

Abstract
We describe a new methodology for genetically labeling single cell lineages in Drosophila called DMARCM. The system offers ultra-low frequency labeling, linear induction, consistent labeling among individuals and virtually no background signal. We compare this technique to an existing approach, which has been widely adopted. We demonstrate how application of DMARCM in the gastrointestinal epithelium permits the effects of labeling frequency on tumorigenic stem cell growth to be distinguished in an established tumor model.
Keywords: Drosophila, cell lineage analysis, monoclonality, stem cells, tumorigenesis
Introduction
Biologists frequently want to determine the contribution of single cells to the adult organism. In Drosophila, genetic mosaic strategies have been used for this purpose because they provide a non-invasive means to stably label cells at any stage of life. Yet, existing methodologies were initially developed to achieve very high rates of cell labeling during development and often lead to polyclonality in adult tissues, thereby hampering efforts to study single cell lineages.
The accuracy of mosaic analysis is, by definition, greatest when the condition of monoclonality is strictly preserved (Fig. 1a). Monoclonality can be achieved by experimental methods that rarely label cells within a tissue. This ensures that the size and composition of an expanded cell lineage will directly reflect the trajectory of a single labeled cell. However, as the frequency of cell labeling increases two common problems emerge (Fig. 1a). First, as the frequency of cell labeling increases, so too, does the probability of polyclonality. Since polyclones consist of two or more cell lineages that have grown together and cannot be discerned retrospectively, the otherwise direct relationship between clone composition and cell lineage is confounded, complicating the interpretation of both wild type and mutant lineage data. A second problem arising from frequent labeling events relates to the potential cell signaling interactions between neighboring marked cell lineages. For wild type lineages, the issue of clone proximity is largely negligible, assuming that the labeling procedure itself exerts no significant influence on cell viability or on endogenous gene expression. However, this is not necessarily the case for mutant cell lineages, which can have non-autonomous effects on neighboring lineages, either promoting or retarding growth and/or differentiation of neighboring clones. The extent to which a given mutation will exert such non-autonomous effects cannot, a prior, be known.
Fig. 1: DMARCM technique.
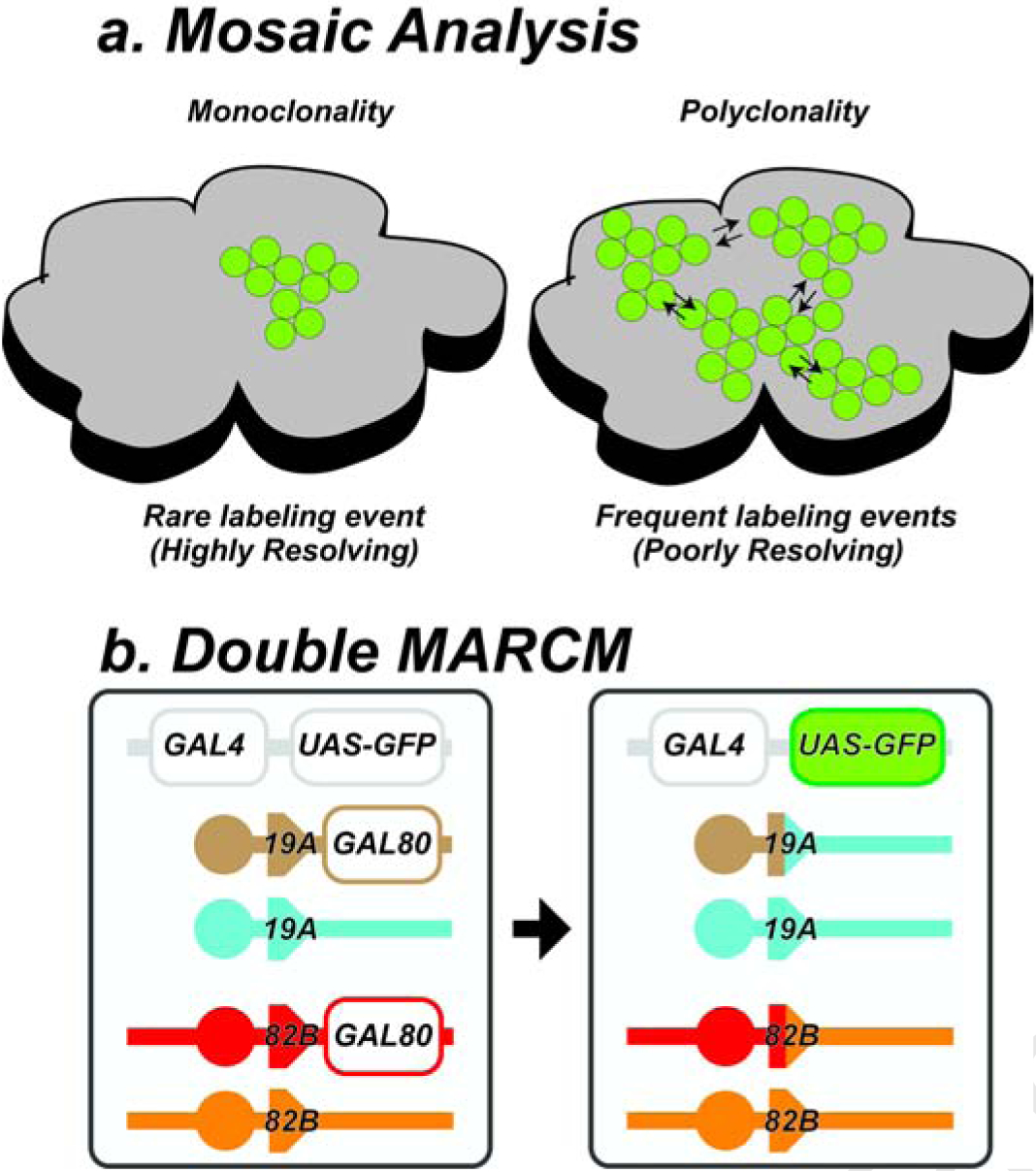
a, Cell labeling using genetic mosaic analysis. Rare labeling events produce high fidelity monoclonal maps of single cell lineages. Frequent labeling events produce polyclones. b, DMARCM strains contain two pairs of FRT sites. For example, a pair of FRT sites on the X chromosome (FRT19A) and second a pair on the right arm of the third chromosome (FRT82B), shown here on the left prior to DNA replication. Labeling induction promotes mitotic recombination between FRT sites on non-sister chromatids. Subsequent G2-X segregation generates a daughter cell lacking both copies of the tubGAL80 repressor, as shown on the right, leading to heritable expression of UAS-GFP under tub-GAL4 control.
Ideally, a cell marking methodology designed to produce ultra-low frequency cell labeling events within an individual, which is titratable, consistent throughout the sample population, and with no background signal would provide the ideal experimental tool for the analysis of single cell lineages. While such benefits lend themselves particularly well to applications where monoclonality is the prime concern (e.g. analysis of stem cell lineages, characterization of tumorigenic mutations or both), they are also of broad experimental utility.
Results and discussion
The FLP recombinase enzyme of yeast is capable of catalyzing site-specific recombination in the Drosophila genome1. Strategic placement of FLP recombination target (FRT) sequences at proximal sites on chromosomal arms using P-element mediated integration in conjunction with a histologically detectable marker gene permits stable cell lineage labeling2. Various implementations of this core methodology have been tailored to meet the needs of specific experimental contexts3–5. Perhaps the most widely adopted of these approaches is called MARCM (mosaic analysis with a repressible cell marker), which results in positive cell marking5. Collectively, these strategies have been developed around the use of a single pair of FRT sites capable of generating very high, or in some cases saturating levels of cell labeling within a tissue6,7.
We reasoned that introducing additional FRT sites into the Drosophila genome would increase the threshold for cell labeling, resulting in fewer labeled cells and lower background signal within a tissue. Fig. 1b summarizes the strategy. The left panel shows the transgenes present in an experimental animal prior to labeling. Here, FRT sites are present on both the X chromosome (FRT19A) and third chromosome (FRT82B). We refer to stocks containing two pairs of FRT sites such as this as ‘double MARCM’ or DMARCM strains (Table 1) to distinguish them from the traditional MARCM strategy. The presence of a ubiquitously expressed GAL80 repressor blocks robust activation of a UAS driven histological marker (e.g. GFP) by GAL4. Only cells that undergo recombination at both pairs of FRT sites, following induction of the flpase enzyme from an hsp70 promoter are capable of transcribing the stably inherited lineage marker.
Table 1:
DMARCM Drosophila Strains
| Genetic Strain | Chromosome | Reference | |
|---|---|---|---|
| 1 | w, hsflp, tub-Gal80, FRT19A; UAS-GFP; tub-Gal4, FRT82B, tub-Gal80 / TM6C Tb, Sb | X ; 3R | This study |
| 2 | w, hsflp, tub-Gal80, FRT19A; UAS-mCherry; tub-Gal4, FRT82B, tub-Gal80 / TM6C Tb, Sb | X ; 3R | This study |
| 3 | w, hsflp, tub-Gal80, FRT19A; UAS-NRNai; tub-Gal4, FRT82B, tub-Gal80 / TM6C Tb, Sb | X ; 3R | This study |
| 4 | w, hsflp, tub-Gal80, FRT19A; Sp/CyO; tub-Gal4, FRT82B, tub-Gal80 / TM6C Tb, Sb | X ; 3R | This study |
| 5 | y,w, FRT19A; FRT82B, hsπM | X ; 3R | This study |
| 6 | y,w, FRT19A; UAS-GFP; FRT82B, hsπM | X ; 3R | This study |
| 7 | y,w, UAS-GFP, hsflp; tub-Gal4, FRT82B, tub-Gal80 / TM6C Tb, Sb | 3R | This study |
| 8 | y,w, UAS-GFP, hsflp; UAS-NRNai; tub-Gal4, FRT82B, tub-Gal80 / TM6C Tb, Sb | 3R | This study |
| 9 | w; FRT82B, hsπM/TM6C Tb, Sb | 3R | Bloomington |
| 10 | y,w, hsflp, UAS-GFP; FRT40A tub-Gal80; tub-Gal4, FRT82B, tub-Gal80 / TM6C Tb, Sb | 2L ; 3R | This study |
| 11 | y,w; FRT40A; FRT82B, hsπM | 2L ; 3R | This study |
| 12 | y,w, hsflp, UAS-GFP; FRT42D tub-Gal80; tub-Gal4, FRT82B, tub-Gal80 / TM6C Tb, Sb | 2R ; 3R | This study |
| 13 | y,w; FRT42D; FRT82B, hsπM | 2R ; 3R | This study |
To test this idea, we first generated marked wild type cell lineages using DMARCM during both development and in the adult (see Methods). Positive cell labeling was noted in a number of different tissues including the developing central nervous system and wing imaginal discs, as well as the adult midgut epithelium (Fig. 2a, c, e). Importantly, many samples were found to contain only a single fluorescently marked cell lineage. By contrast, direct comparison of samples recovered from traditional MARCM labeling experiments using identical induction parameters produced far more frequent labeling events in respective tissue samples (Fig. 2b, d, f).
Fig. 2: Ultra-low frequency cell lineage labeling using DMARCM.
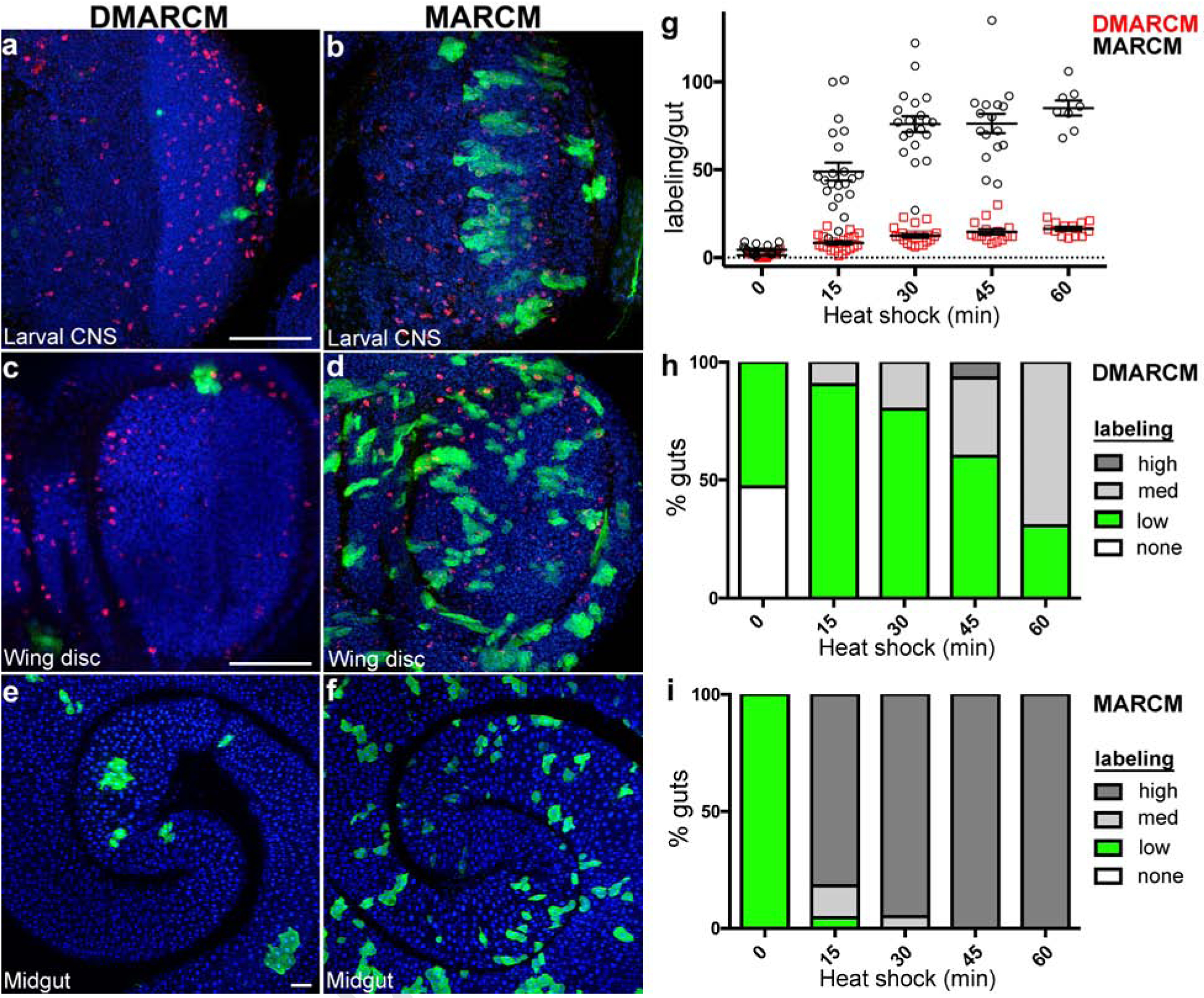
a-f, Confocal micrographs of Drosophila tissues imaged following cell labeling (anti-GFP, green; anti-phospho-histone H3, red; DAPI, blue). a, c, e, DMARCM labeling. b, d, f, MARCM labeling. a, b, Optic lobe of the larval central nervous system. c, d, Larval wing imaginal disc. e, f, Adult midgut. Scale bars, 50 μm. g, h, i, Cell labeling present throughout the entire length of adult midgut analyzed 7 days following a single heat shock. Three independent trials were performed, n ≥ 8 guts per treatment group. g, Total GFP positive cell lineage labeling per gut. h, i, Percentage of guts containing the indicated number of GFP positive cell lineages. High, ≥ 30 cell lineages per gut; Medium, 15–30 cell lineages per gut; low, 1–14 cell lineages per gut. h, DMARCM. i, MARCM.
To quantify the extent of cell labeling using the DMARCM system we focused our analysis on the adult midgut. We first examined the effect of induction duration on the total number of marked wild type cell lineages. Samples were exposed to a single heat shock of varying duration and analyzed one week following treatment (Fig. 2g). A direct linear relationship was observed between the number of marked lineages detected and the duration of the heat shock. The total number of labeling events recovered in each sample was consistent and varied over a narrow range (Fig. 2g; Table 2). To determine the relative levels of induction, DMARCM values were directly compared to the MARCM strategy. By contrast, induction was non-linear with respect to heat shock duration and samples contained far more marked cells (Fig. 2g; Table 2). Overall, DMARCM led to 82% fewer labeling events and 75% less background compared to the MARCM strategy (Table 2).
Table 2:
Analysis of DMARCM cell lineage labeling
| Heat shock minutes | DMARCM labeling/gut ± SD |
MARCM labeling/gut ± SD |
|---|---|---|
| 0 | 1.3 ± 1.5 (n=17) | 4.4 ± 2.7 (n=16) |
| 15 | 8.3 ± 4.6 (n=21) | 48.9 ± 23.7 (n=22) |
| 30 | 12.5 ± 4.9 (n=20) | 76.0 ± 20.2 (n=20) |
| 45 | 14.7 ± 6.0 (n=15) | 76.3 ± 21.9 (n=16) |
| 60 | 16.4 ± 3.9 (n=13) | 85.1 ± 12.0 (n=8) |
To measure how reproducibly the DMARCM technique labels different individuals within a population, we next scored the percentage of midguts containing marked cell lineages following a single heat shock of varying duration. We consistently observed a high percentage of DMARCM samples with low labeling compared to MARCM, irrespective of heat shock duration (Fig. 2h, Fig. 2i; Table 2). While total DMARCM labeling in the midgut was lower than MARCM, we note that DMARCM midgut samples were enriched for actively proliferating cell lineages. This was evident by scoring the number of marked clones, defined as clusters of three or more GFP positive cells within a lineage (Supplementary Fig. 1a; Supplementary Table 1). This suggests that DMARCM labeling may occur when recombination is induced in two successive cell divisions where the Flpase enzyme has perdured. By contrast, MARCM produced greater variations in clone number across the population (Supplementary Fig. 1b; Supplementary Table 1). Thus, despite marking cell lineages at low frequency, DMARCM generated very consistent labeling in the midgut among different individuals in the experimental population.
We next asked if the induction characteristics of DMARCM would permit a direct test of the effects that labeling frequency has on the growth of tumorigenic intestinal stem cell lineages. A number of independent studies have reported that manipulations which reduce or remove Notch (N) function in intestinal stem cells lead to robust tumorigenic growth in the adult gastrointestinal epithelium8,9. The phenotype observed in this tumor model could reflect a cell autonomous requirement for N; alternately the phenotype could arise from a non-autonomous interaction between neighboring clones. If the mechanism driving aberrant growth is strictly cell autonomous, then reducing labeling frequency should not affect the growth of lineages. By contrast, if the mechanism driving growth is a secondary consequence of interactions between clones, then reducing labeling frequency should reduce the rate of stem cell growth.
To distinguish these alternatives, we compared the growth of wild type lineages with lineages lacking N function using both the DMARCM and MARCM labeling methods. To track the growth rate of stem cell lineages in the midgut we scored the number of cells per clone ten days after labeling. As expected, the growth of wild type cell lineages was completely unaffected by labeling frequency (Fig. 3a, b, e). However, rates of clone growth significantly diverged in lineages lacking N function when labeling frequency was reduced using DMARCM (Fig. 3c, d, e). Further, rates of N lineage growth measured using DMARCM did not differ significantly from the growth of marked wild type lineages (Fig. 3e). Thus, the tumorigenic phenotype associated with reductions in N depends critically on the frequency of cell labeling in the gastrointestinal epithelium.
Fig. 3: DMARCM enabled disaggregation of stem cell growth rates.
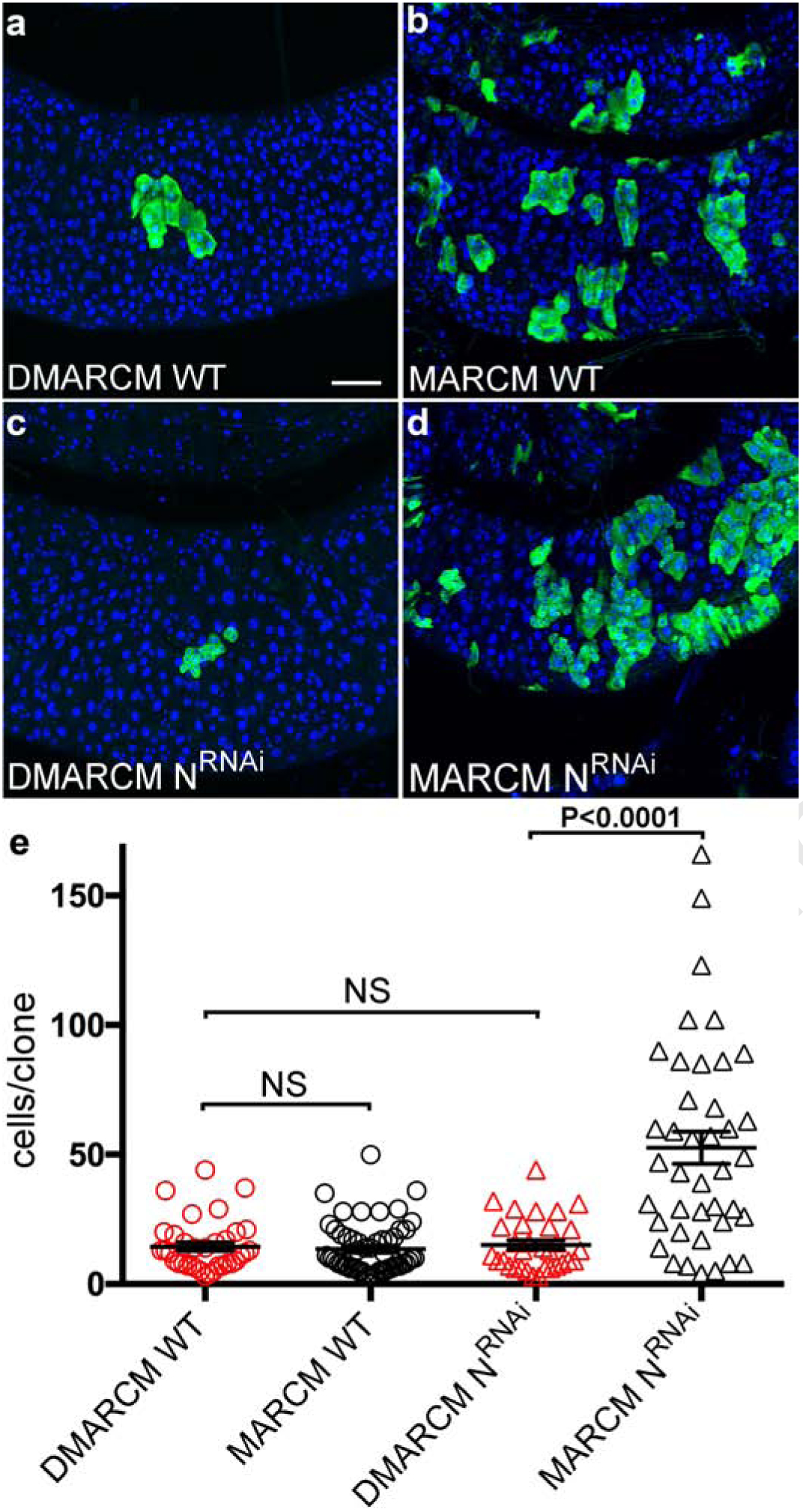
Confocal micrographs of intestinal stem cell lineages in the adult posterior midgut imaged 10 days following labeling (anti-GFP, green; DAPI, blue). a, b, Marked wild type cell lineages. c, d, Marked N RNAi cell lineages. Scale bar, 50 μm. e, Growth rates of marked stem cell lineages in the adult midgut. Three independent trials were performed, n ≥ 32 cell lineages per treatment group. p< 0.0001; t= 6; df = 45. DMARCM, red. MARCM, black.
In summary, this report shows how to consistently generate ultra-low frequency cell labeling events both within samples and between groups of individual experimental animals. DMARCM overcomes the limitations of analyzing polyclonal lineages and can be used to disaggregate the effects of labeling density on tumorigenic intestinal stem cell growth. In principle, DMARCM should also prove valuable for the analysis of imaginal tissues during development which contain large numbers of proliferative cells; mapping single axonal projections in the central nervous system; characterizing mutations that affect cell architecture, movement or force; or overcoming the burden of mutant tissue that affects overall viability of the organism.
Material and methods
Fly strains
y,w, UAS-GFP, hsflp; tub-Gal4, FRT82B, tub-Gal80 / TM6C (CML_174). y,w, UAS-GFP, hsflp; UAS-NRNAi; tub-Gal4, FRT82B, tub-Gal80 / TM6C (CML_333). w; FRT82B, hsπM/TM6C (CML_169). w, hsflp, tub-Gal80, FRT19A; UAS-GFP; tub-Gal4, FRT82B, tub-Gal80 / TM6C (CML_196). w, hsflp, tub-Gal80, FRT19A; UAS-NRNAi; tub-Gal4, FRT82B, tub-Gal80 / TM6C (CML_213). w, hsflp, tub-Gal80, FRT19A; Sp/CyO; tub-Gal4, FRT82B, tub-Gal80 / TM6C (CML_0197). y,w, FRT19A; FRT82B, hsπM (CML_195). y,w, FRT19A; UAS-GFP; FRT82B, hsπM (CML_200). y,w, hsflp, UAS-GFP; FRT40A tub-Gal80; tub-Gal4, FRT82B, tub-Gal80 / TM6C (CML_0214). y,w; FRT40A; FRT82B, hsπM (CML_0215). y,w, hsflp, UAS-GFP; FRT42D tub-Gal80; tub-Gal4, FRT82B, tub-Gal80 / TM6C (CML_0224). y,w; FRT42D; FRT82B, hsπM (CML_0225). Additional information, http://flybase.org.
Cell lineage labeling and counting
All tissue samples in this study were obtained by performing genetic crosses using the stocks listed in Table 1. The following strains were used for the experiments shown in Fig. 2, Table 2, Supplemental Fig. 1, and Supplemental Table 1: wild type DMARCM (stock #1 and #5), wild type MARCM (stocks #5 and #9). The following strains were used for the experiments shown in Fig 3: wild type DMARCM (stock #1 and #5), N RNAi DMARCM (stock #3 and #5), wild type MARCM (stocks #5 and #9), N RNAi MARCM (stocks #8 and #9). Fly crosses were cultured on standard media supplemented with yeast paste at 25 °C.
For adult lineage analysis, newly eclosed females of the appropriate genotype were aged 3–5 days prior to clone induction. Adult labeling was induced by placing vials in a 37 °C water bath for 0–60 minutes. Cotton plugs were pressed into each vial such that flies were confined in the tube below the water line. Larval labeling was induced by placing vials containing larvae seeded at optimal culture density in a 37 °C water bath for 30 minutes. Samples were allowed recover at room temperature and then transferred to a 25 °C incubator.
Cell lineages were scored on either a compound fluorescent or confocal microscope. In Fig. 2 GFP positive cell lineages were scored along the entire length of the adult midgut. In Fig. 3 the number of cells per clone was scored using a confocal microscope. Only clones in the posterior midgut on the epithelial surface closest to the cover glass were scored in this analysis.
Histology
Generation of whole mount samples for immunostaining was performed using standard methodology. Briefly, adult females were in fixed 0.5x PBS and 4% EM-grade formaldehyde (Polysciences) overnight, washed in 1x PBS, 0.1% Triton X-100 (PBST) for a minimum of 2 hours, and incubated in primary antisera overnight. Samples were washed and incubated in secondary antibodies for 3 hours, washed again in PBST, and mounted in Vectashield+ DAPI. All steps were completed at 4 °C. Antisera; Chicken anti-GFP (1:10,000, Abcam); Rabbit anti-phospho-histone H3 (1:2,000, Upstate); Alexa Fluor conjugated secondary antibodies (1:2,000, Molecular Probes).
Microscopy and imaging
Samples were analyzed on a Leica DM5000 compound or Leica TCS SP5 confocal microscope. Images were processed for brightness and contrast in Photoshop CS (Adobe).
Statistical analysis
All statistical analysis was performed using Prism GraphPad Software. The sample size (n) is defined as the total number of independent observations per treatment group. All error bars used denote standard error (SE). An unpaired Welch’s t-test assuming unequal variance was used to compare sample means.
Supplementary Material
Supplementary Fig. 1: Analysis of DMARCM clone number in the adult midgut a, b, Distribution of actively proliferating cell lineages throughout the entire length of adult midgut analyzed 7 days following a single heat shock. Proliferating clones were defined as clusters of three or more GFP positive cells. Three independent trials were performed, n ≥ 8 guts per treatment group. a, b, Percentage of guts containing proliferating clones. High, ≥ 30 clones per gut; Medium, 15–30 clones per gut; low, 1–14 clones per gut. a, DMARCM. b, MARCM.
Highlights:
New method for genetically labeling single cell lineages in Drosophila
Optimized for tracing monoclonal cell lineages in adult tissues
Precisely measure the growth of tumorigenic stem cell lineages in vivo
Funding
This work was supported by grants from the National Institute of Health (R01 DK 108990 and R01 DK117335).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
The authors declare no competing interests.
References
- 1.Golic KG and Lindquist S Cell 59, 499–509 (1989). [DOI] [PubMed] [Google Scholar]
- 2.Blair SS Development 21, 5067–5072 (2003). [Google Scholar]
- 3.Xu T and Rubin GM Development 117, 1223–1237 (1993). [DOI] [PubMed] [Google Scholar]
- 4.Harrison D and Perrimon N Curr Biol. 3, 424–433 (1993). [DOI] [PubMed] [Google Scholar]
- 5.Lee T and Luo L Neuron 22, 451–461 (1999). [DOI] [PubMed] [Google Scholar]
- 6.Newsome TP, Asling B and Dickson BJ Development 127, 851–860 (2000). [DOI] [PubMed] [Google Scholar]
- 7.Stowers RS and Schwarz TL Genetics 152, 1631–1639 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Micchelli CA and Perrimon N Nature 439, 475–479 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Ohlstein B and Spradling A Nature 439, 470–474 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1: Analysis of DMARCM clone number in the adult midgut a, b, Distribution of actively proliferating cell lineages throughout the entire length of adult midgut analyzed 7 days following a single heat shock. Proliferating clones were defined as clusters of three or more GFP positive cells. Three independent trials were performed, n ≥ 8 guts per treatment group. a, b, Percentage of guts containing proliferating clones. High, ≥ 30 clones per gut; Medium, 15–30 clones per gut; low, 1–14 clones per gut. a, DMARCM. b, MARCM.