, t-SNE plots were used for the visualization of differences in the CD3+CD8+ TIL population (figure 4B). Subsequent manual gating confirmed the presence of these subsets and allowed for the quantification of abundance and statistical analysis (figure 4C).
Figure 4.
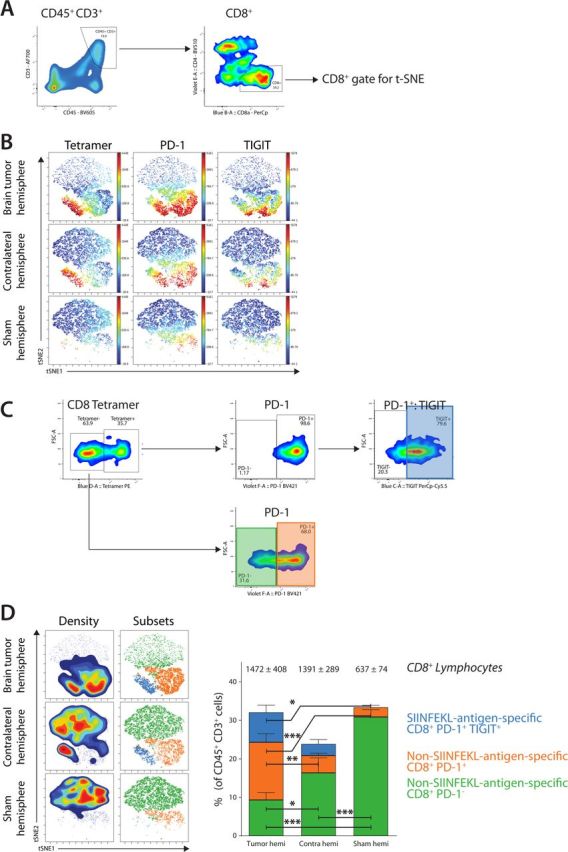
Analysis of the CD8+ T lymphocyte phenotype. (A) Gating strategy for CD45+CD3+ lymphocytes and subsequently CD8+ T lymphocytes. (B) Generated t-SNE plots of CD8+ lymphocytes based on the expression of H-2Kb-SIINFEKL tetramer, programmed cell death 1 (PD-1), TIGIT showed a clear distinction between different subsets and cell abundancies. (C) Gating strategy for different populations of CD8+ T lymphocytes, based on the subsets visualized in t-SNE plots. Gating was used to determine subset abundance and statistics. (D) Density overlaid on t-SNE plot show the relative cell density in each hemisphere. Manual gating overlaid on t-SNE plots show the location of defined subsets. Bar graphs show the frequency of each CD8+ T lymphocyte subset as a percentage of total CD45+CD3+ lymphocytes. Data are represented as mean±SEM. Statistical significance was assessed by analysis of variance followed by Tukey multiple comparisons test (*p<0.05, **p<0.005, ***p<0.001).
Since the GL26 murine GBM cells express the ovalbumin (OVA) model antigen, we aimed to determine whether tumor antigen-specific CD8+ TILs could be found at the site of the tumor. Therefore, we included OVA257–264-H-2Kb (SIINFEKL peptide bound to H-2Kb) tetramer staining to detect SIINFEKL-antigen-specific cells in our CD3+CD8+ T lymphocyte population. Indeed, of all CD3+ cells analyzed in the brain tumor hemisphere, 6.3% were CD8+ TILs positive for tetramer staining (blue; p=0.02 vs sham; figure 4D), indicating that SIINFELK-antigen-specific CD8+ TILs are found in the vicinity of the tumor bulk. When analyzing the contralateral hemisphere for the presence of SIINFEKL-specific CD8+ TILs, a surprising 2.1% of CD3+CD8+ TILs had antigen specificity for SIINFEKL (blue; figure 4D). This indicated that tumor-antigen-specific CD8+ TILs are also present at distant sites. As a control, no SIINFEKL-antigen-specific CD8+ TILs were found in the sham-injected hemisphere. Still, in the brain tumor hemisphere, the majority of CD8+ TILs was not SIINFELK-antigen specific, with 10.9% being CD3+CD8+PD-1- TILs (green; p<0.001 vs sham; p=0.04 vs contra), while a larger subset stained positive for PD-1 (orange, 12.9%; p<0.001 vs sham; p=0.001 vs contra). Instead, the population of CD3+CD8+ lymphocytes in the contralateral hemisphere were largely non-SIINFEKL-antigen-specific CD8+PD-1- TILs (green, 18.4%; p<0.001 vs sham; p=0.04 vs tumor) and a smaller population of PD-1+ non-SIINFEKL-antigen-specific CD8+ TILs was detected (orange, 3.8%). The most dramatic changes were seen in the SIINFEKL-antigen-specific CD3+CD8+ TIL populations, where all these cells in the brain tumor hemisphere and contralateral hemisphere expressed PD-1, of which most also showed coexpression of TIGIT (figure 4C, D). The largest population of CD3+CD8+ TILs in the sham-injected hemisphere did not show coexpression of PD-1 and TIGIT (green, 30.7%), while a small subset did express PD-1 (orange, 2.5%).
Combined, these data show that CD3+CD8+ TILs are present in the brain tumor microenvironment and at distant sites in the contralateral hemisphere. While CD8+ effector cells against the SIINFEKL epitope of our model antigen OVA are primarily found in the brain tumor hemisphere, they are also present in the contralateral hemisphere, located further away from the brain tumor. Furthermore, although these antigen-specific cells are present, they also show high expression of PD-1 and some expression of TIGIT, suggesting an exhausted CD8+ TIL phenotype.28 Moreover, the brain tumor hemisphere had a significantly larger population of PD-1 expressing CD8+ TILs compared with the contralateral (p=0.001) and sham-injected (p<0.001) hemispheres due to the presence of the GBM tumor. Conversely, almost all CD8+ lymphocytes in the sham hemisphere were negative for PD-1.
To analyze myeloid populations, the cell type comprising the bulk of immune cells in the brain (figure 2), gates were manually set for CD45+CD11b+ cells (figure 5A) and further analyzed by t-SNE (figure 5B). Subsequent gating allowed us to find the differences in abundancy of the populations between the brain tumor hemisphere and contralateral hemisphere (figure 5C). Several CD45+CD11b+ populations were identified, and these could further be separated into subpopulations, based on their expression of MHC-II and Ly6C. Analysis of the myeloid subsets could discriminate between brain-resident microglia (CD45loCD11b+, blue and orange) and infiltrating myeloid cells (CD45hiCD11b+, green, red, purple, and brown; figure 5D).
Figure 5.
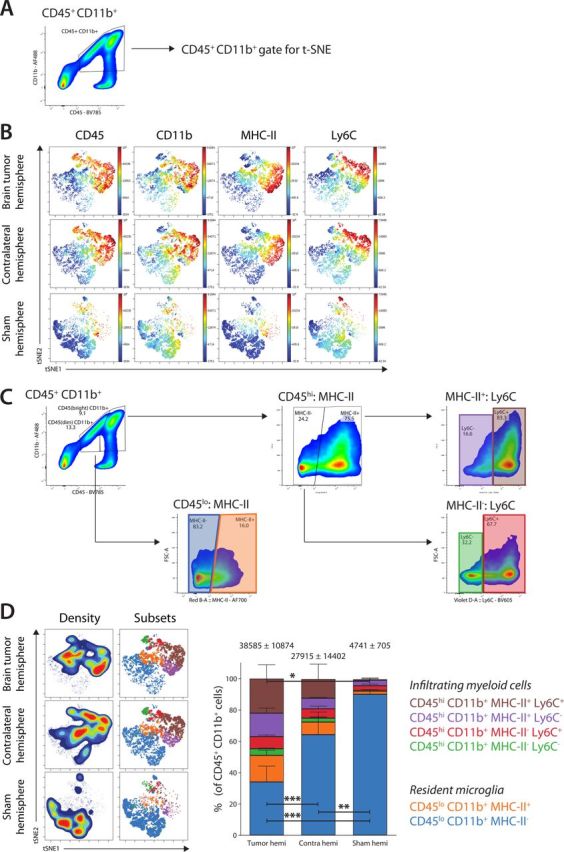
Analysis of the CD11b+ myeloid cell phenotype. (A) Gating strategy for CD45+CD11b+ myeloid cells. (B) Generated t-SNE plots of myeloid cells based on the expression of CD45, CD11b, MHC-II, Ly6C, PD-L1, CD155 showing resident CD45lo microglia and infiltrating CD45hi myeloid cells and their abundancies. (C) Gating strategy for different populations of CD45+CD11b+ myeloid cells, based on the subsets visualized in the t-SNE plots. Gating was used to determine subset abundance and statistics, showing a clear difference between brain resident CD45lo microglia and infiltrating CD45hi myeloid cells. (D) Density overlaid on t-SNE plot shows the relative cell density in each hemisphere. Manual gating overlaid on t-SNE plots shows the location of myeloid cell subsets. Bar graphs show the frequency of each myeloid subset as a percentage of total CD45+CD11b+myeloid cells. Data are represented as mean±SEM. Statistical significance was assessed by analysis of variance followed by Tukey multiple comparisons test (*p<0.05, **p<0.005, ***p<0.001). MHC-II, class II major histocompatibility complexes.
The brain-resident microglia represented only 51.0% of myeloid cells in the brain tumor hemisphere, while their relative abundancy was 72.3% in the contralateral hemisphere and over 92.0% in the sham-injected hemisphere. In addition, the microglia in both the brain tumor hemisphere and contralateral hemisphere showed higher expression of MHC-II, which could be defined as a distinct MHC-II+ population (antigen-presenting microglia), while, in contrast, nearly all microglia in the sham-injected hemisphere were MHC-II- (p=0.006 sham vs contralateral; p<0.001 sham vs tumor; p<0.001 tumor vs contralateral; figure 5D).
When we compared the CD45hiCD11b+ infiltrating cells, we identified similar subsets of myeloid cells infiltrating the brain tumor hemisphere (49.0%) and the contralateral hemisphere (25.0%). However, the frequencies of CD45hiCD11b+MHC-II+Ly6C+ (brown, 22%; p=0.02) and CD45hiCD11b+MHC-II+Ly6C- (purple, 15%) monocytes/macrophages were considerably increased in the brain tumor hemisphere, but also detected at a high frequency in the contralateral infiltrating side (12.3% and 6.8%, respectively) compared with the sham-injected hemisphere. Conversely, the difference between these hemispheres was smaller in the CD45hiCD11b+MHC-II-Ly6C+ (red) and CD45hiCD11b+MHC-II-Ly6C- (green) monocytes/macrophages, totaling 12.8% in the brain tumor hemisphere and 8.5% in the contralateral hemisphere. Very few infiltrating myeloid cells of any of these subsets were found in the sham-injected hemisphere, with the largest populations being the CD45hiCD11b+MHC-II+Ly6C- (purple) monocytes/macrophages (3.5%) and the CD45hiCD11b+MHC-II-Ly6C+ (red) monocytes/macrophages (3.0%; figure 5D).
In the same t-SNE plots of lymphoid and myeloid cells, several immune checkpoint receptors and immune checkpoint ligands, respectively, were taken along to visualize their expression among immune cell populations. We compared the expression of the inhibitory immune checkpoints on CD3+ lymphoid cells against the expression of immune checkpoint ligands on CD11b+ myeloid cells, to find putative interactions between immune checkpoint receptor and ligand pairs (figure 6).
Figure 6.

Expression levels of immune checkpoints and corresponding ligands in the brain tumor hemisphere and contralateral hemisphere. (A) Relative expression of immune checkpoints on analyzed T lymphocyte subpopulations in both hemispheres. (B) Relative expression of immune checkpoint ligands on analyzed brain-resident microglia and infiltrating myeloid cells in both hemispheres. MHC-II, class II major histocompatibility complex; PD-1, programmed cell death 1; TIGIT, T cell immunoreceptor with Ig and ITIM domains; HVEM. Herpes virus entry mediator; Tim-3, T-cell immunoglobulin and mucin-domain containing-3; Lag-3, lymphocyte-activation gene 3; PD-L1, programmed death ligand 1; BTLA, B- and T-lymphocyte attenuator.
Expression of HVEM was confined to the CD4lo population and not detected in any of the other populations we analyzed. The CD4loHVEMhi population also showed high expression of coinhibitory immune checkpoints PD-1, TIGIT, T-cell immunoglobulin and mucin-domain containing-3 (Tim-3) and Lymphocyte-activation gene 3 (Lag-3), suggesting a more exhausted TIL phenotype, which was present not only in the brain tumor hemisphere but also in the contralateral hemisphere (figure 6A). Expression of inhibitory immune checkpoint PD-1 was detected in the entire CD4lo population, as well as a large part of the CD4hi population. Furthermore, the majority cells that expressed PD-1 also expressed the coinhibitory immune checkpoint TIGIT. Of the PD-1+ CD4hi cells that were negative for TIGIT, we still found significant expression of coinhibitory immune checkpoints Tim-3 and Lag-3. In contrast, the CD4hi and CD8+ lymphocytes that did not express PD-1 and TIGIT also did not express Tim-3 and Lag-3, and this was observed in both the brain tumor hemisphere and contralateral hemisphere (figure 6A).
In the myeloid analysis, we observed high expression of PD-L1 and CD155 in different myeloid subsets, most notably the brain-resident MHC-II+ microglia and infiltrating MHC-II+ Ly6C+/− monocytes/macrophages (figure 6B). In general, myeloid cells expressing MHC-II also seemed to have higher expression of inhibitory immune checkpoint ligands. However, curiously, MHC-II- microglia showed clear expression of CD155, B- and T-lymphocyte attenuator (BTLA) and galectin-9 in both the brain tumor and contralateral hemisphere, while there was much lower expression of PD-L1, compared with MHC-II+ microglia (figure 6B).
Taken together, these results show detailed coexpression of myeloid cell immune checkpoint ligands and their potentially inhibitory action when interacting with activated TIL immune checkpoints. High expression of these immune checkpoints indicates a more exhausted TIL phenotype. Homeostatic MHC-II- microglia would be suppressive when interacting with CD8+TIGIT+ or CD8+Tim-3+ TILs, while MHC-II+ microglia expressing PD-L1 are potent suppressors of both CD4+PD-1+ and CD8+PD-1+ TILs. Infiltrating MHC-II+ myeloid cells would be suppressive interacting with CD4+ and CD8+ TILs expressing PD-1, TIGIT, HVEM, Tim-3 or Lag-3. These immune cells were particularly present in the brain tumor hemisphere and, surprisingly, also abundant in the contralateral hemisphere, whose immune checkpoint interaction pairs provide a good rationale for a ‘brain-wide’ therapeutic approach.
Discussion
GBM is the deadliest form of brain cancer and current therapies can insufficiently combat this disease. The invasive nature of GBM, infiltrating both surrounding tissue and distant sites in the brain, makes this tumor inherently difficult to treat, and these infiltrating GBM cells are often responsible for tumor recurrence. Novel therapies are much needed, and due to its success in other tumors, immune checkpoint inhibition is considered.7–10 29 However, preliminary results of clinical trials with anti-PD-1 immunotherapy in GBM are so far inconclusive. Although some trials show no benefit, others suggest that neoadjuvant PD-1 ICB could be beneficial in a subset of patients; however, this has not resulted in significant survival benefit.30–32 The results of these clinical trials show the need to study the immune response in GBM in more detail. Rationally targeted immunotherapy needs to be applied in this group of patients, rather than copying successful trials from other types of cancer. Although PD-1 is an attractive and well-known target on TILs,7 coexpression of other inhibitory molecules complicates the single-cell phenotype and its role in the tumor microenvironment. Resistance to ICB therapy evolves through changes in the tumor and its microenvironment, at least partially by upregulation of additional immune checkpoints.33–35 Therefore, much potential lies in studying other immune checkpoint/ligand pairs beyond PD-1/PD-L1 and CTLA-4/CD80/CD86. Especially in GBM, the tumor microenvironment goes beyond the primary tumor, as tumor-initiating cells invade other parts of the brain and the contralateral hemisphere.4 5 Here, we show in an orthotopic syngeneic GBM mouse model that different subsets of lymphoid and myeloid immune cells are detected in the brain tumor microenvironment, but, more importantly, also at distant sites in the contralateral hemisphere. Furthermore, these immune cells express inhibitory immune checkpoints and immune checkpoint ligands that could be potential targets for immune checkpoint inhibitors.
In our GL26 model, CD4hi TILs in the brain tumor hemisphere showed the highest expression of both PD-1 and TIGIT and these populations were of similar frequency in the contralateral hemisphere. This suggests that exhausted CD4+ TILs are also present at distant sites in the brain. Studies in melanoma have shown that exhausted tumor-specific CD4+ effector TILs also displayed high expression of PD-1 and TIGIT, and their function could be restored by ICB therapy.36 37
The other large CD4+ population in the brain tumor hemisphere had a CD4lo phenotype that could be mostly distinguished by its expression HVEM, in contrast to other subsets. Both CD4lo T lymphocytes that arise on chronic stimulation and CD4loCD25+ Tregs have been described in autoimmune diabetes,38 39 but their role in cancer remains elusive. The effect that high expression of HVEM has in this population is unclear, as signal transduction by HVEM is very dependent on its ligand, with opposing effects.40 Binding of BTLA, assessed here on myeloid cells, would result in T lymphocyte inhibition, while competitive binding of LIGHT or CD160 would result in stimulation.40 In this model, the HVEM-expressing CD4lo TIL population was particularly present in the brain tumor hemisphere, whereas the contralateral hemisphere had a lower frequency of CD4lo TILs and they were almost not detected in sham-injected mice. HVEM seems to be important in the regulation of both effector and regulatory T lymphocytes, with T effector cells downregulating HVEM after stimulation, while HVEM is upregulated in Tregs.41 42 Speculatively, these cells could be Tregs, and an additional experiment including Foxp3 staining provided evidence that most of the CD4loHVEM+ cells indeed expressed the Treg marker Foxp3 (online supplementary figure S1).
jitc-2019-000323supp002.pdf (2.9MB, pdf)
All SIINFEKL-antigen-specific CD8+ TILs and most non-SIINFEKL-antigen-specific CD8 TILs in the brain tumor microenvironment expressed PD-1. In addition, most CD8+PD-1+ cells also coexpressed TIGIT. Although expression of PD-1 is also associated with T lymphocyte activation, sustained expression has been linked to CD8+ T lymphocyte exhaustion and this contributes to a tolerogenic tumor microenvironment.28 43 Interestingly, SIINFEKL-antigen-specific CD8+ TILs were also found to infiltrate brain tissue more distant from the tumor. The presence of antigen-specific CD8+PD1+ TILs in the contralateral hemisphere is critical for the success of ICB at distant sites, where current therapies often fail to make a difference. These cytotoxic CD8+ TILs are potential killers of infiltrating GBM tumor cells that cannot be reached by standard of care (ie, surgery and radiochemotherapy).
Although we found many of the same myeloid subsets in the different analyzed hemispheres, there is a clear shift of the major populations that contribute to the overall total pool of myeloid cells. As expected, the absolute number of microglia in the brain tumor and contralateral hemisphere was similar. There were many infiltrating myeloid cells in the brain tumor hemisphere and the contralateral hemisphere, while only few were found in the sham-injected hemisphere. The microglia in both the brain tumor hemisphere and contralateral hemisphere had an increasingly ‘activated’ phenotype, with observed higher expression of MHC-II, indicative of their ability to present antigens. This was especially apparent in the brain tumor hemisphere, where 35% of microglia were positive for MHC-II, 15% of microglia in the contralateral hemisphere expressed MHC-II, while this was only 5% in the sham-injected hemisphere.
The infiltrating myeloid populations make up the major population in these GBM tumors, representing up to 50% of myeloid cells in the brain tumor hemisphere. Although at a lower frequency, there is also a large population of infiltrating myeloid cells in the contralateral hemisphere (25%). It has been reported that a subset of infiltrating myeloid cells in patients with GBM are myeloid-derived suppressor cells (MDSCs) and higher numbers of MDSCs in the brain tumor microenvironment was associated with poorer prognosis.44 Whether these infiltrating myeloid cells also represent MDSCs remains to be elucidated. Further analysis of immune checkpoint ligands suggests that both microglia and locally accumulated infiltrating myeloid cells appear to suppress immunity. This is particularly apparent in the brain tumor microenvironment, but also at distant sites of infiltration in the contralateral hemisphere. MHC-II+ microglia express intermediate levels of PD-L1, CD155, BTLA and galectin-9, while MHC-II- microglia have much lower expression of PD-L1 but still express the other immune checkpoint ligands that we analyzed. This pattern of checkpoint ligand expression on microglia seems to be identical in the contralateral hemisphere. In contrast, MHC-II+ infiltrating myeloid cells express high levels of PD-L1, CD155, BTLA and galectin-9, while we found very low-to-no expression of these immune checkpoint ligands on MHC-II- infiltrating myeloid cells. Again, this was also observed in the contralateral hemisphere, although here overall expression of immune checkpoint ligands on infiltrating myeloid cells seemed lower. These data show that multiple inhibitory immune checkpoints and immune checkpoint ligands are expressed in the local brain tumor microenvironment as well as in the contralateral hemisphere. Thus, interference with immune checkpoint expression is complex and should focus on multiple targets. Furthermore, depending on expression levels, timing of therapy could be crucial.
In concordance with studies performed in patients with GBM, our GL26 model has a high frequency of infiltrating myeloid cells and some infiltration of TILs, with most TILs expressing PD-1.44–46 Successful ICB requires active checkpoint–ligand interactions, with myeloid cells expressing antigen and antigen-specific CD8+ TILs as effector cells, with the help of CD4+ TILs. GBM recurrence is inevitable because of tumor cell infiltration at sites away from the bulk tumor. The primary tumor is resected during surgery, but infiltrating cells around the tumor bulk and in the contralateral hemisphere remain. A recent clinical study suggests that surgery inhibits the effect of PD-1 ICB, both by taking away the activation signal of TILs and removing most TILs from the brain tumor microenvironment, while treatment with anti-PD-1 could be beneficial in the neoadjuvant setting. Furthermore, partial PD-1 receptor occupancy on CD8+ TILs at the time of surgery suggests that nivolumab can pass the blood–brain barrier.32 However, the contralateral hemisphere or even distant sites in the brain tumor hemisphere have so far not been investigated in patients with GBM. Thus, if the prerequisites for successful checkpoint blockade apply to those distant sites, immunotherapy may be the golden bullet. However, for immunotherapy to be effective, it is paramount to understand which immune checkpoints and ligands are expressed in the tumor microenvironment and if the same principles apply to those distant sites of infiltration. The present results suggest a coinhibitory axis at the primary tumor site and in the contralateral hemisphere, which could cause T lymphocyte exhaustion thereby preventing proper immune responses. While anti-PD-1 therapy could have a potential role in GBM, others have shown limited responses to anti-PD-1 in GBM mouse models, while combination therapy was more effective.47 48 Coexpression of PD-1 and TIGIT has been associated with a viral infection like exhaustion phenotype of CD4+ and CD8+ T lymphocytes.49–52 Moreover, dual blockade of TIGIT and PD-1 or PD-L1 has shown antitumor responses in several tumor models, by increasing proliferating and antigen-specific CD8+ TILs.28 49 53 However, these are only two immune checkpoints, while many more play a role in immune responses. It has been suggested that resistance to anti-PD-1 ICB is associated with specific upregulation of other immune checkpoints, such as Tim-3 or Lag-3.34 35
Finally, in GBM it is still the question whether T lymphocytes are the right targets for ICB, as these tumors also show a strong immune modulatory effect toward tissue resident and infiltrating myeloid cells, microglia and monocytes/macrophages, respectively. The yolk-sac derived microglia and infiltrating myeloid cells are two completely different types of antigen-presenting cells. Based on this analysis, there might be a difference in outcome between TIL and myeloid interactions, depending on the cell type involved, as many concepts about microglia–TIL cross talk remain to be elucidated.54 Further studies are needed to provide additional mechanistic insight in these interactions beyond immune checkpoint expression, especially in the case of the contralateral hemisphere. Only a small number of the infiltrating cells in the tumor are TILs, compared with the entire myeloid population. Perhaps immunotherapy should focus more on targeting and activating myeloid cells against GBM tumors, given their reported abundance in this and other studies.15 16
Conclusions
Using multiparameter FACS analysis of immune cells in a syngeneic orthotopic GBM mouse model, we show that specific subsets of immune cells infiltrate the brain tumor hemisphere and are also present in the contralateral hemisphere at distant sites. High expression of immune checkpoints on CD3+ lymphoid cells and expression of immune checkpoint ligands on CD11b+ myeloid cells in both hemispheres suggests a coinhibitory axis, limiting antitumor responses. Proper activation of TILs or arming myeloid cells against GBM tumors could be an effective strategy, to not only combat the tumor bulk but also target diffusely infiltrating GBM cells at distant sites, such as the contralateral hemisphere, where GBM dissemination often occurs.
Acknowledgments
We thank T Konijn and T O’Toole for technical support at the Microscopy and Cytometry Core Facility at the Amsterdam UMC – Location VUMC. We also thank Dr M G Castro for the GL26-OVA cells and Dr J W Drijfhout for the H-2Kb SIINFEKL tetramers.
Footnotes
Contributors: MHWC, STTS and YvK conceived the study, MHWC, STTS, SAD and YvK designed experiments. MHWC, STTS, SAD and LJWK acquired experimental data, MHWC and STTS analyzed data. MHWC, STTS, SAD, JJG-V and YvK interpreted data. MHWC and YvK drafted the manuscript, MHWC, STTS, SAD, LJWK, JJG-V and YvK provided critical intellectual content. YvK supervised the study. All authors read and approved the final manuscript.
Funding: MHWC, STTS, LK and YvK were financially supported by ERC-Advanced Grant Glycotreat 339977; MC was financially supported by Hersentumorfonds grant DBTF-RP201801; SD was financially supported by Institute of Chemical Immunology ICI-00011.
Competing interests: None declared.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: All data relevant to the study are included in the article or uploaded as supplementary information. All data generated or analyzed during this study are included in this published article and its supplementary information file.
References
- 1.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987–96. 10.1056/NEJMoa043330 [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 2009;10:459–66. 10.1016/S1470-2045(09)70025-7 [DOI] [PubMed] [Google Scholar]
- 3.Bette S, Barz M, Huber T, et al. Retrospective analysis of radiological recurrence patterns in glioblastoma, their prognostic value and association to postoperative infarct volume. Sci Rep 2018;8:4561. 10.1038/s41598-018-22697-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Claes A, Idema AJ, Wesseling P. Diffuse glioma growth: a guerilla war. Acta Neuropathol 2007;114:443–58. 10.1007/s00401-007-0293-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindsay KW, Bone I, Fuller G. Localized neurological disease and its management A. intracranial. Neurol Neurosurg Illus. 5th edn London: Elsevier Ltd, 2010: 217–388. [Google Scholar]
- 6.Vanderbeek AM, Rahman R, Fell G, et al. The clinical trials landscape for glioblastoma: is it adequate to develop new treatments? Neuro Oncol 2018;20:1034–43. 10.1093/neuonc/noy027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54. 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rizvi NA, Hellmann MD, Snyder A, et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science (80-). American Association for the Advancement of. Science 2015;348:124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hugo W, Zaretsky JM, Sun L, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 2016;165:35–44. 10.1016/j.cell.2016.02.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015;372:2509–20. 10.1056/NEJMoa1500596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science 2018;359:1350–5. 10.1126/science.aar4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hui E, Cheung J, Zhu J, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017;355:1428–33. 10.1126/science.aaf1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamphorst AO, Wieland A, Nasti T, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 2017;355:1423–7. 10.1126/science.aaf0683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Linsley PS, Greene JL, Brady W, et al. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1994;1:793–801. 10.1016/S1074-7613(94)80021-9 [DOI] [PubMed] [Google Scholar]
- 15.Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci 2016;19:20–7. 10.1038/nn.4185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graeber MB, Scheithauer BW, Kreutzberg GW. Microglia in brain tumors. Glia 2002;40:252–9. 10.1002/glia.10147 [DOI] [PubMed] [Google Scholar]
- 17.Han S, Zhang C, Li Q, et al. Tumour-infiltrating CD4(+) and CD8(+) lymphocytes as predictors of clinical outcome in glioma. Br J Cancer 2014;110:2560–8. 10.1038/bjc.2014.162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El AA, Lesniak MS. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme1. Neuro Oncol. Narnia 2006;8:234–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitamura T, Doughty-Shenton D, Cassetta L, et al. Monocytes differentiate to immune suppressive precursors of metastasis-associated macrophages in mouse models of metastatic breast cancer. front Immunol. Frontiers 2018;8:2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cassetta L, Kitamura T. Macrophage targeting: opening new possibilities for cancer immunotherapy. Immunology 2018;155:285–93. 10.1111/imm.12976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prionisti I, Bühler LH, Walker PR, et al. Harnessing microglia and macrophages for the treatment of glioblastoma. front Pharmacol. Frontiers 2019;10:506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Darmanis S, Sloan SA, Croote D, et al. Single-Cell RNA-seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep 2017;21:1399–410. 10.1016/j.celrep.2017.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang J, Sanderson NSR, Wawrowsky K, et al. Kupfer-type immunological synapse characteristics do not predict anti-brain tumor cytolytic T-cell function in vivo. Proc Natl Acad Sci U S A 2010;107:4716–21. 10.1073/pnas.0911587107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lagerweij T, Dusoswa SA, Negrean A, et al. Optical clearing and fluorescence deep-tissue imaging for 3D quantitative analysis of the brain tumor microenvironment. Angiogenesis 2017;20:533–46. 10.1007/s10456-017-9565-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amir E-adD, Davis KL, Tadmor MD, et al. ViSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol 2013;31:545–52. 10.1038/nbt.2594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chew GM, Fujita T, Webb GM, et al. Tigit marks exhausted T cells, correlates with disease progression, and serves as a target for immune restoration in HIV and SIV infection. PLoS Pathog 2016;12:e1005349. 10.1371/journal.ppat.1005349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crawford A, Angelosanto JM, Kao C, et al. Molecular and transcriptional basis of CD4⁺ T cell dysfunction during chronic infection. Immunity 2014;40:289–302. 10.1016/j.immuni.2014.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chauvin J-M, Pagliano O, Fourcade J, et al. TIGIT and PD-1 impair tumor antigen–specific CD8+ T cells in melanoma patients. Journal of Clinical Investigation 2015;125:2046–58. 10.1172/JCI80445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caccese M, Indraccolo S, Zagonel V, et al. PD-1/PD-L1 immune-checkpoint inhibitors in glioblastoma: a Concise review. Crit Rev Oncol Hematol 2019;135:128–34. 10.1016/j.critrevonc.2018.12.002 [DOI] [PubMed] [Google Scholar]
- 30.Cloughesy TF, Mochizuki AY, Orpilla JR, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med 2019;25:477–86. 10.1038/s41591-018-0337-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao J, Chen AX, Gartrell RD, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med 2019;25:462–9. 10.1038/s41591-019-0349-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med 2019;25:470–6. 10.1038/s41591-018-0339-5 [DOI] [PubMed] [Google Scholar]
- 33.Riaz N, Havel JJ, Makarov V, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 2017;171:934–49. 10.1016/j.cell.2017.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koyama S, Akbay EA, Li YY, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun 2016;7:1–9. 10.1038/ncomms10501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woo S-R, Turnis ME, Goldberg MV, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res 2012;72:917–27. 10.1158/0008-5472.CAN-11-1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goding SR, Wilson KA, Xie Y, et al. Restoring immune function of tumor-specific CD4+ T cells during recurrence of melanoma. J Immunol 2013;190:4899–909. 10.4049/jimmunol.1300271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carter L, Fouser LA, Jussif J, et al. PD-1:PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. Eur J Immunol 2002;32:634–43. 10.1002/1521-4141(200203)32:3<634::AID-IMMU634>3.0.CO;2-9 [DOI] [PubMed] [Google Scholar]
- 38.Linkes S, Fry C, Quinn A. Antigen-Experienced CD4lo T cells are linked to deficient contraction of the immune response in autoimmune diabetes. Autoimmune Dis 2010;2010:1–14. 10.4061/2010/920148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garg G, Tyler JR, Yang JHM, et al. Type 1 diabetes-associated IL2RA variation lowers IL-2 signaling and contributes to diminished CD4+CD25+ regulatory T cell function. J Immunol 2012;188:4644–53. 10.4049/jimmunol.1100272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cai G, Freeman GJ. The CD160, BTLA, LIGHT/HVEM pathway: a bidirectional switch regulating T-cell activation. Immunol Rev 2009;229:244–58. 10.1111/j.1600-065X.2009.00783.x [DOI] [PubMed] [Google Scholar]
- 41.Jones A, Bourque J, Kuehm L, et al. Immunomodulatory functions of BTLA and HVEM govern induction of extrathymic regulatory T cells and tolerance by dendritic cells. Immunity 2016;45:1066–77. 10.1016/j.immuni.2016.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ward-Kavanagh LK, Lin WW, Šedý JR, et al. The TNF receptor superfamily in Co-stimulating and Co-inhibitory responses. Immunity 2016;44:1005–19. 10.1016/j.immuni.2016.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Josefsson SE, Beiske K, Blaker YN, et al. TIGIT and PD-1 mark intratumoral T cells with reduced effector function in B-cell non-Hodgkin lymphoma. Cancer Immunol Res 2019;7:355–62. 10.1158/2326-6066.CIR-18-0351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alban TJ, Alvarado AG, Sorensen MD, et al. Global immune fingerprinting in glioblastoma patient peripheral blood reveals immune-suppression signatures associated with prognosis. JCI Insight 2018;3:122264. 10.1172/jci.insight.122264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Orrego E, Castaneda CA, Castillo M, et al. Distribution of tumor-infiltrating immune cells in glioblastoma. CNS Oncol 2018;7:CNS21. 10.2217/cns-2017-0037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pinton L, Masetto E, Vettore M, et al. The immune suppressive microenvironment of human gliomas depends on the accumulation of bone marrow-derived macrophages in the center of the lesion. J Immunother Cancer 2019;7:58. 10.1186/s40425-019-0536-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reardon DA, Gokhale PC, Klein SR, et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunology Research 2016;4:124–35. 10.1158/2326-6066.CIR-15-0151 [DOI] [PubMed] [Google Scholar]
- 48.Jahan N, Talat H, Alonso A, et al. Triple combination immunotherapy with GVAX, anti-PD-1 monoclonal antibody, and agonist anti-OX40 monoclonal antibody is highly effective against murine intracranial glioma. Oncoimmunology 2019;8:e1577108. 10.1080/2162402X.2019.1577108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnston RJ, Comps-Agrar L, Hackney J, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 2014;26:923–37. 10.1016/j.ccell.2014.10.018 [DOI] [PubMed] [Google Scholar]
- 50.Bengsch B, Ohtani T, Khan O, et al. Epigenomic-Guided Mass Cytometry Profiling Reveals Disease-Specific Features of Exhausted CD8 T Cells. Immunity 2018;48:1029–45. 10.1016/j.immuni.2018.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Woroniecka K, Chongsathidkiet P, Rhodin K, et al. T-Cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res 2018;24:4175–86. 10.1158/1078-0432.CCR-17-1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fromentin R, Bakeman W, Lawani MB, et al. CD4+ T cells expressing PD-1, TIGIT and LAG-3 contribute to HIV persistence during art. PLoS Pathog 2016;12:e1005761. 10.1371/journal.ppat.1005761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hung AL, Maxwell R, Theodros D, et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology 2018;7:e1466769. 10.1080/2162402X.2018.1466769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schetters STT, Gomez-Nicola D, Garcia-Vallejo JJ, et al. Neuroinflammation: microglia and T cells get ready to tango. Front Immunol 2017;8:1905. 10.3389/fimmu.2017.01905 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2019-000323supp001.pdf (212.5KB, pdf)
jitc-2019-000323supp002.pdf (2.9MB, pdf)