

Abstract
Glioblastoma (GB) is an extremely pugnacious brain cancer originating from neural stem (NS) cell-like cells. Forkhead box G1 (FOXG1; previously recognized as BF-1, qin, Chicken Brain Factor 1, or XBF-1 and renamed FOXG1 for mouse and human, and FoxG1 for other chordates) is an evolutionary preserved transcription factor driven from the forkhead box group of proteins FOXG1 modulates the speed of neurogenesis by maintaining progenitor cells in a proliferative mode as well as obstructing their differentiation into neurons during the initial periods of cortical formation. FOXG1 has been implicated in the formation of central nervous system (CNS) tumors and precisely GBs. Pathophysiologically, joint actions of FOXG1 and phosphatidylinositol- 3-kinases (PI3K) intermediate in intrinsic resistance of human GB cells to transforming growth factor-beta (TGF-β) stimulation of cyclin-dependent kinase inhibitor 1(p21Cip1) as well as growth inhibition. FOXG1 and NOTCH signaling pathways may functionally interrelate at different stages to facilitate gliomagenesis. Furthermore, FoxG1 actively contributed to the formation of transcription suppression complexes with corepressors of the Groucho/transducin-like Enhancer of split (Gro/TLEs). Also, FOXG1 was stimulated by Gro/TLE1 and abridged by Grg6. FOXG1 silencing in brain tumor-initiating cells (BTICs) also resulted in diminished secretion of markers characteristic undifferentiated natural neural stem/progenitor cells (NSPC) states, such as Oligodendrocyte transcription factor (OLIG2), (sex determining region Y)-box 2. (SOX2) and B lymphoma Mo-MLV insertion region 1 homolog (BMI1). This review therefore focuses on the pathogenic and biomarker potentials of FOXG1 in GB.
Key words: FOXG1, glioblastoma, biomarker, BTIC, NSPC, gliomagenesis
Introduction
Glioblastoma (GB) is an extremely pugnacious brain cancer originating from neural stem (NS) cell-like cells.1 Several studies have shown that, the transcriptional as well as epigenetic machineries that regulates the stimulation and continuation of NS as well as progenitor cells are captured and derestricted in GBs.1-3 Forkhead box G1 (FOXG1; previously identified as BF-1, qin, Chicken Brain Factor 1, or XBF-1 and renamed FOXG1 for mouse and human, and FOXG1 for other chordates) is an evolutionary preserved transcription factor driven from the forkhead box group of proteins.4-8 The name forkhead was coined out because these proteins were first observed in Drosophila.6,9 Studies have shown that, in vertebrates, FOXG1 is fundamental for the growth of telencephalon, cell movement, and cerebral cortex modeling as well as layering.6,8 It is proven that, both up-regulation and down-regulation of FOXG1 are interconnected with cancer evolution.4 In 1993, the association between FOXG1 (with the name quin) and cancer was established, exhibiting that FOXG1 is an effective oncogene.6,7
Studies have shown that, in GBs, FOXG1 over-secretion inhibits the transcription of cyclin-dependent kinase inhibitor 1 (p21Cip1) as well as instigates anomalous cellular productions resulting in poor outcomes.4,9,10 Nevertheless, the FOXG1 gene encrypts a growth-related transcription factor with repressor actions and it was secreted at initial phases of telencephalic growth.11-13 Furthermore, it was secreted inadequately by differentiated astroglial cells, and FOXG1 altercation leads to up-regulation of astroglial differentiation genes.14 Also, transcriptomic reporting in The Cancer Genome Atlas (TCGA) GB cohort revealed that elevated FOXG1 mRNA concentrations is prognostic of unfavorable sequels in multivariate investigation.11,15 Another study demonstrated that, secretion of FOXG1 is appreciably poorer in the analytically uncomplimentary K27-mutant midline cancers than in all other glioblastoma subgroups.16 Therefore, FOXG1 secretion is a fundamental consequence in the advent of GBs. This usually transpire either early in gliomagenesis or later, leading to ancillary modification of a low-grade glioma.11 This review focuses on the pathogenic and biomarker potentials of FOXG1 in GBs.
Function of FOXG1
Studies in rats have shown that, during the initial periods of cortical formation, FOXG1 modulates the speed of neurogenesis by maintaining progenitor cells in a proliferative mode as well as obstructing their differentiation into neurons.4,17 It is also proven that, FOXG1 was very vital in exact development of the inner ear, the olfactory gland as well as correct axonal formation in the evolving retina.18-20 Also, FOXG1 was capable of maintaining accurate balance during cell replication and differentiation although the precise mechanisms via which it regulates these fundamental procedures are essentially undetermined.4 Therefore, further studies are needed in this direction.
On the other hand, FOXG1 has been implicated in the formation of central nervous system tumors and precisely GBs.14,15 It was proven that, during typical brain formation in mice, FOXG1 contributes significantly to the formation of the forebrain, more precisely the telecephalon. Also, FOXG1 modulated angiogenesis within the forebrain which typically arise from pial vessels that emerge early in formation of the brain at E9 in the mouse.21-23 Studies have shown that, these may account for the fraction of normal vessels in Tgfbr2-cKO forebrains since FOXG1-driven-secretion is comparatively weak in the initial phases of forebrain formation but intensely upsurges with high E9.5.21,24 Hellbach et al. demonstrated that, Foxg1cre/+; Tgfbr2flox/flox mice can act as a model to innovative comprehension of the interface between neural and angiogenic cells during brain formation.21 Nevertheless, FOXG1 also function as a transcriptional repressor, not only during initial brain formation, but also in the matured brain. Thus, in a matured brain, FOXG1 modulates neuronal survival.25,26 A detailed literature search revealed that no studies have been conducted on the effects of FOXG1 on angiogenesis during the pathogenesis of GBs. Therefore, further research should gear towards this direction.
Post-translational modification of FOXG1
FOXG1 is about 58-kDa and it is mainly located in the nucleus as well as the cytoplasm. It was proven that, the intracellular part of FOXG1 was modifiable post-translationally and it alternates between the nucleus and the cytoplasm.4,27 More Precisely, FOXG1 was restrained mainly in the nucleus and precisely in zones with ongoing neurogenesis during the formation of the mouse brain, while the cytoplasmic portion was associates with initial neuronal differentiation zones.27 Furthermore, in the nucleus, FOXG1 functions as a transcriptional repressor and thus targets fibroblast growth factors (FGFs), sonic hedgehog homolog (SHH), as well as cell-cycle inhibitors like p21Cip1.4,10,28 On the other hand, in the cytoplasm, FOXG1 functions as a transforming growth factor-beta (TGF-β) blocker by binding to receptors like mothers against decapentaplegic homolog 3 (Smad3).10,28 It is shown that, FOXG1 can be imported from the nucleus and cytoplasm into the mitochondria resulting in further proteolization within the matrix.4 Also, the full-length protein can be partly proteolized in the cytoplasm with the production of a 45-kDa fragment that partly remain in the cytoplasm and partly imported into the mitochondria. Nevertheless, this 24-kDa C-terminal fragment of FOXG1 is entirely generated within mitochondria.4 Pancrazi et al. demonstrated that, in isolated mitochondria, cell lines, prime cell cultures, and mouse cortical extracts, the fraction of FOXG1 is confined to the mitochondrial matrix.4 Thus, a distinctive domain sited between amino acids 277 and 302 is accountable for its mitochondrial targeting. They indicated that full-length, mitochondrial, and cytosolic categories of FOXG1 influence cell growth, differentiation, and mitochondrial functions.4
Studies have shown that, mitochondria modulate vital activities during neuron formation as well as neuroplasticity such as differentiation of neurons, formation of axons and dendrites. Also, mitochondria are responsible for the development and reformation of synapse.4,28-31
Nevertheless, the in silico study indicated that FOXG1 is deficient in the typical N-terminal mitochondrial targeting structure but has an inner one sited downstream its forkhead domain.4 Interestingly, FOXG1 undergoes a multifarious as well as comparatively slow post-translational modification, with insignificant disparities based on the cell type. Pancrazi et al. observed an obviously dissimilar proliferation/differentiation-stimulating action of over-secreted both nuclear and mitochondrial (FL-FOXG1), solely mitochondrial (mt-FOXG1) as well as solely cytoplasmic (cyt- FOXG1) exhibiting a dispersed intracellular localization.4 They indicated that While FL-FOXG1 facilitated mitochondrial fission and cellular proliferation, mt-FOXG1 promoted mitochondrial fusion as well as early neuronal differentiation. In literature, little is said about these post-translational changes associated with FOXG1.4 Therefore, further research should focus on these posttranslational roles of FOXG1.
FOXG1 and neural cell apoptosis
FOXG1 possesses an extremely preserved DNA-binding domain, which binds to precise DNA successions and modulates gene communication. Also, FOXG1 over-secretion in vivo was interrelated with neural progenitor cell over development as a result of FOXG1 DNA-binding as well as repressor action.32 Furthermore, FOXG1 acts to preserve the natural neural stem/progenitor cells (NSPC) genre at the expense of neural cell differentiation. Also, its inactivation triggered an intense perturbation of cerebral cortex formation due to premature NSPC differentiation. Nevertheless, FOXG1 protein functions partially by establishing transcription suppression complexes with other modifying proteins.32 Several studies have demonstrated that, FOXG1 oversecretion stimulates overgrowth of NSPC via neutralizing signaling triggered by cytostatic factors like TGF-β and BMP4 through the suppression of transcription cyclin-dependent kinase inhibitors p15Ink4b and p21cip1. It also decrease the rate of normal programmed cell death or apoptosis (Figure 1A).32,33 The apoptotic roles of FOXG1 in neurons as well as GBs still need further investigation. It is proven that, FOXG1 interference blocked glioma cell U118 proliferation and initiated cell apoptosis in time-depend manner.32 Furthermore, Dastidar et al. discovered a pro-survival function of FOXG1 and TLE1 in healthy neurons.34 Dali et al. demonstrated that FOXG1:TLE1 facilitates glioma cell survival partial via the inhibition of the pro-apoptotic roles of ChaC glutathione- specific γ-glutamylcyclotransferase 1(CHAC1) (Figure 1A).35 Several studies have demonstrated that, silencing of FOXG1 in cultured brain tumor initiating cells (BTICs) results in reduced sphere-forming (SF) capability and BrdU amalgamation, with a contemporaneous up-codification genes linked to cell cycle exit as well as replicative senescence like p21Cip1, Growth Arrest and DNA Damage (GADD45A), and β-galactosidase, whose action is recognized to upsurge in senescent cells (Figure 1A).33,36,37 Studies have also demonstrated that, FOXG1 has the ability to restrain cell death in rat cerebellar culture programed to undergo apoptosis, while inhibition of FOXG1 secretion triggers apoptosis in normal neurons.4,34
FOXG1 and Groucho/transducin-like enhancer of split in glioblastoma
Several studies have implicated transcription factors like the Groucho (Gro)/transducin-like Enhancer of split (TLE) family to partakes in several of growth pathways in invertebrates and vertebrates. 38,39 Precisely, Gro/TLE1 was associated with machineries that negatively modulated the production of postmitotic neurons from undifferentiated neural precursors in the telencephalon.40 It is proven that, without ancillary proteins, Gro/TLE proteins cannot function alone because they are deficient in DNA-binding action. Furthermore, they become conscripted to specific gene modulating strains in context-determined fashion by establishing complexes with several DNA-binding transcription factors.38 Several studies have demonstrated that, FOXG1 actively partakes in the formation of transcription suppression complexes with corepressors being the Gro/TLEs.38,41
Studies have proven that, Gro/TLE-related gene product 6 (Grg6), a transcription repression-inept, antagonize the roles of FOXG1: TLE complexes.38,39 Furthermore, Marcal et al. established that, Grg6 exhibites about 60% preservation during the introduction of Gro/TLEs at the level of the WD40 repeat (WDR) domain. This resulted in the facilitation of Gro/TLE binding to FOXG1 but however exhibits only a partial connection at the level of its N-terminal domain.38 Nevertheless, the failure of Grg6 to bind to Gro/TLEs was as a result of its deficiency in two preserved N-terminal leucine zipper-like motifs that are capable of intermediating Gro/TLE oligomerization.42 On the other hand, it is likely that, a solitary recognized leucine zipper-like motif at its N terminus was enough in the intermediation of Grg6 homodimerization but not interface with Gro/TLEs.38 Conversely, the structural components that trigger Grg6 homodimerization is still a matter of debate. Therefore, further studies are warranted in this direction. Also, Grg6 and Gro/TLEs are capable of interacting with FOXG1/BF-1, but only Gro/TLEs bind to hairy and enhancer of split-1(HES1) with high affinity.38
Furthermore, studies have proven that, Grg6 and Gro/TLE1 display analogous biochemical features but intermediate dissimilar operative effects.38,41 However, they both interrelate with FOXG1 transcriptional suppression and their facilitation by FOXG1 is stimulated by Gro/TLE1 and abridged by Grg6.41 Marcal et al. demonstrated that, Grg6 secretion was down-regulated in GBs and up-regulated in normal brain, analogous to the extreme secretion of both FOXG1 and TLE in GBs. They further indicated that, Grg6 binds to FOXG1 via its WD-40 repeat domain with analogous affinity to that of TLE.38 Nevertheless, Grg6 fails to bind to, or interrelates very feebly with numerous other TLE-binding associates as evidence in literature.38 Also, Grg6 does not possess the amino-terminal Gln-rich domain via which TLE is capable exhibiting protein-protein interaction.43 Therefore, Grg6 is not a typical antagonist to the entire roles of TLE. Its limited protein-protein communication competence makes Grg6 a more selective dominant- blocker of transcription multiplexes associated with FOXG1 and, perhaps, other FOXG1-interconnected proteins.14
Several studies have demonstrated that, the ability of FOXG1 to modulate cortical progenitor proliferation is assumed to be intermediated via protein-protein communications, rather than FOXG1’s own DNA-binding capability.10,44 Nevertheless, FOXG1 necessitates a complete DNA-binding domain to block neuronal differentiation of telencephalic precursor cells. Nonetheless, either via its own DNA-binding capability or via communications with other DNA-binding proteins the conscription of FOXG1 to DNA is assumed to result in transcriptional suppression of the targeted genes.10 Several studies have demonstrated that, FOXG1 is cosecreted with Gro/ TLEs in the formation of the telencephalon and Gro/ TLEs actively function as a transcriptional corepressor for FOXG1.41 Therefore, exogenous Gro/TLE1 secretion in cortical progenitor cells results in buildup of proliferating cells as well as reduction in the quantity of progenitors that differentiate into neurons. 40 This further indicate that, FOXG1 works jointly with Gro/TLEs to avert premature precursor cell cycle exit and differentiation in the telencephalon.38
Figure 1.
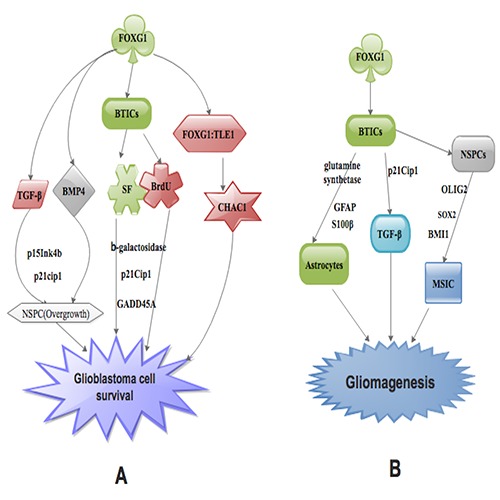
An illustration showing the mechanisms via which FOXG1 facilitates glioblastoma cell survival (A) as well as gliomagenesis (B).
FOXG1 and brain tumor-initiating cells
Many studies have demonstrated that, between the most feebly differentiated GB cells are cells bestowed with stem-like features, explicitly capable of preserving lengthy self-renewal. These cells also propagate precipitously proliferating progenies, hypothetical for multi-lineage differentiation, as well as ability to contribute to cancer proliferation due their resemblance to parental cancers.45-48 GB cells with these physiognomies are hypothesized to function as tumor-forming cells and are usually named brain tumor-initiating cells (BTICs).45,46 Furthermore, BTICs are believed to possess numerous features with NSPCs, such as assiduous self-renewal capacity, pluripotency, as well as tissue repopulating abilities. Nevertheless, they vary from NSPCs with features such as the existence of genetic anomalies and atypical gene secretion repetitions, capacity to proliferate autonomously of mitogens, diminished differentiation ability, as well as tumor-forming capability.49 This therefore means that, the gliomagenic capabilities of BTICs comes from the perturbation of molecular machineries that typically modulates the equilibrium between proliferation and differentiation in NSPCs.49
Also, BTICs have been implicated in GB relapse because of their aptitude to re-amass cancer cells after surgical resection of the primary GB. Thus, BTICs are postulated to epitomize the chemotherapy-resistant cell group inside GBs because of their slow proliferation quotient as well as a more efficient drug resistance ability. This often makes them recalcitrant to anti-mitotic medications.14,47,48 Therefore, BTICs epitomize a curatively striking target for GB management schemes. Several studies have demonstrated that, knockdown of FOXG1 in cultured BTICs led to reduced SF aptitude. Also, BrdU amalgamation with a contemporaneous up-codification of genes linked with cell cycle exit and replicative senescence like p21Cip1, GADD45A, and β-galactosidase resulted in augmentation of senescent cells.14,33,36,50 Nevertheless, FOXG1 silencing in BTICs also resulted in diminished secretion of markers characteristic undifferentiated NSPC states, such as Oligodendrocyte transcription factor (OLIG2), (sex determining region Y)-box 2 (SOX2) and B lymphoma Mo-MLV insertion region 1 homolog (BMI1) (Figure 1B). Also, endogenous FOXG1 was conscripted to the facilitators of both SOX2 and BMI1 in BTICs.14A body of evidence indicated that FOXG1 binds to the BMI1 facilitator in medulloblastoma stem-like cells (MSIC) and silencing of FOXG1 resulted in diminished BMI1 transcription in these cells (Figure 1B).51
Nonetheless, the reduced secretion of NSPC markers as a result of FOXG1 knockdown in BTICs was linked with an inverse up-regulation of three genes normally seen in maturing or matured astrocytes, such as GFAP, S100β, and glutamine synthetase (Figure 1B). Furthermore, endogenous FOXG1 binds to facilitators of these genes, directly involving FOXG1 in the transcriptional modification of GFAP, S100β, and glutamine synthetase in BTICs. Verginelli et al. established participation of mouse FOXG1 in NSPC conservation and blockade of astrocyte differentiation.14 They further indicated that, FOXG1 was associated with conservation of undifferentiated state as well as inhibition of astrocyte cell lineage differentiation in BTICs.14 Also, studies with in vivo orthotopic transplantation revealed that, brain cancers triggered by FOXG1-knockdown BTICs are tinier than cancers triggered by non-knockdown BTICs, leading to persistent host survival. This implies that FOXG1, performs significant functions in BTIC-propagated brain cancers formation.
On the other hand, a study established that, augmentation of FOXG1 secretion in the evolving mouse brain results in forebrain hyper-cellularity leading to the augmentation of progenitor cell expansion as well as deferred differentiation.52 It is proven that, stimulation of p21Cip1 facilitator in a feedback reaction to TGF-β signaling resulted in FOXG1 inhibitory trans-stimulation action and thus antagonized cytostatic consequence of TGF-β (Figure 1B).10 Nonetheless, it appears incongruous that, FOXG1 facilitates BTIC conservation and gliomagenic capability by antagonizing TGF-β signaling (Figure 1B). Furthermore, FOXG1 contributes to TGF-β’s ability to augment proliferation as well as averts differentiation in BTICs.53 This therefore implies that, FOXG1 and TGF-β signaling abilities to conserve BTICs still warrants further studies. Nevertheless, FOXG1 was essential in sustaining BTIC proliferation via inhibiting the secretion of genes that facilitates termination of proliferation as well as replicative senescence.14
Figure 2.
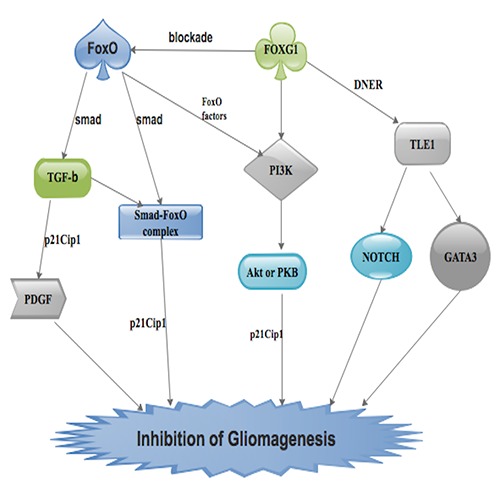
An illustration showing the pathways via which FOXG1 is able to inhibit gliomagenesis.
FOXG1, transducin-like enhancer and brain tumor-initiating cells
TLE proteins are global transcriptional corepressors that partakes in machineries that preserve stem/progenitor cell state as well as restrain differentiation in different tissues.43 It was proven that, FOXG1 was secreted by GBs but its participation in gliomagenesis is still not well studied. It is further proven that, FOXG1 and TLE1 are co-secreted and form a complex with BTICs resulting in brain cell proliferation.10 Functionally, blockade of FOXG1 and TLE resulted in reduction of BTIC ability to trigger gliomagenesis. Correspondingly, both FOXG1 and TLE binds to conjoint domains in the GADD45A and β-galactosidase facilitators. Furthermore, the negative consequence of FOXG1 knockdown on the proliferative capability of BTICs can be phenocopied by TLE1 or TLE2 silencing. This implies that, the roles of FOXG1 in BTICs includes the development of transcription suppression complexes with TLE proteins.13 Nonetheless, FOXG1 and TLE complex was confine to p21Cip1 facilitator on BTICs and knockdown of FOXG1 or TLE resulted in analogous up-regulation of p21Cip1 secretion. Several studies have demonstrated that, FOXG1 forms complexes with the transcriptional corepressor TLE in BTICs during rodent and amphibian forebrain formation.13,38,41 Studies have established that, FOXG1 and TLE1 suppress the secretion of genes linked to the formation astroglial phenotype like GFAP, S100b as well as glutamine synthetase.14 Also, the roles of FOXG1 and TLE1 complexes in GBs proven via the isolation of CHAC1 as a direct FOXG1:TLE1 target gene.35
Signaling pathways FOXG1 and glioblastoma
Seoane et al demonstrated that, Forkhead box O (FoxO) proteins are fundamental associates of Smad3 and Smad4 in the TGF- β-determined production of a p21Cip1 stimulation complex.10 FoxO factors are also associates of the Forkhead box (FOX). They have been implicated in the modulation of cell and organismal growth, development, metabolism, as well as survival.25,54,55 Studies have demonstrated that, FoxO factors are negatively influenced by phosphatidyl inositol 3-kinase (PI3K) growth-stimulating pathway.54,55 It is evidenced that in reaction to mitogenic signals, PI3K triggers Akt or PKB, a protein kinase that phosphorylates FoxO proteins, prohibiting their activities in the nucleus and consequently from target genes.56 Furthermore, FoxO factors functions as Smad associates in p21Cip1 stimulation postulates an interrelationship between the TGF-β/Smad and PI3K/Akt pathways thus implicating FoxO proteins as signal transducers (Figure 2).10
Studies have exhibited that, FoxO-Smad multiplexes are blocked by FOXG18,17. More importantly, joint actions of FOXG1 and PI3K intermediate in intrinsic resistance of human GB cells to TGF-β stimulation of p21Cip1 as well as growth inhibition. Therefore, pathways such as the Smad, PI3K, and FOXG1 pathways congregate on FoxO factors to modulate epithelial and neuronal growth as well as gliomagenesis.10 It is proven that, due to capacity of the PI3K/Akt pathway and the FOXG1 pathway to inhibit p21Cip1 stimulation by a TGF-β-triggered Smad-FoxO complex (Figure 2). This usually results in brain tumor advancement to the most bellicose phase.10 GBs thus arises with a defeat in TGF-β-intermediated p21Cip1 stimulation and cytostasis and an expansion in TGF-β-intermediated PDGF generation as well as cell proliferation (Figure 2).57-59
It is further proven that, transcriptional activities modulated by FOXG1:TLE1 complexes inhibits genes that negatively modulates NOTCH signaling in GBs. This usually results in the conservation of triggered NOTCH pathways as well as GATA3. NOTCH and GATA3 are currently FOXG1:TLE1 transcription suppression targets in GBs (Figure 2). Dali et al. recognized Delta and NOTCHlike epidermal growth factor-related receptor (DNER) as an extra potential transcription suppression target of FOXG1:TLE1.35 They indicated that DNER restrains GB-derived tumor sphere growth and facilitates their differentiation in vivo and in vitro, conflicting with the outcome of FOXG1 and TLE1. They concluded that FOXG1 and NOTCH signaling pathways may functionally interrelate at different stages to facilitate gliomagenesis (Figure 2).35
FOXG1 as biomarker in glioblastoma
Engstrom et al. demonstrated that FOXG1 was one of the most constantly over-secreted genes in their study involving primary cultures of GB-derived NS (GNS) cells and genetically normal NS cells.60 Verginelli et al. also indicated that, FOXG1 was genetically augmented in GB and FOXG1 mRNA concentrations in primary GBs are contrariwise associated with patient outcome.14 Liu et al. established that, the oncogenic EGFR truncation (EGFRvIII) is elevated substantial in typical subtype of GBs as a result of FOXG1 over-secretion.61 Nevertheless, data from 363 evaluated GB samples revealed a substantial upsurge in mean FOXG1 identifying indicators in astrocytic cancers with augmenting WHO grade. This signifies that FOXG1 actively participated in astrocytic malignancy. Verginelli et al. evaluated 58 glioma specimens by FOXG1 immunohistochemistry and found an analogous upsurge in median of up to 50% FOXG1-positive cancer cells in GBs.14 Although this did not correlate well with a specific GB subtype, they depicted FOXG1 positive cells molecularly as inadequately differentiated astroglial cells 11,14
Schafe et al. demonstrated that the quantity of FOXG1 positive nuclei in oligodendroglioma was analogous to IDH mutant astrocytoma (grade II-III) but appreciably decreased compared to the IDH-wildtype GB cohort, again signifying prognostic potentials of FOXG1.11 Furthermore, stratification of brain tumors into significant molecular subgroups indicated that FOXG1 indicators were greater in G34-mutant cancers compared to K27M-mutant gliomas of the midline.16,62,63 Sturm et al. demonstrated that FOXG1+/Olig-2 negative patterns were representative of G34- mutant cancers in their limited immunostained studies.16 Also, FOXG1 over-secretion was implicated in medulloblastoma and interrelated with gliomagenesis as evidenced in the Non- SHH/Non-WNT cohorts.51,64 This markedly support the function of FOXG1 up-regulation as an unfavorable prognostic factor in brain cancers. Schafe et al. detected that cancers with FOXG1 labelling index below partitioning-analysis resolute cutoff (FOXG1 low indices) had a considerably enhanced patient outcome during their univariate study .11
It is proven that, transcriptomic profiling of TCGAdataset confirmed an extreme FOXG1 mRNA concentrations which correlated well with poor survival.15 It was further postulated that FOXG1 maintains cell proliferation via p21CIp1 suppression and thus may aid in gliomagenesis.10,11 Schafe et al. further demonstrated that K27M-mutant cancers possess FoxG1 low/Olig-2 high and G34- mutant cancers display a FoxG1 high /Olig-2 low profile in their H3F3A-mutant glioma cohort.11 Sturm et al. earlier indicated that, in their experiment involving 8 K27M-mutant cancers with Olig- 2+/FoxG1-immunoprofile and 6 G34-mutant cancers with Olig-2- /FOXG1+ profile.16 Schafe et al. concluded that nuclear FOXG1 in glioma correlated well with WHO tumor grade in astrocytic/oligodendroglial cancers. FOXG1 outcomes in univariate analysis (low FOXG1 indices) also correlated with other auspicious prognostic markers like IDH mutation and ATRX secretion.11,65 Further studies are still needed in this direction to further validate FOXG1 as a biomarker in GBs.
Conclusions
This review established that, mutual actions of FOXG1 and PI3K intermediate in intrinsic resistance of human GB cells to TGF-β stimulation of p21Cip1 as well as growth inhibition. Also, FOXG1 and TLE1 are co-secreted and form a complex in BTICs and thus augmented cell proliferation. Furthermore, FoxG1 actively partakes in the formation of transcription suppression complexes with corepressors of the Gro/TLEs. Also, FOXG1 is stimulated by Gro/TLE1 and abridged by Grg6. FOXG1 silencing in BTICs also resulted in diminished secretion of markers characteristic undifferentiated NSPC states, such as OLIG2, SOX2 and BMI1. Moreover, FOXG1 was genetically augmented in GB and FOXG1 mRNA concentrations in primary GBs are contrariwise associated with patient outcome. Transcriptomic profiling of TCG dataset confirmed an extreme FoxG1 mRNA concentrations correlated with poor survival. This review therefore elucidated the pathogenic and biomarker potentials of FOXG1 in GB.
Abbreviation list
ChaC glutathione-specific γ-glutamylcyclotransferase 1= CHAC1, cyclin-dependent kinase inhibitor 1 = p21Cip1, Glioblastoma =GB, Growth Arrest and DNA Damage = GADD45A, Hairy and enhancer of split-1= HES1, Neural stem=NS, Forkhead box G1 =FOXG1, Phosphatidylinositol-3- kinases =PI3K, Transforming growth factor-beta =TGF-β, Groucho/transducin-like Enhancer of split =Gro/TLEs, Gro/TLErelated gene product 6 = Grg6, Brain tumor-initiating cells =BTICs, Neural stem/progenitor cells =NSPC, Fibroblast growth factors =FGFs, Sonic hedgehog homolog =SHH, Mothers against decapentaplegic homolog 3 =Smad3, Nuclear and mitochondrial =FL-FOXG1, Mitochondrial =mt-FOXG1, Cytoplasmic =cyt- FOXG1, Sphere-forming =SF, WD40 repeat =WDR, Medulloblastoma stem-like cells =MSIC, Forkhead box =FOX, Delta and NOTCH-like epidermal growth factor-related receptor =DNER, GB-derived NS =GNS, The Cancer Genome Atlas =TCGA.
References
- 1.Bulstrode H, Johnstone E, Marques-Torrejon MA, et al. Elevated FOXG1 and SOX2 in glioblastoma enforces neural stem cell identity through transcriptional control of cell cycle and epigenetic regulators. Genes & development 2017;31:757-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patel AP, Tirosh I, Trombetta JJ, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014:1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seidu RA, Wu M, Su Z, Xu H. Paradoxical role of high mobility group box 1 in glioma: a suppressor or a promoter? Oncology Reviews 2017;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pancrazi L, Di Benedetto G, Colombaioni L, et al. Foxg1 localizes to mitochondria and coordinates cell differentiation and bioenergetics. Proceedings of the National Academy of Sciences 2015;112:13910-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaestner KH, Knochel W, Martinez DE. Unified nomenclature for the winged helix/forkhead transcription factors. Genes & development 2000;14:142-6. [PubMed] [Google Scholar]
- 6.Kumamoto T, Toma K-i, McKenna WL, et al. Foxg1 coordinates the switch from nonradially to radially migrating glutamatergic subtypes in the neocortex through spatiotemporal repression. Cell reports 2013;3:931-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li J, Vogt PK. The retroviral oncogene qin belongs to the transcription factor family that includes the homeotic gene fork head. Proceedings of the National Academy of Sciences 1993;90:4490-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xuan S, Baptista CA, Balas G, Tao W, Soares VC, Lai E. Winged helix transcription factor BF-1 is essential for the development of the cerebral hemispheres. Neuron 1995;14: 1141-52. [DOI] [PubMed] [Google Scholar]
- 9.Weigel D, Jackle H. The fork head domain: a novel DNA binding motif of eukaryotic transcription factors? Cell 1990; 63:455-6. [DOI] [PubMed] [Google Scholar]
- 10.Seoane J, Le H-V, Shen L, Anderson SA, Massague J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 2004;117:211-23. [DOI] [PubMed] [Google Scholar]
- 11.Schafer S, Behling F, Skardelly M, et al. Low FoxG1 and high Olig 2 labelling indices define a prognostically favourable subset in isocitrate dehydrogenase (IDH)mutant gliomas. Neuropathology and applied neurobiology 2018;44:207-23. [DOI] [PubMed] [Google Scholar]
- 12.Murphy DB, Wiese S, Burfeind P, et al. Human brain factor 1, a new member of the fork head gene family. Genomics 1994;21:551-7. [DOI] [PubMed] [Google Scholar]
- 13.Roth M, Bonev B, Lindsay J, et al. FoxG1 and TLE2 act cooperatively to regulate ventral telencephalon formation. Development 2010;137:1553-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verginelli F, Perin A, Dali R, et al. Transcription factors FOXG1 and Groucho/TLE promote glioblastoma growth. Nature communications 2013;4:2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robertson E, Perry C, Doherty R, Madhusudan S. Transcriptomic profiling of Forkhead box transcription factors in adult glioblastoma multiforme. Cancer Genomics- Proteomics 2015;12:103-12. [PubMed] [Google Scholar]
- 16.Sturm D, Witt H, Hovestadt V, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer cell 2012;22:425-37. [DOI] [PubMed] [Google Scholar]
- 17.Hanashima C, Li SC, Shen L, Lai E, Fishell G. Foxg1 suppresses early cortical cell fate. Science 2004;303:56-9. [DOI] [PubMed] [Google Scholar]
- 18.Pratt T, Tian NM-L, Simpson TI, Mason JO, Price DJ. The winged helix transcription factor Foxg1 facilitates retinal ganglion cell axon crossing of the ventral midline in the mouse. Development 2004;131:3773-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawauchi S, Santos R, Kim J, Hollenbeck PL, Murray RC, Calof AL. The role of foxg1 in the development of neural stem cells of the olfactory epithelium. Annals of the New York Academy of Sciences 2009;1170:21-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hwang CH, Simeone A, Lai E, Wu DK. Foxg1 is required for proper separation and formation of sensory cristae during inner ear development. Developmental Dynamics 2009;238:2725-34. [DOI] [PubMed] [Google Scholar]
- 21.Hellbach N, Weise SC, Vezzali R, et al. Neural deletion of Tgfbr2 impairs angiogenesis through an altered secretome. Human molecular genetics 2014;23:6177-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vasudevan A, Long JE, Crandall JE, Rubenstein JL, Bhide PG. Compartment-specific transcription factors orchestrate angiogenesis gradients in the embryonic brain. Nature neuroscience 2008;11:429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richard SA, Shrestha SS, Zhang C, et al. Successful Treatment of a Child with Ruptured Arteriovenous Malformation Using Onyx Embolization: A Case Report. Open Journal of Modern Neurosurgery 2017;7:153. [Google Scholar]
- 24.Hebert JM, McConnell SK. Targeting of cre to the Foxg1 (BF- 1) locus mediates loxP recombination in the telencephalon and other developing head structures. Developmental biology 2000;222:296-306. [DOI] [PubMed] [Google Scholar]
- 25.Libina N, Berman JR, Kenyon C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell 2003;115:489-502. [DOI] [PubMed] [Google Scholar]
- 26.Danesin C, Houart C. A Fox stops the Wnt: implications for forebrain development and diseases. Current opinion in genetics & development 2012;22:323-30. [DOI] [PubMed] [Google Scholar]
- 27.Regad T, Roth M, Bredenkamp N, Illing N, Papalopulu N. The neural progenitor-specifying activity of FoxG1 is antagonistically regulated by CKI and FGF. Nature cell biology 2007;9:531. [DOI] [PubMed] [Google Scholar]
- 28.Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron 2008;60:748-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gioran A, Nicotera P, Bano D. Impaired mitochondrial respiration promotes dendritic branching via the AMPK signaling pathway. Cell death & disease 2014;5:e1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Su Z, Ni P, She P, et al. Bio-HMGB1 from breast cancer contributes to M-MDSC differentiation from bone marrow progenitor cells and facilitates conversion of monocytes into MDSC-like cells. Cancer Immunology, Immunotherapy 2017;66:391-401. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Richard SA, Jiang Y, Xiang LH, et al. Post-translational modifications of high mobility group box 1 and cancer. American journal of translational research 2017;9:5181-96. [PMC free article] [PubMed] [Google Scholar]
- 32.Zhiwei Shao, Beibei Cong, Aihua Sui, Kai Meng, Yihe Dou. Expression of FOXG1 is associated with the malignancy of human glioma. Chinese-German J Clin Oncol 2014;13:594–9. [Google Scholar]
- 33.Chan D, Liu V, To R, et al. Overexpression of FOXG1 contributes to TGF-β resistance through inhibition of p21 WAF1/CIP1 expression in ovarian cancer. British journal of cancer 2009;101:1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dastidar SG, Landrieu PMZ, D’Mello SR. FoxG1 promotes the survival of postmitotic neurons. Journal of Neuroscience 2011;31:402-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dali R, Verginelli F, Pramatarova A, Sladek R, Stifani S. Characterization of a FOXG1: TLE1 transcriptional network in glioblastoma initiating cells. Molecular oncology 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.E Tamura R, F de Vasconcellos J, Sarkar D, A Libermann T, B Fisher P, F Zerbini L. GADD45 proteins: central players in tumorigenesis. Current molecular medicine 2012;12:634-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Passos JF, Nelson G, Wang C, et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Molecular systems biology 2010;6:347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marcal N, Patel H, Dong Z, et al. Antagonistic effects of Grg6 and Groucho/TLE on the transcription repression activity of brain factor 1/FoxG1 and cortical neuron differentiation. Molecular and cellular biology 2005;25:10916-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen G, Courey AJ. Groucho/TLE family proteins and transcriptional repression. Gene 2000;249:1-16. [DOI] [PubMed] [Google Scholar]
- 40.Nuthall HN, Joachim K, Stifani S. Phosphorylation of serine 239 of Groucho/TLE1 by protein kinase CK2 is important for inhibition of neuronal differentiation. Molecular and cellular biology 2004;24:8395-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yao J, Lai E, Stifani S. The winged-helix protein brain factor 1 interacts with groucho and hes proteins to repress transcription. Molecular and cellular biology 2001;21:1962-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song H, Hasson P, Paroush Ze, Courey AJ. Groucho oligomerization is required for repression in vivo. Molecular and cellular biology 2004;24:4341-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buscarlet M, Stifani S. The ‘Marx’of Groucho on development and disease. Trends in cell biology 2007;17:353-61. [DOI] [PubMed] [Google Scholar]
- 44.Dou C, Lee J, Liu B, et al. BF-1 interferes with transforming growth factor β signaling by associating with Smad partners. Molecular and cellular biology 2000;20:6201-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nature Reviews Cancer 2006;6:425. [DOI] [PubMed] [Google Scholar]
- 46.Park DM, Rich JN. Biology of glioma cancer stem cells. Molecules and cells 2009;28:7-12. [DOI] [PubMed] [Google Scholar]
- 47.Chen J, McKay RM, Parada LF. Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell 2012;149:36-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dirks PB. Cancer: stem cells and brain tumours. Nature 2006;444:687. [DOI] [PubMed] [Google Scholar]
- 49.Kelly JJ, Stechishin O, Chojnacki A, et al. Proliferation of human glioblastoma stem cells occurs independently of exogenous mitogens. Stem cells 2009;27:1722-33. [DOI] [PubMed] [Google Scholar]
- 50.Jurk D, Wang C, Miwa S, et al. Postmitotic neurons develop a p21 dependent senescence like phenotype driven by a DNA damage response. Aging cell 2012;11:996-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Manoranjan B, Wang X, Hallett RM, et al. FoxG1 interacts with Bmi1 to regulate self renewal and tumorigenicity of medulloblastoma stem cells. Stem cells 2013;31:1266-77. [DOI] [PubMed] [Google Scholar]
- 52.Yip DJ, Corcoran CP, Alvarez-Saavedra M, et al. Snf2l regulates Foxg1-dependent progenitor cell expansion in the developing brain. Developmental cell 2012;22:871-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Penuelas S, Anido J, Prieto-Sanchez RM, et al. TGF-β increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer cell 2009;15:315-27. [DOI] [PubMed] [Google Scholar]
- 54.Czech MP. Insulin’s expanding control of forkheads. Proceedings of the National Academy of Sciences 2003;100: 11198-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tran H, Brunet A, Griffith EC, Greenberg ME. The many forks in FOXO’s road. Sci STKE 2003;2003:re5-re. [DOI] [PubMed] [Google Scholar]
- 56.Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. cell 1999;96:857-68. [DOI] [PubMed] [Google Scholar]
- 57.Jennings MT, Pietenpol JA. The role of transforming growth factor β in glioma progression. Journal of neuro-oncology 1998;36:123-40. [DOI] [PubMed] [Google Scholar]
- 58.Rich JN. The role of transforming growth factor-beta in primary brain tumors. Frontiers in bioscience: a journal and virtual library 2003;8:e245-60. [DOI] [PubMed] [Google Scholar]
- 59.Rich JN, Zhang M, Datto MB, Bigner DD, Wang X-F. Transforming Growth Factor-β-mediated p15INK4BInduction and Growth Inhibition in Astrocytes Is SMAD3-dependent and a Pathway Prominently Altered in Human Glioma Cell Lines. Journal of Biological Chemistry 1999;274:35053-8. [DOI] [PubMed] [Google Scholar]
- 60.Engstrom PG, Tommei D, Stricker SH, Ender C, Pollard SM, Bertone P. Digital transcriptome profiling of normal and glioblastoma-derived neural stem cells identifies genes associated with patient survival. Genome medicine 2012;4:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu F, Hon GC, Villa GR, et al. EGFR mutation promotes glioblastoma through epigenome and transcription factor network remodeling. Molecular cell 2015;60:307-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reuss DE, Mamatjan Y, Schrimpf D, et al. IDH mutant diffuse and anaplastic astrocytomas have similar age at presentation and little difference in survival: a grading problem for WHO. Acta neuropathologica 2015;129:867-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Suzuki H, Aoki K, Chiba K, et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nature genetics 2015;47:458. [DOI] [PubMed] [Google Scholar]
- 64.Adesina AM, Nguyen Y, Mehta V, et al. FOXG1 dysregulation is a frequent event in medulloblastoma. Journal of neurooncology 2007;85:111-22. [DOI] [PubMed] [Google Scholar]
- 65.Seidu A., Richard YY, Hao Li, Lu Ma, Chao You. Glioblastoma multiforme subterfuge as acute cerebral hemorrhage: A case report and literature review. Neurology International 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]